Introduction
Esophageal cancer (EC) is one of the most common
types of cancer; it ranks eighth in terms of cancer incidence
worldwide and is the sixth leading cause of cancer-related
mortality worldwide (1). The two
main types of EC are esophageal squamous cell carcinoma (ESCC) and
esophageal adenocarcinoma (EA) (2). A unique feature of EC is its
geographical distribution. An 'EC belt', primarily of ESCC, extends
from North-Central China to the Middle East (3). Early-stage EC does not present with
specific symptoms; the majority of patients present with
advanced/metastatic disease at diagnosis (4) and the 5-year survival rate is
<20% for advanced tumors (5).
However, the precise molecular mechanisms of its pathogenesis
remain largely unknown. Thus, to improve clinical care and the
early detection of EC, it is necessary to gain a deeper
understanding of its molecular mechanisms and genetic networks.
Although an increasing amount of evidence indicates
that EC has a multifactorial etiology which involves numerous
environmental, genetic susceptibility and dietary factors, the
molecular pathogenesis of these tumors is poorly understood
(6). Tumor growth can be
facilitated by genetic alterations in cancer cells that regulate
the phenotypic changes. Genetic alterations play an important role
in EC, and alterations in a large number of oncogenes and tumor
suppressor genes have previously been described in patients with EC
(7). Moreover, emerging evidence
from the molecular characterization of microRNAs (miRNAs or miRs)
suggests that miRNAs may act as tumor suppressors or oncogenes
(8).
miRNAs belong to a class of conserved, endogenous,
non-coding small RNAs that negatively regulate gene expression at
the post-transcriptional level by mainly binding to the 3′
untranslated regions (3′-UTRs) of mRNAs, resulting in mRNA
degradation or the inhibition of translation (9). Many types of human disease,
particularly cancer, have been linked to the deregulation of miRNA
expression. Recent findings have shown that approximately 50% of
human miRNAs are located in cancer-associated genomic regions or
fragile sites and, depending on their targets, can function as
tumor suppressor genes or oncogenes (10).
To date, several study groups have suggested that
human endogenous miRNAs are associated with oncogenesis and tumor
progression in EC. It was previously noted that the expression
levels of 6 miRNAs (miR-21, miR-100, miR-99a, miR-203, miR-143 and
miR-145) were significantly altered in patients with EC (11). miR-210 has been shown to suppress
cell proliferation and regulate apoptosis in EC through the
suppression of fibroblast growth factor receptor-like 1 (FGFRL1)
(12). Moreover, human miRNAs
such as miR-375, miR-129-2, miR-21, miR-143, miR-518b, miR-133 and
miR-29c are aberrantly expressed in cancer, thus contributing to
the development and progression of EC, as well as other types of
cancer such as prostate cancer (13–19).
In a previous study, we used miRNA microarray
analysis to generate specific expression profiles of miRNAs in ESCC
cells, including miR-1 (20),
whose expression levels were significantly altered in ESCC samples.
However, the function of miR-1 in ESCC remains unknown. miR-1 is
believed to act as a tumor suppressor and is known to be
downregulated in human cancers (21,22). However, the roles of miR-1
deregulation in carcinogenesis and cancer progression remain
largely elusive. Hence, in the present study, we focused on miR-1
expression and its role in the development of ESCC. To the best of
our knowledge, the role that miR-1 plays in EC has not previously
been discussed. Therefore, in the present study we aimed to analyze
miR-1 expression and to use in vitro and in vivo
approaches in order to understand the functions and mechanisms of
action of this miRNA in ESCC. We examined the level of miR-1
expression in human ESCC cells and tissues and investigated the
potential role of miR-1 in ESCC tumorigenesis in a murine model. We
also examined its effects on cell growth and apoptosis. Lastly, we
explored the underlying mechanisms of miR-1 functions in ESCC.
In silico analysis further revealed that the key oncogenes,
MET, cyclin D1 (also known as CCND1) and cyclin-dependent
kinase 4 (CDK4), involved in the hepatocyte growth factor
(HGF)/MET signaling pathway, were targets of miR-1. The direct
inhibition of MET, cyclin D1 and CDK4 translation by miR-1, and its
potential involvement as a suppressor of esophageal tumorigenesis,
were validated experimentally. The present study thus provides us
with a better understanding of ESCC pathogenesis.
Materials and methods
Clinical specimens
From 2009 to 2011, 34 pairs of primary ESCC tissues
and the corresponding adjacent non-neoplastic esophageal tissues
were obtained from the 82nd Hospital of the People's Liberation
Army (Huaian, China). All tissue samples were obtained from
untreated patients undergoing tumor resection and were either
snap-frozen in liquid nitrogen for miRNA extraction, or fixed in
10% buffered formalin solution and then paraffin-embedded for
histological analysis. Based on clinicopathological data, the
samples were classified by age (≤60 years, n=18; >60 years,
n=16), gender (male, n=27; female, n=7), grade of differentiation
(well differentiated, n=12; moderately differentiated, n=17; poorly
differentiated, n=5), degree of tumor invasion (submucosa, n=3;
muscularis propria, n=30; adventitia, n=1) and lymph node
metastasis (negative, n=26; positive, n=8). Both tumor and normal
tissues were histologically confirmed by hematoxylin and eosin
(H&E) staining. All participants provided written informed
consent for the use of their samples for research, and the 82nd
Hospital of the People's Liberation Army Ethics Committee approved
the research protocols.
Cell culture
All cell lines [KYSE-150 (ESCC cells), Het-1A
(normal esophageal cells), QBC939 (cholangiocarcinoma cells), HepG2
(hepatocellular carcinoma cells), AGS (gastric adenocarcinoma
cells) and and human 293T cells were purchased from the American
Type Culture Collection (ATCC; Manassas, VA, USA). The cells were
cultured in RPMI-1640 medium (Gibco, Grand Island, NY, USA)
supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco) and
incubated at 37°C in a humidified chamber containing 5%
CO2. When required, the media were supplemented with 2
µg/ml penicillin and 100 mg/ml streptomycin (both from
Invitrogen, Carlsbad, CA, USA).
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA, including miRNA, was isolated from the
cells (or tumor tissues) using a Qiagen miRNeasy kit (Qiagen GmbH,
Hilden, Germany) according to the manufacturer's instructions. RNA
was quantified at 260 nm using a spectrophotometer (Beckman
Coulter, Inc., Brea, CA, USA) at an optical density 260/280 nm at a
ratio of 1.8–2.0 for all samples. Reverse transcription reactions
were carried out using a TaqMan MicroRNA Reverse transcription kit
(Applied Biosystems, Foster City, CA, USA) in a total reaction
volume of 15 µl for 30 min at 16°C, 30 min at 42°C and 5 min
at 85°C. Mature miRNA quantification was performed using TaqMan
miRNA analysis for miR-1. The PCR conditions were as follows: 95°C
for 10 min, and then 40 cycles of 95°C for 15 sec and 60°C for 1
min. The qPCR results were analyzed and expressed as the relative
miRNA expression of the threshold cycle value. The reverse
transcription primers, PCR primers and TaqMan probe for miRNA were
purchased from Applied Biosystems. U6 RNA was used as an internal
control to normalize the miRNA. All qPCR assays were performed in
triplicate in a 96-well plate using an ABI 7500 sequence detector
system (Applied Biosystems) according to the manufacturer's
instructions.
Tumorigenicity assay
To establish a xenograft tumor model, approximately
1×107 KYSE-150 cells were subcutaneously implanted in
the right flanks of female BALB/c athymic nude mice (aged 4–5
weeks; Vital River Laboratory Animal Technology, Beijing, China).
Tumor growth was examined every day for 21 days. Once the cancer
cells had developed into palpable tumors, tumor volumes were
calculated as follows: V = 1/2 × D × d2, where D and d
were the longest and shortest diameter of the tumor, respectively.
When the tumors reached an average volume of 100 mm3,
the mice were randomly divided into 4 groups (n=7 per group) for a
daily intratumoral injection of miR-1 mimics, miR-1 mimics-negative
control (NC) or in vivo transfection agent (Entranster™-in
vivo; Engreen, Inc., Beijing, China) or cisplatin (Qilu
Pharmaceutical Co., Ltd., Jinan, China) as positive controls for 21
days, which has been used as a first-line therapy for patients with
EC (23). For each injection, 5
µg miR-1 mimics or miR-1 mimics-NC were mixed with 8
µl transfection reagent and diluted with phosphate-buffered
saline (PBS) at 50 µg/ml to achieve the desired dose. Growth
curves were plotted using the average tumor volume of each
experimental group every day. Twenty-one days following
implantation, the mice were sacrificed (mice anesthetetized with
0.3% pentobarbital sodium, followed by euthanasia after 1 h) and
the tumors were resected and weighed after necropsy, as previously
described (24). All animal
handling and experimental procedures were approved by the Animal
Experimental Ethics Committee of Joinn Laboratories (Joinn
Laboratories, Inc., Suzhou, China) (Permit number: ACU-12-094).
Cell transfection
The miR-1 gain-of-function experiments using
KYSE-150 cells were performed using miR-1 mimics (100 nM) purchased
from Shanghai GenePharma Co., Ltd., (Shanghai, China) and its
negative control (NC, 100 nM). The loss-of-function experiments
using KYSE-150 cells were performed using miR-1 inhibitor (100 nM)
and its NC (100 nM). The miR-1 inhibitor was 2′-O-methyl
oligoribonucleotides, purchased from GenePharma. For each
experiment, we included a negative control. The cells were
transfected using Lipofectamine™ 2000 (Invitrogen) in Opti-MEM
(Gibco) according to the manufacturer's instructions. The
transfection efficiency was confirmed by the RT-qPCR detection of
miR-1 expression.
Luciferase assay
The 3′-UTRs of MET, cyclin D1 and CDK4 containing
the hsa-miR-1 binding site were amplified by PCR. This portion was
cloned using the XbaI site in a pGL3 Control vector
(Promega, Madison, WI, USA), downstream of the reporter gene.
Control constructs and various 3′-UTR reporter constructs were
co-transfected into 293T cells. The cells were cultured in 24-well
plates and transfected with 500 ng of either 3′-UTR reporter
constructs or pGL3 control vector together with 50 ng of pRL-TK
vector (Promega) and 100 nmol/l of miR-1, anti-miR-1 or negative
controls. At 24 h following transfection, the luciferase activity
was measured by Dual-Luciferase reporter assay (Promega). Each
transfection was repeated twice in triplicate.
MTT assay
KYSE-150 cells (5×103/well) were plated
in 96-well plates in a final volume of 100 ml and transfected with
the miRNAs. Following transfection, the cells were cultured for 0,
24, 36, 48, 60 and 72 h. The effects of miR-1 on cell growth and
viability were then determined. At the indicated time points,
tetrazolium (MTT) reagent was added followed by incubation for 4 h
at 37.5°C. The supernatant was discarded and replaced with dimethyl
sulfoxide to dissolve the formazan product. The absorbance was
measured at 490 nm using a spectrophotometric plate reader (UV-200;
Beckman Coulter, Inc.).
Trypan blue exclusion assay
The KYSE-150 cells (5×103/well) were
plated in 96-well plates in a final volume of 100 ml and
transfected with miR-1 mimics, inhibitors and their respective NCs.
At 0, 24, 36 and 48 h following transfection, the cells were
digested with parenzyme (Invitrogen GmbH, Darmstadt, Germany). The
cells were counted using a hemocytometer; dead cells stained with
trypan blue dye were counted to determine the number of viable
cells.
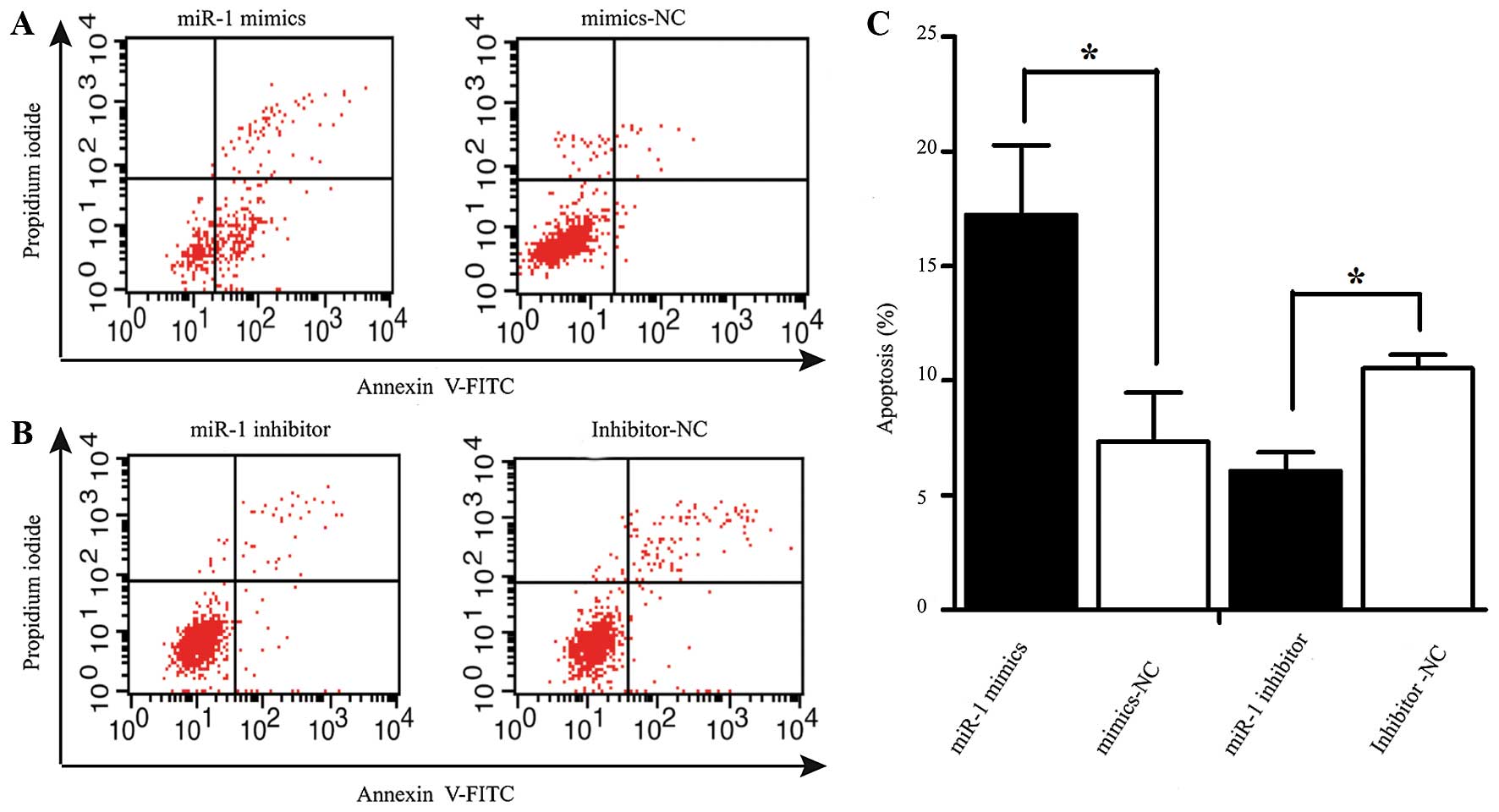
Apoptosis assay
The rate of apoptosis was analyzed using an Annexin
V-fluorescein isothiocyanate (FITC) Apoptosis detection kit
(Beyotime Institute of Biotechnology, Shanghai, China). At 48 h
following transfection, the cells were harvested and resuspended in
binding buffer containing Annexin V-FITC and propidium iodide
according to the manufacturer's instructions. The samples were
analyzed using a flow cytometer (FACScan; BD Biosciences, San Jose,
CA USA). The cells were categorized into viable, necrotic and
apoptotic cells using BD FACSDiva 6.1.3 software (BD Biosciences),
and the percentages of apoptotic cells from each group were
compared. All experiments were repeated in triplicate.
Western blot analysis
The transfected cells were harvested for western
blot analysis following 48 h of incubation. The cells were washed
once in PBS and lysed in protein lysis buffer; protein
concentrations were measured using a bicinchoninic acid (BCA)
protein assay kit (Pierce, Rockford, IL, USA). Total protein was
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis using a 12% polyacrylamide gel and electroblotted
onto a polyvinylidene fluoride membrane (Millipore, Billerica, MA,
USA). The membrane was immunoblotted overnight at 4°C with the
following primary antibodies: rabbit monoclonal anti-human MET
(1:500; D1C2; Cell Signaling Technology, Danvers, MA, USA), mouse
monoclonal anti-human CDK4 (1:500; DCS156; Cell Signaling
Technology) and rabbit monoclonal anti-human cyclin D1 (1:500;
92G2; Cell Signaling Technology) antibodies. The membrane was
incubated with a secondary antibody, horseradish peroxidase
(HRP)-conjugated goat immunoglobulin G (1:1,000; 13E5; Beyotime
Institute of Biotechnology), for 1 h after 3 washes with
Tris-buffered saline containing Triton X-100. Signals were detected
with electrochemiluminescence detection reagent (Beyotime Institute
of Biotechnology). Images were captured on Kodak film and
quantified by Quantity One software (Bio-Rad Laboratories,
Hercules, CA, USA). Western blot analysis of
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 14C10) on the same
membrane was used as a loading control. Densitometric analysis of
protein bands was performed using Image Lab software (Bio-Rad
Laboratories). All experiments were performed in triplicate.
Immunohistochemistry
The resected tumor tissues were fixed in 4%
paraformaldehyde, embedded in paraffin, cut into 4-mm-thick
sections, and mounted on polylysine-coated slides, which were then
deparaffinized, rehydrated and microwave-heated in sodium citrate
buffer (10 mM, pH 6.0) for antigen retrieval. Bovine serum albumin
was used for blocking. The slides were then incubated with MET,
cyclin D1 and CDK4 antibodies (Cell Signaling Technology) overnight
at 4°C at the optimal dilutions and subsequently incubated with an
HRP-conjugated secondary antibody at room temperature for 1 h. All
slides were independently analyzed by two experienced pathologists
blinded to the patient data. Diaminobenzidine was applied for color
development; in cases with ≥30% positive tumor cells, a section was
considered to exhibit positive expression.
Bioinformatics
miR-1 target prediction and analysis were performed
with the algorithms from TargetScan (http://www.targetscan.org/), PicTar (http://pictar.mdc-berlin.de/), miRanda (http://www.microrna.org/) and DIANA LAB TarBase 5.0
(http://diana.imis.athena-innovation.gr/DianaTools/index.php).
Statistical analysis
Unless otherwise stated, data are expressed as the
means ± standard error from at least 3 separate experiments
performed in triplicate. All statistical analyses were performed
using the t-test or Student-Newman-Keuls (SNK)-q test, and unless
otherwise specified; the null hypothesis was rejected at the 0.05
level.
Results
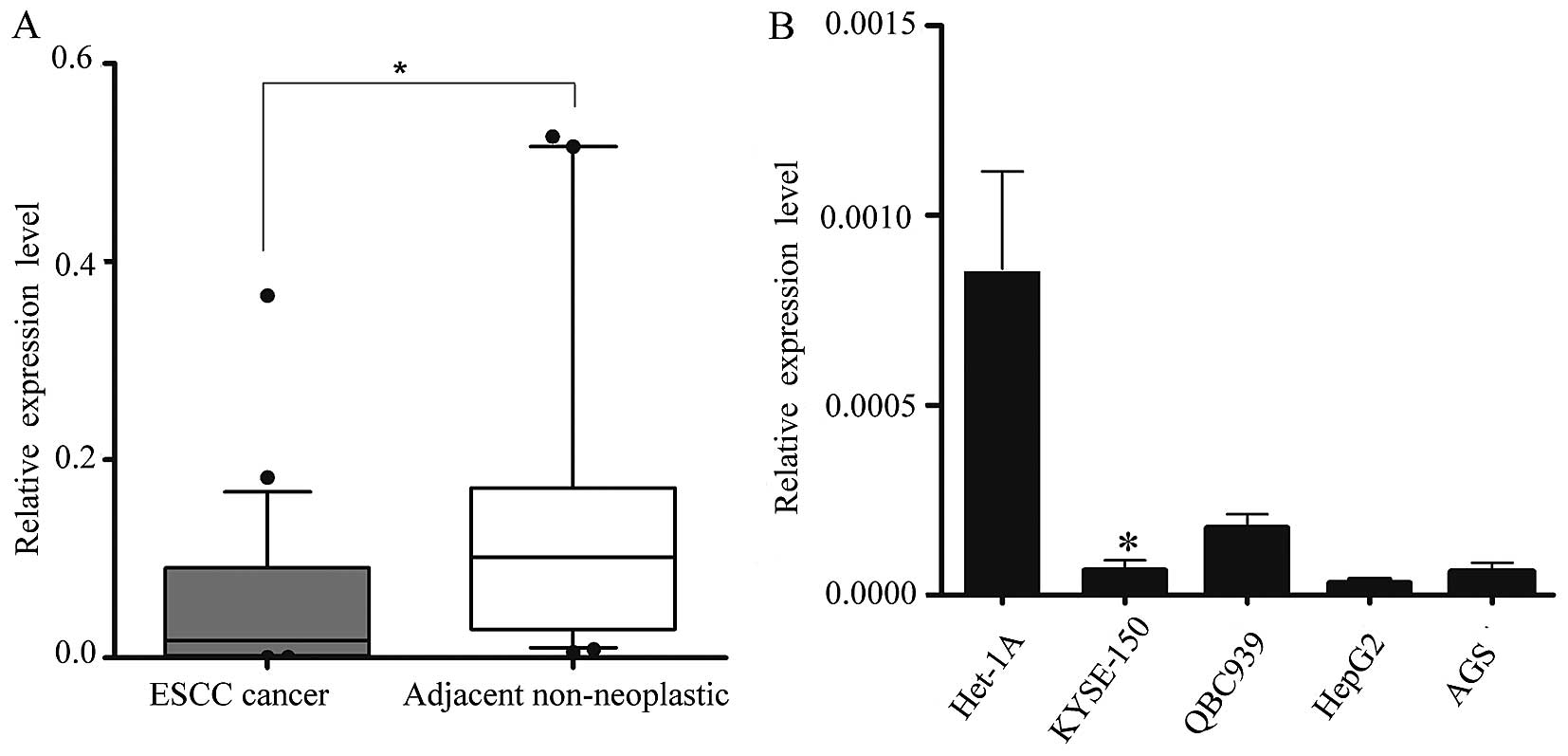
miR-1 is downregulated in human ESCC
tissues
We obtained 34 pairs of ESCC tissue samples and
their corresponding adjacent non-neoplastic esophageal tissue
specimens to examine the level of miR-1 expression using RT-qPCR.
The average miR-1 expression level was significantly lower in the
ESCC specimens than in the adjacent non-neoplastic tissue
specimens, the relative expression level being 0.048±0.079 vs.
0.182±0.280 (Fig. 1A).
Subsequently, we isolated and compared miR-1
expression in malignant and non-malignant esophageal cells
(KYSE-150 vs. Het-1A cells) (25), and confirmed the in vivo
results. Consistent with the data obtained from the ESCC tumor
tissues, miR-1 expression in malignant cells was markedly decreased
when compared with the non-malignant cells. miR-1 expression in
other malignant cells, i.e., QBC939, AGS and HepG2 cells was also
significantly decreased (Fig.
1B).
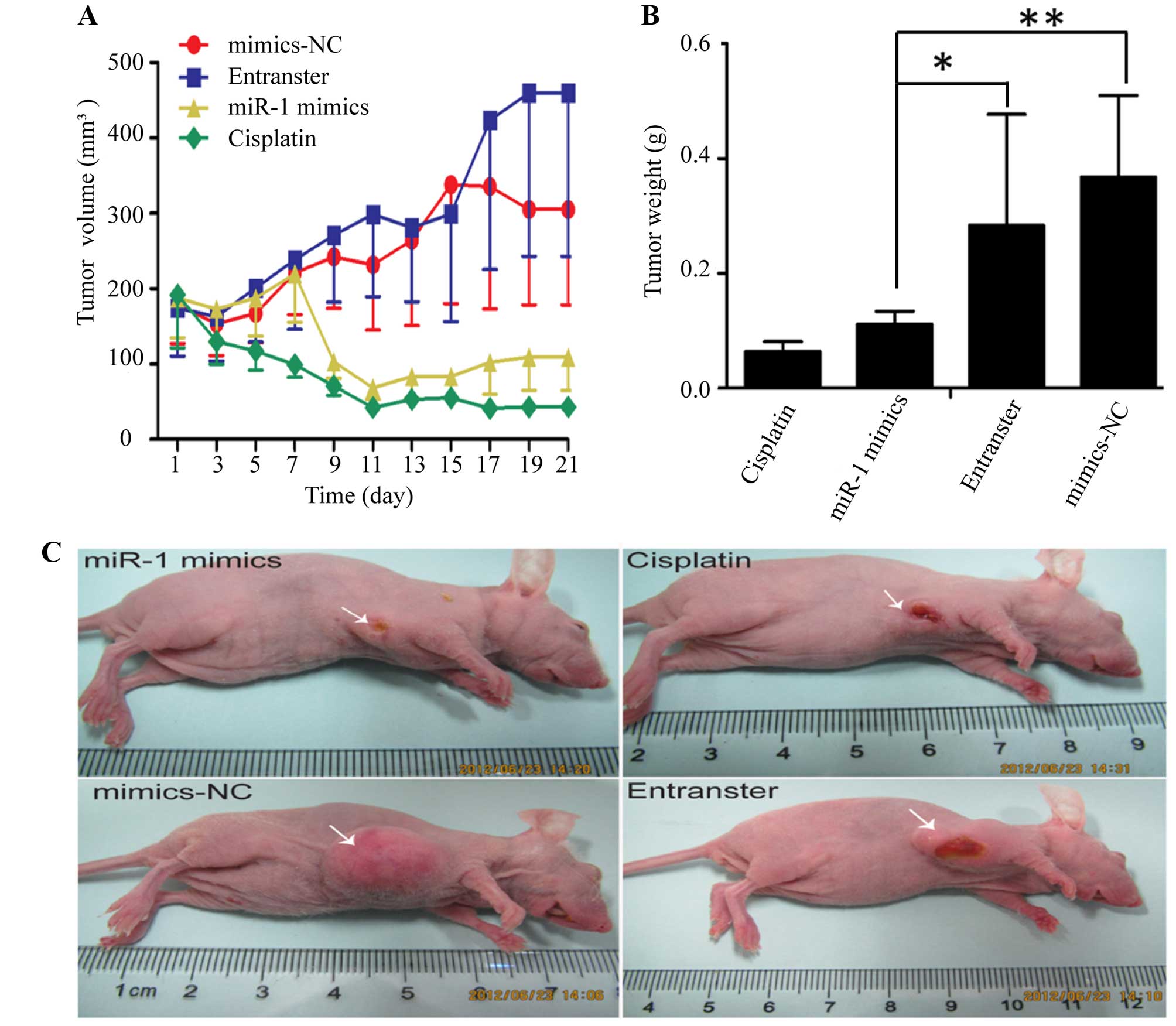
miR-1 suppresses the growth of ESCC
xenograft tumors in nude mice
The KYSE-150 cells were injected subcutaneously into
the right flanks of female nude mice. Tumors became palpable
between 5 and 7 days after inoculation. All mice in the 4 groups
(miR-1 mimics, miR-1 mimics-control, transfection agent and
cisplatin) had developed tumors by the end of the experiment.
Compared with the negative control groups (miR-1 mimics-control and
transfection agent), the average tumor volume in the miR-1 mimics
group was markedly decreased (Fig. 2A
and C), as was the average tumor weight (Fig. 2B). Compared to the negative
control groups, the average tumor volume and weight were also
decreased in cisplatin positive control group.
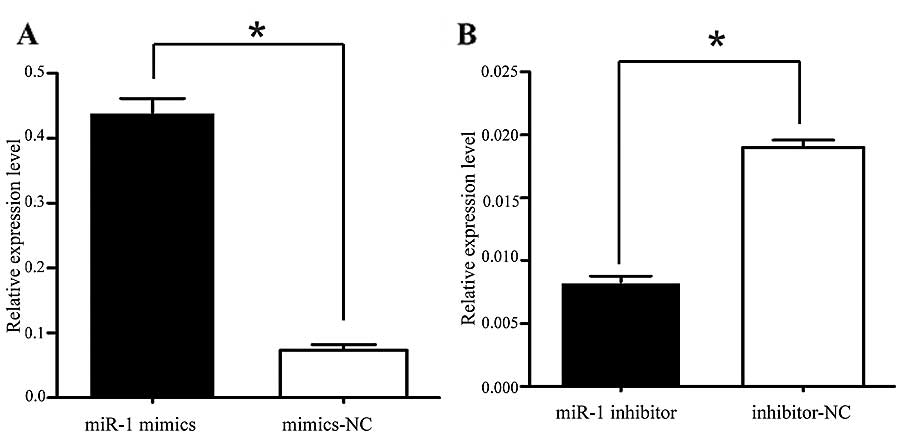
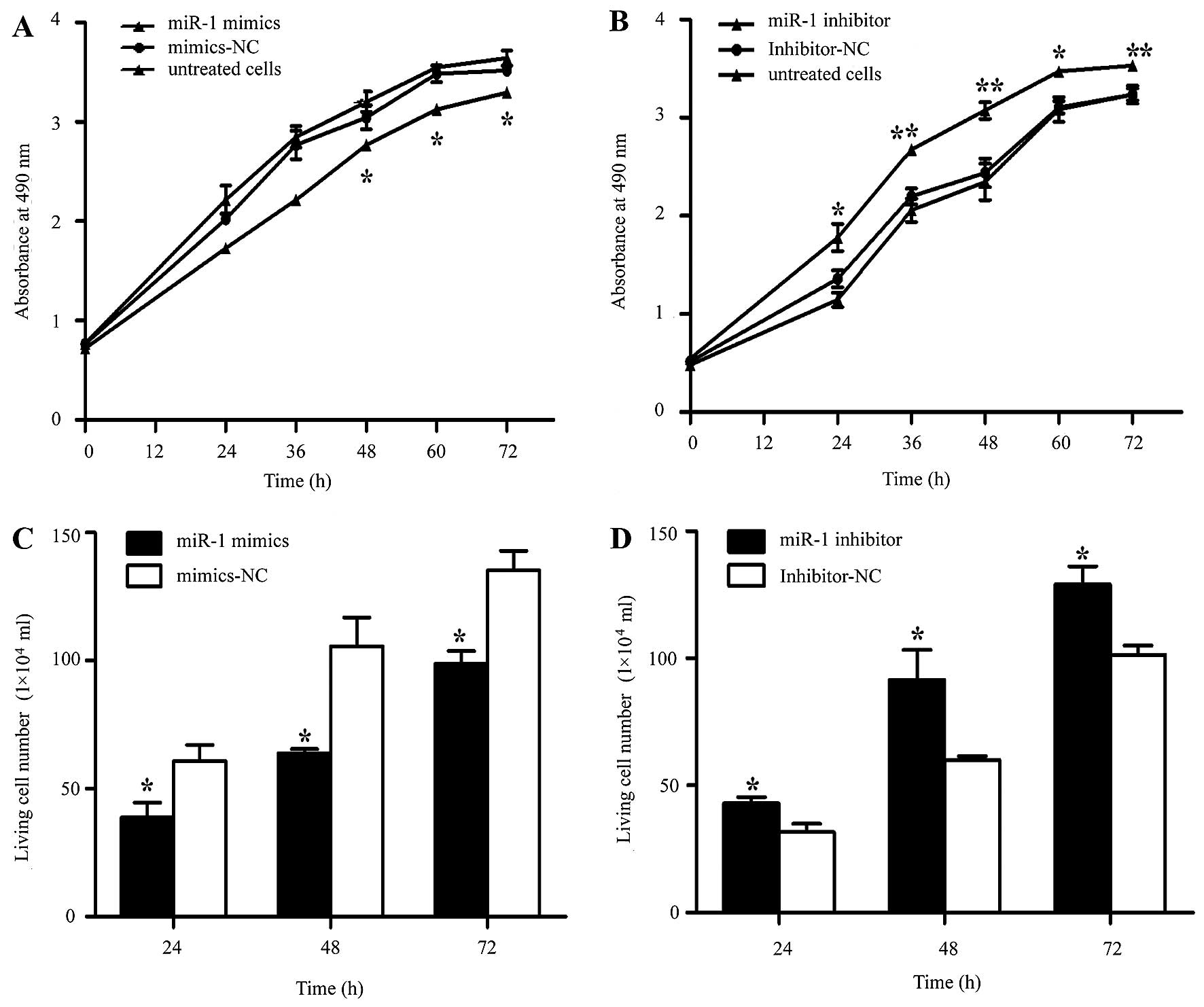
miR-1 suppresses the growth of ESCC cells
in vitro
It was thus proven that miR-1 is downregulated in
ESCC, indicating its potential role in cell biological activities.
We also confirmed that the miR-1 expression level was significantly
increased by transfection with miR-1 mimics, using RT-qPCR
(Fig. 3A). To characterize its
functional importance in ESCC tumorigenesis, we further examined
the effects of miR-1 on ESCC cell proliferation by MTT and trypan
blue exclusion assays. miR-1 overexpression significantly inhibited
cell proliferation at 48 h, whereas the miR-1 inhibitor promoted
proliferation at 24 h after transfection (Fig. 4A and B). Consistent with the
results of MTT assay, the results of trypan blue exclusion assay
also demonstrated that miR-1 overexpression significantly inhibited
ESCC cell viability (Fig. 4C and
D).
miR-1 induces the apoptosis of ESCC
cells
Apoptosis was measured by flow cytometry at 48 h
following transfection with miR-1 or miR-1 inhibitor. The number of
Annexin V-FITC(+) apoptotic cells was significantly increased in
the miR-1 mimics-transfected group compared to the
mimics-NC-transfected group. The percentage of apoptotic cells in
the group treated with miR-1 inhibitor was higher than that of the
inhibitor-NC group (Fig. 5).
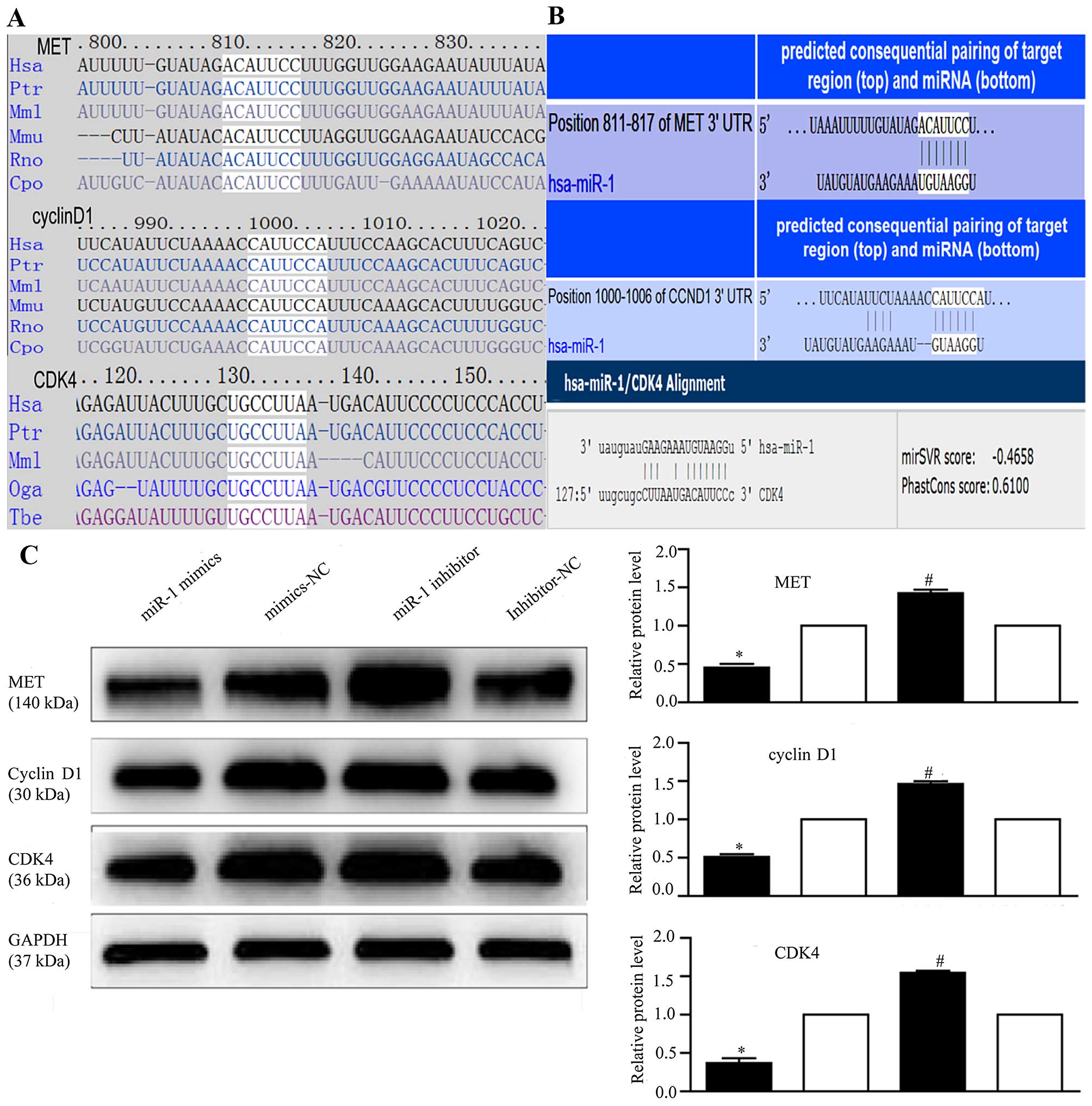
miR-1 regulates MET, cyclin D1 and CDK4
expression
To explore the mechanisms responsible for the growth
inhibitory effects of miR-1, we used 4 miRNA target prediction
programs, TargetScan (http://www.targetscan.org/), PicTar (http://pictar.mdc-berlin.de/), miRanda (http://www.microrna.org/) and DIANA LAB TarBase 5.0
(http://diana.imis.athena-innovation.gr/DianaTools/index.php),
and identified 267 target genes regulated by miR-1 (data not
shown).
Several genes reported to promote cell proliferation
were selected for further analysis (28,33,48,52). Among these, MET, cyclin
D1, and CDK4 were of particular interest, as they are
involved in the cancer and HGF/MET pathways and are closely
associated with cell proliferation. Therefore, we attempted to
describe the mechanisms of action of miR-1 and its target genes,
MET, cyclin D1 and CDK4, in ESCC. Using
TargetScan and miRanda (Fig. 6A),
we identified the putative binding sites for miR-1 in the 3′-UTR of
MET, cyclin D1 and CDK4, which are highly conserved across species
(Fig. 6B). We confirmed that the
miR-1 expression level was significantly decreased following
transfection with miR-1 inhibitor (Fig. 3B), confirming that the miR-1
inhibitor successfully regulated the miR-1 expression level in the
KYSE-150 cells. To confirm that MET, cyclin D1 and CDK4 are
downstream targets of miR-1, miR-1 mimics or inhibitor were
transfected into the KYSE-150 cells. Compared to transfection with
mimics-NC, transfection with miR-1 mimics significantly decreased
MET, cyclin D1 and CDK4 protein expression (Fig. 6C). By contrast, the protein
expression of MET, cyclin D1, and CDK4 was increased in the
loss-of-function experiments following transection with miR-1
inhibitor compared to transfection with miR-1 inhibitor-NC
(Fig. 6C).
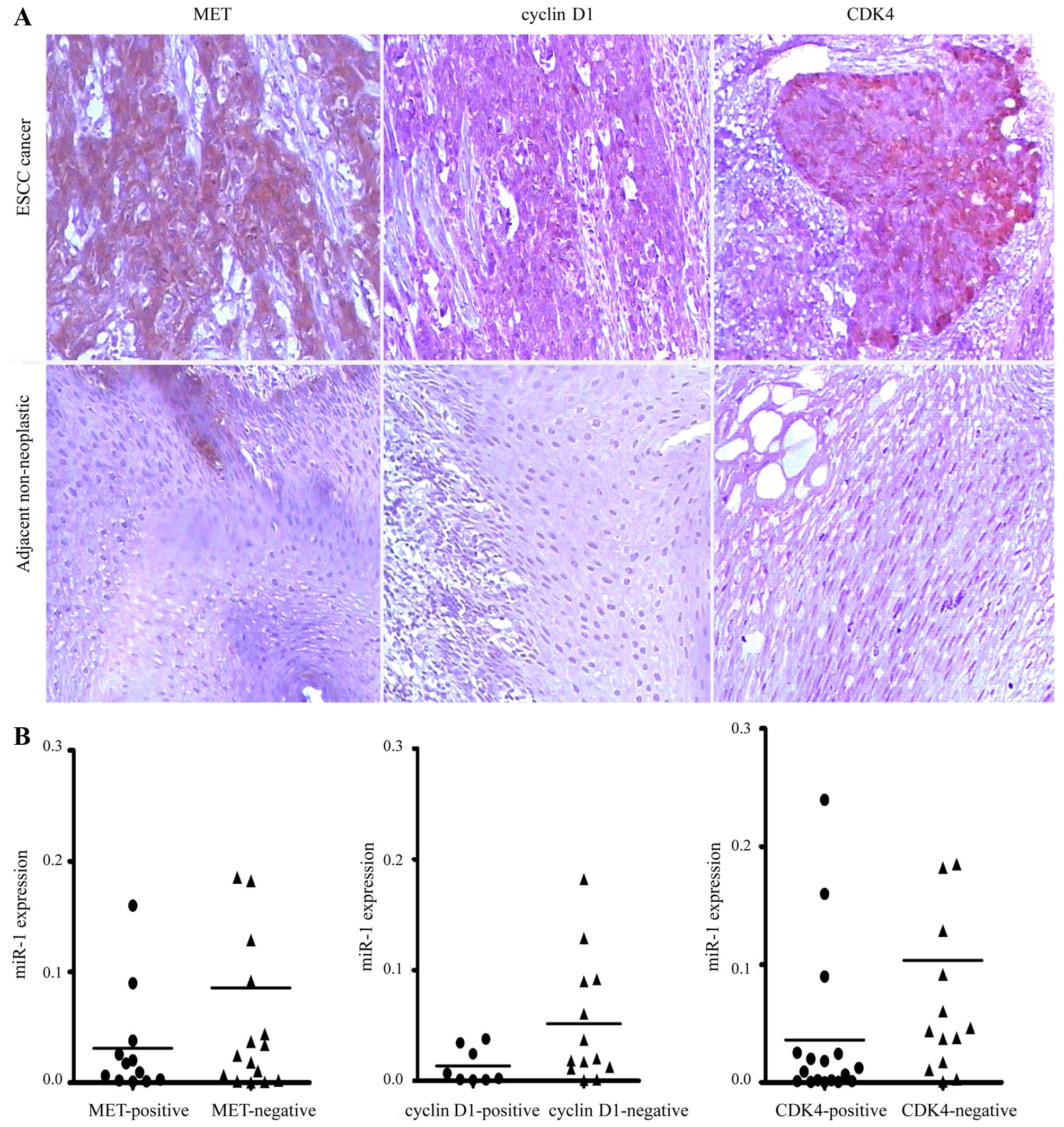
Elevated expression of MET, cyclin D1 and
CDK4 in human ESCC
To investigate whether MET, cyclin D1 and CDK4 are
upregulated in ESCC, we detected their expression levels in ESCC
tissues by immunohistochemical staining. We revealed that MET,
cyclin D1 and CDK4 were commonly overexpressed in ESCC tissues
compared with their paired adjacent non-neoplastic tissues
(Fig. 7A). However, no
association was observed between the miR-1 levels and MET, cyclin
D1 and CDK4 staining in the 34 pairs of ESCC tissues (Fig. 7B).
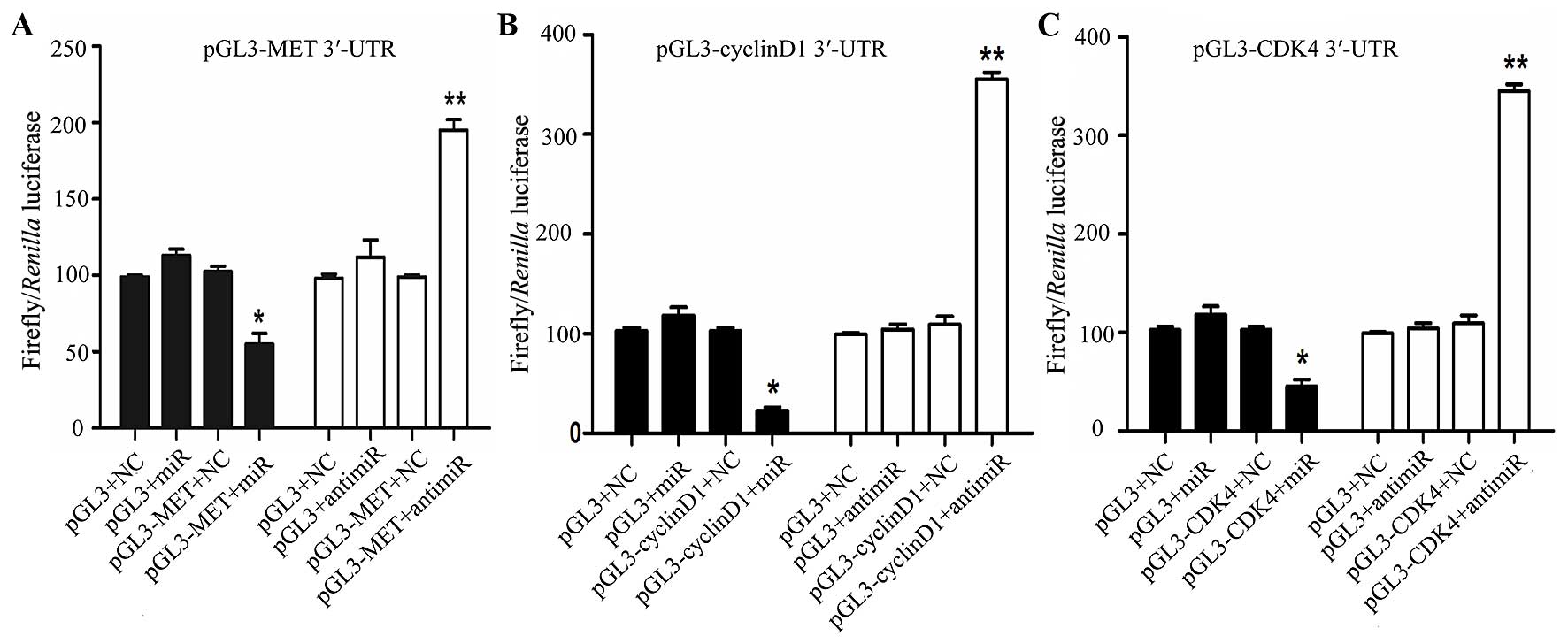
miR-1 directly targets MET, cyclin D1 and
CDK4
In order to determine whether miR-1 directly targets
the 3′-UTR of MET, cyclin D1 and CDK4, a luciferase construct
containing the 3′-UTR of MET, cyclin D1 and CDK4 was transfected
with miR-1, anti-miR-1, or negative controls and then assayed by a
luciferase reporter. MET, cyclin D1 and CDK4 exhibited a 1.2- to
2-fold decrease in luciferase activity when co-transfected with
miR-1, and a significant 1- to 2-fold increase when co-transfected
with anti-miR-1 when compared with the negative control (Fig. 8). Taken together, these findings
indicate a direct interaction between miR-1 and MET, cyclin D1 and
CDK4 mRNA.
Discussion
miRNAs are one of the most important discoveries in
recent years in the field of molecular medicine. Calin et al
(10) found that miRNAs were
mostly localized to tumor-associated fragile sites and are closely
linked to the occurrence and development of tumors. There is
evidence to indicate that miRNAs are involved in the pathogenesis
of EC, and miRNA expression profiles can be used to distinguish
different types of EC tissues. Studies have shown that miRNA
expression in ESCC exhibits both organizational and geographical
specificity. In Beijing, China, Guo et al (26) identified 7 miRNAs that could be
used to distinguish malignant EC lesions from adjacent normal
tissues: miR-25, miR-424 and miR-151 expression in tumor tissues
was significantly upregulated; that of miR-100, miR-99a, miR-29c
and miR-140* was significantly decreased. Researchers
have found that miRNA expression patterns differ between ESCC and
EA; for example, in ESCC, miR-21 and miR-373 were overexpressed and
miR-375 expression was low; in EA however, miR-21, miR-223, miR-192
and miR-194 were upregulated, and miR-203 and miR-31 were
downregulated (27,28). Japanese scholars have also
reported that miR-150, miR-205, miR-218 and miR-203 are involved in
the formation and progression of tumors in ESCC (25,29–31). In the present study, we report on
miR-1 expression in patients with ESCC from Huaian, China, which is
a topic that has never previously been reported in ESCC or EA, to
the best of our knowledge. Accordingly, we speculate that miR-1 may
play a more important role in ESCC in the Huaian region in
China.
A growing body of evidence indicates that miR-1 is
involved in the growth and spread of multiple tumors, and that it
functions as a tumor suppressor (32–34). Using an miRNA microarray, Japanese
scholars (35) constructed miRNA
expression profiles of head and neck squamous cell carcinomas
(SCCs), and reported significantly low miR-1 expression. In the
present study, we also noted the downregulated miR-1 expression in
ESCC. miR-1 suppressed tumorigenesis in an ESCC xenograft murine
model. In addition, we demonstrated that miR-1 was downregulated in
QBC939, AGS and HepG2 cells. Thus, it is highly likely that miR-1
plays a fundamental role in EC tumorigenesis, and our findings are
thus relevant to other tumors in which miR-1 is downregulated.
Under normal circumstances, cell proliferation and
apoptosis are in equilibrium; however, the equilibrium is lost in
malignant cells. Unrestricted proliferation and suppressed
apoptosis are the root causes of tumor development. A number of
studies have shown that miR-1 regulates cell proliferation,
differentiation and apoptosis. Taulli et al (36) confirmed that miR-1 overexpression
promotes the myogenic differentiation of rhabdomyosarcoma cells,
while inhibiting cell proliferation. Wu et al (37) found that nasopharyngeal carcinoma
cells transfected with miR-1 exhibited typical apoptotic metabolic
processes, which were associated with low prothymosin alpha (PTMA)
expression. In addition, Nohata et al (38,39) found that miR-1 inhibited the
proliferation and induced the apoptosis of maxillary sinus SCC
cells by targeting transgelin 2 (TAGLN2) and purine nucleoside
phosphorylase (PNP). Our preliminary study demonstrated that miR-1
was downregulated in ESCC tissues (20), but no data are available, as of
yet, on its functions. The results of the present study demonstrate
that miR-1 overexpression inhibits ESCC cell proliferation and
promotes apoptosis, whereas miR-1 silencing promotes cell
proliferation and inhibits apoptosis, suggesting that miR-1 may
influence the formation and progression of ESCC by regulating cell
proliferation and apoptosis. miR-518 can also inhibit ESCC cell
proliferation (17); yet miR-1
plays a similar role in liver cancer, prostate cancer and other
gastrointestinal tumors (22,33,39,40). Therefore, miR-1 may be a potential
target for ESCC treatment in the future.
Abnormal miRNA expression has been reported in many
types of cancer, and much attention has been focused on
understanding the roles of miRNAs in modulating the process of
cancer development, in which miRNAs exert their biological function
by regulating their target genes (24,25,40,41). Some target genes for miR-1 have
been reported. Using genome-wide expression analysis and luciferase
reporter experiments, Kojima et al (40) confirmed that miR-1 directly
regulated the significantly increased expression of the PNP
gene in prostate cancer tissues. It is well known that the average
miRNA has approximately 100 target sites (41). However, the same gene may also
simultaneously be subject to a number of specific precise temporal
miRNA regulations; consequently, the roles of miRNAs in cancer are
very complex. In the present study, to explore the molecular
mechanisms responsible for the growth inhibitory effects of miR-1,
we first used bioinformatics analysis tools to predict miR-1 target
genes. Further analysis determined that 3 target genes (MET,
cyclin D1 and CDK4) were involved in the HGF/MET
signaling pathway, which is closely associated with cell
proliferation. Western blot analysis confirmed that miR-1
significantly affected the expression of the 3 target proteins,
suggesting that it directly regulates the 3 target genes. However,
in the tissues, no statistical correlation was found through
immunohistochemistry, perhaps too few cases were included.
There is an increasing amount of evidence
demonstrating that, in ESCC cell lines and tissues, the expression
of multiple molecular targets of the HGF/MET signaling pathway,
e.g., MET, cyclin D1 and CDK4, is abnormal (42–44). The HGF/MET signaling pathway plays
an important role in regulating cell proliferation and apoptosis,
and it is closely associated with a variety of tumors (45–49). The HGF/MET signaling pathway
exerts its effects by activating the single receptor MET, which
then binds to a number of intracellular target proteins, triggering
downstream cascades involved in the regulation of a variety of
biological activities, such as cell proliferation and apoptosis
(42,50,51). MET is the encoding oncogene for
the HGF receptor (HGFR). A key protein in the HGF/MET signaling
pathway involved with tyrosine kinase activity, MET affects cell
proliferation, apoptosis, invasion, migration and angiogenesis via
interactions with critical molecules of the Wnt and
phosphatidylinositol-3-kinase (PI3K) signaling pathways. MET
expression is abnormal in ESCC and EA, cancer of the stomach,
pancreas, liver and colon, as well as in other tumors (45–49). miR-1 plays a tumor suppressor role
by directly regulating MET expression in colon cancer,
rhabdomyosarcoma and osteosarcoma (21,52,53). Our results suggest that in ESCC,
miR-1 also inhibits tumor growth through the direct regulation of
MET.
Cyclin D1 and CDK4 are located downstream of MET in
the HGF/MET signaling pathway. Recent studies have shown that the
expression of cyclin D1 and CDK4 is abnormal in a variety of
tumors, including EC, and affects tumorigenesis and tumor
development (54,55). Cyclin D1 overexpression enables
the continuous proliferation of cancer cells, leading to
tumorigenesis (54). CDK4 can
bind with cyclin D1 to form complexes, affecting cell cycle
regulation and cell proliferation (55). Given the results of the present
study, that miR-1 directly regulates MET, cyclin D1 and CDK4, we
hypothesized that miR-1 regulates cell proliferation in two ways:
i) by regulating MET expression, thereby affecting the expression
of the downstream molecules cyclin D1 and CDK4; and ii) by
targeting cyclin D1 and CDK4 directly. We posit that MET, cyclin D1
and CDK4 of the HGF/MET signaling pathway form a regulatory network
around miR-1, which is then involved in the regulation of ESCC
development.
Taken together, our results suggest that miR-1 is a
tumor suppressor miRNA in human ESCC, which acts through the
repression of MET, cyclin D1 and CDK4 expression. Our data provide
further evidence of the pivotal role that miRNAs play in ESCC
tumorigenesis. As miR-1 is downregulated in ESCC, the
re-introduction of this mature miRNA into the tumor tissue may be a
therapeutic strategy that reduces the expression of target genes.
Although miRNA-based therapeutics are still in their infancy, our
findings are encouraging and suggest that miR-1 may be a potential
target for future ESCC treatment.
Acknowledgments
The authors would like to thank Dr Conglin Zuo for
his kind provision of Joinn Laboratories (Suzhou, China) and his
advice for our experiments. The present study was supported by the
Jiangsu Natural Science Foundation (BK2012666).
Abbreviations:
|
miR-1
|
microRNA-1
|
|
EC
|
esophageal cancer
|
|
ESCC
|
esophageal squamous cell carcinoma
|
|
CDK4
|
cyclin-dependent kinase 4
|
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Vizcaino AP, Moreno V, Lambert R and
Parkin DM: Time trends incidence of both major histologic types of
esophageal carcinomas in selected countries, 1973–1995. Int J
Cancer. 99:860–868. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Layke JC and Lopez PP: Esophageal cancer:
a review and update. Am Fam Physician. 73:2187–2194.
2006.PubMed/NCBI
|
|
5
|
Stoner GD and Wang LS: Chemoprevention of
esophageal squamous cell carcinoma with berries. Top Curr Chem.
329:1–20. 2013. View Article : Google Scholar
|
|
6
|
Kamangar F, Chow WH, Abnet CC and Dawsey
SM: Environmental causes of esophageal cancer. Gastroenterol Clin
North Am. 38:27–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hyland PL, Freedman ND, Hu N, Tang ZZ,
Wang L, Wang C, Ding T, Fan JH, Qiao YL, Golozar A, et al: Genetic
variants in sex hormone metabolic pathway genes and risk of
esophageal squamous cell carcinoma. Carcinogenesis. 34:1062–1068.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calin GA, Sevignani C, Dumitru CD, Hyslop
T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M,
et al: Human microRNA genes are frequently located at fragile sites
and genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu BL, Xu LY, Du ZP, Liao LD, Zhang HF,
Huang Q, Fang GQ and Li EM: MiRNA profile in esophageal squamous
cell carcinoma: downregulation of miR-143 and miR-145. World J
Gastroenterol. 17:79–88. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsuchiya S, Fujiwara T, Sato F, Shimada Y,
Tanaka E, Sakai Y, Shimizu K and Tsujimoto G: MicroRNA-210
regulates cancer cell proliferation through targeting fibroblast
growth factor receptor-like 1 (FGFRL1). J Biol Chem. 286:420–428.
2011. View Article : Google Scholar :
|
|
13
|
Kong KL, Kwong DL, Chan TH, Law SY, Chen
L, Li Y, Qin YR and Guan XY: MicroRNA-375 inhibits tumour growth
and metastasis in oesophageal squamous cell carcinoma through
repressing insulin-like growth factor 1 receptor. Gut. 61:33–42.
2012. View Article : Google Scholar
|
|
14
|
Kang M, Li Y, Liu W, Wang R, Tang A, Hao
H, Liu Z and Ou H: miR-129-2 suppresses proliferation and migration
of esophageal carcinoma cells through downregulation of SOX4
expression. Int J Mol Med. 32:51–58. 2013.PubMed/NCBI
|
|
15
|
Wang N, Zhang CQ, He JH, Duan XF, Wang YY,
Ji X, Zang WQ, Li M, Ma YY, Wang T and Zhao GQ: MiR-21
down-regulation suppresses cell growth, invasion and induces cell
apoptosis by targeting FASL, TIMP3, and RECK genes in esophageal
carcinoma. Dig Dis Sci. 58:1863–1870. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ni Y, Meng L, Wang L, Dong W, Shen H, Wang
G, Liu Q and Du J: MicroRNA-143 functions as a tumor suppressor in
human esophageal squamous cell carcinoma. Gene. 517:197–204. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang M, Zhou S, Zhang L, Zhang J, Cai H,
Zhu J, Huang C and Wang J: miR-518b is down-regulated, and involved
in cell proliferation and invasion by targeting Rap1b in esophageal
squamous cell carcinoma. FEBS Lett. 586:3508–3521. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tao J, Wu D, Xu B, Qian W, Li P, Lu Q, Yin
C and Zhang W: microRNA-133 inhibits cell proliferation, migration
and invasion in prostate cancer cells by targeting the epidermal
growth factor receptor. Oncol Rep. 27:1967–1975. 2012.PubMed/NCBI
|
|
19
|
Ding DP, Chen ZL, Zhao XH, Wang JW, Sun J,
Wang Z, Tan FW, Tan XG, Li BZ, Zhou F, et al: miR-29c induces cell
cycle arrest in esophageal squamous cell carcinoma by modulating
cyclin E expression. Carcinogenesis. 32:1025–1032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fu HL, Wu P, Wang XF, Wang JG, Jiao F,
Song LL, Xie H, Wen XY, Shan HS, Du YX and Zhao YP: Altered miRNA
expression is associated with differentiation, invasion, and
metastasis of esophageal squamous cell carcinoma (ESCC) in patients
from Huaian, China. Cell Biochem Biophys. 67:657–668. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Novello C, Pazzaglia L, Cingolani C, Conti
A, Quattrini I, Manara MC, Tognon M, Picci P and Benassi MS: miRNA
expression profile in human osteosarcoma: role of miR-1 and
miR-133b in proliferation and cell cycle control. Int J Oncol.
42:667–675. 2013.
|
|
22
|
Nohata N, Hanazawa T, Enokida H and Seki
N: microRNA-1/133a and microRNA-206/133b clusters: dysregulation
and functional roles in human cancers. Oncotarget. 3:9–21.
2012.PubMed/NCBI
|
|
23
|
Hou G, Zhang Q, Wang L, Liu M, Wang J and
Xue L: mTOR inhibitor rapamycin alone or combined with cisplatin
inhibits growth of esophageal squamous cell carcinoma in nude mice.
Cancer Lett. 290:248–254. 2010. View Article : Google Scholar
|
|
24
|
Wang F, Zhang P, Ma Y, Yang J, Moyer MP,
Shi C, Peng J and Qin H: NIRF is frequently upregulated in
colorectal cancer and its oncogenicity can be suppressed by let-7a
microRNA. Cancer Lett. 314:223–231. 2012. View Article : Google Scholar
|
|
25
|
Matsushima K, Isomoto H, Yamaguchi N,
Inoue N, Machida H, Nakayama T, Hayashi T, Kunizaki M, Hidaka S,
Nagayasu T, et al: MiRNA-205 modulates cellular invasion and
migration via regulating zinc finger E-box binding homeobox 2
expression in esophageal squamous cell carcinoma cells. J Transl
Med. 9:302011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo Y, Chen Z, Zhang L, Zhou F, Shi S,
Feng X, Li B, Meng X, Ma X, Luo M, et al: Distinctive microRNA
profiles relating to patient survival in esophageal squamous cell
carcinoma. Cancer Res. 68:26–33. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leidner RS, Ravi L, Leahy P, Chen Y,
Bednarchik B, Streppel M, Canto M, Wang JS, Maitra A, Willis J, et
al: The microRNAs, MiR-31 and MiR-375, as candidate markers in
Barrett's esophageal carcinogenesis. Genes Chromosomes Cancer.
51:473–479. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee KH, Goan YG, Hsiao M, Lee CH, Jian SH,
Lin JT, Chen YL and Lu PJ: MicroRNA-373 (miR-373)
post-transcriptionally regulates large tumor suppressor, homolog 2
(LATS2) and stimulates proliferation in human esophageal cancer.
Exp Cell Res. 315:2529–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yokobori T, Suzuki S, Tanaka N, Inose T,
Sohda M, Sano A, Sakai M, Nakajima M, Miyazaki T, Kato H and Kuwano
H: MiR-150 is associated with poor prognosis in esophageal squamous
cell carcinoma via targeting the EMT inducer ZEB1. Cancer Sci.
104:48–54. 2013. View Article : Google Scholar
|
|
30
|
Takeshita N, Mori M, Kano M, Hoshino I,
Akutsu Y, Hanari N, Yoneyama Y, Ikeda N, Isozaki Y, Maruyama T, et
al: miR-203 inhibits the migration and invasion of esophageal
squamous cell carcinoma by regulating LASP1. Int J Oncol.
41:1653–1661. 2012.PubMed/NCBI
|
|
31
|
Yamamoto N, Kinoshita T, Nohata N, Itesako
T, Yoshino H, Enokida H, Nakagawa M, Shozu M and Seki N: Tumor
suppressive microRNA-218 inhibits cancer cell migration and
invasion by targeting focal adhesion pathways in cervical squamous
cell carcinoma. Int J Oncol. 42:1523–1532. 2013.PubMed/NCBI
|
|
32
|
Leone V, D'Angelo D, Rubio I, de Freitas
PM, Federico A, Colamaio M, Pallante P, Medeiros-Neto G and Fusco
A: MiR-1 is a tumor suppressor in thyroid carcinogenesis targeting
CCND2, CXCR4, and SDF-1alpha. J Clin Endocrinol Metab.
96:E1388–E1398. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li D, Yang P, Li H, Cheng P, Zhang L, Wei
D, Su X, Peng J, Gao H, Tan Y, et al: MicroRNA-1 inhibits
proliferation of hepatocarcinoma cells by targeting endothelin-1.
Life Sci. 91:440–447. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu YN, Yin JJ, Abou-Kheir W, Hynes PG,
Casey OM, Fang L, Yi M, Stephens RM, Seng V, Sheppard-Tillman H, et
al: MiR-1 and miR-200 inhibit EMT via Slug-dependent and
tumorigenesis via Slug-independent mechanisms. Oncogene.
32:296–306. 2013. View Article : Google Scholar
|
|
35
|
Nohata N, Sone Y, Hanazawa T, Fuse M,
Kikkawa N, Yoshino H, Chiyomaru T, Kawakami K, Enokida H, Nakagawa
M, et al: miR-1 as a tumor suppressive microRNA targeting TAGLN2 in
head and neck squamous cell carcinoma. Oncotarget. 2:29–42.
2011.PubMed/NCBI
|
|
36
|
Taulli R, Bersani F, Foglizzo V, Linari A,
Vigna E, Ladanyi M, Tuschl T and Ponzetto C: The muscle-specific
microRNA miR-206 blocks human rhabdomyosarcoma growth in
xenotransplanted mice by promoting myogenic differentiation. J Clin
Invest. 119:2366–2378. 2009.PubMed/NCBI
|
|
37
|
Wu CD, Kuo YS, Wu HC and Lin CT:
MicroRNA-1 induces apoptosis by targeting prothymosin alpha in
nasopharyngeal carcinoma cells. J Biomed Sci. 18:802011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nohata N, Hanazawa T, Kikkawa N, Sakurai
D, Fujimura L, Chiyomaru T, Kawakami K, Yoshino H, Enokida H,
Nakagawa M, et al: Tumour suppressive microRNA-874 regulates novel
cancer networks in maxillary sinus squamous cell carcinoma. Br J
Cancer. 105:833–841. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nohata N, Hanazawa T, Kikkawa N, Sakurai
D, Sasaki K, Chiyomaru T, Kawakami K, Yoshino H, Enokida H,
Nakagawa M, et al: Identification of novel molecular targets
regulated by tumor suppressive miR-1/miR-133a in maxillary sinus
squamous cell carcinoma. Int J Oncol. 39:1099–1107. 2011.PubMed/NCBI
|
|
40
|
Kojima S, Chiyomaru T, Kawakami K, Yoshino
H, Enokida H, Nohata N, Fuse M, Ichikawa T, Naya Y, Nakagawa M and
Seki N: Tumour suppressors miR-1 and miR-133a target the oncogenic
function of purine nucleoside phosphorylase (PNP) in prostate
cancer. Br J Cancer. 106:405–413. 2012. View Article : Google Scholar :
|
|
41
|
Brennecke J, Stark A, Russell RB and Cohen
SM: Principles of microRNA-target recognition. PLoS Biol.
3:e852005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kato H, Arao T, Matsumoto K, Fujita Y,
Kimura H, Hayashi H, Nishiki K, Iwama M, Shiraishi O, Yasuda A, et
al: Gene amplification of EGFR, HER2, FGFR2 and MET in esophageal
squamous cell carcinoma. Int J Oncol. 42:1151–1158. 2013.PubMed/NCBI
|
|
43
|
Grugan KD, Miller CG, Yao Y, Michaylira
CZ, Ohashi S, Klein-Szanto AJ, Diehl JA, Herlyn M, Han M, Nakagawa
H and Rustgi AK: Fibroblast-secreted hepatocyte growth factor plays
a functional role in esophageal squamous cell carcinoma invasion.
Proc Natl Acad Sci USA. 107:11026–11031. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Leelawat K, Leelawat S, Tepaksorn P,
Rattanasinganchan P, Leungchaweng A, Tohtong R and Sobhon P:
Involvement of c-Met/hepatocyte growth factor pathway in
cholangiocarcinoma cell invasion and its therapeutic inhibition
with small interfering RNA specific for c-Met. J Surg Res.
136:78–84. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ketterer K, Kong B, Frank D, Giese NA,
Bauer A, Hoheisel J, Korc M, Kleeff J, Michalski CW and Friess H:
Neuromedin U is overexpressed in pancreatic cancer and increases
invasiveness via the hepatocyte growth factor c-Met pathway. Cancer
Lett. 277:72–81. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zeng ZS, Weiser MR, Kuntz E, Chen CT, Khan
SA, Forslund A, Nash GM, Gimbel M, Yamaguchi Y, Culliford AT IV, et
al: c-Met gene amplification is associated with advanced stage
colorectal cancer and liver metastases. Cancer Lett. 265:258–269.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ke AW, Shi GM, Zhou J, Wu FZ, Ding ZB, Hu
MY, Xu Y, Song ZJ, Wang ZJ, Wu JC, et al: Role of overexpression of
CD151 and/or c-Met in predicting prognosis of hepatocellular
carcinoma. Hepatology. 49:491–503. 2009. View Article : Google Scholar
|
|
48
|
Drebber U, Baldus SE, Nolden B, Grass G,
Bollschweiler E, Dienes HP, Hölscher AH and Mönig SP: The
overexpression of c-met as a prognostic indicator for gastric
carcinoma compared to p53 and p21 nuclear accumulation. Oncol Rep.
19:1477–1483. 2008.PubMed/NCBI
|
|
49
|
Tuynman JBLS, Lagarde SM, Ten Kate FJ,
Richel DJ and van Lanschot JJ: Met expression is an independent
prognostic risk factor in patients with oesophageal adenocarcinoma.
Br J Cancer. 98:1102–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cecchi F, Rabe DC and Bottaro DP:
Targeting the HGF/Met signaling pathway in cancer therapy. Expert
Opin Ther Targets. 16:553–572. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Peters S and Adjei AA: MET: A promising
anticancer therapeutic target. Nat Rev Clin Oncol. 9:314–326. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Reid JF, Sokolova V, Zoni E, Lampis A,
Pizzamiglio S, Bertan C, Zanutto S, Perrone F, Camerini T, Gallino
G, et al: miRNA profiling in colorectal cancer highlights miR-1
involvement in MET-dependent proliferation. Mol Cancer Res.
10:504–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yan D, Dong XE, Chen X, Wang L, Lu C, Wang
J, Qu J and Tu L: MicroRNA-1/206 targets c-Met and inhibits
rhabdomyosarcoma development. J Biol Chem. 284:29596–29604. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shirali S, Aghaei M, Shabani M, Fathi M,
Sohrabi M and Moeinifard M: Adenosine induces cell cycle arrest and
apoptosis via cyclinD1/Cdk4 and Bcl-2/Bax pathways in human ovarian
cancer cell line OVCAR-3. Tumour Biol. 34:1085–1095. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fong LY, Nguyen VT, Farber JL, Huebner K
and Magee PN: Early deregulation of the the p16ink4a-cyclin
D1/cyclin-dependent kinase 4-retinoblastoma pathway in cell
proliferation-driven esophageal tumorigenesis in zinc-deficient
rats. Cancer Res. 60:4589–4595. 2000.PubMed/NCBI
|