Introduction
Myocardial infarction (MI) is a major cause of
morbidity and mortality worldwide in patients with coronary heart
disease (1). Early reperfusion of
the ischemic region by thrombolysis or primary percutaneous
coronary intervention, although effective for salvaging the damaged
myocardium, can lead to further injury, such as cardiomyocyte death
and cardiac dysfunction, which is known as myocardial
ischemia/reperfusion (I/R) injury (2). Hence, to improve clinical outcomes
in acute MI, it is of pivotal importance to explore the safe and
effective therapeutic intervention for mitigating I/R injury.
It is well established that apoptosis, a main type
of programmed cell death, plays an important role in the
pathogenesis of myocardial I/R injury (3). Caspases are key molecules involved
in signaling cascades of programmed cell death, in which caspase-3
has been recognized as an important initiator and promoter of
apoptosis (3). Among the key
survival signaling transduction pathways, the phosphoinositide
3-kinase/serine/threonine protein kinase (PI3K/Akt) pathway plays
an important role in myocardial I/R injury, and is also essential
for the regulation of proliferation, differentiation and apoptosis
(4). PI3K-activated Akt can
regulate pro-apoptotic proteins and transcription factors, and can
activate endothelial nitric oxide synthase (eNOS) to produce nitric
oxide (NO), thereby promoting cell survival and protecting the
heart against I/R injury (5).
Autophagy, responsible for the degradation and
recycling of cellular components, is also activated in response to
myocardial I/R injury. Research has shown that autophagy is a
'double-edged sword' in I/R: a slight increase in autophagy during
ischemia inhibits apoptosis and promotes cell survival; however, an
abnormal increase in autophagy during the reperfusion period
aggravates myocardial injury (6).
Autophagy is a dynamic process involving the segregation of cargo
within double membrane-bound autophagosomes that fuse with and are
degraded in lysosomes (7). The
above process is termed the autophagic flux. Recent evidence has
indicated that the impairment of the autophagic flux leads to the
progressive consumption of cellular constituents and subsequently,
to cell death in myocardial I/R injury (8). The kinase mammalian target of
rapamycin (mTOR) is a key regulator of autophagy, the activity of
which is enhanced by factors that activate the PI3K/Akt pathway
(9).
Green tea [Camellia sinensis L. Ktze.
(Theaceae)] is an extremely popular beverage that originated in
China several thousands of years ago. An expanding body of evidence
suggests that the consumption of green tea has several health
benefits; namely, it is known to prevent or ameliorate various
diseases, such as obesity, autoimmune disorders and certain types
of cancer, as well as neurodegenerative and cardiovascular diseases
(10,11). These effects of green tea are
mediated by its polyphenols known as catechins. Epigallocatechin
gallate (EGCG) is the most abundant catechin found in green tea
that has been shown to have antioxidant, anti-inflammatory,
anti-mutagenic, anti-proliferative and anti-apoptotic effects in a
variety of experimental models (12). It has been reported that EGCG has
a beneficial effect on coronary artery disease (13). Previous studies have demonstrated
that EGCG protects cardiomyocytes from I/R-induced apoptosis
through the inhibition of signal transducer and activator of
transcription (STAT)1 phosphorylation, the decrease of nuclear
factor-κB (NF-κB) and activator protein 1 expression, as well as
the activation of the reperfusion injury salvage kinase (RISK)
pathway (14–17). However, the mechanism responsible
for the healing effects of EGCG during myocardial I/R injury have
not been completely defined. In particular, the association between
the autophagic flux and the therapeutic effects of EGCG in
myocardial diseases have not been previously investigated, at least
to the best of our knowledge.
Therefore, the aim of the present study was to
investigate whether EGCG mitigates myocardial I/R injury and to
further identify the potential mechanisms involved. It was
hypothesized that the influence of the downstream targets of the
PI3K/Akt pathway activated by EGCG may suppress apoptosis and
restore the autophagic flux, which may contribute to the
cardioprotective effects of EGCG against I/R injury.
Materials and methods
Animals and experimental conditions
The experimental procedures and protocols used in
this study were approved by the Ethics Committee for the
Experimental Use of Animals at Guangxi Medical University (Guangxi,
China) on June 18, 2015 (no. 20150618-08) and carried out in
accordance with their guidelines. A total of 24 male Sprague-Dawley
rats with a body weight of 250–280 g were obtained from the Guangxi
Medical University Laboratory Animal Center. The rats were housed
under standard conditions (20–25°C, 50–60% humidity, with a 12 h
light-dark cycle) and were given standard rodent chow and free
access to water.
Drugs and reagents
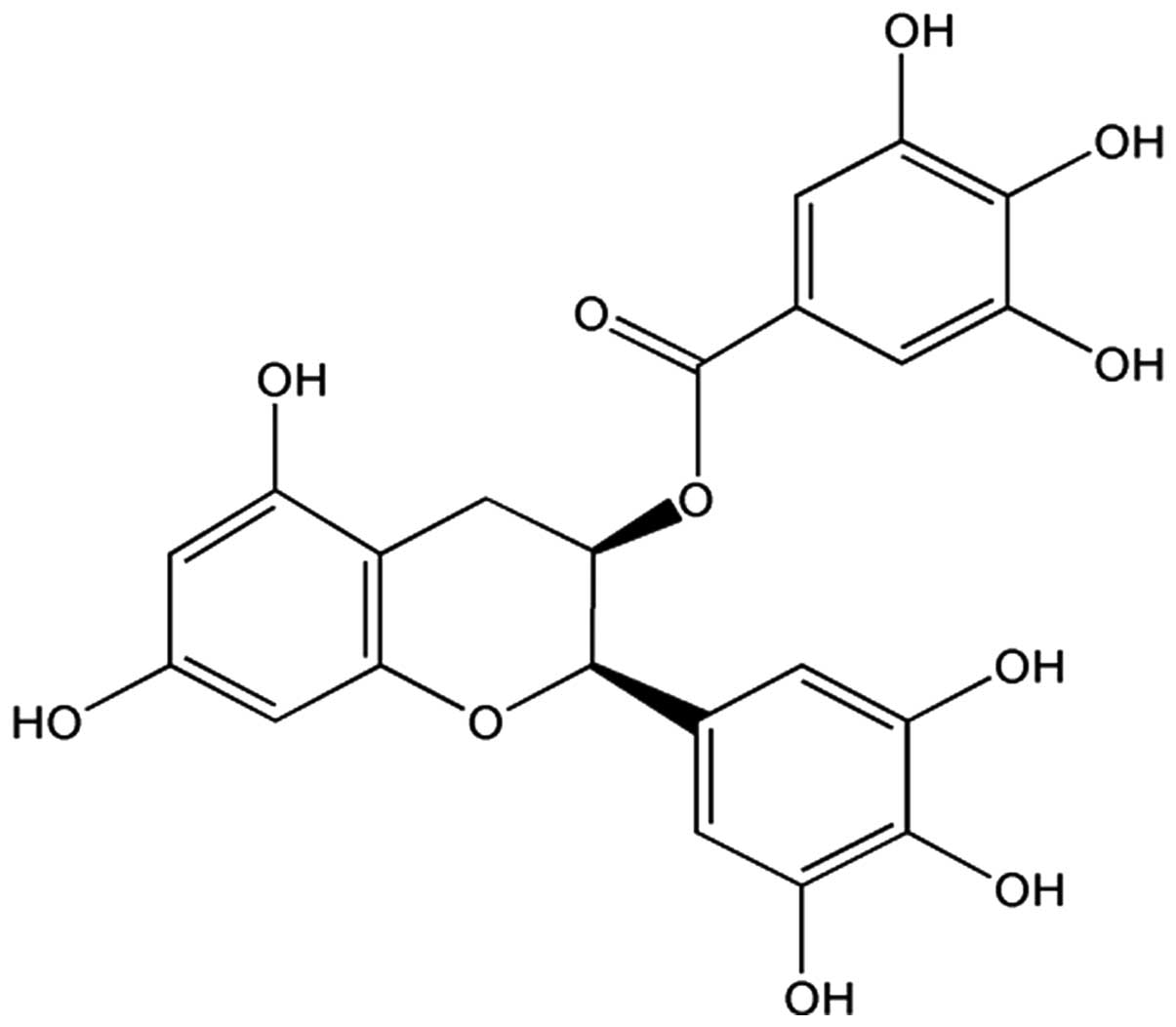
EGCG (Fig. 1;
purity, ≥95%) was obtained from Sigma-Aldrich Inc. (St. Louis, MO,
USA) and diluted with saline to an appropriate concentration as
needed. The creatine kinase-MB (CK-MB), lactate dehydrogenase (LDH)
and NO detection kits were obtained from the Jiancheng
Bioengineering Institute (Nanjing, China). A terminal
deoxynucleotidyl transferase-mediated dUTP nick-end labeling
(TUNEL) apoptosis detection kit was purchased from Roche
Diagnostics (Mannheim, Germany). Antibodies recognizing PI3K
(p110α; #13364), Akt (#9272), phosphorylated (p)-Akt (Ser473;
#9271), eNOS (#9572), p-eNOS (Ser1177; #9571), mTOR (#2983), p-mTOR
(Ser2448; #5536), cleaved caspase-3 (#9661), caspase-3 (#9662),
light chain 3 (LC3; #3868), Beclin1 (#3495), Atg5 (#12994) and p62
(#5114) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Casthepsin D antibody (sc-10725) was purchased
from Santa Cruz Biotechno logy, Inc. (Santa Cruz, CA, USA).
Experimental design
The rats were randomly assigned into 3 groups
(n=8/group): i) the sham-operated group, in which the rats were
subjected to surgical manipulation without ligation of the left
anterior descending (LAD) coronary artery; ii) the I/R group, in
which the rats were subjected to ligation of the LAD artery for 30
min followed by reperfusion for 2 h to induce I/R injury; iii) the
I/R + EGCG group, in which the rats were subjected to I/R injury
and administered EGCG (10 mg/kg in saline) via a sublingual
intravenous injection 10 min prior to the onset of reperfusion. The
rats in the sham-operated and I/R model groups were administered an
identical dose of normal saline to serve as a control.
General surgical procedure
The rats were anesthetized with 20% ethyl carbamate
(5 ml/kg) and placed in the supine position. The chest was opened
through the fourth intercostal space, and the ribs were gently
retracted to expose the heart. After cutting the pericardium, the
LAD coronary artery was positioned between the left atrial
appendage and the pulmonary conus. A 6-0 silk suture was passed
around the LAD artery, and the ends were pulled through a small
vinyl tube to form a snare and then tightened. Oxygen was supplied
through the trachea using an animal ventilator (respiration rate
70/min; respiration-to-expiration ratio, 1:2; tidal volume, 50
ml/kg). Myocardial ischemia for 30 min was confirmed by the visual
inspection of regional cyanosis of the myocardium and ST-segment
elevation on an electrocardiogram. Reperfusion for 2 h was
initiated by releasing the ligation of the LAD artery and confirmed
by a color change in the ventricular surface from cyanosis to
hyperemia, as previously described (18).
Assessment of cardiac function
I/R-induced cardiac dysfunction was determined by
invasive hemodynamic evaluation methods. A micro-catheter connected
to the MS 4000 organism signal quantitative analytical system
(Longfeida Technology Co., Ltd., Shandong, China) was inserted into
the left ventricle through the right common carotid artery to
record left ventricular systolic pressure (LVSP), left ventricular
end-diastolic pressure (LVEDP) and maximum rise/down velocity of
left intraventricular pressure (±dp/dtmax) at
baseline, at 30 min of ischemia, and after 30, 60, 90 and 120 min
of reperfusion.
Determination of infarct size
The myocardial infarct size was measured using
2,3,5-triphenyltetrazolium chloride (TTC) staining as previously
described (19). Briefly, the
hearts were harvested and rinsed with normal saline. The excised
left ventricle was frozen at −80°C for 5 min, and then sectioned
from apex to base into approximately 2-mm-thick slices. The slices
were incubated in a solution of 1% TTC in phosphate buffered saline
(pH 7.4) at 37°C for 20 min in the dark, and then fixed in 10%
formaldehyde. The slices were photographed the following day by a
digital camera (Nikon, Tokyo, Japan). The infarcted (TTC
non-stained) area was isolated from the rest of the cardiac tissue,
which was stained red by TTC. The infarcted and normal tissues were
separately weighed and the infarct size was expressed as a
percentage of the mass of the left ventricle.
Measurement of cardiac enzyme levels and
NO content
Blood samples were collected from the carotid artery
following reperfusion, and the serum was immediately separated from
the blood samples by centrifugation at 3,000 rpm for 10 min. The
levels of CK-MB and LDH content were evaluated by a colorimetric
method using commercial kits (Jiancheng Bioengineering Institute)
according to the manufacturer's instructions, and the absorbance
was measured spectrophotometrically at a wavelength of 340 nm and
490 nm, respectively. Due to its instability in physiological
solutions, most of the NO is rapidly converted into nitrite
(NO2−) and further into nitrate (NO3−). The
serum levels of NO2−/NO3− were measured using
a NO detection kit according to the manufacturer's instructions.
Briefly, nitrate was converted into nitrite using
Aspergillus nitrite reductase, and the total nitrite level
was measured using Griess reagent. The absorbance was determined at
540 nm using a spectrophotometer.
TUNEL assay
Cardiomyocyte apoptosis was determined using an In
Situ Cell Death Detection kit, POD (Roche Diagnostics). TUNEL
staining was used to quantify apoptotic cell nuclei as previously
described (20). Briefly, tissue
sections were washed in phosphate-buffered saline (PBS) and then
fixed in a 4% paraformaldehyde solution prior to incubation in 20
µg/ml proteinase K for 15 min. After washing with PBS, the
tissue sections were immersed in the TUNEL reaction mixture for 1 h
at 37°C in a humid chamber. The reaction was terminated by
transferring the slides to a 2X sodium citrate saline solution.
Endogenous peroxidase activity was quenched by incubation in 0.3%
hydrogen peroxide. Finally, streptavidin horseradish peroxidase was
bound to the biotinylated nucleotides and the peroxidase activity
was visualized in each section by the application of the stable
chromogen, diaminobenzidine. In this method, the apoptotic nuclei
were stained dark brown. Normal nuclei were stained blue with
hematoxylin. The results were scored semi-quantitatively by
averaging the number of apoptotic cells/field at ×400
magnification. Five fields were evaluated per tissue sample, and
cardiomyocyte apoptosis was represented as the apoptotic index (AI)
calculated as follows: AI = the number of TUNEL-positive cells/the
total number of cells counted ×100.
Western blot analysis
At the end of reperfusion, a section of
approximately 70 mg of myocardial tissue was taken from the infarct
area of the left ventricle. Following homogenization and protein
quantification, equal amounts of protein from each sample were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene
difluoride (PVDF)-plus membranes. After blocking with 5% bovine
serum albumin (BSA), the membranes were incubated overnight at 4°C
with the following primary antibodies: PI3K (1:1,000), Akt
(1:2,000), p-Akt (Ser473, 1:1,000), p-eNOS (Ser1177, 1:1,000),
p-mTOR (Ser2448, 1:1,000), Atg5 (1:1,000), Beclin1 (1:1,000), LC3
(1:1,000), p62 (1:1,000) and cathepsin D (1:500). The membranes
were then washed 3 times in Tris-buffered saline with 0.1% Tween-20
(TBST) and incubated with the corresponding secondary antibody
(anti-rabbit IgG, HRP-linked; #7074; 1:5,000; Cell Signaling
Technology, Inc.) conjugated to horseradish peroxidase at room
temperature for 2 h. Finally, the membranes were washed 3 times in
TBST. Relative densitometry was performed using a computerized
software package (NIH Image 1.63 software).
Statistical analysis
Each sample was assayed in triplicate. The results
were averaged and expressed as the means ± SD and data were
evaluated using the Sigma Stat (version 21.0) statistical analysis
program (SPSS Inc., Chicago, IL, USA). A one-way analysis of
variance followed by Bonferroni's multiple comparison test was used
for statistical analysis. P-values <0.05 were considered to
indicate statistically significant differences.
Results
EGCG promotes recovery following
I/R-induced cardiac dysfunction
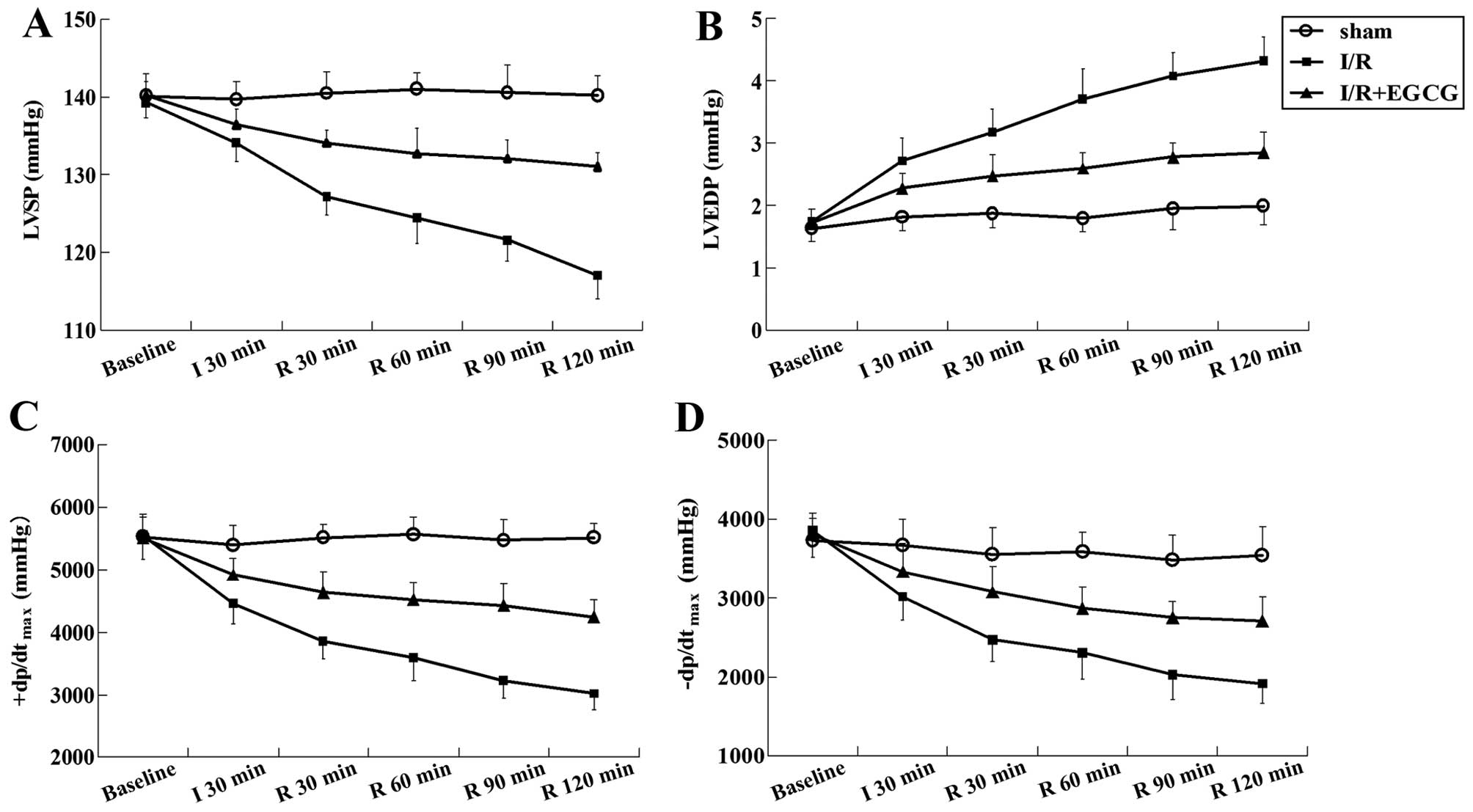
No significant differences in cardiac performance
were observed among the different groups prior to ischemia. During
the occlusion and reperfusion process, myocardial I/R led to left
ventricular dysfunction characterized by a significant increase in
LVEDP, and a decrease in LVSP, +dp/dtmax and
−dp/dtmax (P<0.05 or P<0.01). As expected,
treatment with EGCG preserved cardiac function by decreasing LVEDP,
and increasing LVSP, +dp/dtmax and
−dp/dtmax (P<0.05 or P<0.01) (Fig. 2 and Table I).
 | Table IEffects of EGCG on hemodynamics in a
rat model of I/R injury. |
Table I
Effects of EGCG on hemodynamics in a
rat model of I/R injury.
| Group | Time
(mmHg) | LVSP | LVEDP
(mmHg) |
+dp/dtmax
(mmHg/sec) |
−dp/dtmax
(mmHg/sec) |
|---|
| Sham-operated | Baseline | 140.10±2.87 | 1.64±0.21 | 5529.20±320.59 | 3731.60±283.00 |
| I 30 min | 131.68±2.26 | 1.82±0.22 | 5395.80±309.77 | 3667.80±333.64 |
| R 30 min | 140.48±2.74 | 1.88±0.23 | 5514.20±211.44 | 3557.20±340.45 |
| R 60 min | 139.98±2.11 | 1.80±0.22 | 5470.60±282.39 | 3586.00±252.16 |
| R 90 min | 140.54±3.62 | 1.95±0.35 | 5469.60±329.24 | 3840.20±320.00 |
| R 120 min | 140.14±2.54 | 2.00±0.30 | 5512.60±217.01 | 3543.00±363.41 |
| I/R | Baseline | 139.24±1.96 | 1.74±0.21 | 5547.2±377.60 | 3861.60±342.25 |
| I 30 min | 134.02±2.40a | 2.72±0.36b |
4465.00±329.12b |
3014.00±296.94a |
| R 30 min | 127.18±2.40b | 3.18±0.38b |
3861.80±286.29b |
2475.00±280.36b |
| R 60 min | 124.46±3.25b | 3.72±0.48b |
3593.20±376.92b |
2310.40±334.27b |
| R 90 min | 121.58±2.67b | 3.90±0.55b |
3230.60±284.41b |
2027.80±316.04b |
| R 120 min | 117.00±2.97b | 4.32±0.40b |
3019.60±253.87b |
1915.00±240.77b |
| I/R + EGCG | Baseline | 140.20±1.75 | 1.72±0.23 | 5511.80±381.23 | 3814.20±263.84 |
| I 30 min | 136.40±2.04 | 2.28±0.24 |
4921.80±263.34c |
3333.40±365.40c |
| R 30 min | 134.10±1.62d | 2.48±0.34c |
4643.20±319.91d |
3082.40±313.37c |
| R 60 min | 132.68±3.23d | 2.60±0.25d |
4523.20±283.33d |
2871.80±274.12c |
| R 90 min | 132.00±2.45d | 2.78±0.22d |
4435.20±345.67d |
2758.00±198.41d |
| R 120 min | 131.02±1.83d | 2.86±0.32d |
4249.00±266.26d |
2708.00±303.62d |
EGCG decreases the levels of myocardial
enzyme and increases the NO content
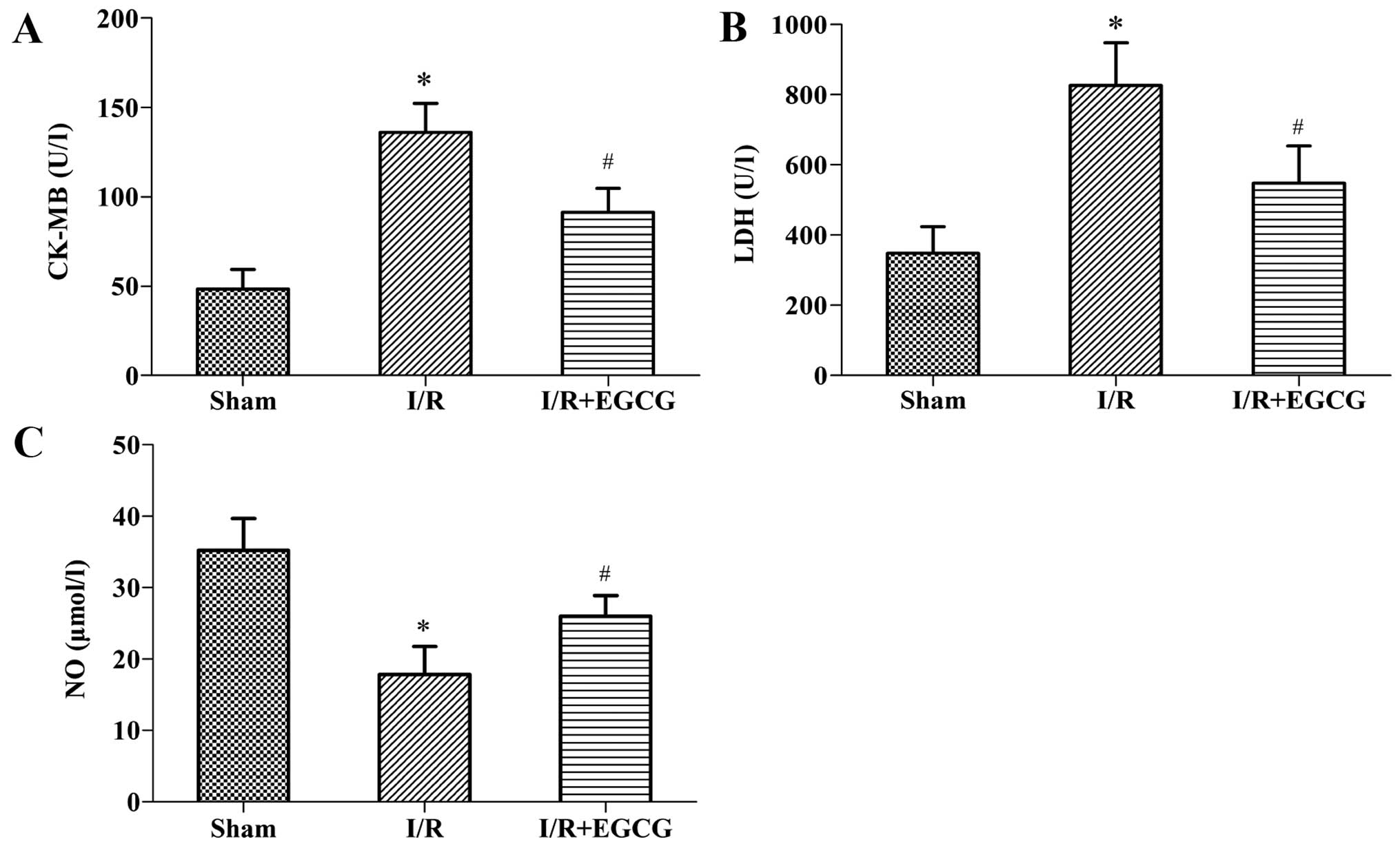
To further evaluate and validate the protective
function of EGCG during I/R injury, we measured the levels of
myocardial enzymes (CK-MB and LDH) in serum (Fig. 3A and B). We found that while the
I/R rats exhibited a significant increase in CK-MB and LDH levels
compared with the sham-operated group (P<0.05), the increase in
these levels was significantly abrogated in the I/R + EGCG group
(P<0.05). In addition, compared with the I/R group, the NO
content in the I/R + EGCG group was markedly elevated (P<0.05;
Fig. 3C).
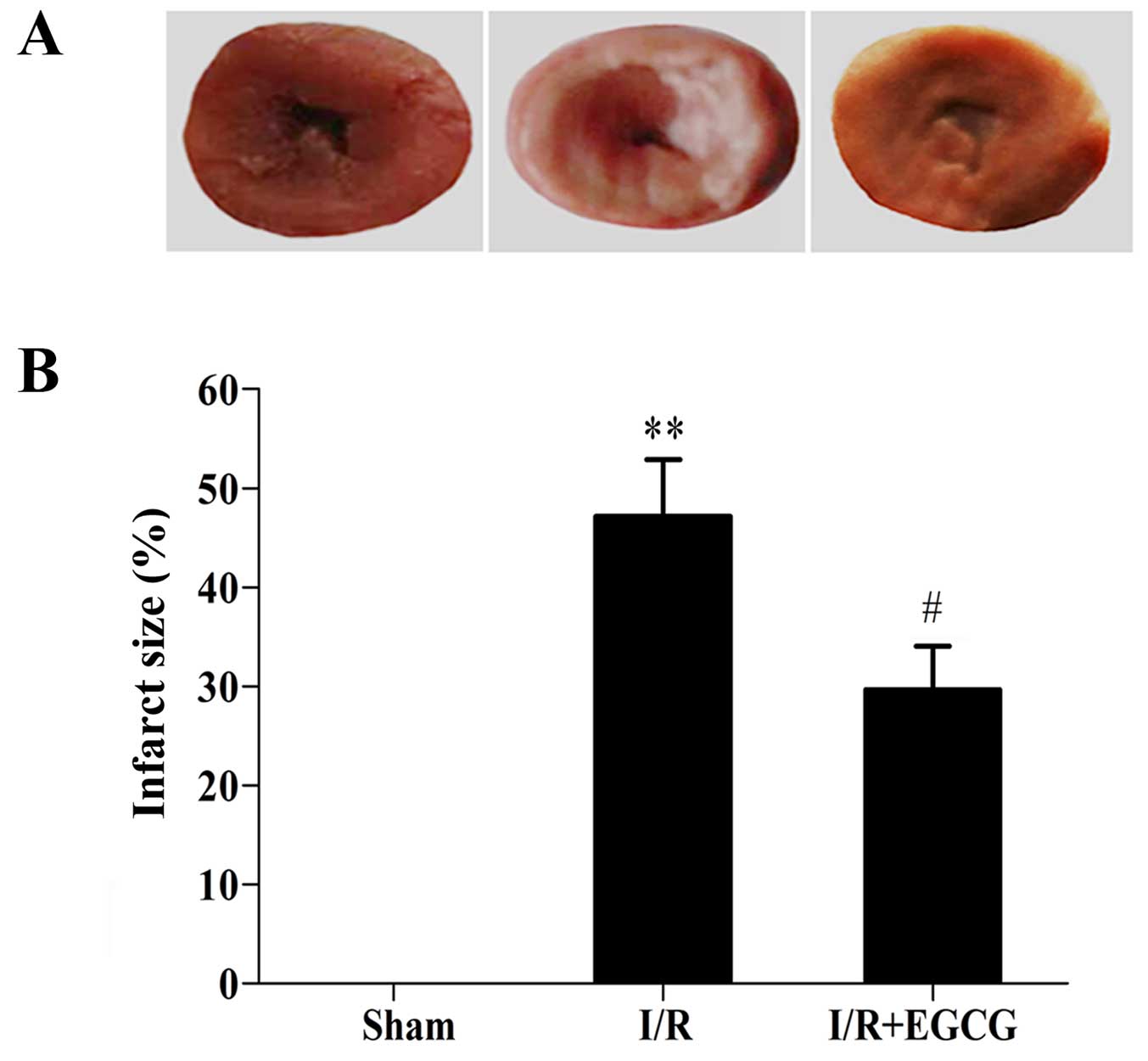
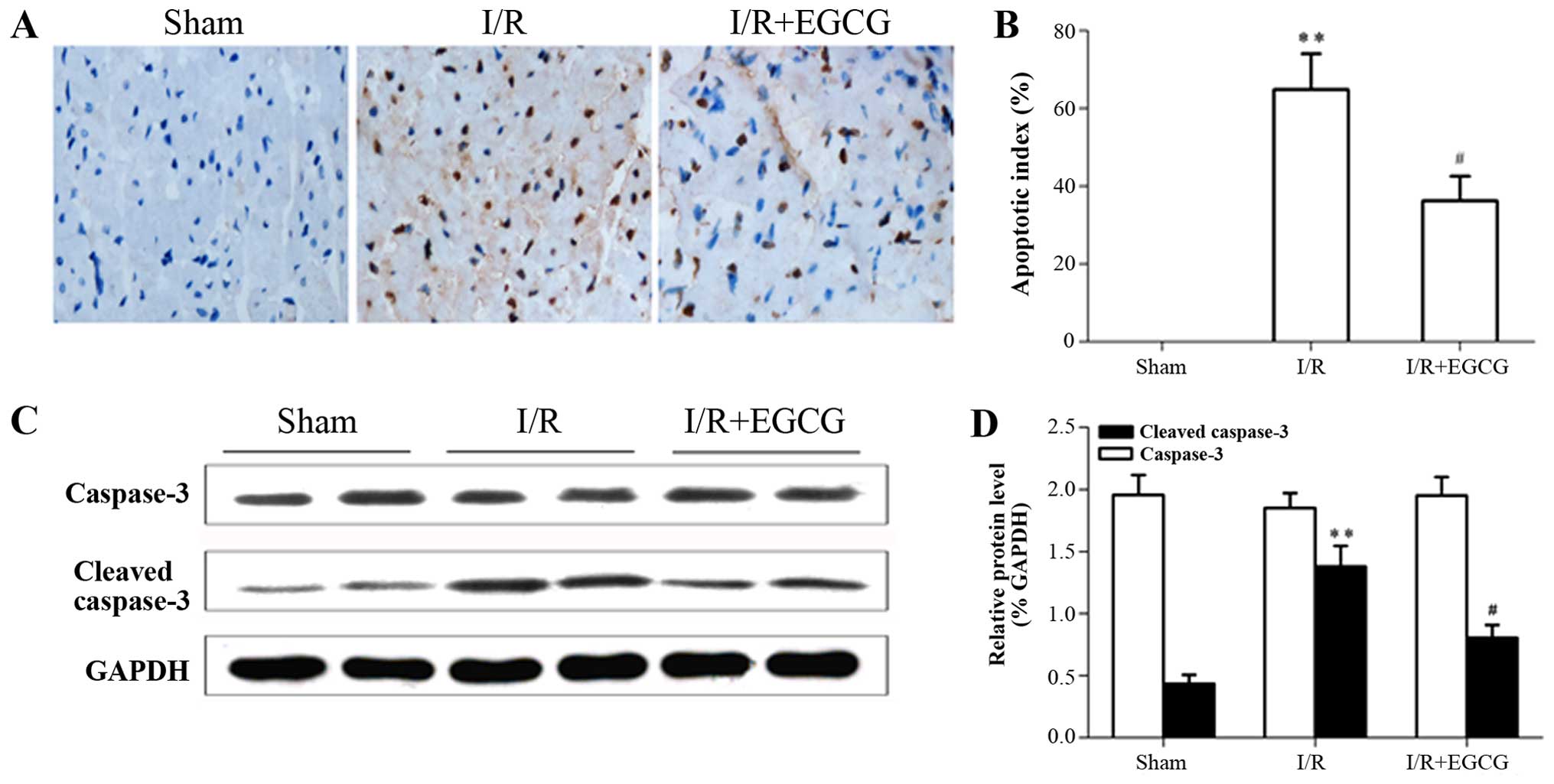
EGCG reduces the infarct size and
prevents cardiomyocyte apoptosis following I/R injury
The myocardial infarct size was enlarged in the I/R
group in comparison with the sham-operated group (P<0.01;
Fig. 4). However, EGCG
post-conditioning evidently decreased the infarct size when
compared with the I/R group (P<0.05). Furthermore, TUNEL
staining revealed that apoptosis was absent in the sham-operated
group. Conversely, the rats in the I/R group exhibited severe
tissue damage that appeared to markedly increase the number of
TUNEL-positive cells (P<0.01). Of note, post-conditioning with
EGCG resulted in a marked reduction in the number of TUNEL-positive
cells compared with the I/R model group (P<0.05; Fig. 5A and B). In order to explore the
underlying mechanisms responsible for the anti-apoptotic effects of
EGCG, the expression of caspase-3 was examined by western blot
analysis. The protein expression of cleaved caspase-3 was
significantly decreased following treatment with EGCG compared to
the myocardial I/R model group (P<0.05; Fig. 5C and D).
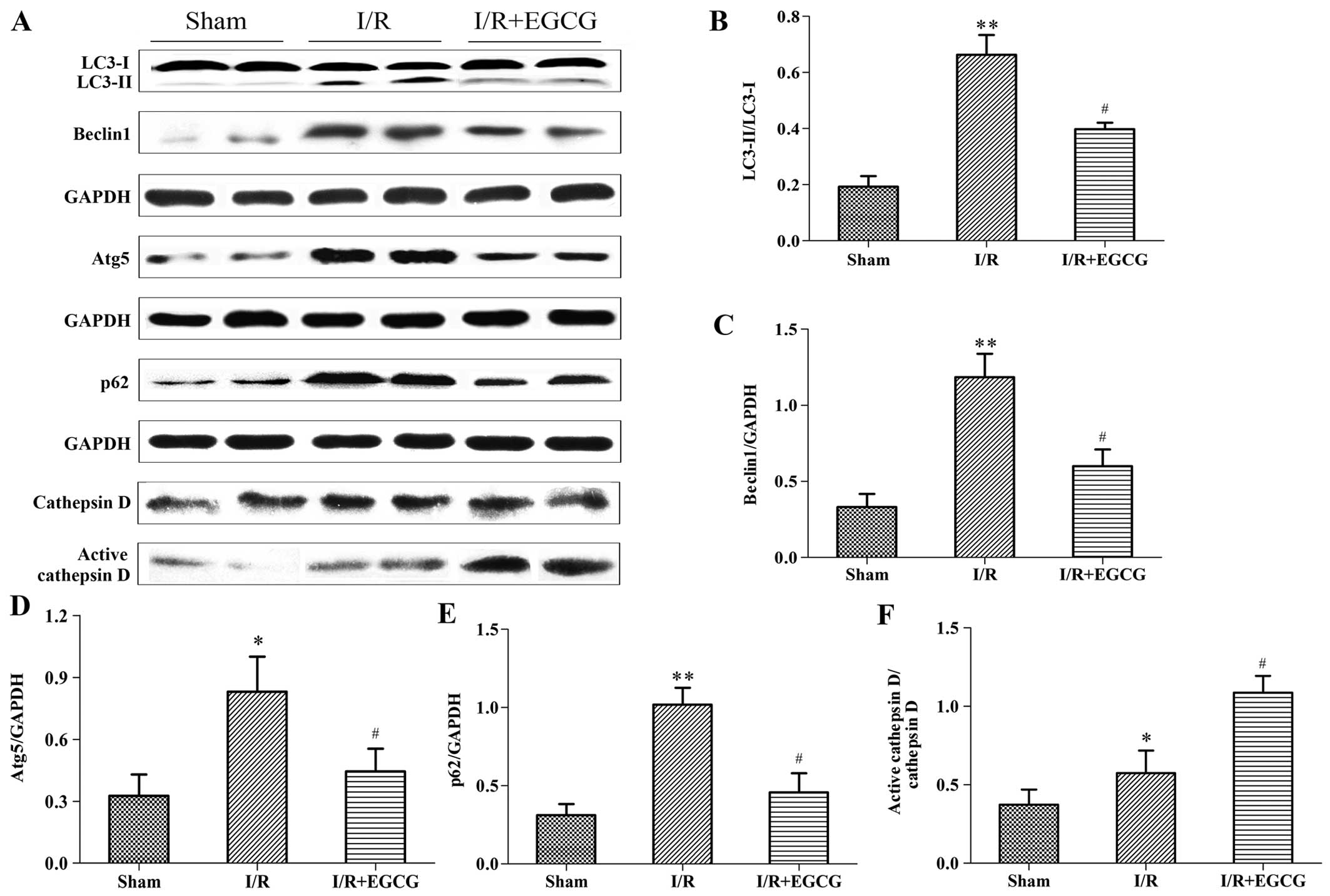
EGCG restores the autophagic flux
following myocardial I/R injury
To investigate whether the I/R-induced myocardial
injury was dependent on the impairment of the autophagic flux, the
expression levels of autophagic markers, including the LC3-II/LC3-I
ratio, Beclin1, Atg5 and p62, as well as the lysosomal protease,
cathepsin D, were measured by western blot analysis. Beclin1 and
Atg5 are essential proteins required for the initiation of
autophagosome formation (21).
The conversion of LC3-I to LC3-II is closely correlated with the
number of autophagosomes (33).
SQSTM1/p62 is an adapter protein that links aggregated proteins in
autophagosomes and promotes degradation in autolysosomes. Increased
p62 levels indicate an impaired autophagic flux (22). I/R significantly increased the
LC3-II/LC3-I ratio, and Beclin1, Atg5 and p62 expression compared
to the sham-operated group (P<0.05 or P<0.01), suggesting
increased autophagic activity or an impaired autophagic flux
(Fig. 6). Following treatment
with EGCG, the increase in the levels of Beclin1, Atg5 and p62, and
the LC3-II/LC3-I ratio were abrogated when compared with the I/R
group (P<0.05), indicating that EGCG prevented excessive
autophagy and promoted the clearance of autophagosomes.
Furthermore, an elevated level of active cathepsin D was noted in
the I/R + EGCG group compared with the I/R group (P<0.05),
indicating that EGCG restores the autophagic flux by enhancing
lysosomal function.
EGCG increases the expression of PI3K,
p-Akt, p-eNOS and p-mTOR in the myocardium following I/R
In order to elucidate the role of the PI3K/Akt
signaling pathway in the protective effects of EGCG against
myocardial I/R injury, the expression of PI3K and p-Akt (Ser473),
as well as that of the downstream targets, p-eNOS (Ser1177) and
p-mTOR (Ser2448) was further investigated (Fig. 7). Compared to the sham-operated
group, a significant decrease in the levels of PI3K, p-Akt, p-eNOS
and p-mTOR was noted in the I/R model group (P<0.05); however, a
marked increase in the levels of these proteins was noted in the
I/R + EGCG group (P<0.01). These results indicate EGCG may
enhance the phosphorylation of eNOS and mTOR via the activation of
the PI3K/Akt pathway.
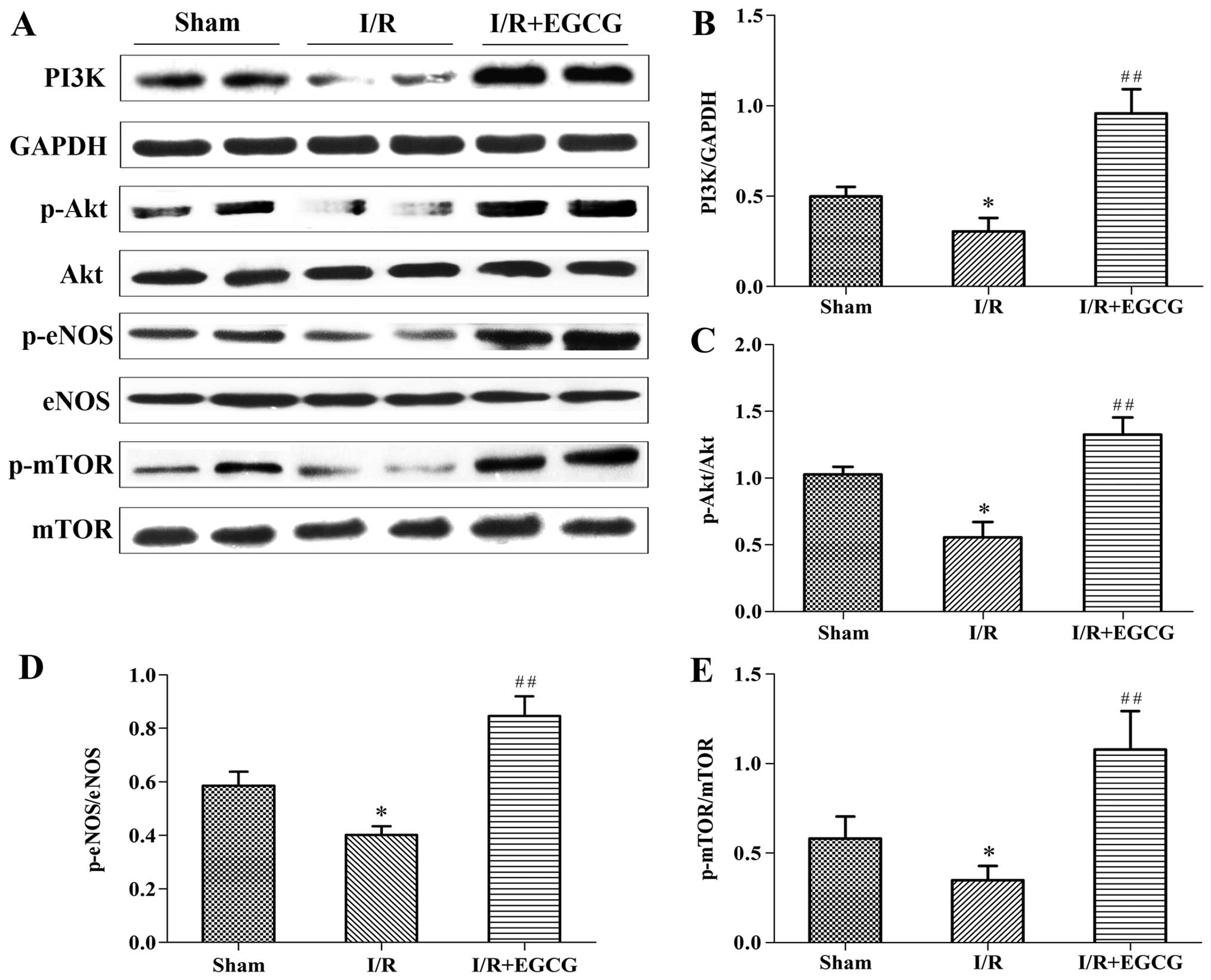 | Figure 7Epigallocatechin gallate (EGCG)
enhances the expression of PI3K, p-Akt, p-endothelial nitric oxide
synthase (eNOS) and p-mammalian target of rapamycin (mTOR) in the
myocardium following ischemia/reperfusion (I/R). (A) Representative
western blots of PI3K, p-Akt, Akt, p-eNOS, eNOS, p-mTOR and mTOR in
the sham-operated (sham_, I/R and I/R + EGCG groups. (B–E)
Quantitative analysis demonstrated the levels of PI3K, p-Akt,
p-eNOS, and p-mTOR. Results were normalized to glyceraldehyde
3-phosphate dehydrogenase (GAPDH), total Akt, eNOS and mTOR,
respectively. The data are presented as the means ± SD (n=8 rats
per group). *P<0.05 vs. sham group;
##P<0.01 vs. I/R group. |
Discussion
It is well known that myocardial I/R injury results
in a high proportion of cardiac dysfunction and heart failure and
that no effective treatment is available to prevent this damage
(2). In the present study, we
confirm that EGCG post-conditioning mitigated myocardial I/R injury
in rats, as manifested by a reduction of the infarct area, a
decrease in the levels of cardiac enzymes and an improvement of
cardiac function. These results are in accordance with those of a
previous study (16).
Additionally, we demonstrated that the role of EGCG in myocardial
I/R recovery was related to the restoration of the autophagic flux
and the inhibition of apoptosis, potentially by affecting the
downstream targets of the PI3K/Akt signaling pathway.
Accumulating evidence suggests that myocardial
apoptosis significantly contributes to cardiomyocyte death during
I/R injury, and blocking the apoptotic process may reduce the loss
of contractile cells, minimizing myocardial I/R injury (23). It has been shown that EGCG
protects cardiomyocytes against I/R-induced apoptosis in
vitro and in vivo (14). Consistent with the findings of
previous studies, we found that apoptosis in the group treated with
EGCG, as indicated by the TUNEL-staining counting result, presented
a marked reduction compared to the I/R group. Furthermore, our data
lend support to the hypothesis that cleaved (activated) caspase-3
plays a pivotal role in promoting the apoptotic signaling cascade.
Caspase-3 is an established member of the caspase family that
participates in the final execution phase of apoptosis (24). As shown in this study, cleaved
caspase-3 in the myocardium was widely expressed in the I/R group,
but failed to exhibit excessive activation in the rats treated with
EGCG (Fig. 5). Therefore, we
concluded that the inhibition of apoptosis may be one of the
mechanisms through which EGCG exerts cardioprotective effects
against I/R injury.
The PI3K/Akt pathway is known to be a target of I/R
injury and plays a key role in pro-survival and anti-apoptosis
(4). The activation of the
PI3K/Akt signaling pathway leads to Akt phosphorylation at the
Ser473 locus, producing p-Akt, and stimulates downstream targets,
such as eNOS, mTOR, NF-κB, the Bcl-2 family of proteins and the
caspase family proteins; these then regulate apoptosis, adjust
transcription factors, affect metabolism, enhance cardiomyocyte
survival, and reduce the morbidity and mortality of I/R injury
(25). PI3K/Akt has been reported
to be activated by EGCG and to mediate EGCG-induced protection
against I/R injury in the heart and kidneys (16,26). Among potential cytoprotective
downstream targets of Akt, the phosphorylation of eNOS with the
subsequent production of NO has been identified to play a critical
role in PI3K/Akt-mediated protection, particularly in the
anti-apoptotic effects of Akt (27). Accordingly, we further
investigated the effects of EGCG post-conditioning on eNOS activity
and NO production. Our data revealed that an increase in eNOS
phosphorylation and NO production occurred concomitantly with the
upregulation of PI3K protein and Akt phosphorylation. Thus, this
suggests that EGCG protects the heart, at least in part, via the
PI3K/Akt/eNOS pathway, resulting in an augmented NO content and
reduced apoptosis.
In addition to apoptosis, the importance of
autophagy in ischemic disease has received much attention in recent
years. Autophagy is a highly regulated process in which cytosolic
proteins and organelles are sequestrated by double-membrane
structures of autophagosomes, transferred to lysosomes and degraded
by proteases therein (28). This
complete autophagic flux is essential for maintaining intracellular
homeostasis and cell survival (29). Research has indicated that the
myocardial I/R-induced impairment of the autophagic flux and
excessive autophagy lead to cardiomyocyte death (8). Despite previous reports on how EGCG
affects autophagy (30,31), the mechanism through which
autophagy, as a dynamic process, participate in the
cardioprotective effets of EGCG remain unknown.
We hypothesized that treatment with EGCG will
restore the autophagic flux during myocardial I/R, which in turn
contributes to its cardioprotective effects. The process of
autophagy involves nucleation, autophagosome formation, the fusion
of autophagosomes to lysosomes, and degradation in lysosomes
(28). Beclin1 forms a complex
with class III PI3K and is localized to the pre-autophagosomal
structure mediating vesicle nucleation (32). The formation of the autophagosome
is initiated by the Atg12-Atg5-Atg16 complex and LC3-I-phospholipid
conjugates (LC3-II) (33). Thus,
the increased ratio of LC3-II/LC3-I, Beclin1 and Atg5 are markers
for autophagosome formation (34). Cathepsin D is a major lysosomal
protease that degrades materials delivered to lysosomes. The
inhibition of cathepsin D activity may compromise lysosomal
function, thus blocking the fusion of autophagosomes with
lysosomes, increasing the accumulation of autophagosomes (35). Moreover, the p62 protein is a
well-known autophagy substrate, which upon direct binding to LC3-II
incorporates into autophagosomes and is efficiently degraded by
autophagy (36). Accordingly,
when the autophagic degradation pathway is blocked, p62
accumulation occurs. In this study, myocardial I/R increased the
LC3-II/LC3-I ratio, and upregulated the protein expression of
Beclin1, Atg5 and p62 compared to the sham-operated rats. These
results indicated that myocardial I/R led to the impairment of the
autophagic flux by inducing autophagosome formation, but impairing
autophagosome clearance. In support of our hypothesis, EGCG
restored the I/R-impaired autophagy flux, which was characterized
by a decrease in the protein expression levels of the autophagy
proteins, Beclin1, Atg5 and p62, and the LC3-II/LC3-I ratio, as
well as an increase in the level of active cathepsin D.
It has been shown that the PI3K/Akt signaling
pathway is involved in the cardioprotection of EGCG (16). The kinase mammalian target of
rapamycin, mTOR, is an important downstream target of the PI3K/Akt
pathway, which is positively regulated by PI3K/Akt and results in
the inhibition of autophagy (37). Previous studies have demonstrated
that phosphorylated mTOR provides cardioprotection by restoring the
impaired autophagic flux and enhancing recovery in myocardial I/R
injury (38). Coincidently, the
present study demonstrated that EGCG inhibited the over-activation
of autophagy and promoted autophagosome clearance, accompanied by
the upregulation of both p-Akt and p-mTOR. Since the activation of
mTOR signaling is known to inhibit autophagic activity and improve
autophagic flux (39,40), our data suggest that EGCG
attenuates I/R-induced excessive autophagy and restores the
autophagic flux through the PI3K/Akt/mTOR pathway.
Autophagy has been demonstrated to engage in a
complex interplay with apoptosis. Recently, interconnections
between the autophagic and the apoptotic pathways, as well as their
alternative functions in cell survival or death have been
demonstrated (7). For instance,
an autophagic flux inhibitor induces the activation of caspases and
the apoptosis of cardiomyocytes (8). Atg5 depletion promotes apoptosis
(41). In the present study, EGCG
restored the autophagic flux and simultaneously inhibited
cardiomyocyte apoptosis. Nevertheless, further investigations are
warranted in order to elucidate the interactions between the
underlying mechanisms responsible for the autophagic flux and
apoptosis.
In conclusion, treatment with EGCG effectively
protected the rat hearts from I/R injury in vivo. This
effect of EGCG may be related to the inhibition of apoptosis and
the restoration of the autophagic flux via the regulation of
several downstream molecules of the PI3K/Akt signaling pathway.
However, there are some limitations to the present
study. Firstly, the autophagic flux needs to be further evaluated
by comparing the extent of autophagosome accumulation in the
presence of inhibitors of lysosomal fusion or degradation or both.
Secondly, our data manifested that the inhibition of apoptosis and
the restoration of the autophagic flux were involved in the
cardioprotective effects of EGCG; however, further studies are
required in order to clarify the causal link between these
mechanisms.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81360041), the Project of Natural
Science Foundation of Guangxi, China (2012GXNSFAA053148) and by the
Guangxi College of Higher Education Outstanding Talent project.
References
|
1
|
Finegold JA, Asaria P and Francis DP:
Mortality from ischaemic heart disease by country, region, and age:
Statistics from World Health Organisation and United Nations. Int J
Cardiol. 168:934–945. 2013. View Article : Google Scholar :
|
|
2
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ganesan R, Mittl PR, Jelakovic S and
Grütter MG: Extended substrate recognition in caspase-3 revealed by
high resolution X-ray structure analysis. J Mol Biol.
359:1378–1388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ha T, Hu Y, Liu L, Lu C, McMullen JR,
Kelley J, Kao RL, Williams DL, Gao X and Li C: TLR2 ligands induce
cardioprotection against ischaemia/reperfusion injury through a
PI3K/Akt-dependent mechanism. Cardiovasc Res. 87:694–703. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ou HC, Lee WJ, Lee SD, Huang CY, Chiu TH,
Tsai KL, Hsu WC and Sheu WH: Ellagic acid protects endothelial
cells from oxidized low-density lipoprotein-induced apoptosis by
modulating the PI3K/Akt/eNOS pathway. Toxicol Appl Pharmacol.
248:134–143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wei K, Wang P and Miao CY: A double-edged
sword with therapeutic potential: An updated role of autophagy in
ischemic cerebral injury. CNS Neurosci Ther. 18:879–886. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang YL, Yao YT, Fang NX, Zhou CH, Gong
JS and Li LH: Restoration of autophagic flux in myocardial tissues
is required for cardioprotection of sevoflurane postconditioning in
rats. Acta Pharmacol Sin. 35:758–769. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiao J, Zhu X, Ji G, Yang Q, Kang B, Zhao
J, Yao F, Wu L, Ni X and Wang Z: Ulinastatin protects
cardiomyocytes against ischemia reperfusion injury by regulating
autophagy through mTOR activation. Mol Med Rep. 10:1949–1953.
2014.PubMed/NCBI
|
|
10
|
Pae M and Wu D: Immunomodulating effects
of epigallocatechin-3-gallate from green tea: Mechanisms and
applications. Food Funct. 4:1287–1303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeng X, Li Q, Zhang M, Wang W and Tan X:
Green tea may be benefit to the therapy of atrial fibrillation. J
Cell Biochem. 112:1709–1712. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen L and Zhang HY: Cancer preventive
mechanisms of the green tea polyphenol
(-)-epigallocatechin-3-gallate. Molecules. 12:946–957. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Widlansky ME, Hamburg NM, Anter E,
Holbrook M, Kahn DF, Elliott JG, Keaney JF Jr and Vita JA: Acute
EGCG supplementation reverses endothelial dysfunction in patients
with coronary artery disease. J Am Coll Nutr. 26:95–102. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Townsend PA, Scarabelli TM, Pasini E,
Gitti G, Menegazzi M, Suzuki H, Knight RA, Latchman DS and
Stephanou A: Epigallocatechin-3-gallate inhibits STAT-1 activation
and protects cardiac myocytes from ischemia/reperfusion-induced
apoptosis. FASEB J. 18:1621–1623. 2004.PubMed/NCBI
|
|
15
|
Aneja R, Hake PW, Burroughs TJ, Denenberg
AG, Wong HR and Zingarelli B: Epigallocatechin, a green tea
polyphenol, attenuates myocardial ischemia reperfusion injury in
rats. Mol Med. 10:55–62. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SJ, Li M, Jeong CW, Bae HB, Kwak SH,
Lee SH, Lee HJ, Heo BH, Yook KB and Yoo KY:
Epigallocatechin-3-gallate, a green tea catechin, protects the
heart against regional ischemia-reperfusion injuries through
activation of RISK survival pathways in rats. Arch Pharm Res.
37:1079–1085. 2014. View Article : Google Scholar
|
|
17
|
Piao CS, Kim DS, Ha KC, Kim HR, Chae HJ
and Chae SW: The protective effect of epigallocatechin-3 gallate on
ischemia/reperfusion injury in isolated rat hearts: An ex vivo
Approach. Korean J Physiol Pharmacol. 15:259–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahmed LA, Salem HA, Attia AS and Agha AM:
Pharmacological preconditioning with nicorandil and pioglitazone
attenuates myocardial ischemia/reperfusion injury in rats. Eur J
Pharmacol. 663:51–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yao C, Shi X, Lin X, Shen L, Xu D and Feng
Y: Increased cardiac distribution of mono-PEGylated Radix
Ophiopogonis polysaccharide in both myocardial infarction and
ischemia/reperfusion rats. Int J Nanomedicine. 10:409–418.
2015.PubMed/NCBI
|
|
20
|
Wu N, Li W, Shu W and Jia D: Protective
effect of picroside II on myocardial ischemia reperfusion injury in
rats. Drug Des Devel Ther. 8:545–554. 2014.PubMed/NCBI
|
|
21
|
Ohsumi Y: Molecular dissection of
autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol.
2:211–216. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bjørkøy G, Lamark T, Pankiv S, Øvervatn A,
Brech A and Johansen T: Monitoring autophagic degradation of
p62/SQSTM1. Methods Enzymol. 452:181–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu H, Guo X, Chu Y and Lu S: Heart
protective effects and mechanism of quercetin preconditioning on
anti-myocardial ischemia reperfusion (IR) injuries in rats. Gene.
545:149–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Giakoustidis DE, Giakoustidis AE, Iliadis
S, Koliakou K, Antoniadis N, Kontos N, Papanikolaou V, Papageorgiou
G, Kaldrimidou E and Takoudas D: Attenuation of liver
ischemia/reperfusion induced apoptosis by
epigallocatechin-3-gallate via down-regulation of NF-kappaB and
c-Jun expression. J Surg Res. 159:720–728. 2010. View Article : Google Scholar
|
|
25
|
Yao H and Han X and Han X: The
cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling
pathway. Am J Cardiovasc Drugs. 14:433–442. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lv J, Feng M, Zhang L, Wan X, Zeng YC,
Liang PF and Xu AP: Protective effect of epigallocatechin gallate,
a major constituent of green tea, against renal
ischemia-reperfusion injury in rats. Int Urol Nephrol.
47:1429–1435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deng C, Sun Z, Tong G, Yi W, Ma L, Zhao B,
Cheng L, Zhang J, Cao F and Yi D: α-Lipoic acid reduces infarct
size and preserves cardiac function in rat myocardial
ischemia/reperfusion injury through activation of PI3K/Akt/Nrf2
pathway. PLoS One. 8:e583712013. View Article : Google Scholar
|
|
28
|
Hariharan N, Zhai P and Sadoshima J:
Oxidative stress stimulates autophagic flux during
ischemia/reperfusion. Antioxid Redox Signal. 14:2179–2190. 2011.
View Article : Google Scholar :
|
|
29
|
Xie H, Xu Q, Jia J, Ao G, Sun Y, Hu L,
Alkayed NJ, Wang C and Cheng J: Hydrogen sulfide protects against
myocardial ischemia and reperfusion injury by activating
AMP-activated protein kinase to restore autophagic flux. Biochem
Biophys Res Commun. 458:632–638. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li W, Zhu S, Li J, Assa A, Jundoria A, Xu
J, Fan S, Eissa NT, Tracey KJ, Sama AE and Wang H: EGCG stimulates
autophagy and reduces cytoplasmic HMGB1 levels in
endotoxin-stimulated macrophages. Biochem Pharmacol. 81:1152–1163.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim HS, Montana V, Jang HJ, Parpura V and
Kim JA: Epigallocatechin gallate (EGCG) stimulates autophagy in
vascular endothelial cells: A potential role for reducing lipid
accumulation. J Biol Chem. 288:22693–22705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Herzog C, Yang C, Holmes A and Kaushal GP:
zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins
but impairs autophagic flux and worsens renal function. Am J
Physiol Renal Physiol. 303:F1239–F1250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weidberg H, Shvets E and Elazar Z:
Biogenesis and cargo selectivity of autophagosomes. Annu Rev
Biochem. 80:125–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tatti M, Motta M, Di Bartolomeo S, Scarpa
S, Cianfanelli V, Cecconi F and Salvioli R: Reduced cathepsins B
and D cause impaired autophagic degradation that can be almost
completely restored by overexpression of these two proteases in Sap
C-deficient fibroblasts. Hum Mol Genet. 21:5159–5173. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang ZG, Wang Y, Huang Y, Lu Q, Zheng L,
Hu D, Feng WK, Liu YL, Ji KT, Zhang HY, et al: bFGF regulates
autophagy and ubiquitinated protein accumulation induced by
myocardial ischemia/reperfusion via the activation of the
PI3K/Akt/mTOR pathway. Sci Rep. 5:92872015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sciarretta S, Volpe M and Sadoshima J:
Mammalian target of rapamycin signaling in cardiac physiology and
disease. Circ Res. 114:549–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu YY, Zhou CH, Dou WH, Tang W, Hu CY, Hu
DM, Feng H, Wang JZ, Qian MJ, Cheng GL and Wang SF: Improved
autophagic flux is correlated with mTOR activation in the later
recovery stage of experimental acute pancreatitis. Pancreatology.
15:470–477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yue W, Hamaï A, Tonelli G, Bauvy C,
Nicolas V, Tharinger H, Codogno P and Mehrpour M: Inhibition of the
autophagic flux by salinomycin in breast cancer
stem-like/progenitor cells interferes with their maintenance.
Autophagy. 9:714–729. 2013. View Article : Google Scholar : PubMed/NCBI
|