Introduction
Hantaviruses belongs to the Bunyaviridae
family (1), and cause a serious
disease in humans named hemorrhagic fever with renal syndrome
(HFRS) which occurs worldwide, particularly in Asia and Europe
(2,3). HFRS is one of the most prevalent
natural focual infectious diseases in China, and due to the severe
symptoms, this virus has the potential to be considered as a
bioterrorism agent. As virus transmission to humans occurs by
inhalation of the aerosolized virus contained in urine, feces and
saliva, by passage through mucous membranes or by bites from
infected animals (4), and no
protective vaccines or effective treatments are currently available
for human use (5), accurate and
reliable diagnostic methods are essential for disease surveillance
in order to implement adequate public health actions.
Hantaviruses are a group of enveloped, negative
sense RNA viruses (6). The genome
organization of hantaviruses is typical of other members of the
Bunyaviridae family, consisting of three negative-stranded
RNA segments with a large (L) segment encoding the viral RNA
polymerase, a medium (M) segment encoding the envelope
glycoproteins G1 and G2, and a small (S) segment encoding the viral
nucleocapsid protein (7).
Thirty genotypes of hantaviruses have been
identified worldwide. In China, seven species have been found, but
only the Hantaan virus (HTNV) and the Seoul virus (SEOV) have been
identified as the most prevalent causative agents of HFRS in this
region (6). Serological and
genetic analyses showed that HTNV and SEOV co-circulate in rodents
(1). It has been reported that
disease severity differs among cases of HFRS caused by different
hantaviruses. The infection of SEOV usually manifests as a mild
form of HFRS and results in lower case fatality rate (8). Compared with the cases caused by
SEOV, HFRS caused by HTNV is more severe with a mortality rate of
5–10% (2,4). Although SEOV-HFRS has a low case
fatality rate, complications and long-term hormonal, renal and
cardiovascular consequences occur. Therefore, it is important to
not only establish a method of diagnosis, but also to detect and
distinguish between these two virus species prior to the
development of symptoms.
Many methods have been developed for the diagnosis
of hantavirus infections. Common methods used for detection involve
immunological techniques (9),
such as the immunofluorescence antibody test (IFAT), enzyme-linked
immunosorbent assays (ELISA) or immunoblot analysis. Recently,
real-time polymerase chain reaction (PCR) assays have also been
developed (10,11). SYBR-Green I-based reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) has
been considered to be technically simpler and more readily
available than the TaqMan probe or molecular beacon-based assays
(12,13). A recent study has also suggested
that there may be an association between the hantavirus RNA load
and disease severity (14).
Furthermore, it has been suggested that quantifying the load of
infectious virions was essential for the effective treatment of
HTNV infection (15).
In the present study, the viral loads in cell
culture supernatant, infected mice blood and clinical serum samples
were quantified by SYBR-Green I-based RT-qPCR, which targeted the S
gene sequence of the HTNV and SEOV genomes. The results obtained
from two different viruses (HTNV and SEOV) were compared. The viral
RNA levels were also evaluated using serum samples obtained from
infected mice at different time points post infection.
Materials and methods
Virus, cells and mice
The virus strains used in this study were the HTNV
strain 76–118 (from our laboratory) and the SEOV strain SR-11 (a
gift from Anhui Medical University, Hefei, China). Vero E6 cells
(ATCC CRL-1586) were cultured in RPMI-1640 (Gibco, Grand Island,
NY, USA) supplemented with L-glutamine and 10% fetal calf serum
(FCS). Sixty BALB/c mice (3–4-days old) were obtained from the
Animal Research Centre at the Fourth Military Medical University
(Xi'an, China).
Design of specific primers
The primers used in this study were designed using
Oligo 6.0 software by multiplying aligning S segment nucleotide
sequences from different strains of HTNV and SEOV viruses,
respectively. The following primer pairs were used: HTNV forward,
5′-GAG CCT GGA GAC CAT CTG-3′ and reverse, 5′-CGG GAC GAC AAA GGA
TGT-3′ (the product was 144 bp); SEOV forward, 5′-GGG AAT ACA CTG
GAC CTG-3′ and reverse, 5′-GTC CTT TGA AGT CTG CCT-3′ (the product
was 159 bp). The primers were synthesized by Augct DNA-Syn
Biotechnology Co., Ltd., (Beijing, China).
RNA extraction
Total RNA from virus-infected cell cultures and mice
serum were extracted using the RNAfast1000 kit (Pioneer Biotech
Co., Ltd., Xi'an, China). The RNA from human serum samples were
extracted using the QIAamp viral RNA mini kit (Qiagen, Hilden,
Germany) according to the manufacturer's instructions. The RNA was
recovered in 20 µl nuclease-free water and stored at
−80°C.
Preparation of cRNA standard
To prepare the HTNV and SEOV cRNA standard for the
quantitative PCR assay, total RNA was extracted from cells infected
with HTNV and SEOV, respectively. The cDNA was derived from the
highly conserved S gene sequence from the HTNV and SEOV genomes,
respectively. One hundred units of SuperScript II reverse
transcriptase was used in 1X First-Strand Buffer (Invitrogen,
Carlsbad, CA, USA) at 20°C for 10 min, 42°C for 90 min, 95°C for 5
min and the reaction was cooled to 4°C. The cDNA (~1.3 kb in
length) were amplified by PCR and cloned into the pMD18T vector
(Takara, Dalian, China). The HTNV S gene sequence in the plasmid
was named HTNV-pMD18T-S and the SEOV S gene sequence in the plasmid
was named SEOV-pMD18T-S. They were confirmed by PCR and subsequent
DNA sequencing. Plasmid isolation was performed using Axygen
Plasmid Mini-Prep kits (Axygen, Union City, CA, USA). Subsequently,
the cRNA was synthesized by in vitro transcription using T7
RNA polymerase (Takara Biotechnology, Dalian, China) in accordance
with the manufacturer's instructions. The cRNA was processed twice
with DNase I (Takara Biotechnology), then the residues were
precipitated using chloroform/isoamyl alcohol and alcohol/ethanol.
The cRNA was finally dissolved in 50 µl RNase-free water and
electrophoresed in a 2% agarose gel (Biowest, Madrid, Spain).
DL3000 DNA Marker (Proteri Biotechnology Co., Ltd., Dalian, China)
was used as molecular weight marker. The cRNA was stored at
−80°C.
The cRNA were assessed for purity using the A260/280
ratio and the concentration was determined from A260 using the
spectrophotometer (6405UV; Jenway, Burlington, NJ, USA) to produce
a best-fit linear regression of the standard curve. The standards
were run in triplicate.
Quantitative PCR
The quantitative PCR step was performed using
SYBR® Premix Ex Taq™ (Takara) on a Stratagene MX3005P
real-time qPCR system (Agilent Technologies, Inc., Santa Clara, CA,
USA). The standard cycling conditions were as follows: 95°C for 10
sec, followed by 40 cycles of 95°C for 5 sec, 60°C for 20 sec and
72°C for 1 min. Quantitative PCR reaction components were set-up in
triplicate according to the manufacturer's instructions. The cycle
threshold (Ct) for each gene was determined by setting the Ct line
at the center of the logarithmic phase of amplification for that
particular amplicon.
Generation of standard curves using
quantitative PCR
The cRNA standards were 10-fold serial diluted over
range from 1.0×105 to 1.0×100
copies/µl using Easy Dilution (Takara Biotechnology). The
mean values were obtained by testing in triplicate. The standard
curves were generated by plotting Ct values versus the log copy
numbers. Regression analysis, standard curve slopes and
amplification efficiencies were calculated using automated software
(Stratagene MX3005P qPCR software).
Specificity and sensitivity
The specificity of the assay was assessed by
processing two different templates from HTNV and SEOV during
RT-qPCR. Additionally, the total cellular RNA extracts were tested
and melting curve analysis of PCR products was performed in order
to exclude the presence of unspecific products or primer dimer
synthesis. Serial dilutions in the range of 1.0×105 to
1.0×100 copies/µl cRNA standards were used in
triplicate to determine the sensitivity of the assay. To calculate
the corresponding number of RNA molecules, a cRNA reference
standard curve was processed in parallel by qPCR.
Mice infected with hantavirus
Suckling mice were infected with viral suspensions
(105 pfu) of HTNV and SEOV, respectively. Any death
occurring 24 h post infection (p.i.) was considered as a traumatic
injury and was excluded from the following experiment. Whole blood,
was extracted and left to stand for 1 h, and centrifuged at 3,000
rpm for 15 min. The serum was obtained from the supernatant. RNA
was extracted from the serum of the mice, obtained on different
days. The study received ethics approval from the Ethics Committee
of the Forth Military Medical University (Xi'an, China). The mice
infected with HTNV died from the disease onset, and the mice
infected with SEOV were sacrificed when the experiment ended.
Clinical specimens
Thirteen patients diagnosed with HFRS and 2 healthy
individuals (normal controls) at the Department of Infectious
Diseases at Tangdu Hospital (Xi'an, China) were enrolled in this
study in 2013. Peripheral blood was collected during the febrile
phase of the illness (3–7 days after the onset of fever) and the
extracted serum was stored at −80°C. The diagnosis of HFRS was
confirmed by the detection of hantavirus IgM/IgG antibodies
according to the manufacturer's instructions for Hantavirus IgG/IgM
Combo Test Card (Boson Biotech, Xiamen, China). The study received
ethics approval from the Ethics Committee of Tangdu Hospital
(Xi'an, China) and written informed consent was obtained from all
enrolled subjects.
Statistical analysis
All analyses were performed using Excel and SPSS
version 13.0 software. Data were analyzed using a
χ2-test.
Results
Preparation of the positive controls
and synthesis of the cRNA
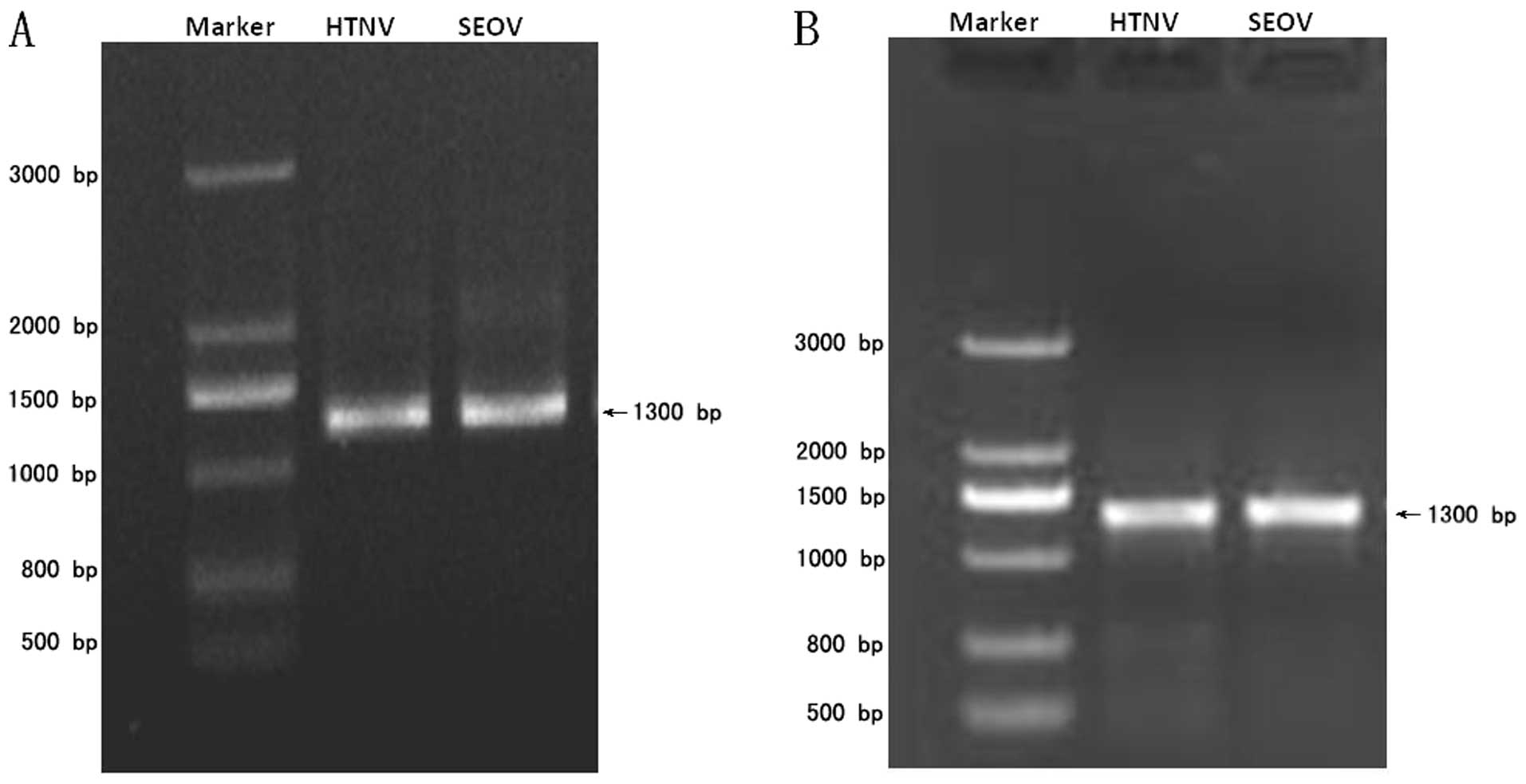
The S gene cDNA for either HTNV or SEOV were
inserted into the pMD18T vector. The successful construction of
both recombinant plasmids was confirmed by DNA sequencing. The
expected fragments of ~1.3 kb were obtained by PCR, respectively
(Fig. 1A). The cRNA were
synthesized in vitro using T7 RNA polymerase and the cDNA as
templates. Two cRNAs of ~1.3 kb were obtained, respectively
(Fig. 1B). The copy number of
cRNA was calculated according to the concentration; HTNV and SEOV
cRNA were diluted to 1×105 copies/µl,
respectively.
Standardization of SYBR-Green I-based
RT-qPCR
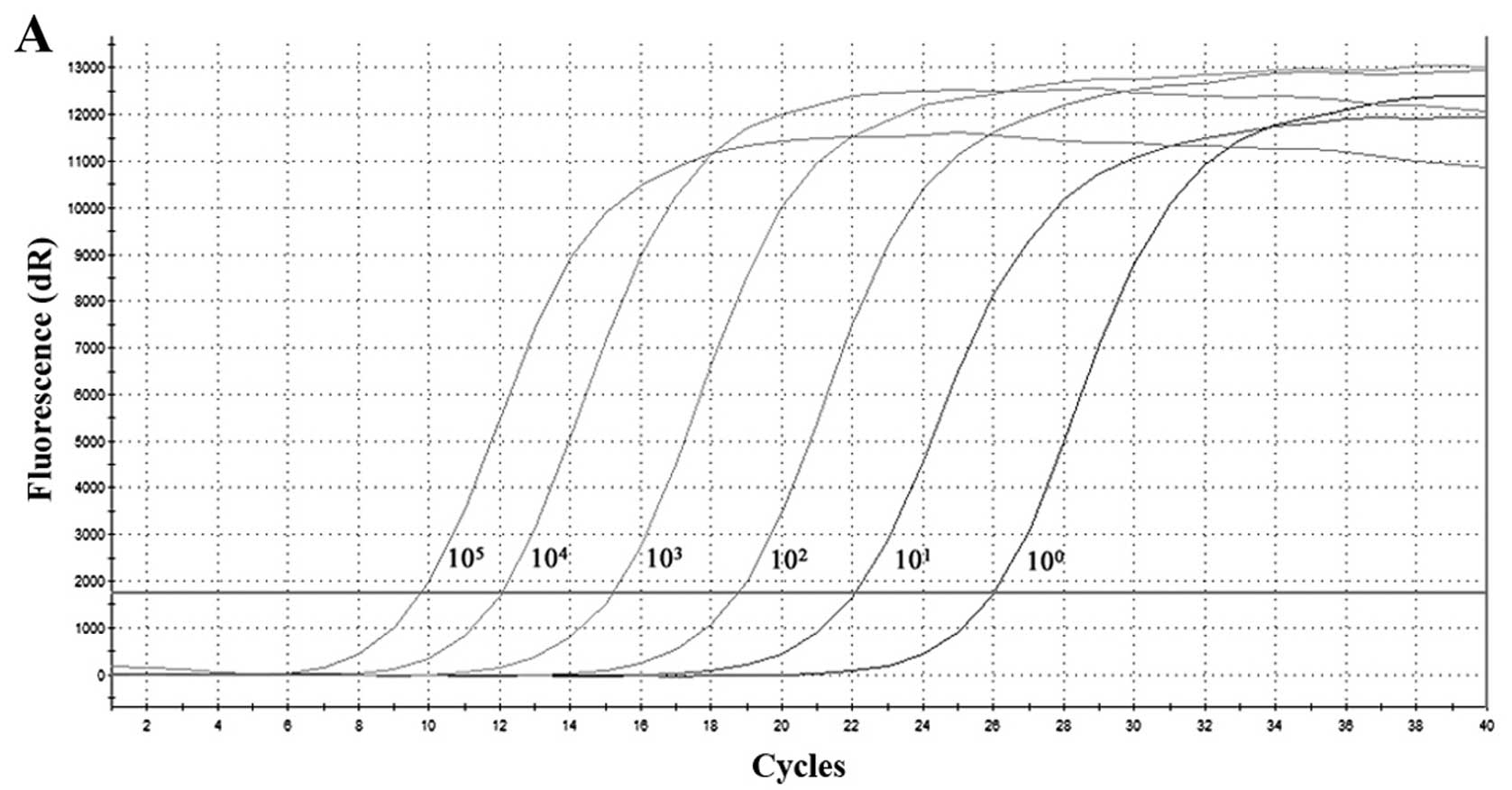
Standard curves for the detection of HTNV and SEOV
RNA by SYBR-Green I-based RT-qPCR were established. The thermal
profile of the PCR were optimized at an annealing temperature of
60°C. A primer concentration of 0.4 µM in 25 µl was
used, in a reaction scale. The RNA standards were both diluted by
10-fold serial dilutions ranging from 1×105 to
1×100 copies/µl, and the standard curves were
generated by plotting Ct versus the copy number in the
amplification chart (Fig. 2). The
mean values were linear over the entire range of HTNV cRNA
dilutions with a correlation coeeficient (R2) of 0.994,
efficiency of amplification (E) of 101.9%, and the slope of −3.278
(Fig. 2A and B). Moreover,
linearity was good for the standard curve of SEOV cRNA dilutions,
with R2 of 0.993, E of 104.8%, and the slope of −3.212
(Fig. 2D and E). The gradients of
the lines were 2–3 Ct/10-fold concentration change.
Sensitivity and specificity of SYBR-Green
I-based RT-qPCR
The standards were diluted by 10-fold serial
dilutions ranging from 1×105 to 1×100
copies/µl. According to the established assay, the minimum
detection limit of RT-qPCR for HTNV and SEOV was 101
copies/µl. Furthermore, SYBR-Green I-based RT-qPCR was
highly specific for HTNV and SEOV, respectively (Fig. 2C and F). No primer dimers or
non-specific products were found by melting curve analysis. Only
one single sharp peak was visible in both the melt peak charts.
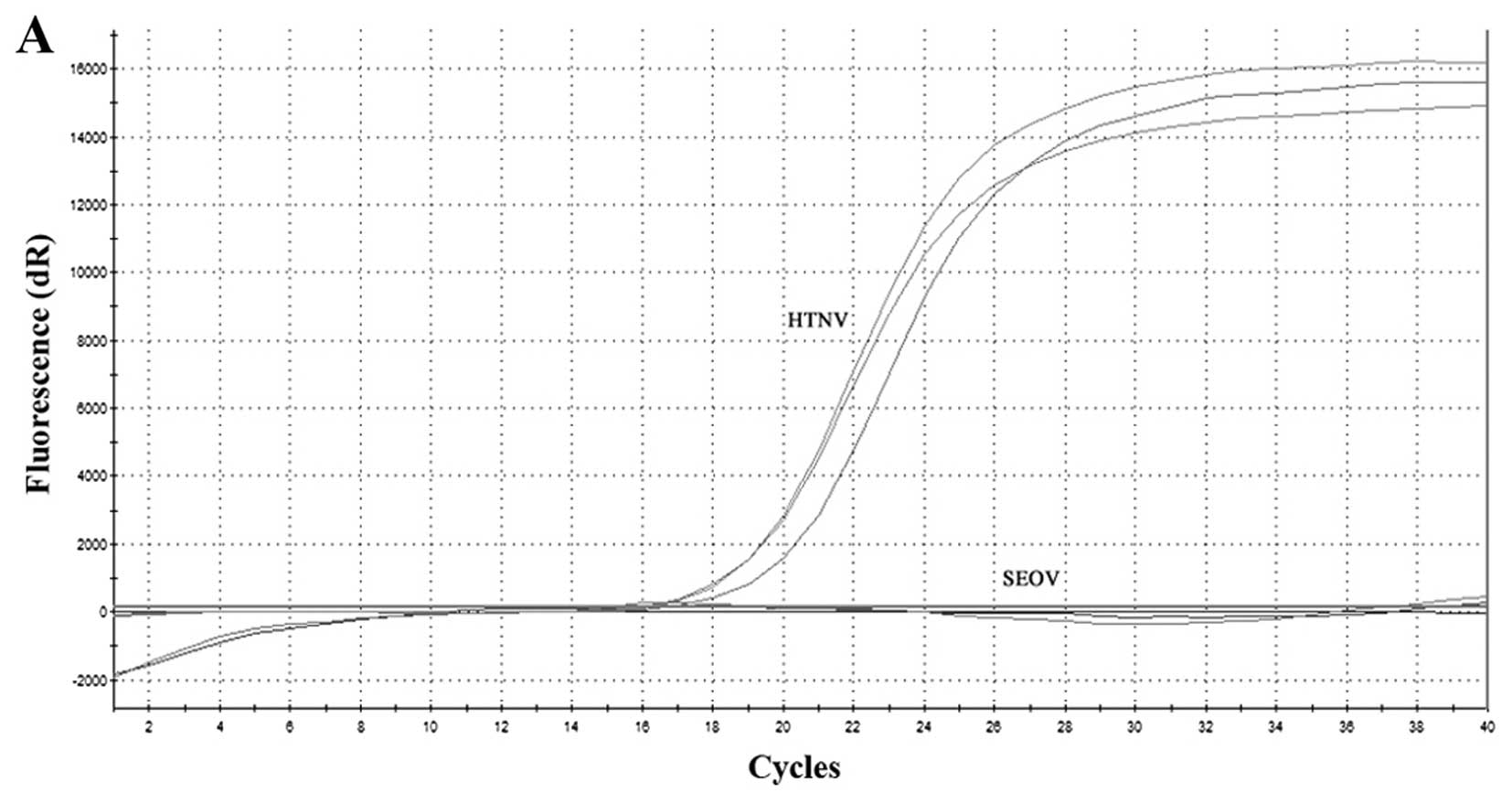
Genotyping of hantaviruses from infected
cells
The Vero E6 cells were infected with HTNV and SEOV,
respectively. There was no evidence of the virus cytopathic effect
(CPE) following infection with either of these two viruses. The
viral RNA was collected from day 4. SYBR-Green I-based RT-qPCR was
conducted according to the previously established assay. Both Ct
values of the amplification curve were obtained. No
cross-reactivity was observed between the HTNV and SEOV (Fig. 3A and C). Negative amplification
were obtained and melting curve analysis showed no sharp peak on
the melting curve chart when using the improper primers (Fig. 3B and D).
Assay performance on samples from
infected mice
In the next experiment, suckling mice were infected
with HTNV and SEOV, respectively. As shown in Fig. 4, the special amplification curve
(the Ct value for HTNV was 25–34) appeared from day 2 to 10 after
infection (Fig. 4A and B), which
demonstrated that HTNV RNA is detectable on the second day after
infection. Furthermore, the load of HTNV RNA increased with
persistent infection of virus until disease onset. The viral load
stablized on the sixth day. Compared with HTNV infection, SEOV
infection was not lethal. The Ct value of SEOV decreased post
infection; however, it started increasing after reaching its lowest
level on day 16 (Fig. 4C and D).
This was consistent with the study that SEOV infection would not
result in exposure.
Assay performance on clinical
samples
In order to evaluate the usefulness of the
SYBR-Green I-based RT-qPCR assay established herein, serum samples
from 13 HFRS patients and two normal controls were collected for
analysis. All the HFRS patients had been diagnosed from clinical
symptoms and laboratory examinations. Peripheral blood was
collected during the febrile phase of the illness (3–7 days after
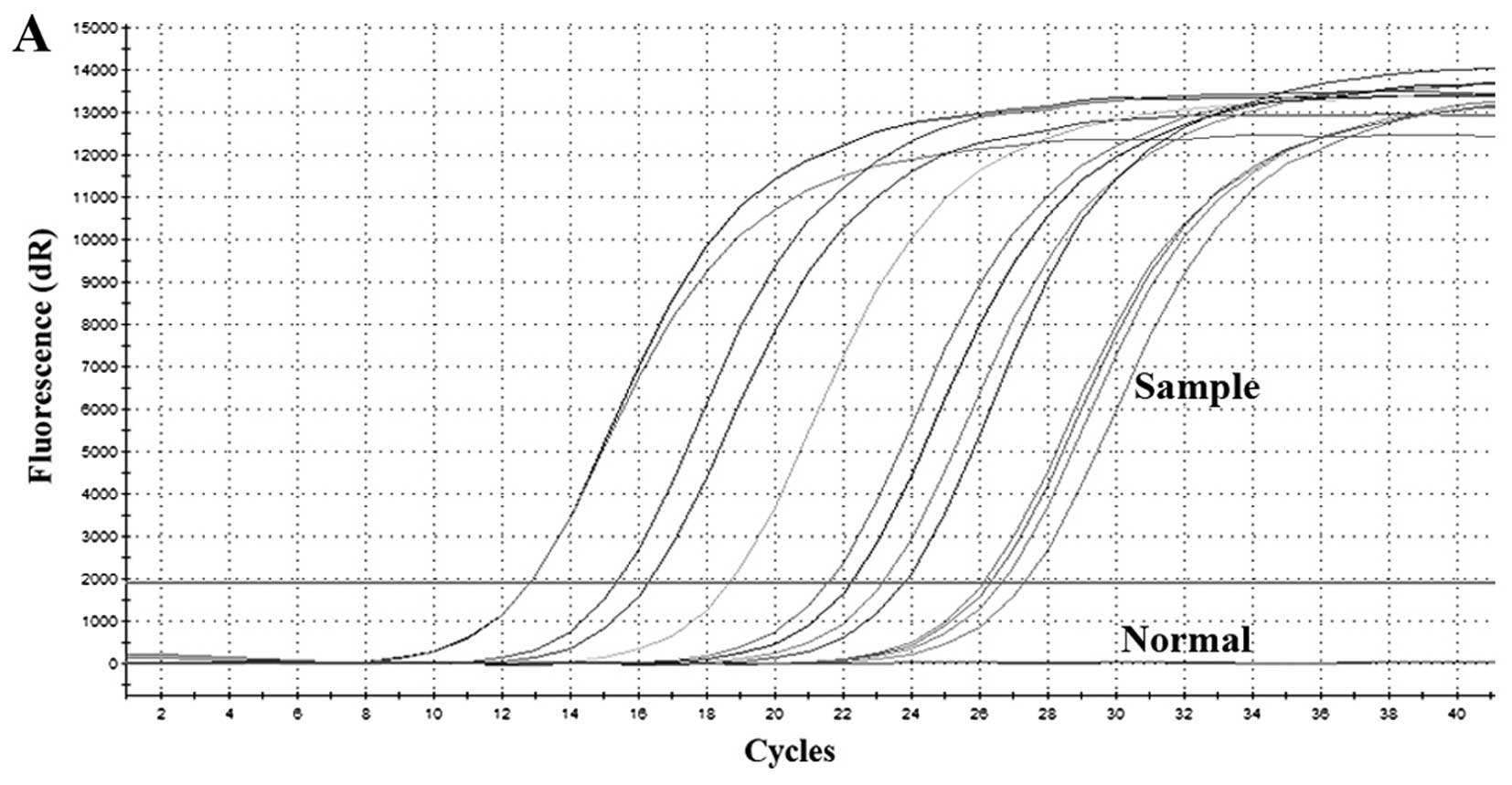
the onset of fever). As shown in Fig.
5, no positive results were obtained from the normal control
samples; however, the copy numbers of HTNV RNA in the serum samples
from HFRS patients ranged from 2.42×102 to
7.53×106. Negative results were obtained when the
samples were analyzed using SEOV primers (Table I).
 | Table IQuantification of HTNV RNA in serum
samples obtained from patients and normal controls using the
SYBR-Green I-based RTq-PCR assay. |
Table I
Quantification of HTNV RNA in serum
samples obtained from patients and normal controls using the
SYBR-Green I-based RTq-PCR assay.
| Sample no. | HTNV-IgM/IgG
antibodies Combo test | Days after onset of
fever | Severity of the HFRS
diseasea | Copies/µl of
the HTNV RNA | Copies/µl of
the SEOV RNA |
|---|
| HFRS 1 |
Positive/positive | 5 | Severe |
7.53×106 | Negative |
| HFRS 2 |
Positive/positive | 7 | Critical |
7.53×106 | Negative |
| HFRS 3 |
Positive/positive | 5 | Critical |
8.67×105 | Negative |
| HFRS 4 |
Positive/positive | 6 | Critical |
5.75×105 | Negative |
| HFRS 5 |
Positive/positive | 4 | Severe |
9.32×104 | Negative |
| HFRS 6 |
Positive/negative | 6 | Moderate |
1.34×104 | Negative |
| HFRS 7 |
Positive/positive | 5 | Severe |
8.55×103 | Negative |
| HFRS 8 |
Positive/positive | 4 | Severe |
4.15×103 | Negative |
| HFRS 9 |
Positive/negative | 3 | Mild |
2.86×103 | Negative |
| HFRS 10 |
Positive/positive | 5 | Moderate |
5.63×102 | Negative |
| HFRS 11 |
Positive/positive | 3 | Moderate |
4.78×102 | Negative |
| HFRS 12 |
Positive/negative | 4 | Mild |
3.74×102 | Negative |
| HFRS 13 |
Positive/positive | 3 | Mild |
2.42×102 | Negative |
| Normal 1 | −/− | – | – | – | – |
| Normal 2 | −/− | – | – | – | – |
Discussion
HFRS caused by hantavirus infection has been
considered to be a serious and often fatal disease, with
approximately 90% of all cases reported in China (7,16).
The genotypes of hantavirus causing HFRS in mainland China are
mainly the HTNV and the SEOV. Disease caused by HTNV is more severe
clinically, whereas SEOV infection results in a longer incubation
period and sometimes, may not lead to the onset of HFRS. Nucleic
acid-based methods, such as RT-PCR, are sensitive and allow for
species-specific virus identification. More recently, RT-qPCR was
used to identify Puumala hantavirus (PUUV) (11) as well as other hemorrhagic
fever-causing agents such as Ebola, Marburg, Lassa, Crimean-Congo
haemorrhagic fever (CCHF), dengue, Rift Valley fever (RVF) and
Yellow fever virus (YFV) (17–19). In the present study, the RT-qPCR
assay was successfully established for the detection of the HTNV
and SEOV. By means of SYBR-Green I-based RT-qPCR, the HTNV and SEOV
can be genotyped precisely without cross-reactivity. Furthermore,
individuals infected with SEOV may be diagnosed which is of value
in the prevention and treatment of SEOV-induced disease.
In real-time PCR, the synthesis of the amplification
product is monitored during the reaction allowing quantification of
the viral RNA in the sample. RT-qPCR has been proved to have
exceptional analytical sensitivity and specificity (13,15). In this study, we have established
a quantitative PCR (qPCR) assay for HTNV and SEOV using cRNAs
generated by the plasmid pMD18T-S, which contained the S gene
sequence of these two viruses. Standard curves of HTNV and SEOV
were described with excellent linear relationships over a range of
1×105 to 1×100 copies/µl cRNA with
R2=0.994 and R2=0.993, respectively. The
limit of sensitivity was 101 cRNA copies/µl. High
E values for HTNV and SEOV were shown as 101.9 and 104.8%,
respectively. These properties of the established SYBR-Green
I-based RT-qPCR assay showed an improved detection limit and that
the test is ideal for use with animal models as well as in clinical
practice.
A major disadvantage of SYBR-Green I-based RT-qPCR
is that SYBR-Green dyes intercalate into any double-stranded DNA
including primer-dimers and non-specific amplification products.
Therefore, well designed primers and melt curve analysis is
required in the SYBR-Green based assay for the discrimination of
specific products from by-products (20,21). In addition, the optimization of
annealing temperatures and primer concentrations were crucial in
preventing the non-specific amplification of other unrelated gene
products. The primers for the S gene sequence of both HTNV and SEOV
are well designed and strictly verified, the reaction assays were
optimized and only one sharp peak showed in each melt curve
analysis, indicating quite good sensitivity and specificity of the
assay and the absence of by-products. Furthermore, the risk of
contamination is greatly reduced as the PCR product is detected
within a closed tube.
Viral RNA from different specimens of infected
individuals is detectable, preferentially during the acute phase of
hantavirus infection or even prior to the signs of infection
becoming apparent. The methods were also applied in the evaluation
of viral load in samples from infected mice prior to disease onset.
Elevated levels of viral RNA have been recorded in mice suffering
from HTNV infection until the onset of disease. However, in the
present study, the level of viral RNA in the serum of mice infected
with SEOV rose first, and then fell and failed to induce the onset
of disease, which is consistent with the findings of previous
experiments performed at our laboratory (data unpublished), that
SEOV almost never induces HFRS. Therefore, patients infected with
SEOV were not included herein for analysis. Although the
pathogenesis of hantavirus infection remains unclear, these
findings indicated that the load and replication of viruses played
a pivotal role in the pathogenesis of hantavirus infection in
suckling mice and potentially during hantavirus infection in humans
based on the results of the RT-qPCR assay.
This study presents the use of the established
SYBR-Green I-based RT-qPCR assay and specific primers for the
evaluation of HTNV and SEOV RNA in vitro and in vivo.
This method proved to be highly sensitive, specific and
reproducible in cRNA positive controls, infected cells, infected
mice and clinical specimens. The low cost and rapid nature of the
technique suggest that this is a useful new tool for monitoring and
evaluating variations in viral RNA levels. The clinical application
of this method may also prove invaluable to clinicians and public
health officials who need to detect this important pathogen in
individuals affected by natural infection or man-made exposure
(bioterrorism).
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 31270978 and
31470890). It was finished at the Department of Microbiology of
Fourth Military Medical University. The authors would like to thank
all the staff at Department of Infectious Disease of Tangdu
Hospital for their support of this study.
References
|
1
|
Schmaljohn CS, Hasty SE, Harrison SA and
Dalrymple JM: Characterization of Hantaan virions, the prototype
virus of hemorrhagic fever with renal syndrome. J Infect Dis.
148:1005–1012. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jonsson CB, Figueiredo LT and Vapalahti O:
A global perspective on hantavirus ecology, epidemiology, and
disease. Clin Microbiol Rev. 23:412–441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaheri A, Henttonen H, Voutilainen L,
Mustonen J, Sironen T and Vapalahti O: Hantavirus infections in
Europe and their impact on public health. Rev Med Virol. 23:35–49.
2013. View
Article : Google Scholar
|
|
4
|
Roda Gracia J, Schumann B and Seidler A:
Climate variability and the occurrence of human puumala hantavirus
infections in Europe: a systematic review. Zoonoses Public Health.
62:465–478. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maes P, Clement J, Gavrilovskaya I and Van
Ranst M: Hantaviruses: Immunology, treatment, and prevention. Viral
Immunol. 17:481–497. 2004. View Article : Google Scholar
|
|
6
|
Kanerva M, Mustonen J and Vaheri A:
Pathogenesis of puumala and other hantavirus infections. Rev Med
Virol. 8:67–86. 1998. View Article : Google Scholar
|
|
7
|
Khaiboullina SF, Morzunov SP and St Jeor
SC : Hantaviruses: molecular biology, evolution and pathogenesis.
Curr Mol Med. 5:773–790. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Clement J, Heyman P, McKenna P, Colson P
and Avsic-Zupanc T: The hantaviruses of Europe: from the bedside to
the bench. Emerg Infect Dis. 3:205–211. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hedman K, Vaheri A and
Brummer-Korvenkontio M: Rapid diagnosis of hantavirus disease with
an IgG-avidity assay. Lancet. 338:1353–1356. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mohamed N, Nilsson E, Johansson P,
Klingström J, Evander M, Ahlm C and Bucht G: Development and
evaluation of a broad reacting SYBR-green based quantitative
real-time PCR for the detection of different hantaviruses. J Clin
Virol. 56:280–285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garin D, Peyrefitte C, Crance JM, Le Faou
A, Jouan A and Bouloy M: Highly sensitive Taqman PCR detection of
Puumala hantavirus. Microbes Infect. 3:739–745. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dash PK, Boutonnier A, Prina E, Sharma S
and Reiter P: Development of a SYBR-Green I based RT-PCR assay for
yellow fever virus: application in assessment of YFV infection in
Aedes aegypti. Virol J. 9:272012. View Article : Google Scholar
|
|
13
|
Jiang W, Yu HT, Zhao K, Zhang Y, Du H,
Wang PZ and Bai XF: Quantification of Hantaan virus with a
SYBR-Green I-based one-step qRT-PCR assay. PLoS One. 8:e815252013.
View Article : Google Scholar
|
|
14
|
Yi J, Xu Z, Zhuang R, Wang J, Zhang Y, Ma
Y, Liu B, Zhang Y, Zhang C, Yan G, et al: Hantaan virus RNA load in
patients having hemorrhagic fever with renal syndrome: correlation
with disease severity. J Infect Dis. 207:1457–1461. 2013.
View Article : Google Scholar
|
|
15
|
Wei F, Li JL, Ling JX, Chen LJ, Li N, Liu
YY, Luo F, Xiong HR, Hou W and Yang ZQ: Establishment of
SYBR-Green-based qPCR assay for rapid evaluation and quantification
for anti-Hantaan virus compounds in vitro and in suckling mice.
Virus Genes. 46:54–62. 2013. View Article : Google Scholar
|
|
16
|
Schmaljohn C and Hjelle B: Hantaviruses: a
global disease problem. Emerg Infect Dis. 3:95–104. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bird BH, Bawiec DA, Ksiazek TG, Shoemaker
TR and Nichol ST: Highly sensitive and broadly reactive
quantitative reverse transcription-PCR assay for high-throughput
detection of Rift Valley fever virus. J Clin Microbiol.
45:3506–3513. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sall AA, Macondo EA, Sène OK, Diagne M,
Sylla R, Mondo M, Girault L, Marrama L, Spiegel A, Diallo M, et al:
Use of reverse transcriptase PCR in early diagnosis of Rift Valley
fever. Clin Diagn Lab Immunol. 9:713–715. 2002.PubMed/NCBI
|
|
19
|
Drosten C, Göttig S, Schilling S, Asper M,
Panning M, Schmitz H and Günther S: Rapid detection and
quantification of RNA of Ebola and Marburg viruses, Lassa virus,
Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus,
dengue virus, and yellow fever virus by real-time reverse
transcription-PCR. J Clin Microbiol. 40:2323–2330. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang W, Wang PZ, Yu HT, Zhang Y, Zhao K,
Du H and Bai XF: Development of a SYBR-Green I based one-step
real-time PCR assay for the detection of Hantaan virus. J Virol
Methods. 196:145–151. 2014. View Article : Google Scholar
|
|
21
|
Chen H, Parimelalagan M, Lai YL, Lee KS,
Koay ES, Hapuarachchi HC, Ng LC, Ho PS and Chu JJ: Development and
evaluation of a SYBR-Green-based real-time multiplex RT-PCR assay
for simultaneous detection and serotyping of dengue and Chikungunya
viruses. J Mol Diagn. 17:722–728. 2015. View Article : Google Scholar : PubMed/NCBI
|