Introduction
Fuchs corneal dystrophy (FCD; MIM 136800), first
described by Ernst Fuchs in 1910 (1), is characterized by bilateral primary
corneal guttae and a reduced endothelial cell density that can
result in corneal edema, discomfort and blurred vision (2). The onset of FCD generally occurs in
the 4th decade of life onwards, and FCD progresses at a slow rate
over the next 2 to3 decades, causing severe impairment of
endothelial cell function (3,4),
ultimately leading to severely impaired vision or blindness
(5,6). Currently, effective methods of
restoring vision in advanced cases are corneal transplantation in
the form of penetrating keratoplasty (PK) (7), Descemet's stripping endothelial
keratoplasty (DSEK) (8) and
Descemet's membrane endothelial keratoplasty (DMEK) (9).
FCD is genetically heterogeneous. To date, 4 loci,
FCD1, FCD2, FCD3 and FCD4, on chromosomes 13, 18, 5 and 9,
respectively, along with numerous linkage peaks and susceptibility
loci, have been localized through linkage analysis. In addition, 4
causal FCD genes, namely collagen, type VIII, alpha 2
(COL8A2) (MIM 12052) (4),
solute carrier family 4, sodium borate transporter, member 11
(SLC4A11) (MIM 610206) (10,11), zinc finger E-box binding homeobox
1 (ZEB1) (MIM 189909) (12) and lipoxygenase homology domains 1
(LOXHD1) (MIM 613072) (13) have been identified, representing a
small proportion of the total genetic load. Furthermore, a single
nucleotide polymorphism (SNP) on chromosome 18q21, rs613872, in an
intron of the transcription factor 4 (TCF4, MIM 602272)
gene, which encodes a member of the E-protein family (E2-2), has
been identified to be significantly associated with FCD; the
association increased the probability of having FCD by a factor of
30 in individuals with 2 copies of the disease variants
(homozygotes) and discriminated between case subjects and control
subjects with approximately 76% accuracy (14). Another study that genotyped 18
SNPs within TCF4 in Singaporean Chinese revealed that the
minor allele of rs613872 was not present in the genotyped cohort; 2
SNPs (rs17089925 and rs17089887) located upstream and in intron 3
of TCF4, respectively, were significantly associated with
FCD; another 3 SNPs (rs1348047, rs1452787 and rs2123392) also
exhibited a marginal association with FCD (15). Another TGC trinucleotide repeat
expansion (rs193922902) within intron 3 of TCF4 has also
been recently identified to be strongly associated with FCD, and a
repeat length of >50 was determined to play a pathogenic role in
the majority of FCD cases and is considered to be a predictor of
disease risk (16).
As the susceptibility of genes to mutations can vary
in different ethnicities and in view of the limited information on
the genetics of FCD in southwestern China, we undertook this study.
We screened for mutations in 4 causal FCD genes (SLC4A11,
ZEB1, LOXHD1 and COL8A2) and genotyped 7 SNPs
within the TCF4 gene to determine whether these known causal
genes are responsible for causing FCD in this specific
multi-generational late-onset (LO) FCD Chinese pedigree.
Subjects and methods
Case presentation
The study protocol was approved by the Ethics
Committee of the First People's Hospital of Yunnan Province and was
in compliance with the Declaration of Helsinki. Written informed
consent was obtained from all study participants or their
guardians. Family members of this LO FCD Chinese family were
recruited through a proband with FCD (a 46-year-old woman) who
presented to the Department of Ophthalmology, the First People's
Hospital of Yunnan Province; extended pedigrees were subsequently
developed through interviews. The age of the affected pedigree
members in generation II and III ranged from 36 to 67 years.
A total of 191 individuals of Chinese ethnicity,
consisting of 104 females and 87 males who ranged in age from 58 to
89 years with an average age of 68.6 (SD 6.9 years), who had a
normal cornea upon ophthalmic examination and no history of any
ocular disease in their family were recruited as healthy
controls.
Clinical evaluation
All family members underwent a complete ophthalmic
examination, including fundoscopy, a slit-lamp examination and
specular microscopy, to document corneal guttae on December 2009.
The diagnosis of FCD and the severity grading were based on a
modified Krachmer scale, with grade 0 indicating an absence of
guttae, grade 1 representing 12 or more central guttae, grade 5
denoting confluent central guttae with corneal edema, and grade 6
corresponding to disease severe enough to require keratoplasty
(3). A detailed history was
recorded for all subjects, including any family history and the
duration of symptom onset.
DNA sample preparation
After obtaining written informed consent, peripheral
blood samples were collected from the proband and her family
members. Genomic DNA was extracted from the leukocytes of the
peripheral blood using a cell/tissue genomic DNA extraction kit
(BioTeke Corp., Beijing, China) according to the manufacturer's
instructions. In addition, genomic DNA from 191 healthy individuals
was extracted and used as the control DNA.
Sequencing analysis of SLC4A11, ZEB1,
LOXHD1, COL8A2, and TCF4
A total of 69 sets of primers (Table I) were designed to completely
incorporate the exon and intron boundaries of the SLC4A11
(NM_032034.3), ZEB1 (NM_030751.5), LOXHD1
(NM_144612.6) and COL8A2 (NM_005202.3) genes, and all
primers were designed such that they would be positioned on
intronic segments at least 80 nucleotides on either side of the
intron-exon boundary, to ensure complete reading of the exons.
Primers were also designed to amplify the fragment of the
TCF4 gene containing 7 SNPs. Polymerase chain reaction (PCR)
was carried out with 25 ng of genomic DNA as a template in a
mixture of PCR buffer, 2.5 mM MgCl2, 0.2 mM of each
dNTP, 0.4 µM of each primer, and 0.75 units of rTaq DNA
polymerase (Takara Bio, Dalian, China). After an initial
denaturation step at 95°C for 5 min, 35 PCR cycles were performed
as follows: 95°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min,
followed by a final extension at 72°C for 5 min. The PCR-amplified
products were purified and sequenced on an ABI 3130 Genetic
Analyzer using the BigDye Terminator Cycle Sequencing v3.1 kit
(Applied Biosystems Life Technologies, Foster City, CA, USA)
according to the manufacturer's instructions. Sequence assembly and
analysis were performed using the DNASTAR Lasergene.v7.1 program
(DNAstar Inc., Madison, WI, USA).
 | Table IPrimer sequences used to amplify
exons of the SLC4A11, ZEB1, LOXHD1 and
COL8A2 genes, and 7 fragments containing 7 SNPs within the
TCF4 gene. |
Table I
Primer sequences used to amplify
exons of the SLC4A11, ZEB1, LOXHD1 and
COL8A2 genes, and 7 fragments containing 7 SNPs within the
TCF4 gene.
| Gene | Exon | Forward primer
(5′→3′) | Reverse primer
(5′→3′) | Product size
(bp) | Coding region size
(bp) |
|---|
| SLC4A11 | 1 |
GCCCGGTCCCTTCCTCTC |
GCCAAAAGCATTCCAGCACTAG | 553 | 136 |
| 2, 3 |
CGGCTAGGGAATGCTGGAGA |
GGAGCAGCGGGAGGATTCT | 631 | 153, 50 |
| 4, 5 |
CCCGCTGCTCCCCTCTTC |
GCAGTGCTCCAGCCCTCTTC | 720 | 232, 82 |
| 6 |
GGCGGCCCAACCAACTT |
CCGCGTGTTTGAATAGGGATAG | 556 | 124 |
| 7, 8, 9A |
GGGGAGAGCACCTTCACCTG |
CCCCGTCTGTGTTCTCGTCA | 688 | 219, 94, 126 |
| 9B, 10, 11 |
TCCCCAGCAAACCCTCTCTC |
TGGGGCAGCAATATGGTGG | 687 | 114, 133 |
| 12 |
TGCGCTTTATGCCTTTTTCAA |
CACGGGCACACACTCAGCTT | 493 | 74 |
| 12, 14, 15A |
CCCCCTGGAGCCCTTTCT |
GGCGGCCACCAAGTTCTG | 755 | 253, 107, 169 |
| 15B, 16, 17A |
TCCGGGAAATCGAGAGTGAGT |
AGCGCGATGTAGAGGAAGAGG | 791 | 174, 196 |
| 17B, 18 |
GCCGTGGACCCTGAGGAGT |
CCCGCCCATTCTCCACAC | 617 | 170 |
| 19 |
TGGGCTGGGATGGGTGTC |
GGCAGTAGCAGGGACACAGGT | 537 | 70 |
| ZEB1 | 1 |
CCGCCCGGTCCCTAGCAACAAG |
CGGAGGGGGGCAGAGAGCACTACTT | 413 | 58 |
| 2 |
TTGCTGTTAAAATCCTGGCTCTG |
TCCTTCCACTCAGCCATACTTTG | 1,115 | 20 |
| 3 |
TCCTTTTCAGATTTCGGGAAGTT |
TGATTCTCGTTTGCTGTGACATG | 789 | 60 |
| 4 |
GGGGCTGTCTATTGTCCAACTTT |
AAGGCAGATTCAGGAAAACCACT | 994 | 162 |
| 5 |
AGCCCGTATTTGAACCCTGACT |
TTCCATTGAGGGCTGAGTTGTC | 512 | 203 |
| 6 |
CAAAACAACCATCAGGCTCACA |
TTCACTCCCTCTCATTGCCTCTA | 674 | 106 |
| 7A |
CAGTTCTGTCACAAGCATGCATG |
TGGCTAGGCTGCTCAAGACTGT | 791 | 1,811a |
| 7B |
CCCATTACAGGCAACCAGTTCT |
TGGGGTTCATTTGCATTTGC | 876 | |
| 7C |
TGAAAAGATGCAAGCTGGACAG |
GGCTGGATCACTTTCAAGGGT | 702 | |
| 7D |
GGCCATTGCTGACCAGAACA |
GGTTCACAGCCACACTTCCTCAT | 743 | |
| 8 |
TTCGGTGTCCTTGCTTTCTTTC |
GCCGAGATTGAGATTGCGTG | 640 | 181 |
| 9 |
AACCCTCCCCTTTCTACAACATG |
GGCACACCCGGATTTATTTTG | 900 | 593 |
| LOXHD1 | 1 |
AGAAGGCAGAGGGAACA |
ATGGGATAATCAGTGAGGAA | 439 | 130 |
| 2 |
GTTGTGCTGGAAAGATTAC |
CTGGTCCCTGGTGAGA | 844 | 115 |
| 3 |
TTCCTCCTCCATCCAC |
CAATCCCTCACTTTCATC | 508 | 81 |
| 4 |
ACCGAGGTTCAGGAGA |
GCAAGACAGGCACGA | 405 | 185 |
| 5 |
GGGGTAAGTGTAGATGGTG |
TTCTTGCTTTCCCTGTG | 650 | 99 |
| 6 |
AAGGAAGTCTGTAGGCTGAA |
CTGGCTTAGGTAGAAGAGTGG | 657 | 149 |
| 7 |
AAGGTAATCGCCAGTCA |
TTCAGGAGCAGGAGGA | 473 | 124 |
| 8 |
ATTCTTAGCCAACCCG |
GGATAATCATAACCACCAA | 822 | 251 |
| 9, 10 |
TGGGTGATACCTACTTTG |
ATCCCTTCCTCCTTTC | 1,116 | 136, 161 |
| 11, 12 |
GTTTATTGCTTGGAGGAT |
ACTTGGAGATGGCTTTT | 1,127 | 87, 136 |
| 13 |
GGAAGGTCAGCCCAGAT |
TCCCAGGAGTCCAACAG | 546 | 155 |
| 14 |
GAGCAGGATGTTGTGG |
AGTAGGGCTGGGTCTT | 913 | 161 |
| 15 |
TCCAATCTCAGCCAAAC |
GCACAGGCAGGAACTCT | 452 | 77 |
| 16 |
ATTTACACGCTTTCCTG |
TCTTAGTCCTCCCTTCTC | 857 | 197 |
| 17 |
CCCCTCTTGTTTCTCAC |
CATTGGGTCCTCAGTTT | 677 | 193 |
| 18 |
GCTGGTATGTGACTCCTC |
GATTTGCCTGGTATGG | 737 | 161 |
| 19, 20 |
CTGGCTCTTTGTTGGG |
TGTTTGTTCCTGTGGGT | 1,423 | 463, 155 |
| 21 |
TCCAGCAAACCTATCTTC |
GTCTTCTTCCAGGACTACC | 513 | 134 |
| 22 |
CAGGCAAATGACTAATGG |
GAGGGAAGGAAGATGGA | 428 | 164 |
| 23 |
GGGCTCACAGATACAGG |
GACCCAACACTAAACACC | 640 | 105 |
| 24, 25 |
AACTCACCCATGTAACCA |
CAGGGATGAAAGACCAA | 1,441 | 129, 165 |
| 26 |
GGGATACGGAGAAGAGTG |
AGGAGGAGCAGGGTGAG | 897 | 182 |
| 27, 28 |
GGCAAGACAGGAGCAT |
GGACGAGGATAAACCAG | 1,984 | 117, 163 |
| 29 |
TGGCAGGTAGATTTAGTGA | AGGGCAAGGGCAGA | 661 | 155 |
| 30, 31 |
GAGTGGGTTGAGTGGG |
ATCGGTGATGGTGGG | 608 | 210, 136 |
| 32 |
GAAACCTACCCAACAATG |
GTGGCTCACCCAAAGT | 1,003 | 209 |
| 33 |
GCCTGGACTTAGGTTGG |
GAAGAAATGTTATGGGTAGA | 657 | 128 |
| 34 |
GCAGCATTACCTTCTATTT |
AAGCCAGAGGAACCAG | 786 | 118 |
| 35 |
GCAAATGTCAGCGTTCT |
GTTGGAGTGGTAAGGGA | 601 | 175 |
| 36 |
AACTTCCCTGCTTCCT |
AACACTGGCTCTTTCATAC | 743 | 186 |
| 37 |
CTCTGAGACCACCTAACC |
ACAAGCCTCTTCCAATC | 879 | 171 |
| 38 |
CTACCTACAACGCCTCAA |
TGGGCATCCGAACAG | 620 | 133 |
| 39 |
AAATGCTTACCTGCTTCA |
TCTGGTCCCTACTCTGC | 646 | 159 |
| 40 |
CCAAGTAGCAGGAGGG |
TTGCCACTGGGTTTATT | 1,449 | 699 |
| COL8A2 | 1 |
CCCCGCGACTTTGAAAATTG |
GGGCGCCTGAGGATCTGAT | 470 | 193 |
| 2A |
CCCATTCTTCCTCTCCCGTGTA |
TTGCCTAAGCCAGCTGGACC | 630 | 1,919a |
| 2B |
GGCCTCAAGGGGGATAATGG |
TTTCCCAGCCAGGCCACTAG | 606 | |
| 2C |
GGGCTTCCTGGCAGACGTG |
CGGTGTGGCATGGGCAGA | 627 | |
| 2D |
TGGGGCCTTCGATGAGACTG |
GCCGCCTCTGTTCAGCTTTT | 624 | |
| TCF4 | Including
rs613872 |
CCCAGGCACTCCCCATTTACT |
GGACGTTGAACAGCTTGACAGG | 579 | |
| Including
rs17089925 |
TTCCTGCTTCTGACCC |
AGTGACCTGCTTGCTC | 924 | |
| Including
rs17089887 |
GCATAGAAGGCAAGA |
GTAAGGAAGAGGCAAT | 972 | |
| Including
rs1348047 |
GGGAATCATAAGCACG | GGCGAAAGGTAGCG | 576 | |
| Including
rs1452787 |
GAATGGGAATCAAATAG |
GCAAACTGTGGGAGG | 760 | |
| Including
rs2123392 |
AAGAATGTCAGGGAAAG |
CAGAATCACTGCGAAA | 960 | |
| Including
rs193922902 (TGC repeat) |
CAGATGAGTTTGGTGTAAGATG |
ACAAGCAGAAAGGGGGCTGCAA |
230+(TGC)12-100 | |
| 5-FAM-TCF-Fuchs,
including rs193922902 (TGC repeat) |
FAM-CAGATGAGTTTGGTGTAAGATG |
ACAAGCAGAAAGGGGGCTGCAA | | |
Strand-specific sequencing
To confirm the indels we observed as mixed
sequences, the 2 alleles were cloned so they could be sequenced
separately. Fragments containing insertion or deletion alleles were
amplified as described above and sub-cloned within the
pZeroBack/blunt vector by using ZeroBack Fast Ligation kit (Tiangen
Biotech Co., Ltd., Beijing, China), according to the instructions
provided by the manufacturer. Plasmid DNA was isolated using the
MiniPrep kit (Qiagen China Co., Ltd., Shanghai, China), followed by
bidirectional sequencing according to the method described above,
with a pZeroBack/blunt forward primer,
5′-CGACTCACTATAGGGAGAGCGGC-3′ and reverse,
5′-AAGAACATCGATTTTCCATGGCAG-3′.
Short tandem repeat (STR) assay
For TGC trinucleotide repeat expansion analysis of
the TCF4 gene, a 5′-FAM-conjugated forward primer
corresponding to a location upstream of the STR region was used in
PCR as previously described (16)
(Table I). Following PCR, 2
µl of DNA were mixed with 12 µl of diluted Map Marker
1000 (BioVentures, Inc., Murfreesboro, TN, USA). The gene scan was
carried out using an ABI 3130 Genetic Analyzer (Applied Biosystems
Life Technologies).
Statistical analysis
Statistical analysis was performed using the SPSS 16
software package. A χ2 test and Fisher's exact test were
performed to compare the minor allele frequency (MAF) between data
from the 1000 Genomes database and the Chinese healthy controls
tested in the present study.
Results
Findings on ocular examination
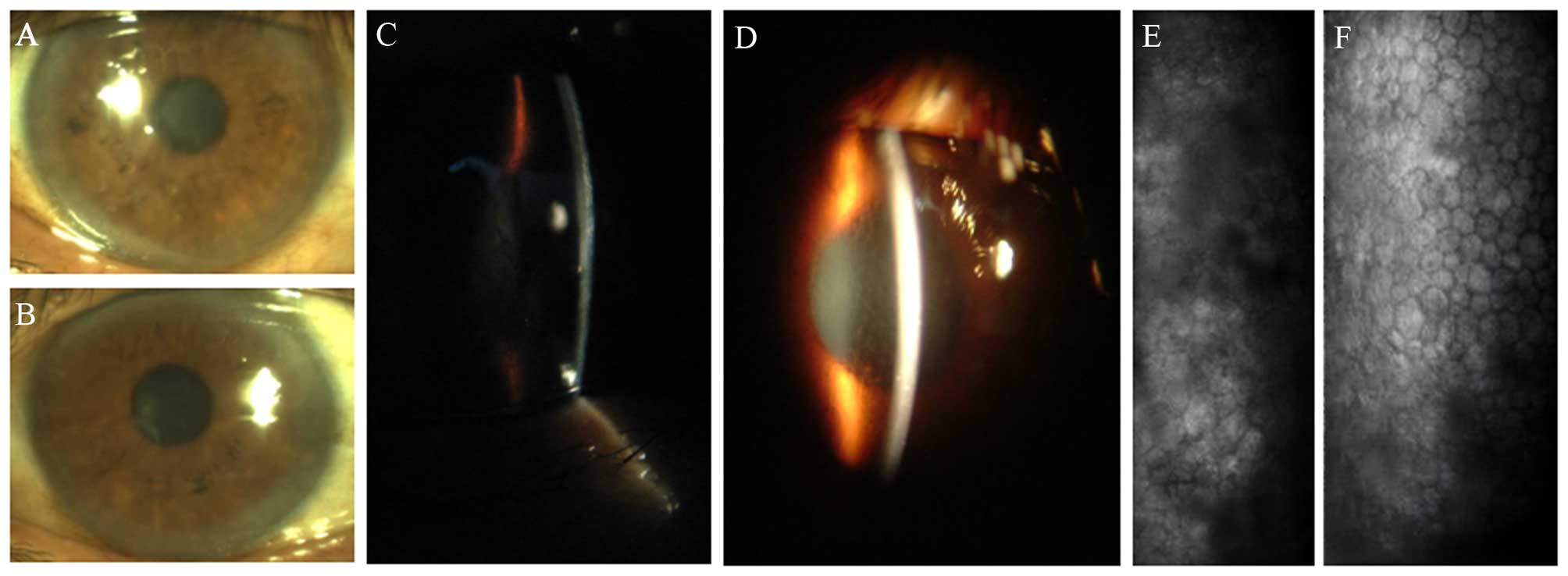
Microscopic investigation of the proband II-9, a
46-year-old woman, revealed the pleomorphism of corneal endothelial
cells and the presence of corneal guttae in both eyes of the
proband at her first presentation to our hospital on December 2009
(Fig. 1). A 5-generation Chinese
pedigree with 8 affected individuals was subsequently assembled
through interviews with the initial proband (Fig. 2, arrow). FCD was diagnosed using
slit-lamp biomicroscopy and assigned severity grades as described
in the Subjects and methods (Fig.
2). The presence of an age-severity profile in this family was
found to be generally consistent with that of LO FCD, which
typically progresses from onset to end-stage disease over a period
of approximately 2 decades (17,18). Those affected in generation II,
whose aged ranged from 56 to 67 years, all exhibited advanced
advanced FCD (II-1, II-3, and II-5 all had grade 6 FCD; II-7 had
grade 5 FCD), whereas in generations II and III, the affected
individuals ranged in age from 36 to 46 years and typically had
grades 3 and 4 disease (II-9 had grade 4 FCD; III-7, III-9, and
III-19 all had grade 3 FCD) (Fig.
2).
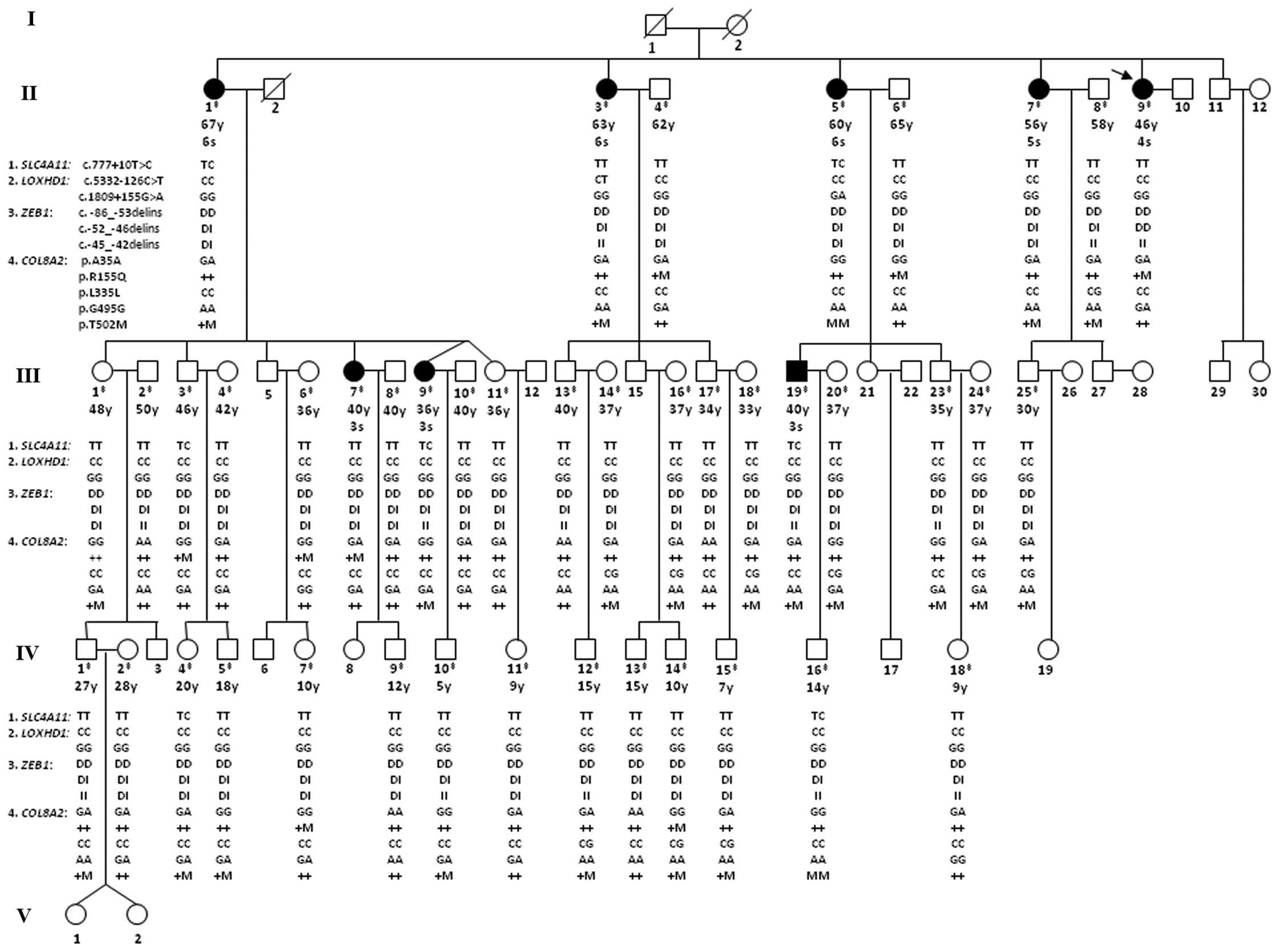 | Figure 2Pedigree of a Chinese family with
late-onset (LO) Fuchs corneal dystrophy (FCD) with genotypes of 11
variantions identified across the SLC4A11, LOXHD1,
ZEB1 and COL8A2 genes. Squares, males; circles,
females; diagonal lines, deceased; filled symbols, affected
individuals; unfilled symbols, unaffected individuals or not known
to be affected; arrowhead, the proband. The double cross symbol
indicates individuals in whom DNA collection and genetic analysis
were performed, age is presented in years (y) and a severity grade
is indicated for affected and unaffected individuals examined in
detail in December 2009. Genotypes of 11 variantions identified
across the SLC4A11, LOXHD1, ZEB1 and
COL8A2 genes are shown below the individual symbols in the
following order: i) SLC4A11: c.777+10T>C (rs372201212);
ii) LOXHD1: c.5332-126C>T and c.1809+155G>A; iii)
ZEB1: Indel1, Indel2 and Indel3; and iv) COL8A2: p.A35A,
p.R155Q, p.L335L, p.G495G and p.T502M. Genotypes of 3 intronic
variations (SLC4A11: c.777+10T>C; and LOXHD1:
c.5332-126C>T and c.1809+155G>A) and 3 synonymous variations
of COL8A2 (p.A35A, p.L335L and p.G495G) are shown in
alleles. Three indels of ZEB1 (Indel 1, Indel 2 and Indel 3)
are showed as: D, deletion; I, insertion. Two missense variations
of COL8A2 (p.R155Q and p.T502M) are shown as: +, wild-type;
M, missense mutation. |
Genetic analysis
Analysis of SLC4A11 gene
A total of 14 known variants (3 coding and 11
non-coding variants) from the Single Nucleotide Polymorphism
Database (dbSNP) were detected in our analysis of the
SLC4A11 gene (Table II).
The 3 coding variants were synonymous variants that have been
previously reported in Asian FCD cases and controls, namely,
p.R161R (rs3827075, MAF: G=0.4798/2402), p.S213S (rs3803956, MAF:
T=0.1663/833) (19,20), and p.T833T (rs58757394, MAF:
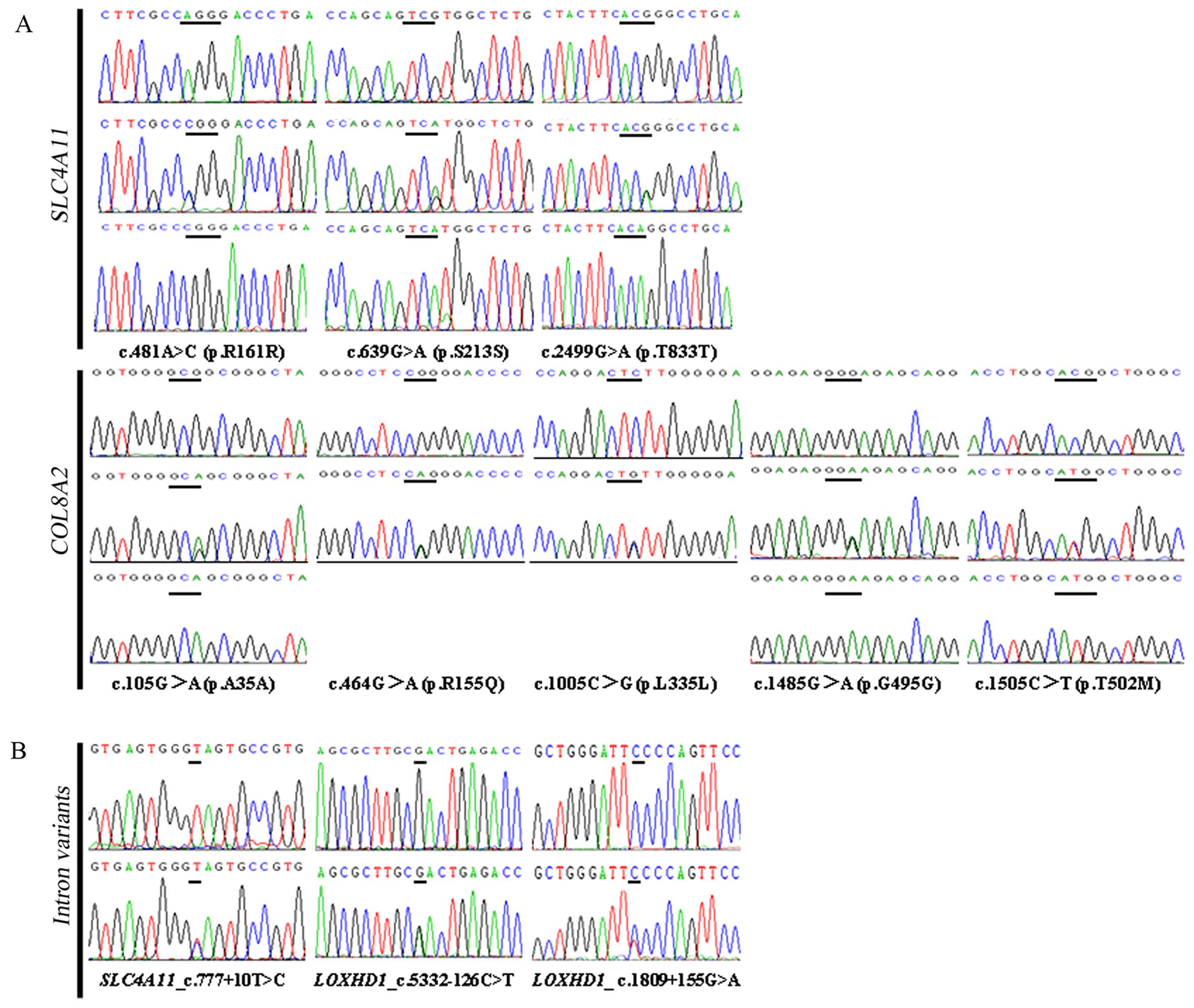
T=0.0901/450) (20) (Fig. 3A). All 3 variants were detected in
both the affected members of this FCD pedigree (p.R161R, 5/16;
p.S213S, 2/16; and p.T833T, 5/16) and in 14 unaffected spouses who
married into this family (the 14 spouses in whom DNA collection and
genetic analysis were performed; p.R161R, 13/28; p.S213S, 5/28; and
p.T833T, 6/28), as well as in unrelated, ethnically matched,
healthy control subjects (n≥100) (Table II).
| Table IISequence variants identified across
the SLC4A11, ZEB1, LOXHD1 and COL8A2
genes, and genotypes of 7 SNPs within the TCF4 gene in 8
cases of this FCD pedigree, 14 unrelated spouses married into the
family, and ethnically matched healthy controls (n≥100). |
Table II
Sequence variants identified across
the SLC4A11, ZEB1, LOXHD1 and COL8A2
genes, and genotypes of 7 SNPs within the TCF4 gene in 8
cases of this FCD pedigree, 14 unrelated spouses married into the
family, and ethnically matched healthy controls (n≥100).
| Gene | Chr position | rs ID | mRNA | Amino acid
change | Functional
consequence | MAF
|
|---|
| 1000 Genomes | 8 Cases | 14 Unrelated
spouses | Healthy
controls |
|---|
| SLC4A11 | | | NM_032034.3 | NP_114423.1 | | | n=16 | n=28 | n≥100 |
|
| 20:3237709 | rs3827076 | c.-30G>C | | nc | G=0.4503/2255 | 5/16 (0.3125) | 6/28 (0.2143) | |
| 20:3235025 | rs3803958 |
c.137-131A>G | | nc | C=0.0038/19 | 1/16 (0.0625) | 3/28 (0.1071) | |
| 20:3234400 | rs6139040 | c.340-86G>C | | nc | G=0.1697/849 | 1/16 (0.0625) | 5/28 (0.1786) | |
| 20:3234173 | rs3827075 | c.481A>C | p.R161R | syn | G=0.4798/2402 | 5/16 (0.3125) | 13/28 (0.4643) | 108/280
(0.3857)a |
| 20:3233935 | rs3803956 | c.639G>A | p.S213S | syn | T=0.1663/833 | 2/16 (0.1250) | 5/28 (0.1786) | 45/274
(0.1642) |
| 20:3233504 | rs372201212 | c.777+10T>
C | | nc | G=0.0014/7 | 4/16 (0.2500) | 0/28 (0.0000) | 0/382 (0.0000) |
| 20:3233480 | rs3803955 | c.777+34G>A | | nc | T=0.2498/1250 | 9/16 (0.5625) | 11/28 (0.3929) | |
| 20:3233374 | rs2144771 |
c.777+140C>A | | nc | G=0.4477/2242 | 0/16 (0.0000) | 16/28 (0.5714) | |
| 20:3231077 | rs3803954 |
c.1091-19T>C | | nc | G=0.0669/335 | 3/16 (0.1875) | 9/28 (0.3214) | |
| 20:3231073 | rs3803953 |
c.1091-15A>C | | nc | G=0.4006/2005 | 2/16 (0.1250) | 6/28 (0.2143) | |
| 20:3230418 | rs3810561 |
c.1463+97T>G | | nc | A=0.2117/1059 | 5/16 (0.3125) | 12/28 (0.4286) | |
| 20:3228711 | rs10048856 | c.2241-4G>A | | nc | T=0.1160/580 | 8/16 (0.5000) | 6/28 (0.2143) | |
| 20:3228437 | rs41281858 | c.2437-9C>T | | nc | A=0.1591/796 | 5/16 (0.3125) | 4/28 (0.1429) | |
| 20:3228366 | rs58757394 | c.2499G>A | p.T833T | syn | T=0.0901/450 | 5/16 (0.3125) | 6/28 (0.2143) | 18/264
(0.0682) |
|
| ZEB1 | | | NM_030751.5 | | | | | | |
|
| 10:31319149 | NA |
c.-86_-53delinsgggaggggtggaggcggaggggtGGGGGGGAAGG | | utr 5 prime,
ex | NA | Del=16/16
(1.0000) | 28/28 (1.0000) | 368/370
(0.9946) |
| 10:31319183 | NA |
c.-52_-46delinsGGGAGGG | | ex | NA | Del=9/16
(0.5625) | 14/28 (0.5000) | 183/370
(0.4946) |
| 10:31319190 | NA |
c.-45_-42delinsAGGG | | ex | NA | Del=5/16
(0.3125) | 12/28 (0.4286) | 162/370
(0.4378) |
| 10:31502731 | rs220057 |
c.481+222C>T | | in | C=0.2524/1264 | 0/16 (0.0000) | 2/28 (0.0714) | |
| 10:31504588 | rs220060 | c.685-15G>A | | in | G=0.0787/394 | 0/16 (0.0000) | 1/28 (0.0357) | |
|
| LOXHD1 | | | NM_144612.6 | | | | | | |
|
| 18:46507838 | NA |
c.5332-126C>T | | in | NA | T=1/16
(0.0625) | 0/28 (0.0000) | 0/382 (0.0000) |
| 18:46579407 | rs16939650 |
c.1809+223G>A | | in | T=0.2584/1294 | 7/16 (0.4375) | 9/28 (0.3214) | |
| 18:46579475 | NA |
c.1809+155G>A | | in | NA | A=1/16
(0.0625) | 0/28 (0.0000) | 0/382 (0.0000) |
|
| COL8A2 | | | NM_005202.3 | NP_005193.1 | | | | | |
|
| 1:36100138 | rs57985157 | c.105G>A | p.A35A | in, syn | T=0.0966/484 | 5/16 (0.3125) | 12/28 (0.4286) | 128/364
(0.3516)a |
| 1:36099217 | rs75864656 | c.464G>A | p.R155Q | mis | T=0.0377/188 | 3/16 (0.1875) | 3/28 (0.1071) | 23/364
(0.0632) |
| 1:36098676 | rs79833067 | c.1005C>G | p.L335L | syn | C=0.0413/207 | 0/16 (0.0000) | 6/28 (0.2143) | 88/364
(0.2418)a |
| 1:36098196 | rs35495320 | c.1485G>A | p.G495G | syn | T=0.1815/909 | 12/16 (0.7500) | 19/28 (0.6786) | 202/364
(0.5549)a |
| 1:36098176 | rs117860804 | c.1505C>T | p.T502M | mis | A=0.0587/294 | 7/16 (0.4375) | 6/28 (0.2143) | 79/364
(0.2170)a |
|
| TCF4 | | | NM_003199.2 | | | | n=16 | n=28 | n≥100 |
|
| 18:55382827 | rs1348047 |
c.369+20627C>A | | in | T=0.2780/1392 | 10/16 (0.6250) | 16/28 (0.5714) | |
| 18:55539976 | rs1452787 |
c.145+45304t>c | | in | G=0.2708/1356 | 8/16 (0.5000) | 14/28 (0.5000) | |
| 18:55541025 | rs17089887 |
c.l45+44255A>G | | in | C=0.1689/846 | 3/16 (0.1875) | 6/28 (0.2143) | |
| 18:55543071 | rs613872 |
c.145+42209c>a | | in | G=0.0697/348 | 0/16 (0.0000) | 0/28 (0.0000) | 1/382
(0.0026)a |
| 18:55547634 | rs2123392 |
c.145+37646a>g | | in | C=0.3005/1504 | 13/16 (0.8125) | 11/28 (0.3929) | |
|
18:55586156:55586227 | rsl93922902 |
c.72+818_73-804CTG(10_37) | | STR | NA | TGC(11)=8/16 (0.5000) | 9/28 (0.3214) | |
| 18:55733603 | rs17089925 | NA | | in | T=0.1569/786 | 1/16 (0.0625) | 10/28 (0.3571) | |
Among the 11 non-coding variants identified in this
FCD pedigree, 9 variants (rs3827076, rs3803958, rs6139040,
rs3803955, rs3803954, rs3803953, rs3810561, rs10048856 and
rs41281858) were detected in both the affected members of this FCD
pedigree and in 14 unaffected individuals who married into this
family. As for rs2144771, it was absent in the 8 affected members
of this FCD pedigree (0/16), whereas it was detected in the 14
unaffected individuals who married into this family (16/28)
(Table II). For an intronic
variant (rs372201212, MAF: G=0.0014/7), its minor allele (G) was
detected in 4 of the 8 affected members of this FCD pedigree, II-1,
II-5, III-9 and III-19 (4/16), and in 3 of 20 healthy descendants
in this family (in whom DNA collection and genetic analysis were
performed), III-3, IV-4 and IV-16 (3/40). This variant was absent
in the other 4 affected members of this FCD pedigree (II-3, II-7,
II-9 and III-7). As this variant was not identified in the 14
unaffected individuals who married into this family (0/28)
(Table II) (Fig. 2), this variant was further tested
in unrelated ethnically matched controls (n≥100), and the results
revealed that it was also absent in the 191 healthy samples we
tested (0/382) (Table II).
Analysis of ZEB1 gene
Bidirectional sequencing of the PCR product
encompassing the 5′-UTR region and exon 1 of the ZEB1
genomic DNA (GenBank reference ID: NC_000010.11) of the proband
(II-9) of this pedigree revealed a homozygous 34 bp deletion
involving 23 bp of the 5′-UTR region and the adjacent 11 bp at the
5′ end of exon 1 (GenBank reference ID: NM_030751.5):
c.-86_-53delins gggaggggtggaggcgg aggggtGGGGGGGAAGG (exon and
5′-UTR sequences are depicted by capital and lower case letters,
respectively), as well as a heterozygous 7 bp indel in exon 1:
c.-52_-46delins GGGAGGG. Follow-up screening of the other family
members of this specific FCD pedigree revealed that there was
another 4 bp indel: c.-45_-42delinsAGGG (Fig. 4A). These 3 indels were named Indel
1, Indel 2 and Indel 3; however, these 3 indels were present in
both the affected members of this FCD pedigree (Indel 1: Del,
16/16; Indel 2: Del, 9/16; and Indel 3: Del, 5/16) and in the 14
unaffected individuals who married into this family (Indel 1: Del,
28/28; Indel 2: Del, 14/28; Indel 3: Del, 12/28) (Table II) (Fig. 2), as well as in the unrelated,
ethnically matched, healthy control subjects (Indel 1: Del,
368/370; Indel 2: Del, 183/370; Indel 3: Del, 162/370) (Table II).
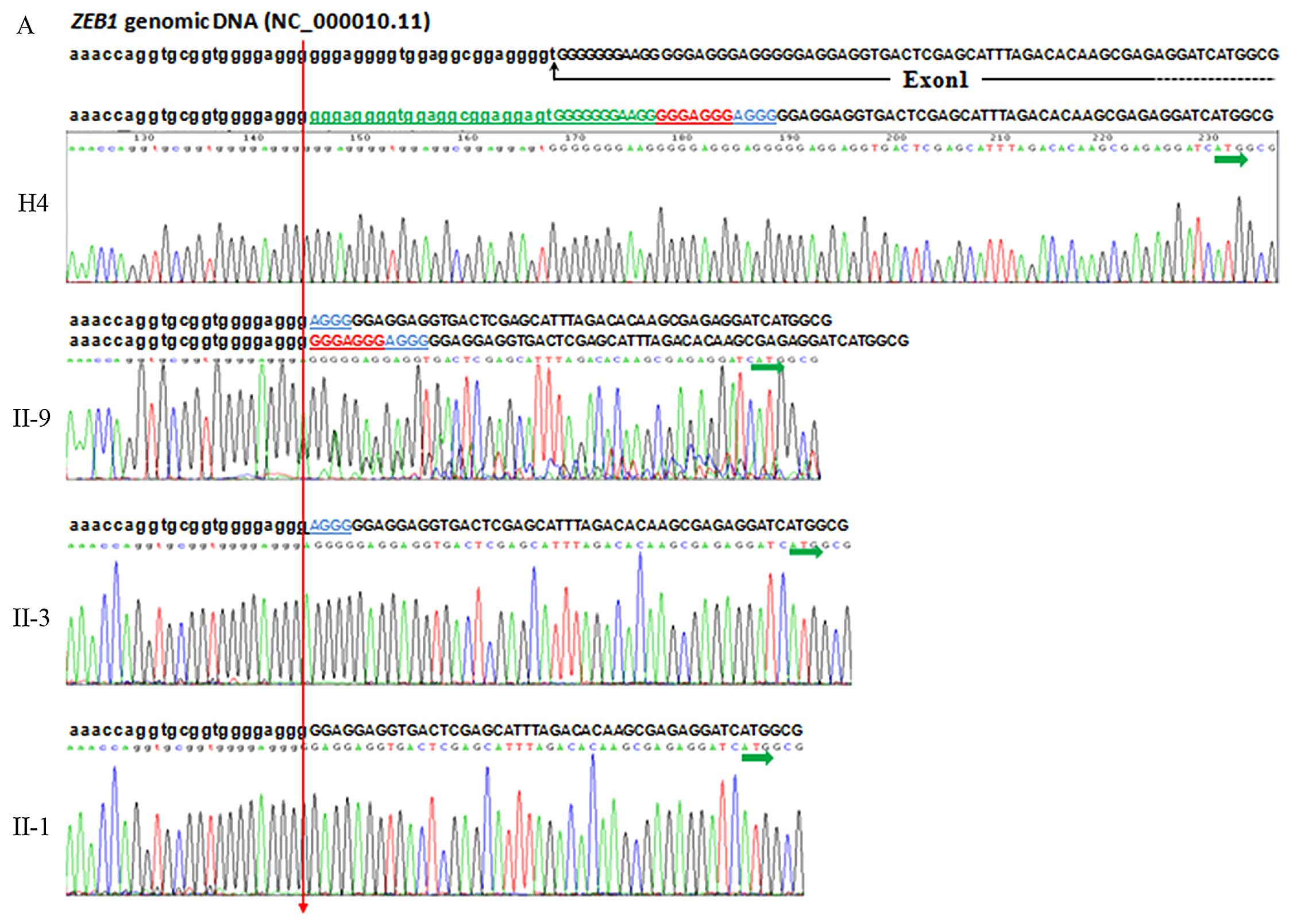 | Figure 4Sequencing analysis of the
ZEB1 gene. (A) Sequence electropherograms of PCR products
encompassing the 5′-UTR region and exon 1 of the ZEB1
genomic DNA. Sequences and sequencing chromatograms of PCR products
encompassing 5′-UTR region and exon 1 of the ZEB1 genomic
DNA from H4 (healthy control), II-9 (proband), II-3 (FCD case), and
II-1 (FCD case) are shown from top to bottom (homozygous is shown
in only one sequence, heterozygous is shown in both sequences).
ZEB1 genomic DNA sequence (GenBank reference ID:
NC_000010.11) was show above (exon and 5′-UTR seqences are depicted
by upper and lower case letters, respectively) The breakpoint is
indicated by the red arrow. Three indels detected in the present
study are indicated in different colors: green for Indel 1, 34 bp
indel containing 23 bp of the 5′-UTR region and 11 bp 5′ end of
exon 1 (NM_030751.5:
c.-86_-53delinsgggaggggtggaggcggaggggtGGGGGGGAAGG); red for Indel
2, 7 bp (NM_030751.5:c.-52_-46delinsGGGAGGG); blue for Indel 3, 4
bp indel (NM_030751.5:c.-45_-42delinsAGGG). Transcription start
site (TSS) is indicated by a horizontal green arrow underneath ATG.
The numbering system used for sequence variations is based on cDNA
sequence with +1 corresponding to the A of the ATG TSS (GenBank
Reference ID: NM_030751.5). FCD, Fuchs corneal dystrophy.
Sequencing analysis of ZEB1 gene. (B) DNA sequencing results
of the four haplotypes subcloned into pZeroBack/blunt vector. Three
indels were underlined in different colors: green for Indel 1; red
for Indel 2; and blue for Indel 3. Sequencing chromatograms of 4
haplotypes (ordered as Indel 1/Indel 2/Indel 3), I/I/I, D/I/I,
D/D/I, D/D/D, are shown from top to bottom. The 5′ and 3′
boundaries of indel are shown by a vertical red and a blue arrow,
respectively. TSS is indicated by a horizontal green arrow
underneath ATG. (C) Schematic illustration of the ZEB1 genomic DNA
and distribution of 3 continuous Indels (Indel 1, 2 and 3) relative
to exon 1. The numbering system used for sequence variations is
based on cDNA sequence with +1 corresponding to the A of the ATG
TSS (GenBank Reference ID: NM_030751.5). FCD, Fuchs corneal
dystrophy. |
To further characterize the indels observed, 8 cases
(II-1, II-3, II-5, II-7, II-9, III-7, III-9 and III-19), 3 healthy
individuals who married into the family (II-4, II-6 and II-8), 2
healthy descendants of the family (III-11 and III-23) and 7 healthy
control subjects (H4, H22, H31, H67, C8, C30 and C52) were enrolled
in the validation set. PCR products of the segment encompassing the
5′-UTR region and exon 1 of the ZEB1 gene were then
subcloned into a pZeroBack/blunt vector. Plasmids were extracted
from 10-20 positive colonies in each sample and sequenced
bidirectionally by ABI 3130, according to the method described
above. Subsequent sequence analysis demonstrated that 4 haplotypes
(ordered as Indel 1/Indel 2/Indel 3), I/I/I, D/I/I, D/D/I and
D/D/D, were detected in the present study (Fig. 4B), and these observations were
consistent with our bidirectional sequencing results. A schematic
illustration of the ZEB1 genomic DNA and the position of the
3 continuous indels (Indel 1, Indel 2 and Indel 3) relative to exon
1 is shown in Fig. 4C. Although
these 3 indels have not been previously reported in patients with
FCD and are absent from dbSNP, they were detected in both the cases
and healthy controls (n≥100) (Table
II) (Fig. 2), leading to the
conclusion that these 3 indels have no pathogenic correlation with
FCD.
Another 2 known dbSNP intron variants were detected
in our analysis of the ZEB1 gene (rs220057, MAF:
C=0.2524/1264 and rs220060, MAF: G=0.0787/394), both of them were
detected in 14 healthy spouses who married into the family
(rs220057, 2/28; rs220060, 1/28) (Table II).
Analysis of LOXHD1 gene
Only one known dbSNP intron variant was detected in
our analysis of the LOXHD1 gene (rs16939650, MAF:
T=0.2584/1294), and it was detected in both the cases in this FCD
pedigree (7/16) and in the 14 healthy spouses who married into the
family (9/28) (Table II).
Another 2 intron variants were identified in the cases in this FCD
pedigree that have not been previously reported in patients with
FCD, namely, c.5332-126C>T and c.1809+155G>A (GenBank
reference ID: NM_144612.6) (Table
II) (Fig. 2). Both of these
variants were absent from dbSNP and were not identified in the 14
unaffected spouses who married into this family (0/28) or in the 20
healthy descendants of this family (0/40) (Table II and Fig. 2). Therefore, these 2 variants were
further tested in unrelated, ethnically matched controls (n≥100),
and the results revealed that both of the variants were also absent
from the 191 healthy samples we tested (0/382) (Table II). Heterozygous alterations in
each variant were only identified in a single case each in this FCD
pedigree (c.5332-126C>T was found in II-3, and c.1809+155G>A
was found in II-5; Fig. 2) and
are likely examples of de novo mutations, the pathological
consequences of which are uncertain.
Analysis of COL8A2 gene
Five known dbSNP variants were detected in our
analysis of the COL8A2 gene, including 3 synonymous
variants, p.A35A (rs57985157, MAF: T=0.0966/484), p.L335L
(rs79833067, MAF: C= 0.0413/207) and p.G495G (rs35495320, MAF:
T=0.1815/909), and 2 missense variants, p.R155Q (rs75864656, MAF:
T=0.0377/188) and p.T502M (rs117860804, MAF: A=0.0587/294)
(Table II) (Fig. 3). Four of these SNP coding
variants from dbSNP (p.A35A, p.G495G, p.R155Q and p.T502M) have
been reported in patients with FCD and unaffected individuals
previously (21-23) and were present in both the
affected members of this FCD pedigree (p.A35A, 5/16; p.G495G,
12/16; p.R155Q, 3/16; and p.T502M, 7/16) and in the 14 healthy
spouses who married into the family (p.A35A, 12/28; p.G495G, 19/28;
p.R155Q, 3/28; and p.T502M, 6/28) (Table II) (Fig. 2), as well as in the unrelated,
ethnically matched, healthy control subjects (p.A35A, 128/364;
p.G495G, 202/364; p.R155Q, 23/364; and p.T502M, 79/364) (Table II).
In addition, the synonymous variant p.L335L, which
has been previously reported in 2 patients with posterior
polymorphous corneal dystrophy (PPCD; MIM 122000) (18), was present in 10 unaffected family
members, 6 of whom were unrelated spouses who married into this
family, and none of them displayed any clinical features of FCD
(Table II) (Fig. 2). Furthermore, the finding of this
synonymous variant in 182 healthy control individuals (p.L335L,
88/364) (Table II), along with
the absence of the p.L335L synonymous change in any of the 8
affected individuals in this Chinese FCD family (Table II) (Fig. 2) and the detection of this silent
variant in 1 out of 116 healthy controls previously reported
(18), leads to the conclusion
that this substitution is a known polymorphism (dbSNP: rs79833067),
and it has no association with FCD.
TCF4 genotype
The PCR products of the TCF4 gene, which
contains 7 previously reported SNPs significantly associated with
LO FCD, were sequenced. An analysis of an intronic SNP in the
TCF4 gene, rs613872 (MAF: G=0.0697/348), the risk allele (G)
that has been identified to be significantly associated with FCD
among Europeans through genome-wide association studies (GWAS)
(14), revealed that the risk
allele (G) was not present in any subject in our FCD pedigree
(0/84; 84 refers to the total number of chromosomes detected for
the 42 members of the pedigree in whom DNA collection and genetic
analysis were performed), and only one individual was heterozygous
for the risk allele (G) out of the 191 unrelated healthy controls
we tested (1/382) (Table II).
This result was consistent with a previous study in which rs613872
was not present in Singaporean Chinese (15). The detection of 2 other SNPs
(rs17089887, MAF: C=0.1689/846 and rs17089925, MAF: T=0.1569/786),
which had been found to be significantly associated with FCD in
Singaporean Chinese (15),
revealed that only 3 out of 8 cases carried the heterozygous risk
allele (C) of rs17089887 (3/16) and that only 1 out of 8 cases
carried the heterozygous risk allele (T) of rs17089925 (1/16). Both
of these risk alleles were also present in the 14 healthy
individuals who married into this family (rs17089887: 6/28;
rs17089925: 10/28) (Table II).
The analysis of another 3 SNPs that exhibited a marginal
association with FCD in Singaporean Chinese (rs1348047, MAF:
T=0.2780/1392; rs1452787, MAF: G=0.2708/1356; and rs2123392, MAF:
C=0.3005/1504) (15) revealed
that 7 out of 8 cases carried the risk allele (T) of rs1348047 (3
homozygous and 4 heterozygous for the risk allele, 10/16), 7 out of
8 cases carried the risk allele (G) of rs1452787 (1 homozygous and
6 heterozygous for the risk allele, 8/16), and all 8 cases carried
the risk allele (C) of rs2123392 (5 homozygous and 3 heterozygous
for the risk allele, 13/16). While these 3 risk alleles were also
present in the 14 healthy individuals who married into this family
(rs1348047, 16/28; rs1452787, 14/28; and rs2123392, 11/28)
(Table II), none of these 6 SNPs
from dbSNP co-segregated with the disease.
One TGC trinucleotide repeat expansion (rs193922902)
of TCF4, a repeat length >50 of which is known to play a
pathogenic role in the majority of FCD cases and is considered to
be a predictor of disease risk (16), was also detected in the present
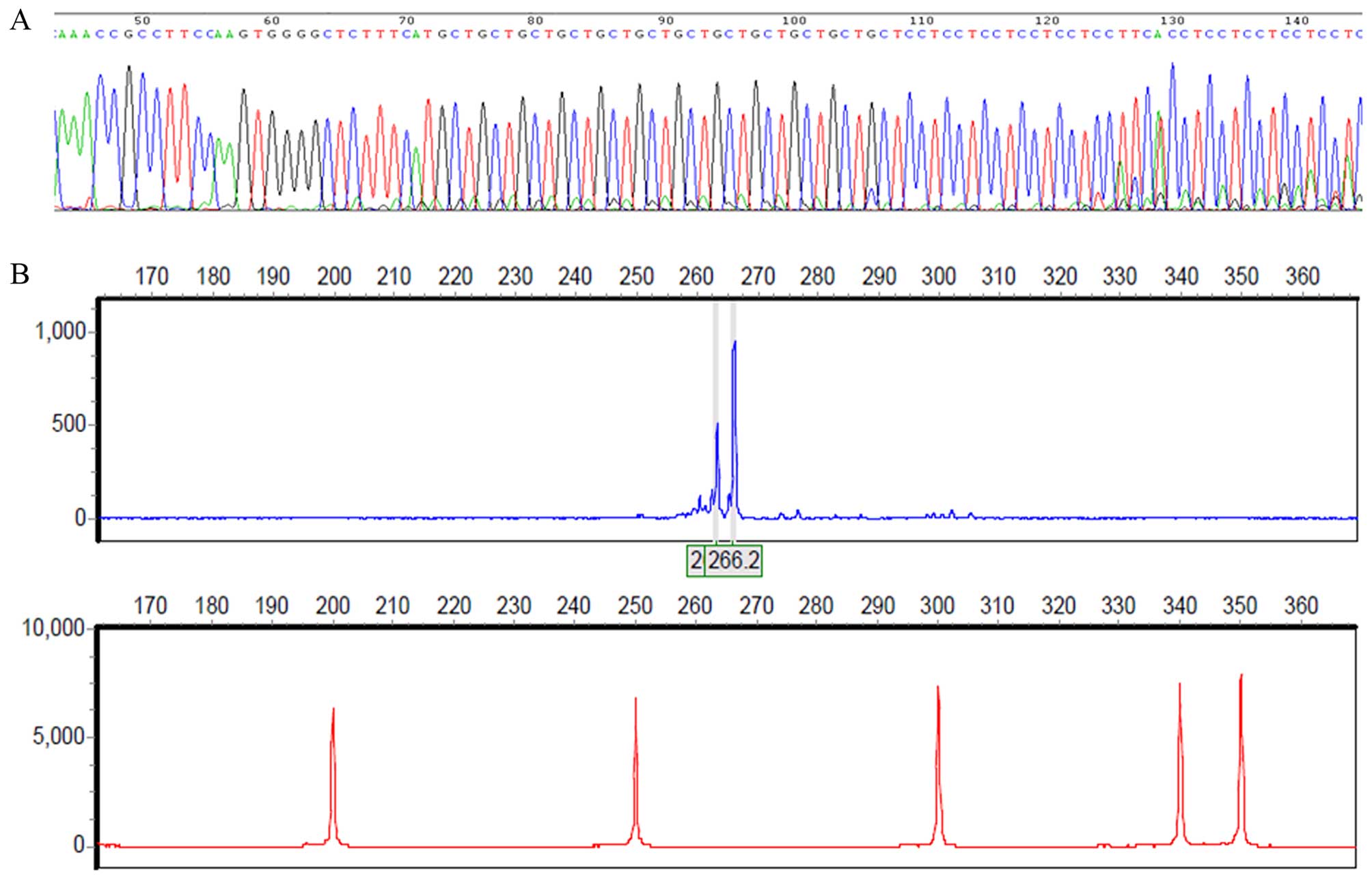
study. The direct sequencing of the proband indicated that it
contained one 11- and one 12-repeat allele (Fig. 5A), and this result was further
confirmed by STR analysis (Fig.
5B). The expanded repeat was not found in any of the subjects
in our pedigree (0/84) (Table
II), which indicated that this TGC trinucleotide expansion did
not play a pathogenic role in this specific FCD family.
Discussion
To date, progress toward identifying the underlying
genetic components of FCD has been limited to the analysis of a few
genes, including SLC4A11, ZEB1, LOXHD1 and
COL8A2 (4,10,12,13,20,21). Several genome-wide linkage studies
have additionally provided evidence of linkage to several different
chromosomal loci, namely FCD1, FCD2, FCD3 and FCD4, on chromosomes
13, 18, 5 and 9, respectively (10,24-26), that appear to influence familial
FCD. Thus, it appears that locus heterogeneity may exist for FCD,
whereby mutations in several genes on different chromosomes may
produce a common disease phenotype. Significant progress toward
understanding non-familial FCD was made using GWAS. Baratz et
al identified an SNP on chromosome 18q21, rs613872, in an
intron of a gene encoding (TCF4) (MIM 602272) and showed a
significant genome-wide association with FCD susceptibility in
Europeans (14). This finding was
further validated by Li et al (27) in another independent study.
Although rs613872 was not found to be present in Singaporean
Chinese FCD subjects, 2 other SNPs (rs17089887 and rs17089925)
(15) and one TGC trinucleotide
repeat expansion (rs193922902) (16) of the TCF4 gene were
reported to be significantly associated with FCD in the Chinese
subjects.
Despite these insights, knowledge regarding the
genetic basis of FCD in the China mainland population has remained
limited, possibly due to the varying prevalence of FCD in different
ethnic populations. The prevalence of FCD is generally considered
to be approximately 4% in individuals above 40 years of age in the
United States and accounts for the second most common indication
for corneal transplants performed in the United States in patients
over the age of 60 years (28,29). The prevalence of FCD in other
countries and areas has been confirmed by studies that have
examined indications for PK at various institutions worldwide;
prevalences of 15.4, 7.1 and 4.7% have been reported in populations
from the UK (30), Singapore
(31) and Australia (32), respectively, while studies in
China suggest a relatively lower prevalence of FCD, namely, 4.5% in
Taiwan (33) and <3.9% in both
the northern and eastern mainland of China (34,35). Combined with clinical experience
in the US that suggests a significantly decreased prevalence of FCD
among individuals of African-American, Latin-American, or Asian
origin, a greater understanding of the genetic basis of FCD in
patients of different ethnic origins will shed more light on the
molecular mechanisms of the disease.
To the best of our knowledge, the present study is
the first study on a Chinese mainland population to focus on the
genetic basis of the multi-generational Chinese pedigree with LO
FCD that was previously reported by our group (36). In the present study, we performed
a sequence analysis of the SLC4A11, ZEB1,
LOXHD1, COL8A2 and TCF4 genes in this LO FCD
pedigree.
Screening of the SLC4A11 gene revealed 14
known dbSNP variants; among these, an intronic variant, is a known
SNP from dbSNP (rs372201212, MAF: G=0.0014/7), its minor allele (G)
was detected in 4 of 8 affected members of this FCD pedigree, II-1,
II-5, III-9 (II-1's daughter), and III-19 (II-5′ son) (4/16), and 3
of 20 healthy descendants in this family, III-3 (II-1's son,
III-9's older brother), IV-4 (II-1's granddaughter, III-3's
daughter) and IV-16 (II-5's grandson, III-19's son) (3/40).
Although this variant was not identified in any unaffected
individuals who married into this family (0/28) or in the unrelated
healthy controls (0/382), it may not be considered pathogenic as it
did not co-segregate with the disease in this FCD pedigree.
However, we cannot rule out the possibility that this variant has
an association with FCD; if we consider the late onset of the
disease and the fact that IV-4 and IV-16 were 20 and 14 years old,
respectively, when the blood samples were collected in 2009, the
disease status in these younger individuals is uncertain, as they
may not have been old enough to manifest the disorder and may not
clinically exhibit the disease, suggesting that this variant may be
correlated with FCD in this family. Further analysis of this known
SNP from dbSNP (rs372201212) in the SLC4A11 gene in larger
numbers of Chinese patients with FCD may elucidate the significance
of this gene in corneal endothelial dystrophies.
Similarly, 2 intron variants of the LOXHD1
gene were identified in this FCD pedigree: c.5332-126C>T and
c.1809+155G>A (GenBank reference ID: NM_144612.6). These 2
variants have not been previously reported in FCD patients and were
absent from dbSNP. Neither of these variants was identified in the
unaffected family members or in the healthy controls (n≥100). As
these variants were each found in only a single case in this FCD
pedigree (c.5332-126C>T in II-3, and c.1809+155G>A in II-5),
they are likely examples of de novo mutations.
ZEB1 is a zinc finger E-box binding homeobox
1 gene (MIM 189909) and is also known as human zinc finger
TCF8, which maps to chromosome 10p11.2, comprises 9 exons
and encodes a transcription factor that is organized into multiple
functional domains starting with N-terminal zinc finger clusters
(172-292), followed by a homeodomain (581-640), a repression domain
(754-901), C-terminal zinc finger clusters (905-981) and an acidic
activation domain (1011-1124) (37). The structure of ZEB1 allows
for a wide range of functions as each zinc finger has different
DNA-binding specificities and effects on gene expression (38). Mutations in the ZEB1
transcripts have been shown to produce a wide range of ocular
phenotypes (39). It was
estimated that changes in this gene may account for approximately
50% of all PPCD cases (40), and
its mutations also account for LO FCD (12). Through ZEB1 screening, we
identified 3 continuous indels located at the junction of the
5′-UTR and the adjacent 5′ end of exon 1 of the ZEB1 gene in
the cases in this FCD pedigree, and these 3 indels covered the
region from -86 to -42 (numbering system based on the cDNA sequence
with +1 corresponding to the A of the ATG TSS in the Ref Seq:
NM_030751.5). A schematic illustration of the ZEB1 genomic
DNA and the location of these 3 continuous indels relative to exon
1 is shown in Fig. 4C, and these
3 continuous indels, including 34 bp Indel 1 (containing 23 bp of
the 5′-UTR region and 11 bp of the 5′ end of exon 1), 7 bp Indel 2
(containing 7 bp of exon 1), and 4 bp Indel 3 (containing 4 bp of
exon 1). In addition, according to the UCSC Genome Browser
(http://genome.ucsc.edu/cgi-bin/hgGateway?hgsid=488683917_eMaJPXBAmxFX5D7tqTNSeva4dT39),
the bases affected by these 3 indels are relatively well conserved
through evolution and also lie within transcription factor binding
sites, and these regions are also enriched with H3K27AC, which is
often found near active regulatory elements. In the case of Indel
1, the splice site variation affects the first splice site and may
likely cause mis-splicing of the pre-mRNA transcript. This
variation would lead to either exon skipping or intron retention,
which would consequently result in an altered protein structure.
Therefore, we hypothesized that a different haplotype of
ZEB1 will alter the mRNA structure or influence splicing
efficiency. Further studies are needed to determine what effect, if
any, these indels may have on ZEB1 gene function and FCD
pathogenesis; however, the presence of these 3 indels in the 14
healthy spouses who married into this family, the unaffected family
members, and the healthy controls (n≥100) suggests that these 3
indels are not likely to be pathogenic.
In the screening of the COL8A2 gene, neither
of the previously reported pathogenic mutations of COL8A2
(p.L450W and p.Q455K or Q455V) (4,21,23) was observed in this family, and
none of the novel mutations were identified in the COL8A2
gene in this LO FCD pedigree of Chinese descent. Our results are
not surprising, as several studies have been published
demonstrating a lack of COL8A2 mutations in LO FCD (22).
Variations in candidate genes of FCD, deemed
pathogenic on the basis of their absence in control chromosomes,
were later identified as common polymorphisms in other ethnic
populations (21-23), due to the fact that frequency of
gene variant may depend greatly on the population screened. The MAF
of 8 coding variants across SLC4A11 (p.R161R, p.S213S and
p.T833T) and COL8A2 (p.A35A, p.R155Q, p.L335L, p.G495G and
p.T502M) genes in healthy controls with Chinese ancestry (n≥100)
were compared with MAF data from the 1000 Genomes database
(Table II). The statistical
analysis demonstrated that 4 MAF of COL8A2 gene (p.A35A,
p.L335L, p.G495G and p.T502M) in Chinese healthy controls tested in
the present study were all significantly higher than the data from
1000 Genomes (P<0.01) (Table
II), indicated that these varations were rare in occidental
populations (21), which may
account for the absence of these minor allele distribution in
control chromosomes in previous study (21-23).
Since the publication of the initial GWAS results
indicating a significant association of an intronic SNP in
TCF4, rs613872, with FCD (14), several studies across different
cohorts from diverse populations have demonstrated that
polymorphisms near TCF4 were consistently linked to an
increased prevalence of FCD (15,27). These SNPs of TCF4 were
detected in this Chinese LO FCD pedigree, and the analysis revealed
that the SNP rs613872, which is most highly associated with FCD in
Caucasians, was not found to be present in this Chinese FCD family
(0/84), and only one individual carrying the heterozygous variant
of rs613872 was detected in the healthy controls (1/382), the MAF
of healthy controls tested in the present study is significantly
lower when compared with the data from 1000 Genomes (P<0.01)
(Table II). This result is
consistent with previous research (15), in which rs613872 was not found to
be present in Chinese FCD subjects, as well as with the data from
the Human Genome Diversity Project, in which the minor (risk)
allele, G, of rs613872 was found to be rare in populations from
Africa, Eastern Asia, and Central and South America and more
frequent in European, Middle Eastern, and Southern Asian
populations (41). Two other SNPs
(rs17089887 and rs17089925), which have been reported to be
significantly associated with FCD in Singaporean Chinese (15), and a TGC trinucleotide repeat
expansion (rs193922902) of TCF4, a repeat length >50 of
which plays a pathogenic role in the majority of FCD cases
(16), were also detected in this
Chinese LO FCD pedigree. The results revealed that none of these 3
SNPs co-segregated with the disease. Therefore, we investigated
whether the three SNPs that were marginally associated with FCD in
Singaporean Chinese (rs1348047, rs1452787 and rs2123392) (15) had some association with FCD in
this Chinese LO FCD pedigree, and the results revealed that all
three risk alleles were present in both the 8 cases and the 14
healthy individuals who married into this family. This finding led
to the conclusion that none of these known SNPs provide strong
evidence of pathogenesis in this specific muti-generational LO FCD
Chinese family; however, because only seven fragments containing 7
SNPs were sequenced in the present study, we still cannot rule out
the possibility that additional variants in other regions of the
TCF4 gene that were not assessed in the present study may be
present that could confer phenotypic changes.
In conclusion, to identify and exclude known
mutations and SNPs associated with FCD, we screened our LO FCD
pedigree for all known exons and adjacent splice sites in the
previously reported FCD genes that were associated with either LO
FCD (SLC4A11, ZEB1, LOXHD1 and TCF4) or
EO FCD (COL8A2). Twenty-seven variants (including 22 known
dbSNP vari ants and 5 variants absent from dbSNP) were detected.
None of these variants provided strong evidence of pathogenesis,
making it unlikely that SNPs or mutations in them caused FCD in
this specific pedigree. The possibility of pathogenic changes
occurring within the promoter, intronic, or untranslated non-coding
regions of these genes playing a role in the pathogenesis of FCD
has not been excluded in this study. The fact that we did not
detect any pathogenic variants in these genes in our pedigree is
likely a combination of the fact that these genes carry a low
genetic load in FCD and that we screened only one LO FCD pedigree,
a small sample that is underpowered for detecting variants that
occur at relatively low frequencies. Clearly, a genome-wide linkage
scan to identify linkage to one of the previously described FCD
loci or to identify a novel locus for FCD will need to be performed
in this multi-generational Chinese pedigree with LO FCD. Our
observation, nevertheless, expands the current knowledge regarding
the genetic status of Chinese ancestry patients with FCD.
Acknowledgments
The authors would like to thank all the family
members and other people who generously participated in the study.
The study was supported by the joint grands from the Yunnan
Scientific and Technology Committee and Kunming Medical University,
P.R. China (no. 2012FB090).
References
|
1
|
Fuchs E: Fuchs E Dystrophia epithelialis
corneae. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 76:478–508.
1910. View Article : Google Scholar
|
|
2
|
Adamis AP, Filatov V, Tripathi BJ and
Tripathi RC: Fuchs' endothelial dystrophy of the cornea. Surv
Ophthalmol. 38:149–168. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krachmer JH, Purcell JJ Jr, Young CW and
Bucher KD: Corneal endothelial dystrophy. A study of 64 families.
Arch Ophthalmol. 96:2036–2039. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gottsch JD, Sundin OH, Liu SH, Jun AS,
Broman KW, Stark WJ, Vito EC, Narang AK, Thompson JM and Magovern
M: Inheritance of a novel COL8A2 mutation defines a distinct
early-onset subtype of fuchs corneal dystrophy. Invest Ophthalmol
Vis Sci. 46:1934–1939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Waring GO III, Bourne WM, Edelhauser HF
and Kenyon KR: The corneal endothelium. Normal and pathologic
structure and function. Ophthalmology. 89:531–590. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bergmanson JP, Sheldon TM and Goosey JD:
Fuchs' endothelial dystrophy: A fresh look at an aging disease.
Ophthalmic Physiol Opt. 19:210–222. 1999. View Article : Google Scholar
|
|
7
|
Thompson RW Jr, Price MO, Bowers PJ and
Price FW Jr: Long-term graft survival after penetrating
keratoplasty. Ophthalmology. 110:1396–1402. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anshu A, Price MO, Tan DT and Price FW Jr:
Endothelial keratoplasty: a revolution in evolution. Surv
Ophthalmol. 57:236–252. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ham L, Dapena I, van Luijk C, van der Wees
J and Melles GR: Descemet membrane endothelial keratoplasty (DMEK)
for Fuchs endothelial dystrophy: review of the first 50 consecutive
cases. Eye (Lond). 23:1990–1998. 2009. View Article : Google Scholar
|
|
10
|
Riazuddin SA, Vithana EN, Seet LF, Liu Y,
Al-Saif A, Koh LW, Heng YM, Aung T, Meadows DN, Eghrari AO, et al:
Missense mutations in the sodium borate cotransporter SLC4A11 cause
late-onset Fuchs corneal dystrophy. Hum Mutat. 31:1261–1268. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vithana EN, Morgan P, Sundaresan P,
Ebenezer ND, Tan DT, Mohamed MD, Anand S, Khine KO, Venkataraman D,
Yong VH, et al: Mutations in sodium-borate cotransporter SLC4A11
cause recessive congenital hereditary endothelial dystrophy
(CHED2). Nat Genet. 38:755–757. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riazuddin SA, Zaghloul NA, Al-Saif A,
Davey L, Diplas BH, Meadows DN, Eghrari AO, Minear MA, Li YJ,
Klintworth GK, et al: Missense mutations in TCF8 cause late-onset
Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am
J Hum Genet. 86:45–53. 2010. View Article : Google Scholar :
|
|
13
|
Riazuddin SA, Parker DS, McGlumphy EJ, Oh
EC, Iliff BW, Schmedt T, Jurkunas U, Schleif R, Katsanis N and
Gottsch JD: Mutations in LOXHD1, a recessive-deafness locus, cause
dominant late-onset Fuchs corneal dystrophy. Am J Hum Genet.
90:533–539. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baratz KH, Tosakulwong N, Ryu E, Brown WL,
Branham K, Chen W, Tran KD, Schmid-Kubista KE, Heckenlively JR,
Swaroop A, et al: E2-2 protein and Fuchs's corneal dystrophy. N
Engl J Med. 363:1016–1024. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thalamuthu A, Khor CC, Venkataraman D, Koh
LW, Tan DT, Aung T, Mehta JS and Vithana EN: Association of TCF4
gene polymorphisms with Fuchs' corneal dystrophy in the Chinese.
Invest Ophthalmol Vis Sci. 52:5573–5578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wieben ED, Aleff RA, Tosakulwong N, Butz
ML, Highsmith WE, Edwards AO and Baratz KH: A common trinucleotide
repeat expansion within the transcription factor 4 (TCF4, E2-2)
gene predicts Fuchs corneal dystrophy. PLoS One. 7:e490832012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gottsch JD, Zhang C, Sundin OH, Bell WR,
Stark WJ and Green WR: Fuchs corneal dystrophy: Aberrant collagen
distribution in an L450W mutant of the COL8A2 gene. Invest
Ophthalmol Vis Sci. 46:4504–4511. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yellore VS, Rayner SA, Emmert-Buck L,
Tabin GC, Raber I, Hannush SB, Stulting RD, Sampat K, Momi R,
Principe AH and Aldave AJ: No pathogenic mutations identified in
the COL8A2 gene or four positional candidate genes in patients with
posterior polymorphous corneal dystrophy. Invest Ophthalmol Vis
Sci. 46:1599–1603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hemadevi B, Srinivasan M, Arunkumar J,
Prajna NV and Sundaresan P: Genetic analysis of patients with Fuchs
endothelial corneal dystrophy in India. BMC Ophthalmol. 10:32010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vithana EN, Morgan PE, Ramprasad V, Tan
DT, Yong VH, Venkataraman D, Venkatraman A, Yam GH, Nagasamy S, Law
RW, et al: SLC4A11 mutations in Fuchs endothelial corneal
dystrophy. Hum Mol Genet. 17:656–666. 2008. View Article : Google Scholar
|
|
21
|
Biswas S, Munier FL, Yardley J,
Hart-Holden N, Perveen R, Cousin P, Sutphin JE, Noble B, Batterbury
M, Kielty C, et al: Missense mutations in COL8A2, the gene encoding
the alpha2 chain of type VIII collagen, cause two forms of corneal
endothelial dystrophy. Hum Mol Genet. 10:2415–2423. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kobayashi A, Fujiki K, Murakami A, Kato T,
Chen LZ, Onoe H, Nakayasu K, Sakurai M, Takahashi M, Sugiyama K and
Kanai A: Analysis of COL8A2 gene mutation in Japanese patients with
Fuchs' endothelial dystrophy and posterior polymorphous dystrophy.
Jpn J Ophthalmol. 48:195–198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mok JW, Kim HS and Joo CK: Q455V mutation
in COL8A2 is associated with Fuchs' corneal dystrophy in Korean
patients. Eye (Lond). 23:895–903. 2009. View Article : Google Scholar
|
|
24
|
Sundin OH, Jun AS, Broman KW, Liu SH,
Sheehan SE, Vito EC, Stark WJ and Gottsch JD: Linkage of late-onset
Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Invest
Ophthalmol Vis Sci. 47:140–145. 2006. View Article : Google Scholar
|
|
25
|
Sundin OH, Broman KW, Chang HH, Vito EC,
Stark WJ and Gottsch JD: A common locus for late-onset Fuchs
corneal dystrophy maps to 18q21.2-q21.32. Invest Ophthalmol Vis
Sci. 47:3919–3926. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Riazuddin SA, Eghrari AO, Al-Saif A, Davey
L, Meadows DN, Katsanis N and Gottsch JD: Linkage of a mild
late-onset phenotype of Fuchs corneal dystrophy to a novel locus at
5q33.1-q35.2. Invest Ophthalmol Vis Sci. 50:5667–5671. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y-J, Minear MA, Rimmler J, Zhao B,
Balajonda E, Hauser MA, Allingham RR, Eghrari AO, Riazuddin SA,
Katsanis N, et al: Replication of TCF4 through association and
linkage studies in late-onset Fuchs endothelial corneal dystrophy.
PLoS One. 6:e180442011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Klintworth GK: Corneal dystrophies.
Orphanet J Rare Dis. 4:72009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mannis MJ, Holland EJ, Beck RW, Belin MW,
Goldberg MA, Gal RL, Kalajian AD, Kenyon KR, Kollman C, Ruedy KJ,
et al Cornea Donor Study Group: Clinical profile and early surgical
complications in the Cornea Donor Study. Cornea. 25:164–170. 2006.
View Article : Google Scholar
|
|
30
|
Rahman I, Carley F, Hillarby C, Brahma A
and Tullo AB: Penetrating keratoplasty: indications, outcomes, and
complications. Eye (Lond). 23:1288–1294. 2009. View Article : Google Scholar
|
|
31
|
Tan DT, Janardhanan P, Zhou H, Chan YH,
Htoon HM, Ang LP and Lim LS: Penetrating keratoplasty in Asian
eyes: The Singapore Corneal Transplant Study. Ophthalmology.
115:975–982.e1. 2008. View Article : Google Scholar
|
|
32
|
No authors listed. The Australian Corneal
Graft Registry: 1990 to 1992 report. Aust N Z J Ophthalmol.
21(Suppl 2): 1–48. 1993.
|
|
33
|
Chen WL, Hu FR and Wang IJ: Changing
indications for penetrating keratoplasty in Taiwan from 1987 to
1999. Cornea. 20:141–144. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xie L, Song Z, Zhao J, Shi W and Wang F:
Indications for penetrating keratoplasty in north. China Cornea.
26:1070–1073. 2007. View Article : Google Scholar
|
|
35
|
Zhang C and Xu J: Indications for
penetrating keratoplasty in East China, 1994–2003. Graefes Arch
Clin Exp Ophthalmol. 243:1005–1009. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huo L, Hui T, Tao S, Hong D and Yan X: A
pedigree of Fuchs Corneal Dystrophy. Zhonghua Yi Xue Yi Chuan Xue
Za Zhi. 27:231–232. 2010.In Chinese.
|
|
37
|
Ikeda K, Halle JP, Stelzer G, Meisterernst
M and Kawakami K: Involvement of negative cofactor NC2 in active
repression by zinc finger-homeodomain transcription factor AREB6.
Mol Cell Biol. 18:10–18. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ikeda K and Kawakami K, Stelzer G,
Meisterernst M and Kawakami K: DNA binding through distinct domains
of zinc-finger-homeodomain protein AREB6 has different effects on
gene transcription. Eur J Biochem. 233:73–82. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Moroi SE, Gokhale PA, Schteingart MT,
Sugar A, Downs CA, Shimizu S, Krafchak C, Fuse N, Elner SG, Elner
VM, et al: Clinicopathologic correlation and genetic analysis in a
case of posterior polymorphous corneal dystrophy. Am J Ophthalmol.
135:461–470. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Krafchak CM, Pawar H, Moroi SE, Sugar A,
Lichter PR, Mackey DA, Mian S, Nairus T, Elner V, Schteingart MT,
et al: Mutations in TCF8 cause posterior polymorphous corneal
dystrophy and ectopic expression of COL4A3 by corneal endothelial
cells. Am J Hum Genet. 77:694–708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : Article published
online before print in May 2002. PubMed/NCBI
|