Introduction
Bisphosphonates (BPs) are analogues of pyrophosphate
that are known to prevent bone resorption by inhibiting osteoclast
activity (reviewed in ref. 1).
BPs are used for the treatment of various bone diseases, including
osteoporosis and bone metastasis in cancer with or without
hypercalcemia (2).
Nitrogen-containing BPs, such as zoledronic acid (ZA), are used in
the treatment of osteoporosis and bone metastasis (reviewed in ref.
3), as well as in the treatment
of other bone diseases, such as Erdheim-Chester disease (4). In addition, they are more potent
inhibitors of bone resorption than non-nitrogen-containing BPs
(reviewed in ref. 5). However,
despite their several benefits, ZA induces BP-related osteonecrosis
of the jaw (BRONJ) in cancer patients (6,7).
BRONJ is defined as necrotic bone exposure in the oral cavity
continuing for >8 weeks in BP-treated patients who have not had
received head and neck radiation therapy (8). The molecular mechanisms responsible
for the development of BRONJ remain to be elucidated; however, some
risk factors, including periodontitis with bacterial plaque in the
oral cavity have been associated with this symptom (reviewed in
ref. 7). Previous studies have
suggested that infectious agents, including Actinomyces,
play important roles in the etiology and progression of BRONJ
(9–11). Interestingly, Kobayashi et
al reported that ZA promoted the adherence of Streptococcus
mutans to hydroxyapatite and the proliferation of oral bacteria
obtained from healthy individuals, suggesting that ZA increases
bacterial infection (12). Wound
closure by oral epithelial cells (ECs) and gingival fibroblasts
(GFs) is important, not only for successful wound healing, but also
for the protection of the socket from oral bacterial infection
following tooth extraction. Thus, it is possible that an oral
bacterial infection may induce BRONJ in cooperation with other risk
factors, such as diabetes mellitus with steroid intake (13) or with microvascular disease
(14), smoking, prosthetic
trauma, and implant treatment (15). In addition, the ability of GFs to
migrate and synthesize type I collagen is essential for the
formation of rigid gingival connective tissue to cover the
intraoral bone exposure, which protects the alveolar bone of the
maxilla and the jaw from infection with oral bacteria. However, the
mechanisms through which ZA affects the ability of human GFs (hGFs)
to migrate and synthesize the connective tissue at the molecular
level remain to be elucidated.
When tissue injury occurs, the blood coagulation
cascade is initiated, resulting in the formation of fibrous
connective tissue, which functions as a scaffold for the influx of
inflammatory cells. Platelets also aggregate at the site of injury,
and become activated by binding to the negatively charged
extravascular fibrous tissue, and release various growth factors,
including transforming growth factor-β (TGF-β) (reviewed in ref.
16). TGF-β1 is known to be
expressed in serveral types of cells, including hGFs, and is
involved in the proliferation and differentiation of these cells
(17,18). Thus, the functions of TGF-β seem
to be autocrine or paracrine as regards the regulation of hGFs
during oral inflammation and the wound healing processes at the
site of injury. The TGF-β superfamily consists of two families: the
TGF-β/activin/Nodal family and the bone morphogenetic protein
(BMP)/growth and differentiation factor (GDF)/Mullerian inhibiting
substance (MIS) family (reviewed in ref. 19).
The TGF-β superfamily ligands initiate a cascade of
signaling events by binding to their respective type I and type II
receptors in the extracellular space. Following this, two type I
and two type II receptors form a tetrameric complex. In this
ligand-bound complex of type I and type II receptors, the type II
receptor kinase activates the type I receptor kinase. The type I
receptor induces intracellular signal transduction by
phosphorylating the receptor-regulated Smads (R-Smads) (reviewed in
refs. 20–23). Smads are central signal
transducers of TGF-β superfamily and are composed of 3 groups. The
first group comprises the R-Smads; Smad1, Smad5 and Smad8 are
primarily activated by the BMP-specific type I receptors, whereas
Smad2 and Smad3 are activated by TGF-β-specific type I receptors.
The second group contains the common mediator Smad (Co-Smad; e.g.,
Smad4). The third group comprises the inhibitory Smads (I-Smads;
e.g., Smad6 and Smad7). Activated R-Smads form complexes with the
Co-Smad, which enter the nucleus, and, together with other
cooperative proteins, positively or negatively control the
transcription of specific target genes. I-Smads suppress the
activation of R-Smads by competing with R-Smads for type I receptor
interaction and by recruiting specific ubiquitin ligases, resulting
in their proteasomal degradation (reviewed in ref. 24).
TGF-β has the ability to induce the differentiation
of various types of cells into myofibroblasts (MFs), which
typically exhibit the formation of F-actin stress fibers (reviewed
in ref. 25). We have previously
demonstrated that TGF-β induces the expression of the MF markers,
α-smooth muscle actin (α-SMA) and type I collagen, in fibroblastic
cells derived from periodontal ligament (26). Sobral et al reported that
TGF-β induced the differentiation of hGFs into MFs in a
Smad-dependent manner (27).
Thus, TGF-β is now known to induce MF differentiation from
fibroblasts, which preferentially form a fibrous tissue. TGF-β is
also known to induce migratory activity in various types of cells,
including fibroblastic cells in a Smad-dependent manner (28–30). In addition, Bakin et al
reported that TGF-β regulates the migratory activity of ECs in a
p38 mitogen-activated protein kinase (MAPK)-dependent manner
(31), suggesting that TGF-β
induces cell migratory activity through MAPKs as opposed to Smads.
Moreover, TGF-β plays important roles in physiological wound
closure by promoting cell migration and type I collagen synthesis.
On the other hand, the abnormal and persistent appearance of MFs
causes scar formation or fibrosis (32,33). The abnormal potentiation of TGF-β
signaling in MFs possibly causes fibrogenic diseases. Therefore,
MFs represent key players in the physiological reconstruction of
connective tissue following injury and in generating the
pathological tissue deformations that characterize fibrosis
(34).
In this study, we investigated the mechanisms
through which ZA, at its maximum concentration in serum (Cmax),
which clinically, is usually found after the intravenous
administration of 4 mg ZA, which is the appropriate amount for the
usual treatment of bone metastasis (35,36) or bone diseases such as
Erdheim-Chester disease (4),
affects TGF-β-induced intracellular signal transduction and MF
differentiation of hGFs, which are important processes for the
progression of fibrogenesis during inflammation.
Materials and methods
Reagents
Recombinant TGF-β was purchased from Peprotech, Inc.
(Rocky Hill, NJ, USA). The TGF-β type I receptor inhibitor,
SB-431542, which preferentially suppresses the activation of the
intracellular signal transduction of Smad2 by this receptor
(37,38), and sometimes broadly suppresses
the TGF-β1-induced activation of p38 MAPK, and extracellular
signal-regulated kinase (ERK) as opposed to Smad2 (30) was purchased from Calbiochem (La
Jolla, CA, USA). Zometa® obtained from Novartis
Pharmaceuticals (Tokyo, Japan) was used as ZA in all our
experiments. The Cmax of ZA in an adult human body is approximately
1.47 µM within 15 min after its intravenous administration
(4 mg/5 ml) according to the user instructions provided with
Zometa®. This is the appropriate amount of ZA for the
treatment of cancer bone metastasis (32,33), and is referred in the internal
document ZOMU00007 belonging to Novartis Pharmaceuticals.
Cell culture
hGFs were isolated and cultured as described in our
previous study (39). Briefly,
the cells were isolated from the fresh gingival tissue biopsy
samples of 3 volunteers, and were maintained in a Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum (FBS) and penicillin-streptomycin (both from Invitrogen,
Gaithersburg, MD, USA). Informed consent was obtained from all the
volunteers prior to obtaining the samples, and the Ethics Committee
of Iwate Medical University approved the research protocol
(approval no. 1126). Subsequently, one hGF culture retaining a high
proliferative potential among the 3 cultures was used. NIH3T3 mouse
embryonic fibroblasts (used as a standard control for the
fibroblasts), obtained from RIKEN Cell Bank (Tsukuba, Japan), were
cultured with DMEM supplemented with 10% FBS and
penicillin-streptomycin (both from Invitrogen).
Cell viability assay
The status of cell viability was evaluated using an
alamarBlue assay (AbD Serotec, Oxon, UK) according to the
manufacturer's instructions. This assay reagent includes an
indicator that fluoresces and undergoes colorimetric changes when
reduced by mitochondrial respiration, which is proportional to the
number of living cells. For the viability assay, the cells were
seeded in 96-well plates at a density of 2.48×103
cells/well and cultured for 48 h in medium containing 10% FBS, with
or without ZA at the indicated concentrations (0.0147-147
µM). Some of the cells were subsequently treated with TGF-β1
(1–5 ng/ml) for 24 h following treatment with ZA. The medium was
replaced with DMEM containing 10% alamarBlue solution to evaluate
the viability of the cells, and the cells were cultured for an
additional 4.5 h. The absorbance in each well was measured using an
ELISA plate reader (Tosoh Corp., Tokyo, Japan). The data were
presented as values of Abs570 − Abs600. Each
experiment was repeated 3 times, with 5-wells dedicated for each
time point.
RNA isolation and RT-qPCR
Total RNA from the hGFs and NIH3T3 cells was
isolated using ISOGEN II reagent (Nippon Gene, Toyama, Japan)
according to the manufacturer's instructions. First-strand cDNA was
synthesized from total RNA using the PrimeScript RT reagent kit
(Takara Bio, Shiga, Japan). PCR was subsequently performed on a
Thermal Cycler Dice Real-Time system using SYBR Premix Ex Taq II
(both from Takara Bio) with specific oligonucleotide primers (human
α-SMA forward, 5′-ATACAACATGGCATCATCACCAA-3′ and reverse,
5′-GGGCAACACGAAGCTCATTGTA-3′; mouse α-SMA forward,
5′-CAGATGTGGATACAGCAAACAGGA-3′ and reverse,
5′-GACTTAGAAGCATTTGCGGTGGA-3′; human TGF-β type I receptor forward,
5′-GCTGCTCCTCCTCGTGCT-3′ and reverse, 5′-TTGTCTTTTGTACAGAGGTGGC-3′;
human TGF-β type II receptor forward, 5′-CTGCACATCGTCCTGTGG-3′ and
reverse, 5′-GGAAACTTGACTGCACCGTT-3′; and human glyceraldehyde
3-phosphate dehydrogenase (GAPDH) forward,
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′-ATGGTGGTGAAGACCCCACT-3′;
and mouse GAPDH forward, 5′-TGTGTCCGTCGTGGATCTG-3′ and reverse,
5′-TTGCTGTTGAAGTCGCAGGAG-3′). The mRNA expression levels of α-SMA,
TGF-β type I receptor and TGF-β type II receptor were normalized to
those of GAPDH, and the relative expression levels were presented
as the fold increase or decrease relative to the control.
Western blot analysis
The cells were lysed in RIPA buffer [50 mM Tris-HCl
(pH 7.2), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1%
SDS] or lysis buffer [20 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA
and 1% Triton® X-100] containing protease and
phosphatase inhibitor cocktails (both from Sigma, St. Louis, MO,
USA). The protein content of the samples was measured using BCA
reagent (Pierce, Rockford, IL, USA). Samples containing equal
amounts of protein were separated on 10% SDS-polyacrylamide gels
and transferred onto a polyvinylidenedifluoride membrane (Millipore
Corp., Bedford, MA, USA). After being blocked with 1% BSA or 1%
skim milk in T-TBS (50 mM Tris-HCl, pH 7.2, 150 mM NaCl and 0.05%
Tween-20), the membrane was incubated with primary antibodies
including anti-α-SMA rabbit polyclonal antibody (1:1,000; ab5694;
Abcam, Cambridge, UK), anti-Smad2/3 purified mouse monoclonal
antibody (1:1,000; 610842; BD Transduction Laboratories™, Franklin
Lakes, NJ, USA), anti-phospho-Smad2/3 (#8828), anti-p38 MAPK
(#9212), anti-phospho-p38 MAPK (#9211), anti-c-Jun N-terminal
kinase (JNK; #9251) and anti-phospho-JNK polyclonal antibodies
(#9252) (1:1,000; all from Cell Signaling Technology, Inc.,
Beverly, MA, USA) and anti-α-tubulin mouse monoclonal antibody
(1:25,000; #3873, Cell Signaling Technology, Inc.) as a loading
control for normalization. The proteins of interest were then
detected using appropriate horseradish peroxidase-conjugated
secondary antibodies (Cell Signaling Technology, Inc.) and an
Amersham ECL™ Prime Western Blotting Detection reagent (GE
Healthcare Bio-Sciences, Pittsburgh, PA, USA).
Immunofluorescence analysis of cultured
cells
For immunofluorescence analysis of the cultured
cells, the hGFs were subcultured on non-coated cover glass slips
(Matsunami Glass Ind., Ltd. Kishiwada, Japan) at a density of
2.5×104 cells/glass slip (BD Biosciences, Franklin
Lakes, NJ, USA) and maintained in DMEM supplemented with 10% FBS,
with or without ZA (1.47 µM) for 48 h. The culture medium
was replaced with DMEM without FBS, supplemented with or without
TGF-β1 (5 ng/ml). The cells were maintained in this medium for 4 or
5 days, fixed in 4% paraformaldehyde (Nacalai Tesque, Inc., Kyoto,
Japan) for 15 min and permeabilized with Triton X-100 (Sigma).
Following background reduction with normal goat serum, the cells
were labeled with anti-α-SMA rabbit polyclonal antibody (1:100;
ab5694; Abcam), or anti-collagen type I rabbit polyclonal antibody
(1:100; 600-401-103-0.1; Rockland, Inc., Rockland, ME, USA) at room
temperature for 1 h. After being washed with phosphate-buffered
saline (PBS) to remove the excess primary antibody, the cells were
incubated with Alexa Fluor® 488-conjugated goat
anti-rabbit IgG (1:1,000; A-11034; Molecular Probes, Leiden, The
Netherlands) and DAPI (1:1,000; KPL, Gaithersburg, MD, USA) for 30
min at room temperature. After being washed with PBS to remove the
excess secondary antibody, the fluorescent signal was detected
using an Olympus IX70 fluorescence microscope with the LCPIanFI 20
objective lens (Olympus Co., Tokyo, Japan).
Evaluation of the migratory activities of
hGFs and NIH3T3 cells
Cell migration assays were performed using
Transwell® membrane cell culture inserts (8 µm
pore size; Corning Inc., Corning, NY, USA) according to the
manufacturer's instructions. The hGFs were cultured with or without
ZA (1.47 µM) in DMEM supplemented with 10% FBS for 48 h at a
cell density of 6.0×105 cells in 10 cm culture dish. The
cells were then placed at a density of 1.0×105 cells in
the cell culture inserts in the each well of a 24-well cell culture
plate in DMEM with 0.1% BSA. The cells were allowed to migrate
through the porous membrane that bordered the upper cell culture
insert and the lower well of 24-well culture plate which contained
DMEM in the presence or absence of TGF-β (5 ng/ml) for 6 h at 37°C.
TGF-β was added to the culture medium in the lower well of 24-well
culture plate. In some cases, SB-431542 was added to both the upper
and lower culture media. The cells on the upper side of the
membrane were wiped, and the membrane was fixed in 4%
paraformaldehyde in PBS. After being washed with PBS, the cells
that had migrated onto the underside of the membrane were labeled
with DAPI (1:1,000; KPL) and counted. The values were shown as
average of those from 3 wells. The evaluation of the migratory
activity of the NIH3T3 cells was performed as described above
without pre-treatment of the cells with ZA.
Flow cytometry
The hGFs were cultured with or without ZA (1.47
µM) in DMEM supplemented with 10% FBS for 48 h at a density
of 6.0×105 cells in a 10-cm culture dish. The cells
(1.0×105 cells) were suspended in PBS containing 0.5%
FBS and 2 mM EDTA and incubated with anti-TGF-β receptor type I
(1:100; ab30103) or type II (1:50; ab78419) (both from Abcam)
primary antibodies for 1 h at 4°C. For the negative control
experiments, the cells were incubated with the same protein amount
of normal control IgG (sc-2028; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) as each specific antibody. The cells were then
incubated with PE-conjugated secondary antibodies (#732988 for
ab31013; #732970 for ab78419) for 30 min in the dark. Acquisition
was performed using an EPICS XL EXPO 32 ADC system (Beckman
Coulter, Fullerton, CA, USA).
Statistical analysis
The data are presented as the mean ± SD (n=3 or 5
experiments). The data were statistically analyzed using the
Student's t-test, and P<0.01 (indicated by an asterisk) was
considered significant. The results shown in all experiments are
representative of at least 3 separate experiments.
Results
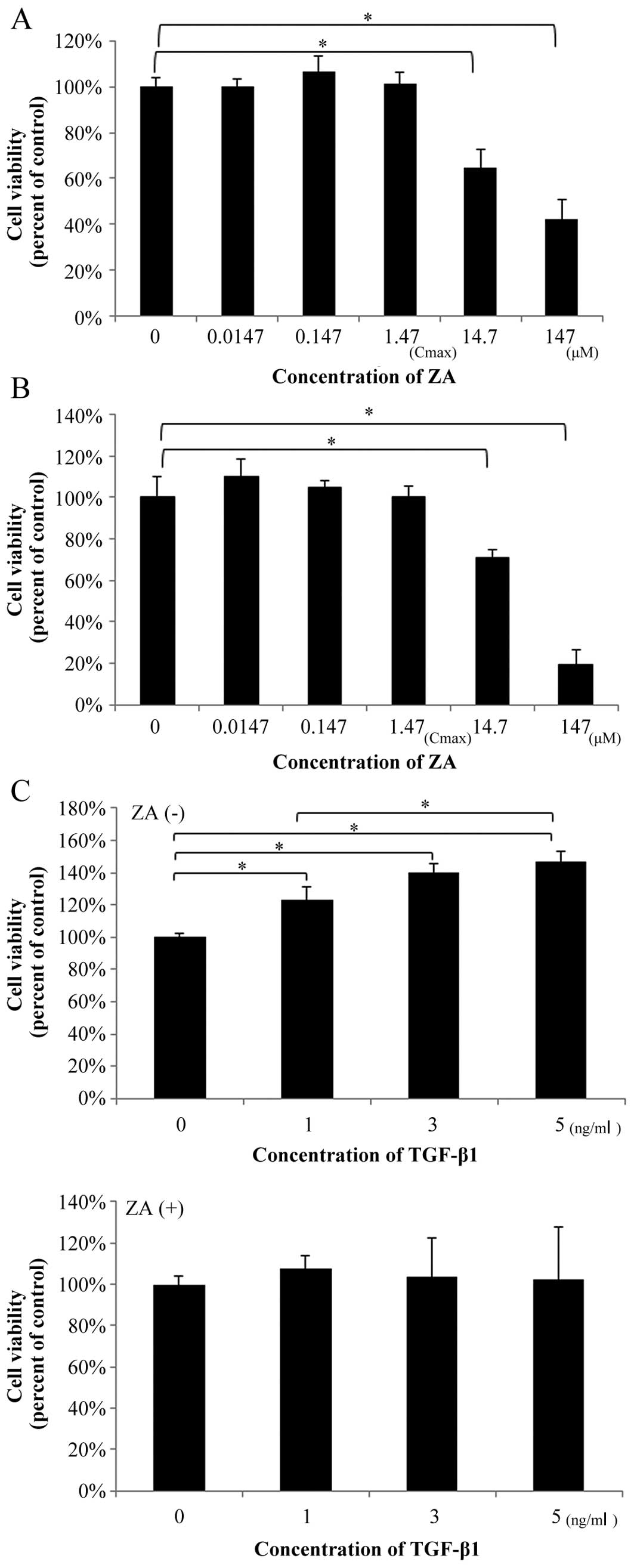
ZA suppresses the TGFβ1-induced increase
in the viability of hGFs
Treatment with ZA for 48 h in medium containing 10%
FBS at the concentrations of 0.0147 µM (1/100 of Cmax),
0.147 µM (1/10 of Cmax) and 1.47 µM (Cmax) did not
affect the viability of the hGFs (Fig. 1A) and the NIH3T3 cells which were
used as a standard control for the fibroblasts (Fig. 1B). However, at 10-100-fold higher
concentrations (14.7–147 µM) than the Cmax, ZA significantly
suppressed the viability of the hGFs (35–58% suppression of the
control) and that of the NIH3T3 cells (29–80% suppression of the
control) in a dose-dependent manner. Thus, ZA (Cmax) does not
affect the viability of hGFs and normal standard fibroblasts at 48
h following administration.
In order to elucidate the mechanisms through which
ZA affects TGF-β-induced fibrogenesis by hGFs, the hGFs were
pre-treated with ZA (Cmax) for 48 h and subsequently treated with
TGF-β1 at the indicated concentrations for the indicated periods of
time. Finally, the effect of pre-treatment with ZA (Cmax) on
TGF-β-induced functions in hGFs was investigated using the
following experiments:
We examined whether ZA (Cmax) affects the viability
of hGFs stimulated with TGF-β1 (1–5 ng/ml). As shown in Fig. 1C (upper graph), TGF-β1 (1–5 ng/ml)
increased the viability of the hGFs (23–47% promotion of the
control). However, ZA (Cmax) suppressed the TGFβ1-induced increase
in the viability of the hGFs (Fig.
1C, lower graph; compare same TGF-β1 concentrations between
graphs).
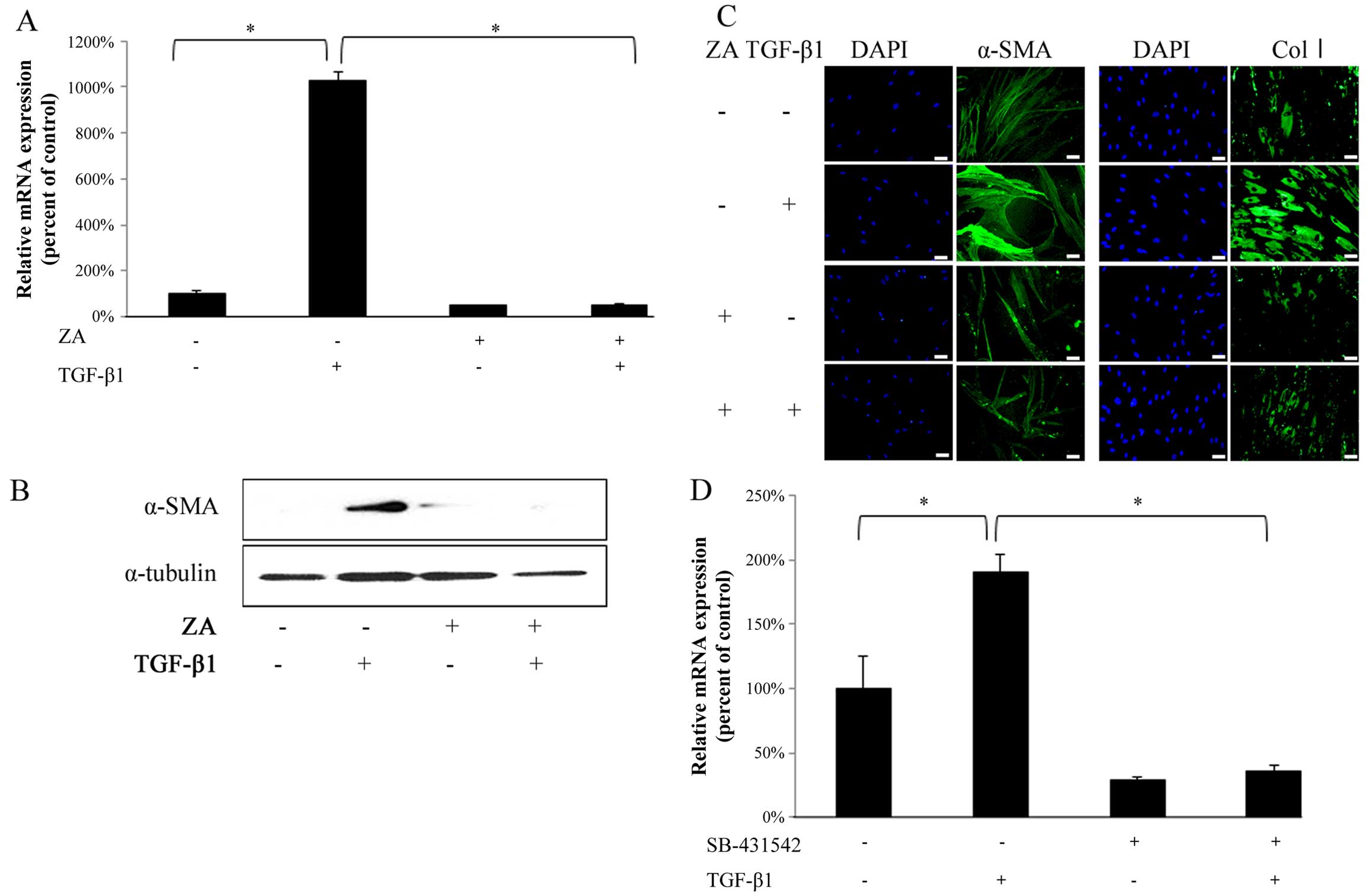
ZA (Cmax) suppresses the TGF-β-induced MF
differentiation of hGFs
TGF-β1 (5 ng/ml) significantly upregulated the α-SMA
mRNA expression level in the hGFs (Fig. 2A, bars 1 and 2 from left). ZA
(Cmax) completely suppressed the TGF-β1-induced increase in the
mRNA expression of α-SMA in the hGFs (Fig. 2A, bars 2 and 4 from left). In
addition, western blot analysis revealed that ZA (Cmax) clearly
suppressed the TGF-β1-induced upregulation of α-SMA expression at
the protein level in the hGFs (Fig.
2B). Immunofluorescence staining also revealed that ZA (Cmax)
clearly suppressed the TGF-β1-induced upregulation of α-SMA
expression (Fig. 2C, left panels)
and type I collagen expression (Fig.
2C, right panels) at the protein level in the hGFs. We
confirmed that the TGF-β1-induced increase in the mRNA expression
of α-SMA in the hGFs was significantly inhibited by SB-431542, an
inhibitor of TGF-β type I receptor (Fig. 2D), indicating that TGF-β1
specifically induced the MF differentiation of hGFs through
ligand-receptor interaction. Thus, these results indicate that ZA
(Cmax) suppresses the TGF-β-induced MF differentiation of hGFs.
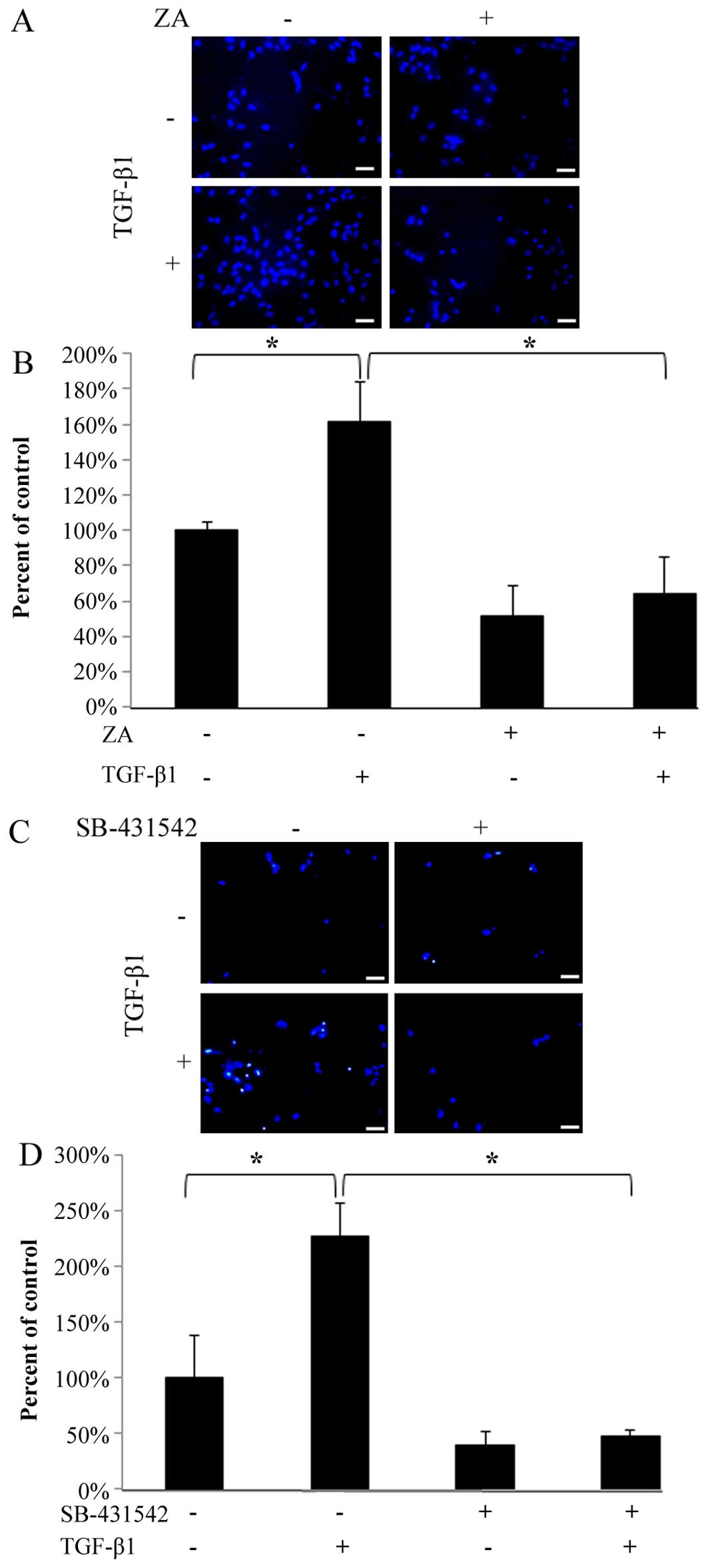
ZA (Cmax) inhibits the TGF-β-induced
migratory activity of hGFs
As shown in Fig. 3A
and C, the migration of the hGFs through the porous membrane
that bordered the upper and lower chambers was significantly
enhanced by stimulation with TGF-β1 (5 ng/ml). However, ZA (Cmax)
completely suppressed the TGF-β1-induced migratory activity
(Fig. 3A and B). In addition, we
confirmed that the TGF-β-induced migratory activity was markedly
inhibited by SB-431542 (Fig. 3C and
D), indicating that TGF-β1 specifically induced the migration
of hGFs through ligand-receptor interaction. These results indicate
that ZA (Cmax) inhibits the TGF-β-induced migratory activity of
hGFs.
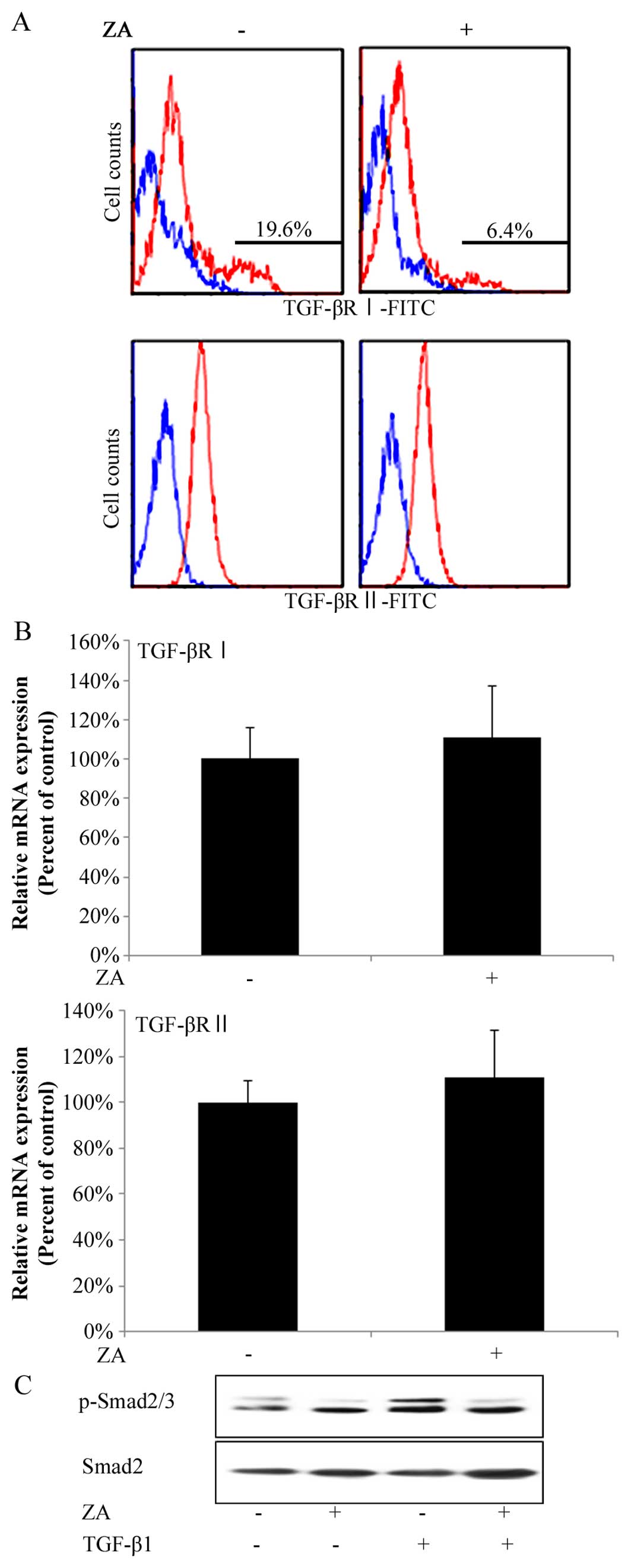
ZA (Cmax) suppresses the expression of
TGF-β type I receptor on the surfaces of hGFs and the TGF-β-induced
phosphorylation of Smad2/3 in hGFs
In order to gain insight into the molecular
mechanisms underlying the suppressive effects of ZA (Cmax) on
TGF-β-induced fibrogenesis by hGFs, we investigated whether ZA
affects the expression of TGF-β type I and II receptors on the
surface of hGFs. In flow cytometric analysis, hGFs had two peaks in
the histogram of TGF-β type I receptor-positive cells, that is, 2
groups of hGFs weakly or strongly expressed TGF-β type I receptors,
respectively (Fig. 4A, upper left
panel). Intriguingly, ZA (Cmax) lowered the ratio of the number of
cells strongly expressing TGF-β type I receptors on their surface
against that of total hGFs by 19.6 to 6.4% (Fig. 4A, upper panels). By contrast, hGFs
had only one peak in the histogram of TGF-β type II
receptor-positive cells (Fig. 4A,
lower left panel). ZA (Cmax) did not change the position of the
peak in the histogram of TGF-β type II receptor-positive cells
(Fig. 4A, lower panels).
Moreover, RT-qPCR analysis revealed that ZA (Cmax) did not decrease
the total amount of both TGF-β receptor type (types I and II)
expression at the mRNA level (Fig.
4B, upper and lower graphs, respectively), suggesting that ZA
(Cmax) suppressed the expression of TGF-β type I receptor on the
surface of hGFs. In addition, ZA (Cmax) suppressed the TGF-β1 (5
ng/ml)-induced phosphorylation of Smad2/3 (Fig. 4C). We confirmed these results by
reproducing them in independent experiments.
Discussion
Wang et al reported that TGF-β1 increased the
viability of human dermal fibroblasts (40). We found that ZA (Cmax) reduced the
TGF-β1-induced increase in the viability of hGFs (Fig. 1C). These results suggest that ZA
(Cmax) possibly suppresses the TGF-β1-induced fibrogenesis in oral
gingival tissue. Lu et al reported that TGF-β1 promoted the
viability of pulmonary artery endothelial cells through
Smad2-mediated signal transduction (41). We found that ZA (Cmax) suppressed
the TGF-β1 (5 ng/ml)-induced phosphorylation of Smad2/3 (Fig. 4C), implicating that ZA (Cmax) may
possibly suppress the TGF-β1-induced promotion of the viability of
hGFs through suppression of Smad2/3 activities.
Pan et al or Koch et al reported that
5 or 50 µM ZA induced the osteoblastic differentiation of
osteoblast precursor cells (42,43). In addition, Chen et al
reported that 10 µM ZA induced the dendritic cell
differentiation of monocytes (44). However, the mechanisms through
which ZA (Cmax) affected the differentiation ability of cells
derived from the oral cavity remained to be elucidated. In this
study, we demonstrated that 1.47 µM ZA (Cmax) significantly
suppressed the TGF-β1-induced MF differentiation of hGFs (Fig. 2A, B, and C). These results
strongly suggest that ZA (Cmax) inhibits wound healing in the oral
cavity by the attenuation of type I collagen synthesis in MFs
differentiated from hGFs following TGF-β1 stimulation. In addition,
it is generally known that MFs play an important role in generating
a contractile force, facilitating wound closure (reviewed in ref.
16), suggesting that ZA (Cmax)
also interferes with this process through the suppression of the
TGF-β1-induced MF differentiation of hGFs. Of note, we found that
ZA (Cmax) clearly suppressed the TGF-β1-induced phosphorylation of
Smad2/3 in hGFs (Fig. 4C).
Smad2/3 are known to be key signaling molecules inducing the MF
differentiation of fibroblasts (27). These results suggest that ZA
(Cmax) suppressed TGF-β1-induced MF differentiation of hGFs through
inhibition of Smad2/3 phosphorylation by TGF-β1.
Pabst et al reported that ZA at 50 µM,
a much higher concentration than Cmax, significantly decreased the
migratory activity of human oral keratinocytes at 72 h following
treatment with ZA (45). However,
it remained to be clarified whether ZA (Cmax) affected the
migratory activity of various cells derived from the oral cavity,
including hGFs. On the other hand, TGF-β is known to induce
migratory activity in various types of cells, including
fibroblastic cells, in a Smad-dependent manner (28–30). In this study, we demonstrated that
ZA (Cmax) significantly suppressed the TGF-β1-induced migratory
activity of hGFs (Fig. 3A and B).
Wound healing or mucous re-epithelialization is a complex process
involving the proliferation and migration of mesenchymal cells,
such as fibroblasts and MFs, which construct granulation tissue for
wound closure and then induce mucous re-epithelialization on it
(reviewed in ref. 46). As
described above, ZA (Cmax) suppressed TGF-β1-induced
phosphorylation of Smad2/3 (Fig.
4C). These results suggest that ZA (Cmax) suppresses wound
closure with granulation tissue in the oral cavity by the
inhibition of the TGF-β1-induced migration of mesenchymal cells,
such as hGFs by suppressing Smad2/3 phosphorylation by TGF-β1. On
the other hand, Ozdamar et al (47) reported that the phosphorylation of
polarity protein Par6 is required for TGF-β1-dependent
epithelial-to-mesenchymal transition (EMT) in mammary gland ECs and
controls the interaction of Par6 with the E3 ubiquitin ligase
Smurf1. Smurf1, in turn, targets the guanosine triphosphatase RhoA
for degradation, thereby leading to a loss of tight junctions
(47). In addition, RhoA is
generally known to be a regulator of cell migratory activity in
various types of cells by promoting stress fiber assembly and the
resultant cell adhesion (48).
Intriguingly, serine-to-alanine point mutation [Par6(S345A)] of the
TGF-β1-induced phosphorylation site in Par6 suppressed the
TGF-β1-induced EMT in a Smad-independent manner. It remains to be
clarified whether ZA (Cmax) suppresses the TGF-β1-induced migratory
activity of hGFs through affecting Par6/Smurf1/RohA signal
transduction pathway in a Smad-independent manner.
Saito et al demonstrated the negative effects
of ZA on the re-epithelialization of oral mucosa in a
three-dimensional in vitro oral mucosa wound healing model
(49). They demonstrated
histologically that ZA downregulated the expression of TGF-β type I
and II receptors and Smad3 phosphorylation in oral keratinocytes.
However, they used ZA at 10 µM, which was a much higher
concentration than Cmax, for evaluating the effects on
TGF-β1-induced signal transduction in human oral keratinocytes.
Moreover, they did not elucidate how TGF-β-induced intracellular
signals affect the function of human oral keratinocytes at the
cellular and molecular levels. In this study, we demonstrated that
ZA (Cmax) lowered the ratio of the number of cells strongly
expressing TGF-β type I receptors on their surface against that of
total hGFs, but did not affect the expression status of TGF-β type
II receptors on hGFs (Fig. 4A).
This finding suggests that ZA (Cmax) preferentially suppresses the
expression of TGF-β type I receptor over that of TGF-β type II
receptor on the surfaces of hGFs. By contrast, as described above,
a high concentration of ZA (10 µM) seemed to downregulate
the expression of both TGF-β receptor types (types I and II). In
addition, we found that ZA (Cmax) suppressed the TGF-β1-induced
phosphorylation of Smad2/3 in hGFs (Fig. 4C), implicating that the
suppression of the TGF-β1-induced phosphorylation of Smad2/3 in
hGFs by ZA (Cmax) may be caused by the inhibition of TGF-β type I
receptor expression on the surfaces of hGFs. However, it remains to
be clarified whether ZA (Cmax) directly suppresses the
TGF-β1-induced phosphorylation of Smad2/3, or indirectly suppresses
that through the inhibition of the TGF-β type I receptor expression
on the surface of hGFs. On the other hand, Okamoto et al
demonstrated that ZA (30-50 µM) induced apoptosis and
S-phase arrest in mesothelioma by inhibiting the functions of Rab
family proteins (50). The Rab
family is generally known to control the endosomal trafficking of
membrane molecules, such as growth factor receptors (reviewed in
ref. 51). It is also generally
known that Rab5, activated by a guanine nucleotide exchange factor
(GEF), directs TGF-β-activated receptors into the endocytic pathway
that promotes TGF-β-induced Smad2/3-dependent signals (52). In addition, Kardassis et al
suggested that TGF-β receptors may be recycled on the cell membrane
in a Rab-11-dependent manner following clathrin-dependent
internalization of TGF-β receptors, which may positively affect the
activities of Smad2/3-dependent TGF-β-induced intracellular signals
mediated by these receptors (53). Our laboratory is currently
investigating whether ZA (Cmax) suppresses the function of Rab
family members that may upregulate the activity of TGF-β1-induced
Smad2/3-dependent intracellular signals in hGFs at the receptor
level, thereby resulting in downregulating TGF-β1-induced
fibrogenic activity of the cells.
As described above, it has been reported that the
TGF-β-induced activation of p38 MAPK positively regulates migratory
activity (31). In fact, TGF-β1
upregulated the phosphorylation of p38 MAPK in hGFs; however, ZA
(Cmax) did not suppress the TGF-β1 (5 ng/ml)-induced
phosphorylation of p38 MAPK in hGFs (data not shown), suggesting
that p38 MAPK activity is not related to the suppressive effects of
ZA (Cmax) on TGF-β1-induced fibrogenic activity of hGFs. It is
plausible that the suppression of type I receptor expression on the
surface of hGFs by ZA (Cmax), as shown in Fig. 4A, may not be sufficient to
suppress the TGF-β1-induced p38 MAPK phosphorylation. On the other
hand, the activation of MAPKs such as JNK and p38 MAPK, but not
ERK, is necessary for the progression of the hypoxia-induced MF
differentiation of fibroblasts (54). Although, in western blot analysis,
the phosphorylation of JNK was not detectable in hGFs before/after
TGF-β1 (5 ng/ml) stimulation (data not shown), it remains to be
clarified whether JNK affects the TGF-β1-induced fibrogenic and
migratory activities of hGFs. In addition, the mechanisms through
which p38 MAPK affects the TGF-β1-induced fibrogenic and migratory
activities of hGFs also remains to be clarified.
In conclusion, in the present study, it was
suggested that ZA (Cmax) attenuates TGF-β1-induced wound closure by
inhibiting the formation of granulation tissue by hGFs stimulated
with TGF-β1 that was derived from inflammatory tissue, possibly
through the suppression of Smad2/3 signaling. Our findings partly
clarify the molecular mechanisms underlying BRONJ and would benefit
research into drug targets at the molecular level for the treatment
of this symptom.
Acknowledgments
This study was supported in part by Grants-in-Aid
for Scientific Research (KAKENHI) (grant nos. 24791981 awarded to
M.I., 25463053 awarded to N.C., 26462823 awarded to S.K., 22592076
awarded to M.K., 26293426 awarded to T.S., 24593002 awarded to
Y.S., and 26670852 awarded to A.I.) from the Ministry of Education,
Culture, Sports, Science and Technology of Japan; Grant-in-Aid from
the Dental Society of Iwate Medical University; and Grant-in-Aid
for Strategic Medical Science Research Centre from the Ministry of
Education, Culture, Sports, Science, and Technology of Japan,
2010–2014.
Abbreviations:
|
BPs
|
bisphosphonates
|
|
ZA
|
zoledronic acid
|
|
BRONJ
|
BPs-related osteonecrosis of the
jaw
|
|
ECs
|
epithelial cells
|
|
GFs
|
gingival fibroblasts
|
|
hGFs
|
human GFs
|
|
TGF-β
|
transforming growth factor-β
|
|
BMP
|
bone morphogenetic protein
|
|
GDF
|
growth and differentiation factor
|
|
MIS
|
Mullerian inhibiting substance
|
|
R-Smads
|
receptor-regulated Smads
|
|
Co-Smad
|
common mediator Smad
|
|
I-Smads
|
inhibitory Smads
|
|
MFs
|
myofibroblasts
|
|
α-SMA
|
α-smooth muscle actin
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
ERK
|
extra-cellular signal-regulated
kinase
|
|
Cmax
|
maximum concentration in serum
|
|
FBS
|
fetal bovine serum
|
|
PBS
|
phosphate-buffered saline
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
SDS-PAGE
|
SDS-polyacrylamide gel
electrophoresis
|
|
JNK
|
c-Jun N-terminal kinase
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
GEF
|
guanine nucleotide exchange factor
|
References
|
1
|
Gong L, Altman RB and Klein TE:
Bisphosphonates pathway. Pharmacogenet Genomics. 21:50–53. 2011.
View Article : Google Scholar :
|
|
2
|
Boonyapakorn T, Schirmer I, Reichart PA,
Sturm I and Massenkeil G: Bisphosphonate-induced osteonecrosis of
the jaws: prospective study of 80 patients with multiple myeloma
and other malignancies. Oral Oncol. 44:857–869. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fliefel R, Tröltzsch M, Kühnisch J,
Ehrenfeld M and Otto S: Treatment strategies and outcomes of
bisphosphonate-related osteonecrosis of the jaw (BRONJ) with
characterization of patients: a systematic review. Int J Oral
Maxillofac Surg. 44:568–585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manaka K, Makita N and Iiri T:
Erdheim-Chester disease and pituitary involvement: a unique case
and the literature. Endocr J. 61:185–194. 2014. View Article : Google Scholar
|
|
5
|
Rogers MJ, Crockett JC, Coxon FP and
Mönkkönen J: Biochemical and molecular mechanisms of action of
bisphosphonates. Bone. 49:34–41. 2011. View Article : Google Scholar
|
|
6
|
Vahtsevanos K, Kyrgidis A, Verrou E,
Katodritou E, Triaridis S, Andreadis CG, Boukovinas I, Koloutsos
GE, Teleioudis Z, Kitikidou K, et al: Longitudinal cohort study of
risk factors in cancer patients of bisphosphonate-related
osteonecrosis of the jaw. J Clin Oncol. 27:5356–5362. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marx RE, Sawatari Y, Fortin M and Broumand
V: Bisphosphonate-induced exposed bone
(osteonecrosis/osteopetrosis) of the jaws: risk factors,
recognition, prevention, and treatment. J Oral Maxillofac Surg.
63:1567–1575. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ruggiero SL, Dodson TB, Assael LA,
Landesberg R, Marx RE and Mehrotra B; American Association of Oral
and Maxillofacial Surgeons: American Association of Oral and
Maxillofacial Surgeons position paper on bisphosphonate-related
osteonecrosis of the jaws. J Oral Maxillofac Surg. 67(Suppl 5):
2–12. 2009.PubMed/NCBI
|
|
9
|
Hansen T, Kunkel M, Weber A and James
Kirkpatrick C: Osteonecrosis of the jaws in patients treated with
bisphosphonates-histomorpholoic analysis in comparison with
infected osteoradionecrosis. J Oral Pathol Med. 35:155–160. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoefert S, Wierich W, Eufinger H and
Krempien B: BP-associated vascular necrosis (AN) of the jaws:
histological findings. Bone. 38(suppl 1): 762006. View Article : Google Scholar
|
|
11
|
De Ceulaer J, Tacconelli E and
Vandecasteele SJ: Actinomyces osteomyelitis in
bisphosphonate-related osteonecrosis of the jaw (BRONJ): the
missing link? Eur J Clin Microbiol Infect Dis. 33:1873–1880. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kobayashi Y, Hiraga T, Ueda A, Wang L,
Matsumoto-Nakano M, Hata K, Yatani H and Yoneda T: Zoledronic acid
delays wound healing of the tooth extraction socket, inhibits oral
epithelial cell migration, and promotes proliferation and adhesion
to hydroxyapatite of oral bacteria, without causing osteonecrosis
of the jaw, in mice. J Bone Miner Metab. 28:165–175. 2010.
View Article : Google Scholar
|
|
13
|
Berti-Couto SA, Vasconcelos AC, Iglesias
JE, Figueiredo MA, Salum FG and Cherubini K: Diabetes mellitus and
corticotherapy as risk factors for alendronate-related
osteonecrosis of the jaws: a study in Wistar rats. Head Neck.
36:84–93. 2014. View Article : Google Scholar
|
|
14
|
Molcho S, Peer A, Berg T, Futerman B and
Khamaisi M: Diabetes microvascular disease and the risk for
bisphosphonate-related osteonecrosis of the jaw: A single center
study. J Clin Endocrinol Metab. 98:E1807–E1812. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nisi M, La Ferla F, Karapetsa D, Gennai S,
Miccoli M, Baggiani A, Graziani F and Gabriele M: Risk factors
influencing BRONJ staging in patients receiving intravenous
bisphosphonates: a multivariate analysis. Int J Oral Maxillofac
Surg. 44:586–591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Valluru M, Staton CA, Reed MWR and Brown
NJ: Transforming growth factor-β and endoglin signaling orchestrate
wound healing. Front Physiol. 2:892011. View Article : Google Scholar
|
|
17
|
Tipton DA and Dabbous MK: Autocrine
transforming growth factor β stimulation of extracellular matrix
production by fibroblasts from fibrotic human gingiva. J
Periodontol. 69:609–619. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cotrim P, Martelli-Junior H, Graner E,
Sauk JJ and Coletta RD: Cyclosporin A induces proliferation in
human gingival fibroblasts via induction of transforming growth
factor-beta1. J Periodontol. 74:1625–1633. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu MY and Hill CS: TGF-β superfamily
signaling in embryonic development and homeostasis. Dev Cell.
16:329–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goumans MJ, Liu Z and ten Dijke P: TGF-β
signaling in vascular biology and dysfunction. Cell Res.
19:116–127. 2009. View Article : Google Scholar
|
|
21
|
Heldin CH, Landström M and Moustakas A:
Mechanism of TGF-β signaling to growth arrest, apoptosis, and
epithelial-mesenchymal transition. Curr Opin Cell Biol. 21:166–176.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu T and Feng XH: Regulation of TGF-β
signalling by protein phosphatases. Biochem J. 430:191–198. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meulmeester E and Ten Dijke P: The dynamic
roles of TGF-β in cancer. J Pathol. 223:205–218. 2011. View Article : Google Scholar
|
|
24
|
Song B, Estrada KD and Lyons KM: Smad
signaling in skeletal development and regeneration. Cytokine Growth
Factor Rev. 20:379–388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sandbo N and Dulin N: Actin cytoskeleton
in myofibroblast differentiation: ultrastructure defining form and
driving function. Transl Res. 158:181–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kimura H, Okubo N, Chosa N, Kyakumoto S,
Kamo M, Miura H and Ishisaki A: EGF positively regulates the
proliferation and migration, and negatively regulates the
myofibroblast differentiation of periodontal ligament-derived
endothelial progenitor cells through MEK/ERK- and JNK-dependent
signals. Cell Physiol Biochem. 32:899–914. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sobral LM, Montan PF, Zecchin KG,
Martelli-Junior H, Vargas PA, Graner E and Coletta RD: Smad7 blocks
transforming growth factor-β1-induced gingival
fibroblast-myofibroblast transition via inhibitory regulation of
Smad2 and connective tissue growth factor. J Periodontol.
82:642–651. 2011. View Article : Google Scholar
|
|
28
|
Motizuki M, Isogaya K, Miyake K, Ikushima
H, Kubota T, Miyazono K, Saitoh M and Miyazawa K: Oligodendrocyte
transcription factor 1 (Olig1) is a Smad cofactor involved in cell
motility induced by transforming growth factor-β. J Biol Chem.
288:18911–18922. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakano N, Maeyama K, Sakata N, Itoh F,
Akatsu R, Nakata M, Katsu Y, Ikeno S, Togawa Y, Vo Nguyen TT, et
al: C18 ORF1, a novel negative regulator of transforming growth
factor-β signaling. J Biol Chem. 289:12680–12692. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiao Y-Q, Liu K, Shen J-F, Xu G-T and Ye
W: SB-431542 inhibition of scar formation after filtration surgery
and its potential mechanism. Invest Ophthalmol Vis Sci.
50:1698–1706. 2009. View Article : Google Scholar
|
|
31
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFbeta-mediated fibroblastic transdifferentiation and cell
migration. J Cell Sci. 115:3193–3206. 2002.PubMed/NCBI
|
|
32
|
Sarrazy V, Billet F, Micallef L, Coulomb B
and Desmoulière A: Mechanisms of pathological scarring: role of
myofibroblasts and current developments. Wound Repair Regen.
19(Suppl 1): s10–s15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Van De Water L, Varney S and Tomasek JJ:
Mechanoregulation of the myofibroblast in wound contraction,
scarring, and fibrosis: Opportunities for new therapeutic
intervention. Adv Wound Care (New Rochelle). 2:122–141. 2013.
View Article : Google Scholar
|
|
34
|
Hinz B: Formation and function of the
myofibroblast during tissue repair. J Invest Dermatol. 127:526–537.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshinami T, Yagi T, Sakai D, Sugimoto N
and Imamura F: A case of acquired Fanconi syndrome induced by
zoledronic acid. Intern Med. 50:1075–1079. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kurishima K, Ohara G, Kagohashi K,
Takayashiki N, Tamura T, Shiozawa T, Miyazaki K, Kawaguchi M, Satoh
H and Hizawa N: Ossification and increased bone mineral density
with zoledronic acid in a patient with lung adenocarcinoma: a case
report. Exp Ther Med. 8:1267–1270. 2014.PubMed/NCBI
|
|
37
|
DeMaio L, Buckley ST, Krishnaveni MS,
Flodby P, Dubourd M, Banfalvi A, Xing Y, Ehrhardt C, Minoo P, Zhou
B, et al: Ligand-independent transforming growth factor-β type I
receptor signalling mediates type I collagen-induced
epithelial-mesenchymal transition. J Pathol. 226:633–644. 2012.
View Article : Google Scholar
|
|
38
|
Xu X, Wan X, Geng J, Li F, Wang C and Dai
H: Kinase inhibitors fail to induce mesenchymal-epithelial
transition in fibroblasts from fibrotic lung tissue. Int J Mol Med.
32:430–438. 2013.PubMed/NCBI
|
|
39
|
Sawada S, Chosa N, Ishisaki A and Naruishi
K: Enhancement of gingival inflammation induced by synergism of
IL-1β and IL-6. Biomed Res. 34:31–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang X, Chu J, Wen CJ, Fu SB, Qian YL, Wo
Y, Wang C and Wang DR: Functional characterization of TRAP1-like
protein involved in modulating fibrotic processes mediated by
TGF-β/Smad signaling in hypertrophic scar fibroblasts. Exp Cell
Res. 332:202–211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lu Q: Transforming growth factor-beta1
protects against pulmonary artery endothelial cell apoptosis via
ALK5. Am J Physiol Lung Cell Mol Physiol. 295:L123–L133. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pan B, To LB, Farrugia AN, Findlay DM,
Green J, Gronthos S, Evdokiou A, Lynch K, Atkins GJ and Zannettino
AC: The nitrogen-containing bisphosphonate, zoledronic acid,
increases mineralisation of human bone-derived cells in vitro.
Bone. 34:112–123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Koch FP, Merkel C, Al-Nawas B, Smeets R,
Ziebart T, Walter C and Wagner W: Zoledronate, ibandronate and
clodronate enhance osteoblast differentiation in a dose dependent
manner - a quantitative in vitro gene expression analysis of Dlx5,
Runx2, OCN, MSX1 and MSX2. J Craniomaxillofac Surg. 39:562–569.
2011. View Article : Google Scholar
|
|
44
|
Chen YJ, Chao KS, Yang YC, Hsu ML, Lin CP
and Chen YY: Zoledronic acid, an aminobisphosphonate, modulates
differentiation and maturation of human dendritic cells.
Immunopharmacol Immunotoxicol. 31:499–508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pabst AM, Ziebart T, Koch FP, Taylor KY,
Al-Nawas B and Walter C: The influence of bisphosphonates on
viability, migration, and apoptosis of human oral keratinocytes -
in vitro study. Clin Oral Investig. 16:87–93. 2012. View Article : Google Scholar
|
|
46
|
Tarnawski AS: Cellular and molecular
mechanisms of gastrointestinal ulcer healing. Dig Dis Sci. 50(Suppl
1): S24–S33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ozdamar B, Bose R, Barrios-Rodiles M, Wang
HR, Zhang Y and Wrana JL: Regulation of the polarity protein Par6
by TGFbeta receptors controls epithelial cell plasticity. Science.
307:1603–1609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tojkander S, Gateva G and Lappalainen P:
Actin stress fibers - assembly, dynamics and biological roles. J
Cell Sci. 125:1855–1864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Saito T, Izumi K, Shiomi A, Uenoyama A,
Ohnuki H, Kato H, Terada M, Nozawa-Inoue K, Kawano Y, Takagi R and
Maeda T: Zoledronic acid impairs re-epithelialization through
down-regulation of integrin αvβ6 and transforming growth factor
beta signalling in a three-dimensional in vitro wound healing
model. Int J Oral Maxillofac Surg. 43:373–380. 2014. View Article : Google Scholar
|
|
50
|
Okamoto S, Jiang Y, Kawamura K, Shingyoji
M, Tada Y, Sekine I, Takiguchi Y, Tatsumi K, Kobayashi H, Shimada
H, et al: Zoledronic acid induces apoptosis and S-phase arrest in
mesothelioma through inhibiting Rab family proteins and
topoisomerase II actions. Cell Death Dis. 5:e15172014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aloisi AL and Bucci C: Rab GTPases-cargo
direct interactions: fine modulators of intracellular trafficking.
Histol Histopathol. 28:839–849. 2013.PubMed/NCBI
|
|
52
|
Hu H, Milstein M, Bliss JM, Thai M,
Malhotra G, Huynh LC and Colicelli J: Integration of transforming
growth factor beta and RAS signaling silences a RAB5 guanine
nucleotide exchange factor and enhances growth factor-directed cell
migration. Mol Cell Biol. 28:1573–1583. 2008. View Article : Google Scholar
|
|
53
|
Kardassis D, Murphy C, Fotsis T, Moustakas
A and Stournaras C: Control of transforming growth factor β signal
transduction by small GTPases. FEBS J. 276:2947–2965. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Short M, Nemenoff RA, Zawada WM, Stenmark
KR and Das M: Hypoxia induces differentiation of pulmonary artery
adventitial fibroblasts into myofibroblasts. Am J Physiol Cell
Physiol. 286:C416–C425. 2004. View Article : Google Scholar
|