Introduction
Interleukin-1β (IL-1β) is a pivotal pro-inflammatory
cytokine that has been linked to the pathogenesis of a broad
spectrum of acute and chronic inflammatory diseases (1). IL-1β is synthesized as a precursor
in the cytosol in response to various stimuli. A low level of IL-1β
in vivo can evoke various mediators, which induce an
inflammatory response (2). IL-1β
is considered an important pro-inflammatory cytokine in the brain
and plays a critical role in the progression of neuroinflammation
(3). Neuroinflammation is a
well-known factor in the pathogenesis of neurodegenerative
diseases, such as Alzheimer's disease (AD), Parkinson's disease
(PD) and multiple sclerosis (MS) (4). IL-1β is produced by
lipopolysaccharide (LPS)-stimulated BV2 microglia. LPS induces
neuroinflammation by activating inflammatory cells, including
astrocytes and microglial cells (5). In a previous study, it was shown
that the systemic injection of LPS led to neuroinflammatory
responses in the brain, which then led to amyloid-β accumulation
(6).
As regards IL-1β biological activity, mature IL-1β
is regulated through cytosolic multi-protein complexes referred to
as inflammasomes, such as the NLR family, pyrin domain containing 3
(NLRP3) inflammasome, which contains a nucleotide binding domain
leucine-rich repeat with a pyrin-domain containing 3 sensor, an
apoptosis-associated speck-like protein containing a
caspase-recruitment domain (ASC) adaptor and a caspase-1 enzyme
(7,8). For the secretion of IL-1β, pro-IL-1β
must be cleaved by activated caspase-1. Caspase-1 is activated by
an inflammasome assembly with NLRP3, ASC and pro-caspase-1. LPS
must induce IL-1β for caspase-1 activation in response to adenosine
triphosphate (ATP), which is a well-characterized danger-associated
molecular pattern (DAMP) (9,10).
Extracellular ATP promotes NLRP3 inflammasome activation by
stimulating purinergic receptor P2X ligand-gated ion channel 7
(P2X7) (11,12). Pro-caspase-1 association with
NLRP3 binds the adaptor molecule ASC, leading to pro-caspase-1
activation, which, in turn, triggers pro-IL-1β processing to mature
IL-1β, in LPS- and ATP-stimulated cells.
Isothiocyanates are found abundantly in cruciferous
or 'cabbage family' vegetables, such as garden cress, broccoli,
cabbage, kale, cauliflower and radish, and have been used as diet
components with potent chemopreventive and/or anticancer properties
(13,14). Certain isothiocyanates, such as
sulforaphane (SFN), phenethyl isothiocyanate (PEITC), and benzyl
isothiocyanate (BITC), are derived from glucosinolates in
cruciferous vegetables (15).
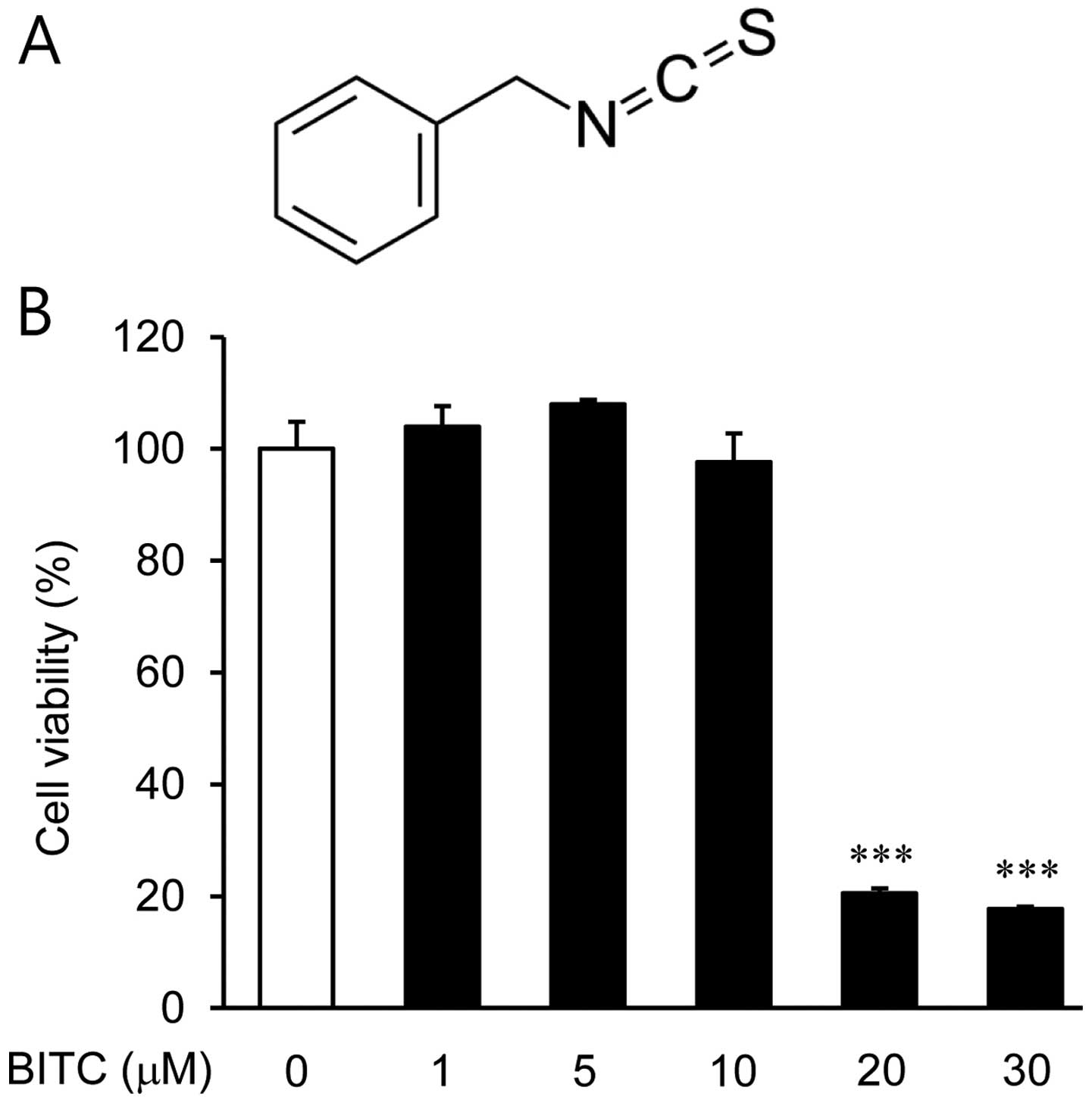
Among several isothiocyanates, BITC (C8H7NS)
(Fig. 1A) is an effector molecule
in many cruciferous vegetable defense systems with antioxidant,
antitumor and anti-inflammatory activity (16–18). It has been indicated that BITC
exhibits anti-inflammatory activities (18); however, to the best of our
knowledge, no studies to date have examined the effects of BITC on
neuroinflammation, and in particular, through inflammasome
mediation. In the present study, we examined the inhibitory effects
of BITC against IL-1β-induced expression in LPS-stimulated BV2
microglial cells, as well as its effects on intracellular signaling
pathways, specifically inflammasome components.
Materials and methods
Reagents
We purchased LPS, BITC, diphenyleneiodonium (DPI)
and N-acetyl-L-cysteine (NAC) from Sigma Chemical Co. (St. Louis,
MO, USA); YCG 063 was obtained from Millipore (Billerica, MA, USA).
An antibody against nuclear factor-κB (NF-κB) (cat. no. 14-6731)
was obtained from eBioscience (San Diego, CA, USA). The antibody
against NLRP3 (cat. no. AG-20B-0014) was purchased from AdipoGen
(San Diego, CA, USA). An antibody against IL-1β (cat. no.
AF-401-NA) was purchased from R&D Systems (Minneapolis, MN,
USA). An antibody against caspase-1 (cat. no. sc-514) was purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
CellTiter-Glo® Luminescent assay was purchased from
Promega (Madison, WI, USA).
Cell culture
The murine BV2 cell line, obtained from Professor
Eun-Hye Joe (Ajou University School of Medicine, Suwon, Korea), was
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 µg/ml streptomycin at 37°C in a humidified incubator
with 5% CO2. Confluent cultures were passed using
trypsinization. For the experiments, the cells were washed twice
with warm DMEM (without phenol red) and cultured in serum-free
medium for 16 h prior to the treatments. In all the experiments,
the cells were treated with various concentrations (1, 5 and 10
µM) of BITC for various periods of time prior to stimulation
with LPS (1 µg/ml) for the indicated periods of time.
Determination of cell viability
Cell viability was assessed using the Cell Counting
kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan). Briefly,
wells containing 2×105 cells/ml were treated with BITC
(0, 1, 5, 10, 20 and 30 µM). Following incubation for 24 h,
the cells were washed twice with phosphate-buffered saline (PBS).
CCK-8 was then added to each well followed by incubation at 37°C
for 1 h followed by an analysis at 450 nm using a microplate reader
(Model EL800; Bio-Tek Instruments, Winooski, VT, USA).
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA). Total RNA (1.0 µg) which
was obtained from the cells was reverse transcribed using M-MLV
reverse transcriptase (Promega) to produce cDNA. The RT-generated
cDNA encoding the IL-1β, NLRP3 and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) genes was amplified using PCR and selective
primers (Table I). Following
amplification, portions of the PCR reactions were subjected to
agarose gel electrophoresis.
 | Table IInformation on primers used in
RT-PCR. |
Table I
Information on primers used in
RT-PCR.
| Genes | NCBI no. | Primer sequences
(5′–3′) | Size
(bp) |
|---|
| IL-1β | NM_008361 | F:
CTCGTGCTGTCGGACCCATAT
R: TTGAAGACAAACCGC TTTTCCA | 254 |
| NLRP3 | NM_145827 | F:
CTGTGTGTGGGACTGAAGCAC
R: GCAGCCCTGCTGTTTCAGCAC | 543 |
| GAPDH | NM_001289726 | F:
TTCACCACCATGGAGAAGGC
R: GGCATGGACTGTGGTCATGA | 237 |
Measurement of ATP levels
The total ATP content was measured using the
CellTiter-Glo® Luminescence assay kit following the
manufacturer's instructions. Briefly, the assay buffer and
substrate were equilibrated to room temperature. The buffer was
transferred and gently mixed with the substrate to obtain a
homogeneous solution. Twenty microliters of the culture medium and
20 µl of the assay reagent were added to each well (384-well
plate), and the content was gently mixed under light protection on
an orbital shaker. After 10 min, the luminescence was measured
using a Microplate Reader (SpectraMax L; Molecular Devices, Devon,
UK) at 570 nm.
Enzyme-linked immunosorbent assay
(ELISA)
The level of IL-1β expression was measured using an
ELISA kit (R&D Systems). The cells were treated with various
concentrations of NAC, DPI and YCG 063 for 1 h prior to LPS
stimulation (1 µg/ml). Following incubation for 24 h, the
culture supernatants were collected, and the IL-1β quantity was
measured. The results of ELISA were quantified using an ELISA plate
reader (Model EL800; Bio-Tek Instruments) at 450 nm, which was
corrected for absorbance at 540 nm in accordance with the
manufacturer's instructions.
Western blot analysis
The cells were washed 3 times with PBS and lysed
with lysis buffer (Mammalian Cell-PE LB; G-Biosciences, St. Louis,
MO, USA). Equal quantities of protein were separated on 10% sodium
dodecyl sulfate (SDS)-polyacrylamide minigels and transferred onto
nitrocellulose membranes. Following incubation with the appropriate
primary antibody (IL-1β, NLRP3, caspase-1 and NF-κB), the membranes
were incubated for 1 h at room temperature with a secondary
antibody [goat anti-rabbit IgG (cat. no. 31460; Pierce, Rockford,
IL, USA) goat anti-mouse IgG (cat. no. sc-2031; Santa Cruz
Biotechnology, Inc.)] conjugated to horseradish peroxidase.
Following 3 washes in Tris-buffered saline Tween-20 (TBST), the
immunoreactive bands were visualized using the ECL detection
system.
Electrophoretic mobility shift assay
(EMSA)
Nuclear extract was prepared using the NE-PER
nuclear extraction reagent (Pierce). An oligonucleotide containing
the immunoglobulin κ-chain binding site (κB, 5′-GATCTCAGAGGGGACTTT
CCGAGAGA-3′) was synthesized as a probe for the gel retardation
assay. A non-radioactive method in which the 3′ end of the probe
was labeled with biotin was used (Pierce). The binding reactions
contained 5 µg of nuclear extract protein, buffer (10 mM
Tris, pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM
dithiothreitol, 0.05% Nonidet P-40, and 2.5% glycerol), 50 ng of
poly(dI-dC) and 20 fM of the biotin-labeled DNA. The reactions were
incubated for 20 min at room temperature in a final volume of 20
µl. The competition reactions were performed by the addition
of a 100-fold excess of unlabeled κB to the reaction mixture. The
mixture was then separated using electrophoresis on a 5%
polyacrylamide gel in 0.5X Tris-borate buffer and transferred to
nylon membranes. The biotin-labeled DNA was detected using a
LightShift chemiluminescent EMSA kit (Pierce).
Statistical analysis
Data values represent the means ± standard deviation
(SD). To analyze the data produced from the experiments with 2
independent variables, one-way analysis of variance (ANOVA) was
performed using GraphPad Prism software (GraphPad Software, La
Jolla, CA, USA). Values of p<0.05, p<0.01 and p<0.001 were
considered to indicate statistically significant differences.
Results
Effects of BITC on BV2 microglial cell
viability
Initially, we examined the viability of the BV2
microglial cells treated with BITC (1, 5, 10, 20 and 30 µM)
by CCK-8 assay. Treatment of the BV2 microglial cells with up to 10
µM BITC did not produce any cytotoxic effects, whereas cell
viability was significantly decreased by 80 and 82% following
treatment with 20 and 30 µM BITC, respectively (Fig. 1B). Based on these results, BITC at
concentrations of 1, 5 and 10 µM was used in the subsequent
experiments.
Effects of BITC on IL-1β expression in
LPS-stimulated BV2 microglial cells
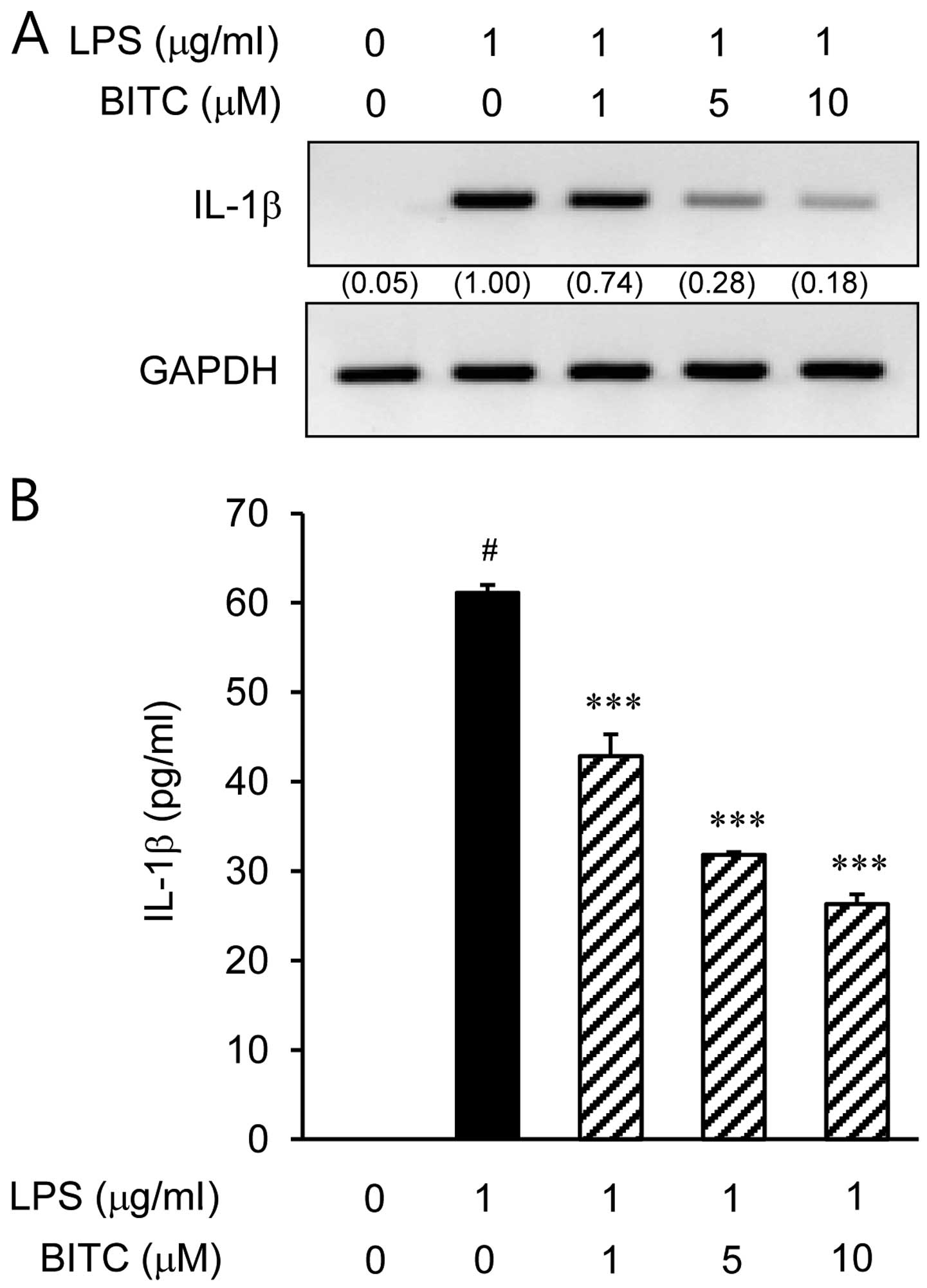
The IL-1β expression levels increased considerably
following the stimulation of BV2 microglial cells with LPS
(Fig. 2). The inhibitory effects
of BITC on IL-1β mRNA and protein expression were determined using
RT-PCR and ELISA, respectively. The IL-1β mRNA levels were markedly
upregulated after 3 h of LPS (1 µg/ml) stimulation, and BITC
significantly decreased the IL-1β mRNA expression levels in the
LPS-stimulated BV2 microglial cells in a concentration-dependent
manner (Fig. 2A). To evaluate the
effects of BITC on IL-1β protein expression in the LPS-stimulated
BV2 microglial cells, the cells were treated with BITC (1, 5 and 10
µM) for 1 h prior to LPS stimulation for 48 h. Treatment
with BITC suppressed the LPS-induced increase in IL-1β protein
expression in a concentration-dependent manner (Fig. 2B). The results of ELISA revealed
that the reduction in IL-1β protein levels correlated with a
reduction i the corresponding mRNA levels.
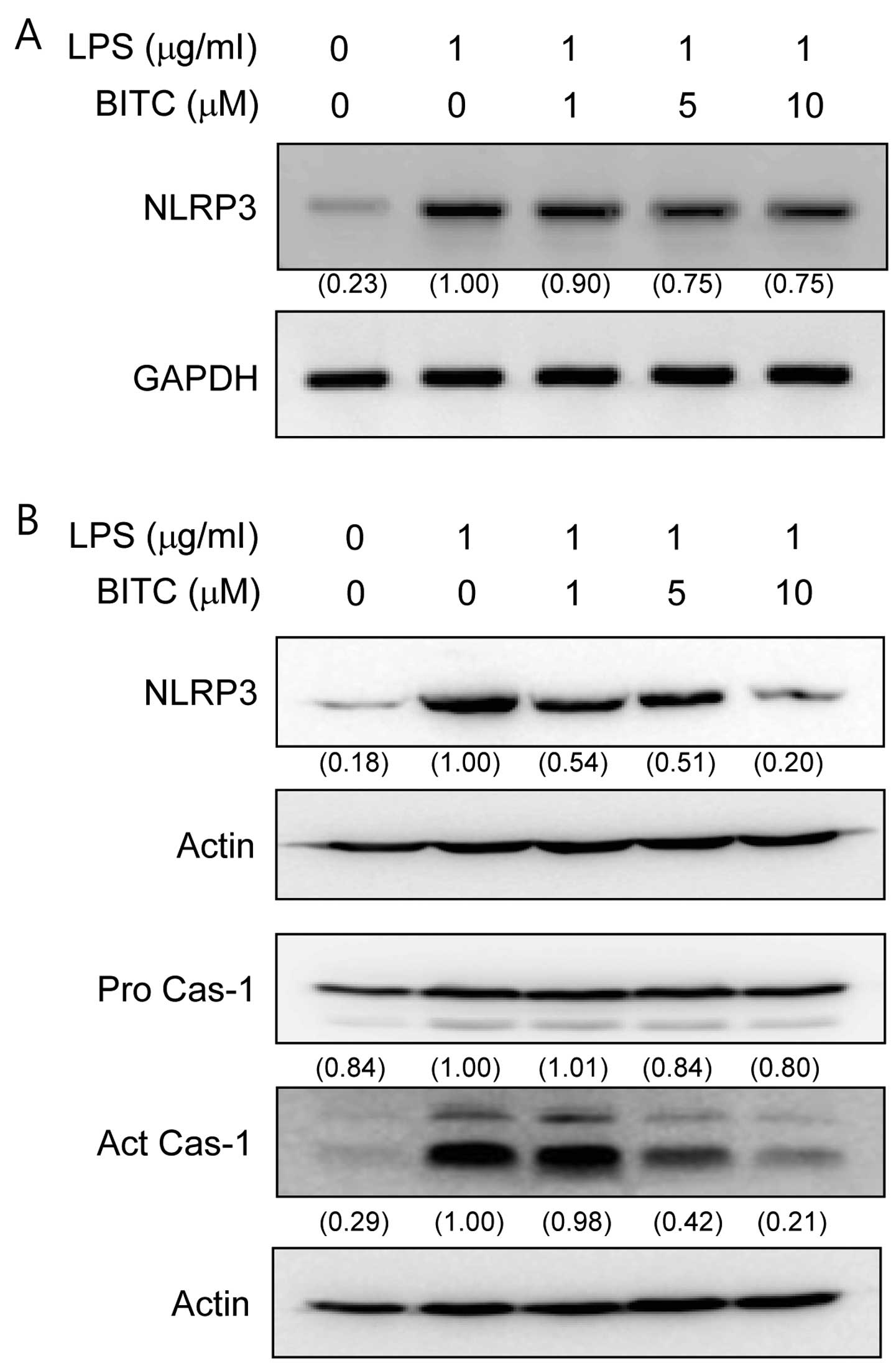
Effects of BITC on NLRP3 and caspase-1
activation in LPS-stimulated BV2 microglial cells
To determine whether BITC affects NLRP3 and
caspase-1 activation, the BV2 microglial cells were stimulated with
LPS in the presence or absence of BITC. LPS significantly increased
NLRP3 mRNA expression (Fig. 3A).
However, treatment with BITC attenuated the increase in NLRP3 mRNA
expression. To evaluate the effects of BITC on NALP3 and caspase-1
protein expression in the LPS-stimulated BV2 microglial cells, we
pre-treated the cells with BITC (1, 5 and 10 µM) prior to
stimulation with LPS. Treatment with BITC suppressed the
LPS-induced production of NLRP3 and caspase-1 (the subunit p10)
activation in a concentration-dependent manner (Fig. 3B).
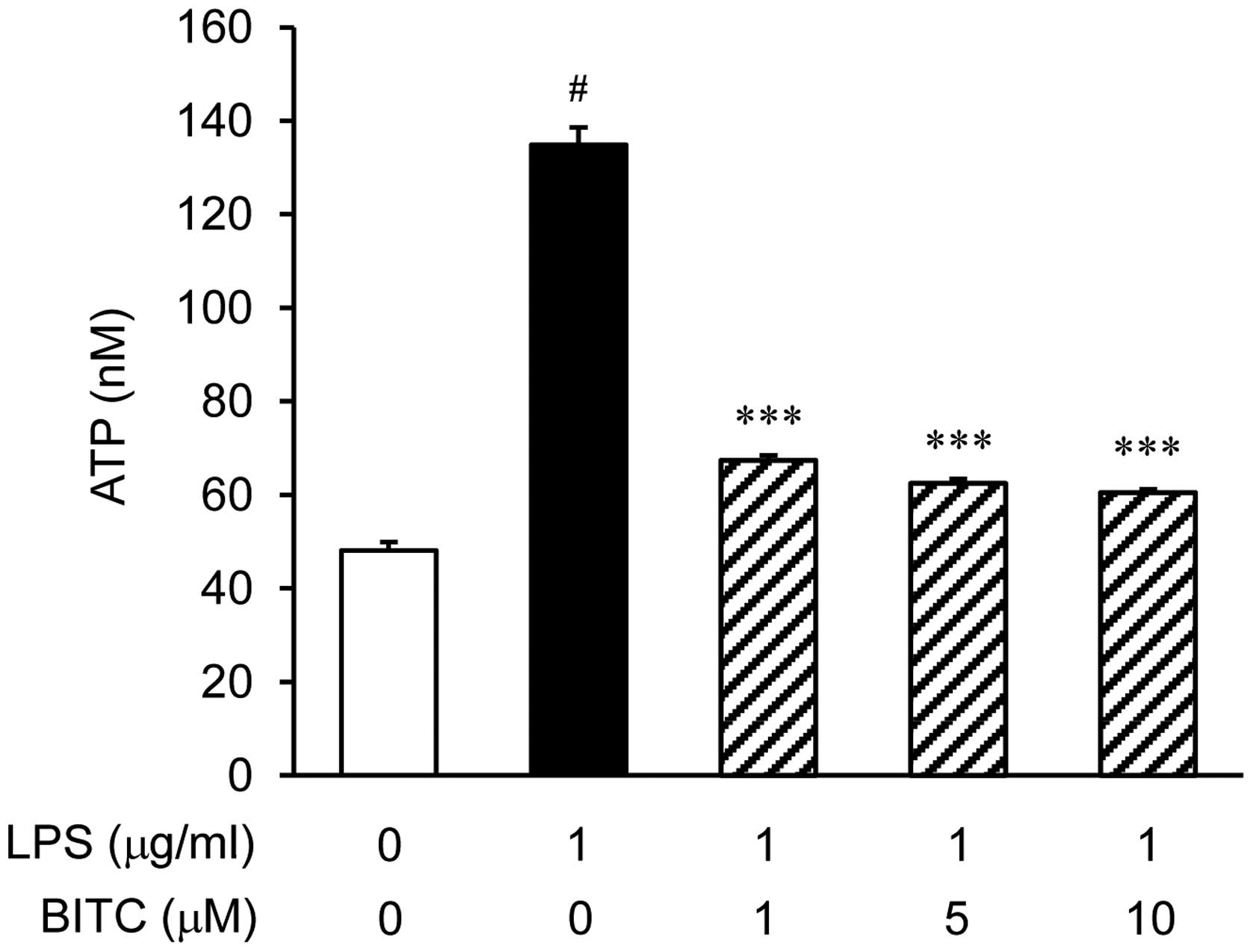
Effects of BITC on ATP levels in
LPS-stimulated BV2 microglial cells
To quantify the total extracellular ATP levels, the
BV2 microglial cells were stimulated with LPS in the presence or
absence of BITC. LPS significantly increased the ATP levels
(Fig. 4). To examine the effects
of BITC on the ATP levels in LPS-stimulated BV2 microglial cells,
we pre-treated the cells with BITC (1, 5 and 10 µM) prior to
stimulation with LPS. Treatment with BITC prevented the LPS-induced
increase in ATP levels.
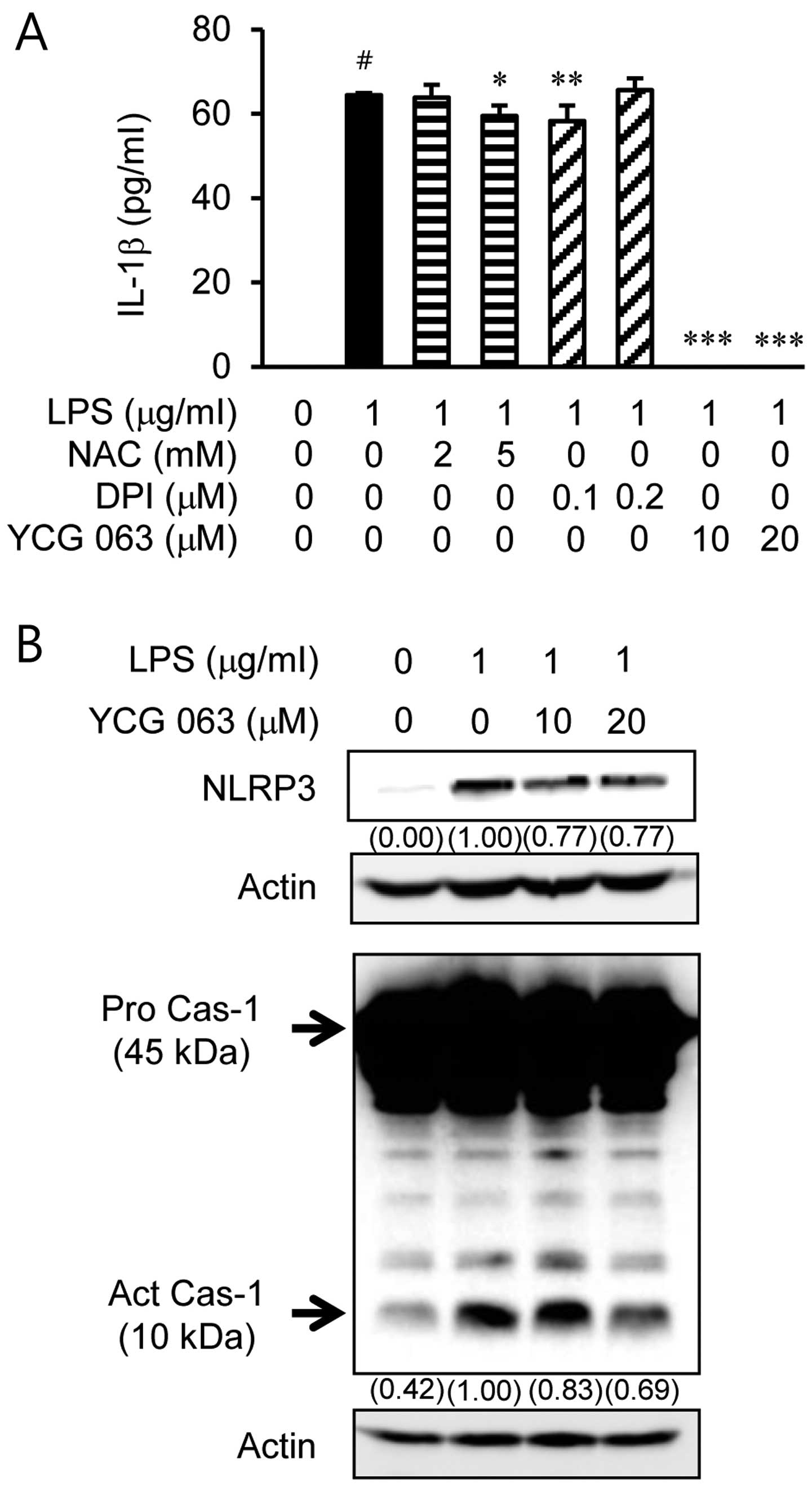
Involvement of mitochondrial ROS in NLRP3
and caspase-1 activation
We examined whether the LPS-induced IL-1β production
is associated with ROS generation (Fig. 5). First, the BV2 microglial cells
were stimulated with LPS for 24 h in the presence or absence of ROS
inhibitors, such as the ROS scavenger, NAC (2 or 5 mM), the NADPH
oxidase inhibitor, DPI (0.1 or 0.2 µM), and the
mitochondrial ROS inhibitor, YCG 063 (10 or 20 µM) (Fig. 5A). YCG 063 significantly inhibited
the LPS-induced production of IL-1β. However, IL-1β expression was
not altered in the cells pre-treated with NAC and DPI. To further
determine the involvement of mitochondrial ROS in the inflammasome
pathway, we analyzed inflammasome activation in the BV2 microglial
cells. The immunoblot data showed that treatment with YCG 063
suppressed inflammasome activation, including NLRP3 and the active
form of caspase-1 (Fig. 5B).
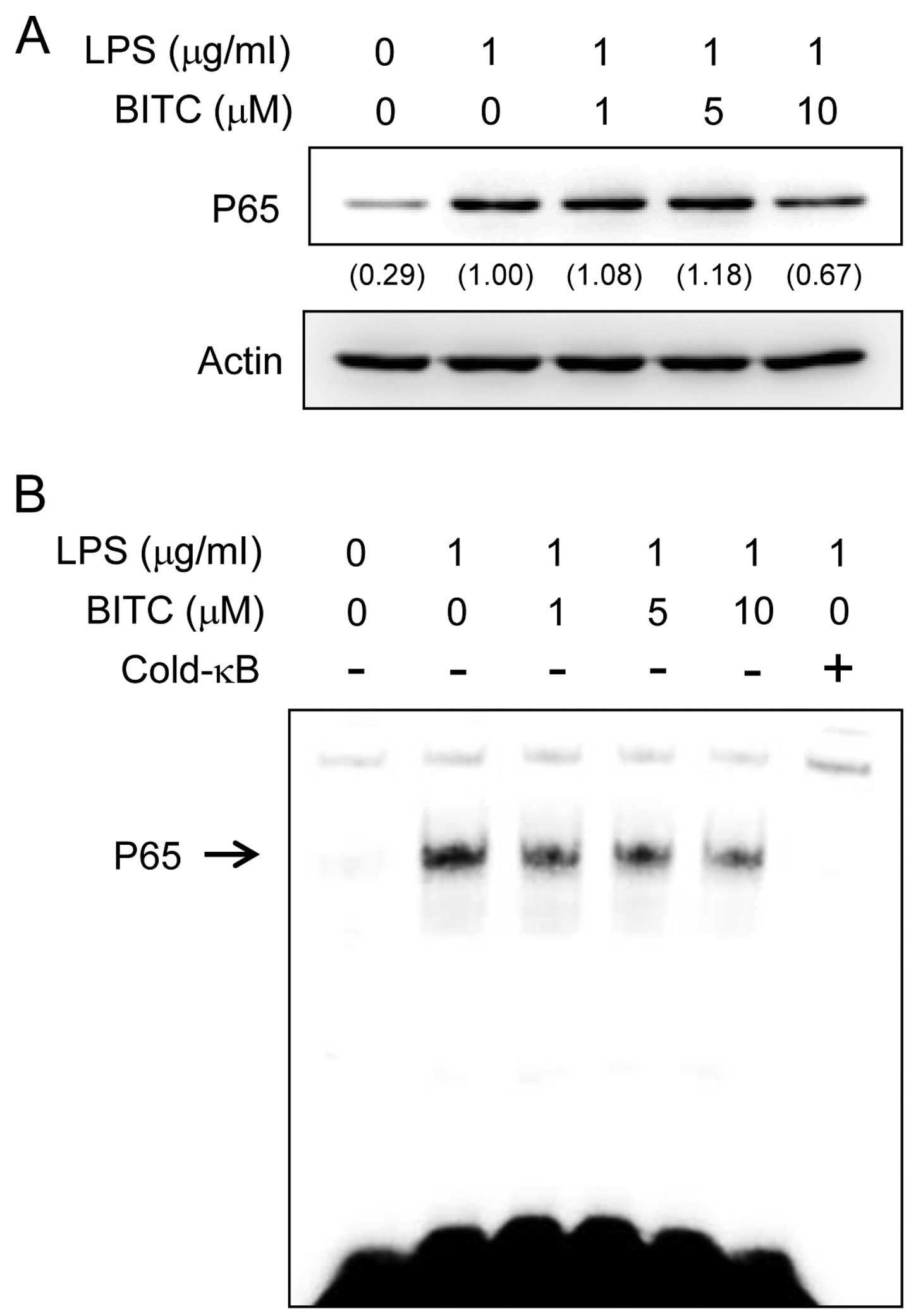
Effects of BITC on NF-κB activation in
LPS-stimulated BV2 microglial cells
The production of IL-1β is regulated by the
transcription factor, NF-κB. Furthermore, NF-κB plays a critical
role in priming the NLRP3 inflammasome (19). Therefore, to elucidate the
mechanisms through which BITC affects IL-1β expression, we examined
the effects of BITC on NF-κB activation. We examined the effects of
BITC on LPS-induced NF-κB p65 nuclear translocation as NF-κB
translocation to the nucleus is required for NF-κB-dependent
transcription following LPS stimulation. The nuclear localization
of NF-κB p65 was examined by western blot analysis. The results
revealed that stimulation of the BV2 microglial cells LPS strongly
induced NF-κB p65 nuclear localization. The LPS-induced NF-κB p65
translocation was abolished by pre-treatment of the cells with BITC
(Fig. 6A). We then examined the
effects of BITC on the DNA-binding activity of NF-κB by EMSA
(Fig. 6B); stimulation with LPS
significantly increased the DNA-binding activity of NF-κB, whereas
pre-treatment wiht BITC reduced the LPS-induced NF-κB DNA-binding
activity.
Discussion
Neuroinflammation may be a common characteristic of
various neurological and neurodegenerative disorders through
release of proinflammatory cytokines and chemokines (20). A recent study demonstrated that
inflammasome-mediated inflammation is involved in infectious
diseases affecting the central nervous system (CNS) (21). In this respect, the regulation of
inflammasome-mediated inflammatory pathways involved in infectious
diseases affecting the CNS has gained attention. In this study, we
investigated the inhibitory effects of BITC on IL-1β production in
E. coli LPS-stimulated BV2 microglia. Additionally, we
explored the several mechanisms involved in the inhibitory effects
of BITC, specifically inflammasome-associated pathways.
Microglia are the resident marcrophage cells and are
widely distributed in the brain. In response to brain
neuroinflammatory stimuli, activated microglia can overproduce
pro-inflammatory and/or neurotoxic factors, including
pro-inflammatory cytokines [IL-1, IL-6 and tumor necrosis factor-α
(TNF-α)], nitric oxide (NO), prostaglandin E2
(PGE2) and ROS (22).
These factors are involved in pathological conditions of various
neurodegenerative diseases, such as AD, PD, MS, trauma and cerebral
ischemia (23,24). Thus, reducing pro-inflammatory
mediators in microglia may attenuate the severity of
neurodegenerative disorders (25,26). Activated microglia are major
cellular sources of the pro-inflammatory and/or cytotoxic factors
that lead to neuronal damage in the CNS. Among the pro-inflammatory
cytokines involved in neuroinflammation, IL-1β results from the
inflammasome activation pathway. In this study, we demonstrate the
inhibitory effects of BITC on low levels of IL-1β production in
ultrapure E. coli LPS-stimulated BV2 microglia without the
addition of extracellular ATP.
LPS induces neuroinflammation by activating
inflammatory cells, including astrocytes and microglial cells
(27,28). In a previous study, systemically
injecting LPS led to neuroinflammatory responses in the brain,
leading to amyloid-β accumulation (Aβ), which is toxic to neurons
(29). IL-1β is produced in
LPS-stimulated BV2 microglia. The present study demonstrated that
BITC inhibited IL-1β production in LPS-stimulated BV2 microglia
(Fig. 2). The inflammasome is
required for IL-1β maturation in LPS-stimulated BV2 microglia. In
this regard, we investigated whether BITC inhibits IL-1β expression
by suppressing inflammasome activation. The cells were stimulated
with LPS followed by the assembly and activation of the
inflammasome, which facilitated caspase-1 activation. Without
extracellular ATP, the expression of NLRP3 and active caspase-1
markedly increased in response to LPS stimulation; however,
treatment with BITC ameliorated this increase in a dose-dependent
manner (Fig. 3). Therefore, the
regulation of IL-1β production by BITC may be an effective means of
inhibiting neuroinflammation through the attenuation of
inflammasome activation.
Cell priming with LPS is necessary for ATP binding
to surface-expressed P2X7 purinergic receptors (P2X7Rs), which are
ATP-gated non-selective cation channels, to induce inflammasome
activation, which results in IL-1β secretion (30). A recent study showed that P2X7Rs
contribute to various CNS pathologies (31). ATP, a damage associated molecular
pattern, is released by any type of cell injury. ATP binding to
P2X7R facilitates K+ efflux, which then activates the
NLRP3 inflammasome (32). To
determine whether BITC inhibits IL-1β production by altering ATP
secretion, we measured the levels of secreted ATP in LPS-stimulated
BV2 microglia. BITC dereased ATP secretion in a
concentration-dependent manner (Fig.
4).
Although certain scholars debate this point, the
field generally accepts the in vitro macrophage studies
indicating that the activation and release of IL-1β via an
NLRP3-inflammasome-dependent response requires two distinct
signals: i) first, a priming signal can be triggered by
pathogen-associated molecular pattern (PAMP) molecules, such as
LPS, that target toll-like receptors and NF-κB, which leads to
pro-IL-1β synthesis; ii) second, a signal can be derived from P2X7R
activation, which leads to caspase-1 activation and the release of
IL-1β (33,34). Therefore, LPS, the first signal,
combined with ATP, the second signal, are commonly used to induce
IL-1β production in microphages in vitro. However, LPS
stimulation alone upregulated IL-1β production in our in
vitro study (Fig. 2). In
addition, we found that stimulation with LPS alone induced NLRP3
and caspase-1 activation (Fig.
3). Therefore, we examined whether LPS stimulation alone can
induce ATP secretion. Of note, in our in vitro study, LPS
stimulation alone significantly upregulated ATP secretion (Fig. 4). ATP acted in an autocrine mode
when LPS stimulation alone was used for stimulation. Furthermore,
while P2X7R did not induce expression with LPS stimulation, at
almost basal levels, the BV2 cells constitutively expressed P2X7R
(data not shown). However, BITC decreased ATP secretion in a
concentration-dependent manner in the LPS-stimulated BV2 microglia
(Fig. 4).
NLRP3 inflammasome activation has been widely
implicated in ROS and NF-κB signaling (19,35). Therefore, NF-κB signaling and ROS
levels were examined in this study. It has been proposed that ROS
are an actual trigger for NLRP3 inflammasome assembly (36). Furthermore, potassium efflux
triggers ROS production in human granulocytes (37). To investigate whether ROS
production was responsible for the enhanced IL-1β production, we
stimulated the cells with LPS in the presence of a mitochondrial
ROS inhibitor or total ROS scavenger. In the present study, YCG
063, a mitochondrial ROS inhibitor, inhibited IL-1β production.
However, NAC or DPI did not attenuate ROS production. In previous
studies, mitochondrial-derived ROS were shown to be associated with
NLRP3 activation (38–40). These data suggest that BITC
attenuates NLRP3 activation via mitochondria-generated ROS
inhibition. Furthermore, a previous study reported that NF-κB is
involved in IL-1β, NLRP3 and caspase-1 expression (41). In previous studies, LPS-induced
neuroinflammation was associated with upregulated NF-κB expression
(42,43). Consistent with these studies, we
investigated whether the treatment of LPS-stimulated BV2 microglia
with BITC inhibits NF-κB activation. We found that BITC inhibited
NF-κB activation. Collectively, based on these previous
observations and our results, BITC likely attenuates IL-1β
production and NLRP3 inflammasome activation by inhibiting
mitochondrial ROS generation and NF-κB activation. Our results are
consistent with those of previous publications, showing that LPS or
NF-κB prime NLRP3 complex formation and ROS activate the NLRP3
complex (19,30,44).
In conclusion, our data demonstrate that BITC
decreases IL-1β production in activated BV2 microglia. Our data
also show that the decreased IL-1β production and the inhibition of
NLRP3 inflammasome activation is associated with the attenuation of
mitochondrial ROS generation and NF-κB activation by treatment with
BITC. Thus, treatment with BITC may be an effective novel
therapeutic strategy with which to combat inflammation-associated
pathological damage which occurs due to LPS by targeting
inflammasome-mediated signaling pathways. Further studies are
warranted however, to determine whether BITC suppresses LPS-induced
inflammasome-related neuroinflammation in vivo.
Acknowledgments
This study was supported by the Basic Science
Research program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(no. 2013R1A1A4A01011649).
References
|
1
|
Dinarello CA: A clinical perspective of
IL-1β as the gatekeeper of inflammation. Eur J Immunol.
41:1203–1217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li L, Fei Z, Ren J, Sun R, Liu Z, Sheng Z,
Wang L, Sun X, Yu J, Wang Z, et al: Functional imaging of
interleukin 1 beta expression in inflammatory process using
bioluminescence imaging in transgenic mice. BMC Immunol. 9:492008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Allan SM, Tyrrell PJ and Rothwell NJ:
Interleukin-1 and neuronal injury. Nat Rev Immunol. 5:629–640.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cappellano G, Carecchio M, Fleetwood T,
Magistrelli L, Cantello R, Dianzani U and Comi C: Immunity and
inflammation in neurodegenerative diseases. Am J Neurodegener Dis.
2:89–107. 2013.PubMed/NCBI
|
|
5
|
Lu X, Ma L, Ruan L, Kong Y, Mou H, Zhang
Z, Wang Z, Wang JM and Le Y: Resveratrol differentially modulates
inflammatory responses of microglia and astrocytes. J
Neuroinflammation. 7:462010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sheng JG, Bora SH, Xu G, Borchelt DR,
Price DL and Koliatsos VE:
Lipopolysaccharide-induced-neuroinflammation increases
intracellular accumulation of amyloid precursor protein and amyloid
beta peptide in APPswe transgenic mice. Neurobiol Dis. 14:133–145.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cassel SL, Joly S and Sutterwala FS: The
NLRP3 inflammasome: A sensor of immune danger signals. Semin
Immunol. 21:194–198. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Nardo D and Latz E: NLRP3 inflammasomes
link inflammation and metabolic disease. Trends Immunol.
32:373–379. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Englezou PC, Rothwell SW, Ainscough JS,
Brough D, Landsiedel R, Verkhratsky A, Kimber I and Dearman RJ:
P2X7R activation drives distinct IL-1 responses in dendritic cells
compared to macrophages. Cytokine. 74:293–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mariathasan S, Weiss DS, Newton K, McBride
J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM and
Dixit VM: Cryopyrin activates the inflammasome in response to
toxins and ATP. Nature. 440:228–232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schroder K, Zhou R and Tschopp J: The
NLRP3 inflammasome: A sensor for metabolic danger? Science.
327:296–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferrari D, Pizzirani C, Adinolfi E, Lemoli
RM, Curti A, Idzko M, Panther E and Di Virgilio F: The P2X7
receptor: A key player in IL-1 processing and release. J Immunol.
176:3877–3883. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lam TK, Gallicchio L, Lindsley K, Shiels
M, Hammond E, Tao XG, Chen L, Robinson KA, Caulfield LE, Herman JG,
et al: Cruciferous vegetable consumption and lung cancer risk: A
systematic review. Cancer Epidemiol Biomarkers Prev. 18:184–195.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang L, Paonessa JD, Zhang Y, Ambrosone CB
and McCann SE: Total isothiocyanate yield from raw cruciferous
vegetables commonly consumed in the United States. J Funct Foods.
5:1996–2001. 2013. View Article : Google Scholar
|
|
15
|
Wu X, Zhou QH and Xu K: Are
isothiocyanates potential anticancer drugs? Acta Pharmacol Sin.
30:501–512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee Y, Kim YJ, Choi YJ, Lee JW, Lee S and
Chung HW: Enhancement of cisplatin cytotoxicity by benzyl
isothiocyanate in HL-60 cells. Food Chem Toxicol. 50:2397–2406.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lai KC, Huang AC, Hsu SC, Kuo CL, Yang JS,
Wu SH and Chung JG: Benzyl isothiocyanate (BITC) inhibits migration
and invasion of human colon cancer HT29 cells by inhibiting matrix
metalloproteinase-2/-9 and urokinase plasminogen (uPA) through PKC
and MAPK signaling pathway. J Agric Food Chem. 58:2935–2942. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee YM, Seon MR, Cho HJ, Kim JS and Park
JH: Benzyl isothiocyanate exhibits anti-inflammatory effects in
murine macrophages and in mouse skin. J Mol Med Berl. 87:1251–1261.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bauernfeind FG, Horvath G, Stutz A,
Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks
BG, Fitzgerald KA, et al: Cutting edge: NF-kappaB activating
pattern recognition and cytokine receptors license NLRP3
inflammasome activation by regulating NLRP3 expression. J Immunol.
183:787–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hagberg H, Mallard C, Ferriero DM,
Vannucci SJ, Levison SW, Vexler ZS and Gressens P: The role of
inflammation in perinatal brain injury. Nat Rev Neurol. 11:192–208.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Rivero Vaccari JP, Dietrich WD and
Keane RW: Therapeutics targeting the inflammasome after central
nervous system injury. Transl Res. 167:35–45. 2016. View Article : Google Scholar
|
|
22
|
Jung WK, Lee DY, Park C, Choi YH, Choi I,
Park SG, Seo SK, Lee SW, Yea SS, Ahn SC, et al: Cilostazol is
anti-inflammatory in BV2 microglial cells by inactivating nuclear
factor-kappaB and inhibiting mitogen-activated protein kinases. Br
J Pharmacol. 159:1274–1285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McGeer PL and McGeer EG: The inflammatory
response system of brain: Implications for therapy of Alzheimer and
other neurodegenerative diseases. Brain Res Brain Res Rev.
21:195–218. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
González-Scarano F and Baltuch G:
Microglia as mediators of inflammatory and degenerative diseases.
Annu Rev Neurosci. 22:219–240. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu B and Hong JS: Role of microglia in
inflammation-mediated neurodegenerative diseases: Mechanisms and
strategies for therapeutic intervention. J Pharmacol Exp Ther.
304:1–7. 2003. View Article : Google Scholar
|
|
26
|
Eikelenboom P and van Gool WA:
Neuroinflammatory perspectives on the two faces of Alzheimer's
disease. J Neural Transm Vienna. 111:281–294. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pascual-Lucas M, Fernandez-Lizarbe S,
Montesinos J and Guerri C: LPS or ethanol triggers clathrin- and
rafts/caveolae-dependent endocytosis of TLR4 in cortical
astrocytes. J Neurochem. 129:448–462. 2014. View Article : Google Scholar
|
|
28
|
Min KJ, Choi K and Kwon TK: Withaferin A
down-regulates lipopolysaccharide-induced cyclooxygenase-2
expression and PGE2 production through the inhibition of
STAT1/3 activation in microglial cells. Int Immunopharmacol.
11:1137–1142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lull ME and Block ML: Microglial
activation and chronic neurodegeneration. Neurotherapeutics.
7:354–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Franchi L, Eigenbrod T and Núñez G:
Cutting edge: TNF-alpha mediates sensitization to ATP and silica
via the NLRP3 inflammasome in the absence of microbial stimulation.
J Immunol. 183:792–796. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sperlágh B and Illes P: P2X7 receptor: An
emerging target in central nervous system diseases. Trends
Pharmacol Sci. 35:537–547. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Choi AJ and Ryter SW: Inflammasomes:
Molecular regulation and implications for metabolic and cognitive
diseases. Mol Cells. 37:441–448. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mariathasan S and Monack DM: Inflammasome
adaptors and sensors: Intracellular regulators of infection and
inflammation. Nat Rev Immunol. 7:31–40. 2007. View Article : Google Scholar
|
|
34
|
Guo H, Callaway JB and Ting JP:
Inflammasomes: Mechanism of action, role in disease, and
therapeutics. Nat Med. 21:677–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martinon F: Signaling by ROS drives
inflammasome activation. Eur J Immunol. 40:616–619. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tschopp J and Schroder K: NLRP3
inflammasome activation: The convergence of multiple signalling
pathways on ROS production? Nat Rev Immunol. 10:210–215. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fay AJ, Qian X, Jan YN and Jan LY: SK
channels mediate NADPH oxidase-independent reactive oxygen species
production and apoptosis in granulocytes. Proc Natl Acad Sci USA.
103:17548–17553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
39
|
Wen H, Gris D, Lei Y, Jha S, Zhang L,
Huang MT, Brickey WJ and Ting JP: Fatty acid-induced NLRP3-ASC
inflammasome activation interferes with insulin signaling. Nat
Immunol. 12:408–415. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar
|
|
41
|
Budai MM, Varga A, Milesz S, Tőzsér J and
Benkő S: Aloe vera downregulates LPS-induced inflammatory cytokine
production and expression of NLRP3 inflammasome in human
macrophages. Mol Immunol. 56:471–479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang F, Qian L, Flood PM, Shi JS, Hong JS
and Gao HM: Inhibition of IkappaB kinase-beta protects dopamine
neurons against lipopolysaccharide-induced neurotoxicity. J
Pharmacol Exp Ther. 333:822–833. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hang CH, Shi JX, Tian J, Li JS, Wu W and
Yin HX: Effect of systemic LPS injection on cortical NF-kappaB
activity and inflammatory response following traumatic brain injury
in rats. Brain Res. 1026:23–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Land WG: Transfusion-related acute lung
injury: The work of DAMPs. Transfus Med Hemother. 40:3–13. 2013.
View Article : Google Scholar : PubMed/NCBI
|