Introduction
Drug-induced liver injury is a serious issue for
patients and physicians, and hence an area of intense
investigations for pharmaceutical companies. Since its introduction
60 years ago, alanine aminotransferase (ALT) activity in serum
still remains the gold standard biomarker of liver injury both
preclinically and clinically (1).
Although, the protein expression of ALT is mainly observed in the
liver, the ALT enzyme is expressed in many other organs and
tissues, such as the kidneys, heart, skeletal muscle and pancreas
(2,3). Furthermore, the two genes,
glutamic-pyruvate transaminase 1 and 2 (GPT1 and
GPT2), located on chromosomes 8 and 16, encode the human ALT
proteins, ALT1 and ALT2, and their protein products show similar
enzymatic activities (4,5). ALT1 is the dominant isoform
expressed in the liver and constitutes the majority of the basal
ALT activity of normal human serum (6,7).
Of note, several reports and a large amount of empirical data from
the pharmaceutical industry have shown that increases in serum ALT
activity can occur without apparent liver damage, as detected in
histopathological assessments. For example, the exposure of rats to
dexamethasone has been shown to increase both liver and serum ALT
activity >4-fold, without any histopatological evidence of
hepatocellular damage (8,9). Likewise, it has been demonstrated
that fibrates, which are peroxisome proliferator-activated receptor
(PPAR)α agonists, have been shown to increase ALT activity in the
serum of rats and humans (10,11). In humans, fibrates have been shown
to cause transient elevations in serum aspartate aminotransferase
(AST) and ALT levels in approximately 10% of patients, without any
other evidence of hepatotoxicity (11). It has also been demonstrated that
the gene expression of AST and ALT in human hepatoma cells and
primary human hepatocytes increases following treatment with
fibrates, thus suggesting an alternative 'non-toxic' mechanism for
the elevation of AST and ALT enzyme levels in serum (12). The hypothesis states that moderate
increases (2–4-fold) in AST and ALT activity in serum could in some
cases, be due to the induction of the expression of respective
genes, resulting in elevated protein expression levels in the
liver. The transport of the increased amounts of protein from the
liver into the serum is a poorly understood process; however, it is
believed to be due to normal hepatocyte turnover.
In a previous study, we described a clinical trial
of a selective PPARα agonist; AZD4619, in which moderately
increased levels of serum ALT and AST activity were observed in
human volunteers, without any other signs of hepatic injury
(13). We identified a functional
PPAR response element (PPRE) in the proximal ALT1 promoter and
concluded that PPAR agonists are inducers of the human ALT1 gene.
In the present study, we demonstrate that treatment with AZD4619
induces serum ALT activity in humans, but not in rats, and provide
a hypothesis to explain this observed species-related difference.
We also suggest that future drug candidates be screened for their
capacity to induce the human and rat ALT1 gene, which will help
drug projects in early discovery in removing compounds with this
liability, as even a benign increase in serum ALT levels would
hinder the progression of the compound to further clinical
development.
Materials and methods
Compounds
AZD4619, potassium salt with a molecular mass of
554.7 g/mol had a purity of 99.5% and was dissolved in tap water
(in vivo, rat study). AZD4619 has a high binding affinity
for albumin of 99.97%. Fenofibric acid and dexamethasone were
obtained from Sigma-Aldrich (St. Louis, MO, USA). Fenofibric acid
and dexamethasone have a molecular weight of 318.7 and 392.5 g/mol,
respectively. All compounds were dissolved in dimetyl sulfoxide
(DMSO) on the same day of treatment of the cell cultures.
Rat study
Four groups of Wistar rats (Taconic M&B A/S,
Denmark), each consisting of 10 males and 10 females, were
administered AZD4619 orally by gavage once a day for 32 days. The
dose levels were 5, 30, 180 and 1,000 µmol/kg (3, 15, 93,
517 mg/kg), respectively. One control group was included in the
study and was administered an appropriate amount of tap water. The
animals were approximately 8 weeks old when the dosing started and
the weight ranges were 180–320 g for the males and 130–210 g for
the females. The animals had free access to food (RM1.E.SQC low
protein food for rats and mice; Special Diets Services Ltd.,
England) and to municipal tap water for human consumption. The
systemic exposure to AZD4619 was investigated by analyzing the
concentration of the test compound in blood plasma after a single
oral dose or after 32 days of repeated oral dosing. Blood chemistry
measurements were analysed by using Cobas Integra 400, (Tegimenta
Ltd., Roche Diagnostics Instrument Center, Basel, Switzerland) with
appropriate test kits. The animals were euthanised on the day after
final day of dose administration. The study was conducted within
the framework of the Swedish National Animal Welfare Act including
a review and approval of the study protocol by the local laboratory
animal ethics committee. It was also conducted in compliance with
the Organisation for Economic Co-operation and development (OECD)
Good Laboratory Practice.
Clinical trial
The study was a phase I study, to assess the safety,
tolerability, effect on lipids and pharmacokinetics of repeated
oral doses of AZD4619. A total of 109 subjects was enrolled in
order to procure 40 randomised subjects aged between 20 and 29
years, inclusively. The subjects were divided into 2 groups with 20
subjects in each. In the first group, through random selection, 15
subjects were administered AZD4619 (5 mg daily) and 5 subjects were
administered the placebo. In the second group, through random
selection, 15 subjects were administered AZD4619 (0.5 mg daily) and
5 subjects the placebo. The dosing of AZD4619 occurred from days
1–21 and the follow-up serum measurements proceeded for 31 days.
Additional information about the clinical trial of AZD4619 has been
published previously (13).
Cell culture
Cryopreserved male human hepatocytes from one donor
(Lot GIU) were obtained from In Vitro Technologies (Baltimore MD,
USA) and cryopreserved male rat hepatocytes (Lot Rs704) were
obtained from Life Technologies/Gibco/Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). The cells were maintained in Dulbecco's
Modified Eagle's Medium (DMEM, 1 g/l glucose) with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin,
1% non-essential amino acids (Gibco/Thermo Fisher Scientific, Inc.)
in a humidified atmosphere of 95% air, 5% CO2 at 37°C.
The hepatoma cell lines, HuH-7 and MH1C1, were maintained using the
culture conditions described above. The human osteosarcoma U2OS
cells were cultivated in DMEM supplemented with
resin-charcoal-stripped FBS for the GAL4 binding assays. All cell
lines were from ATCC (Manassas, VA, USA).
Analysis of protein expression
Primary human hepatocytes (GIU) and primary rat
hepatocytes (Rs704) were seeded out in collagen-coated 6-well
plates (500,000 cells/well) and treated with 0.5% DMSO as a
control, or with 10, 100, 1,000 and 10,0000 nM AZD4619 and 250
µM fenofibric acid for 48 h. After washing the cells in
phosphate-buffered saline (PBS), they were lysed with RIPA lysis
buffer [1X PBS, 150 mM sodium chloride, 1% NP-40, 0.5% sodium
deoxycholate, 0.1% SDS, protease inhibitors (Complete, Roche
Diagnostics, Basel, Switzerland)] and cell debris was removed by
centrifugation. The protein concentration was determined by Bio-Rad
protein assay (Bio-Rad, Hercules, CA, USA). A total of 5 µg
of the human and 10 µg of the rat whole cell lysates were
subjected to SDS-polyacrylamide gel electrophoresis, performed
under reducing conditions on 4–12% Nupage Novex Bis-Tris gels with
MES-SDS running buffer (both from Invitrogen/Thermo Fisher
Scientific, Inc). The resolved proteins were transferred onto a
nitrocellulose sheet and subjected to Ponceau staining
(Sigma-Aldrich). The staining intensity was consistently identical
in all lanes. The filters were subsequently incubated for 2 h with
either a rabbit polyclonal antibody against human (h)ALT1 (cat no.
Ab1399, 0.1 µg/ml; Abcam, Cambridge, UK) or a goat
polyclonal antibody against rat (r)ALT1 (cat. no. sc-47024, 0.2
µg/ml; Santa Cruz Biotechnology, CA, USA). The filters
incubated with primary antibody were then probed with the
corresponding secondary antibodies to IgG (goat α rabbit, cat. no.
31464, 1:30,000 dilution; Pierce, Thermo Fisher Scientific, Inc.;
or rabbit α goat, cat. no. PO449, 1:10,000 dilution; Dako,
Glostrup, Denmark), conjugated to horseradish peroxidase (HRP). The
chemiluminescent HRP substrate (Immobilon Western, Millipore,
Billerica, MA, USA) was used according to the manufacturer's
instructions and chemiluminescence was detected using a Fuji Film
Las-3000 mini image analyser (Science Imaging Scandinavia, Nacka,
Sweden). The resulting bands were compared with the size of known
molecular markers (MagicMark XP; Invitrogen/Thermo Fisher
Scientific, Inc.).
Promoter constructs
The human ALT1 promoter construct has been described
elsewhere (13). To obtain the
rat ALT1 promoter construct, approximately 2,000 bp of the proximal
ALT1 promoter were amplified from rat genomic DNA using the primers
gpt1f (5′-atccacttcagcacataccc) and gpt1r
(5′-tgggaatgggaaaatctgcg). The PCR-product was subcloned into the
PCR 2.1 vector (Invitrogen/Thermo Fisher Scientific, Inc.) and
further introduced into the multiple cloning site (MCS) of the
pGL3b vector (Promega, Madison, WI, USA) using the KpnI and
XhoI restriction sites. The DNA used in the transfection
experiments was purified using the column-based Endofree Plasmid
Maxi kit (Qiagen, Valencia, CA, USA). The gluco-corticoid receptor
(GR) constructs were a kind gift from Dr Ann-Charlotte Wikström,
Karolinska Institutet, Stockholm, Sweden.
Transfection and luciferase assay
The HuH-7 and MH1C1 cells (5×106) were
seeded in a Petri dish with culture medium and incubated at 37°C
for 24 h. After washing with PBS, the cells were incubated at 37°C
for 4 h with 16 µg of plasmid DNA in transfection medium
(DMEM medium only) and 40 µl Lipofectamine™ 2000, according
to the manufacturer's instructions (Invitrogen/Thermo Fisher
Scientific, Inc.). A total of 100,000 cells/well was seeded in a
24-well plate with full growth culture medium (containing 10% FBS)
and incubated overnight at 37°C. The culture medium was removed and
replaced with the compounds to be tested (AZD4619, fenofibric acid
or dexamethasone). The concentration of the vehicle (DMSO) was
>0.1% in both the control and treated cells. Every control and
treatment was run in 4 replicates on the same plate (n=4). The
cells were incubated 24 h at 37°C and subsequently lysed (Promega
lysis buffer 1X with the addition of 1% Triton X-100). After 20 min
on ice, the cell lysates were transferred onto a non-transparent
96-multiwell plate and the luciferase activity was measured using a
luminometer (Luminoskan Ascent, Thermo Electron Corp./Thermo Fisher
Scientific Inc.).
PPARα-GAL4 assay
The U2OS cells were transiently transfected via
electroporation with a luciferase reporter gene under control of
the yeast UAS sequence and a mouse, rat, monkey or human PPARα
construct, where the PPARα ligand binding domain had been cloned in
frame downstream of the DNA binding domain from yeast GAL4
(14). The cells were seeded in
96-well plates and incubated for 3 h at 37°C, 5% CO2,
prior to the addition of the compounds and additional incubation
for 40 h. The signal was developed using the SteadyLiye kit
(Perkin-Elmer Inc., Waltham, MA, USA) and resulting luminiscence
was measured after 10 min of incubation at room temperature in a
Victor plate reader (Wallach, FI; Perkin-Elmer Inc.). The
concentration response curves and half maximal effective
concentration (EC50) values were generated using Xlfit
(ID Business Solutions, Ltd., Guildford, UK).
In silico analysis
Approximately 2 kb of the ALT1 promoter sequences
corresponding to Homo sapiens (NC_000008 REGION:
145698231.145700231) and Rattus norvegicus (NC_005106
REGION: 114744054.114746054) were analysed using the online tools,
ECRbrowser (15) and zPicture
(16). The EMBL-EBI T-Coffee
multiple sequence alignment program (http://www.ebi.ac.uk/Tools/msa/tcoffee/) was used for
alignment of the protein sequences for Homo sapiens
(NP_001001928.1), Macaca mulatta (NP_001028201.1) and
Rattus norvegicus (NP_037328.1) PPARα.
Statistical analysis
In order to establish differences in variables
measured over time from the clinical data, a paired t-test was used
to calculate significant changes. In the animal study, a Pairwise
Shirley's test was used. In addition, one-way Anova with Dunnett's
test was used to analyse differences in the transfection
experiments between the control and treatment groups. Statistical
differences were tested at the confidence level of 95% and are
represented in bar diagrams using asterisks to indicate P-values
<0.05. Bar diagrams represent the mean values ± SD.
Results
AZD4619 causes elevations in ALT levels
in the phase I clinical trial
AZD4619 was administered orally at 0.5 and 5 mg
daily to healthy individuals for 21 days. The results from this
study revealed an increase in aminotransferase levels in 1 out of
15 subjects in the 0.5 mg dose group and 5 out of 15 subjects in
the 5 mg dose group. AZD4619 was rapidly absorbed both after single
and repeated dosing, and the Cmax was generally reached within 1 h.
The pharmacokinetic steady state was achieved within 4 days and the
steady state Cmax at day 21 was on average 0.40 µM (AUC 0–24
h: 0.83 µM × h) for the 5 mg dose (Table I). In addition, a decrease in
serum triglyceride (TG) levels, which is a pharmacological marker
of PPARα activation, was observed in both the 0.5 and 5 mg
treatment groups compared to the pre-dose sample [TG decreased by
about 30% (P<0.01) from day 1 to day 21].
 | Table ISerum ALT, AST and concentrations
following the repeated oral administration of AZD4619 (0.5 or 5 mg)
or the placebo to human volunteers. |
Table I
Serum ALT, AST and concentrations
following the repeated oral administration of AZD4619 (0.5 or 5 mg)
or the placebo to human volunteers.
| Plasma parameter | Placebo | 0.5 mg dose | 5 mg dose |
|---|
| ALT, U/l |
| Pre-dose | 26±16 | 19±7 | 21±7 |
| ALT, U/l |
| At day 31,
follow-up | 28±14 | 23±13 | 41±26a |
| AST, U/l |
| Pre-dose | 26±7 | 22±4 | 23±5 |
| AST, U/l |
| At day 31,
follow-up | 32±12 | 26±5 | 39±30 |
| Triglycerides,
mmol/l |
| Pre-dose | 0.84±0.28 | 0.88±0.48 | 0.78±0.23 |
| Triglycerides,
mmol/l |
| At day 21,
follow-up | 1.05±0.64 | 0.63±0.23 | 0.54±0.15 |
| Plasma |
| Cmax,
µM | n/a | 0.05±0.02 | 0.40±0.20 |
| Plasma
exposure |
| AUC, 0–24 h
µM × h | n/a | n/a | 0.83±0.40 |
AZD4619 does not cause elevations in ALT
levels in the 1-month rat study
During the preclinical toxicological evaluations of
AZD4619, 4 groups of rats were administered AZD4619 orally by
gavage once a day for 1 month. A control group was also included in
the study and received tap water without the compound. The dose
levels for the treatment group were 3, 15, 93 and 517 mg/kg
bodyweight. At day 32, the compound concentration in the plasma
ranged between 0.64 to 742 µM for the low- to high-dose
groups, respectively (Table II).
Blood chemical analysis did not reveal any elevations in ALT levels
in any of the treatment groups, and only a marginal elevation of
AST levels was observed in the male high-dose group. In addition,
serum TG levels decreased only in the high-dose group (517 mg/kg)
from 2.30 to 0.94 mmol/l (P<0.001) in males and from 1.39 to
0.78 mmol/l (P<0.01) in females. As expected, histopathological
analysis demonstrated that AZD4619 had a clear-cut effect on the
liver, comprising a diffuse hepatocyte hypertrophy and diffuse
eosinophilic cytoplasm. The changes occurred in a dose-dependent
manner and closely correlated with the observed increase in liver
weights (data not shown). These diffuse liver changes were
considered to be a known effect of PPARα agonists mediated by the
proliferation of peroxisomes in rodent hepatocytes.
| Table IISerum ALT, AST and AZD4619
concentrations in rats after 32 days of daily oral
administration. |
Table II
Serum ALT, AST and AZD4619
concentrations in rats after 32 days of daily oral
administration.
| Gender | Control | 3 mg/kg | 15 mg/kg | 93 mg/kg | 517 mg/kg |
|---|
| ALT (U/l) | M | 79±18 | 76±14 | 71±10 | 67±6 | 81±20 |
| F | 55±21 | 47±10 | 51±6 | 56±8 | 52±6 |
| AST (U/l) | M | 87±15 | 93±15 | 93±15 | 93±23 | 103±15a |
| F | 110±48 | 87±15 | 87±14 | 88±10 | 84±10 |
| Triglycerides,
mmol/l | M | 2.30±0.76 | 2.40±0.67 | 2.03±0.67 | 1.87±0.48 | 0.94±0.22c |
| F | 1.39±0.62 | 1.24±0.39 | 0.90±0.37 | 1.20±0.56 | 0.78±0.22b |
| Cmax,
µM | M | n/a | 0.64±0.15 | 5.84±4.48 | 26.0±5.6 | 536±91 |
| F | n/a | 1.35±0.56 | 11.9±5.0 | 65.3±36.9 | 742±357 |
| Exposure | M | n/a | 3.31
(2.64–4.95) | 15.5
(10.8–20.7) | 157 (81–278) | 1,870
(1,020–3,040) |
| AUC (0–24 h)
µM × h | F | n/a | 6.16
(3.71–10.1) | 32.1
(23.9–38.9) | 149 (112–190) | 1,730
(921–3,410) |
AZD4619 induces ALT1 expression in human,
but not in rat hepatocytes
To elucidate the mechanisms responsible for the
increase in ALT levels by AZD4619 in humans, but not in rats,
primary human and rat hepatocytes were treated with AZD4619. Cell
extracts from treated human and rat cells were subjected to western
blot analysis and the results revealed that the expression of the
dominant liver ALT isoform (ALT1) was increased in human
hepatocytes following treatment with AZD4619 (10–10 000 nM)
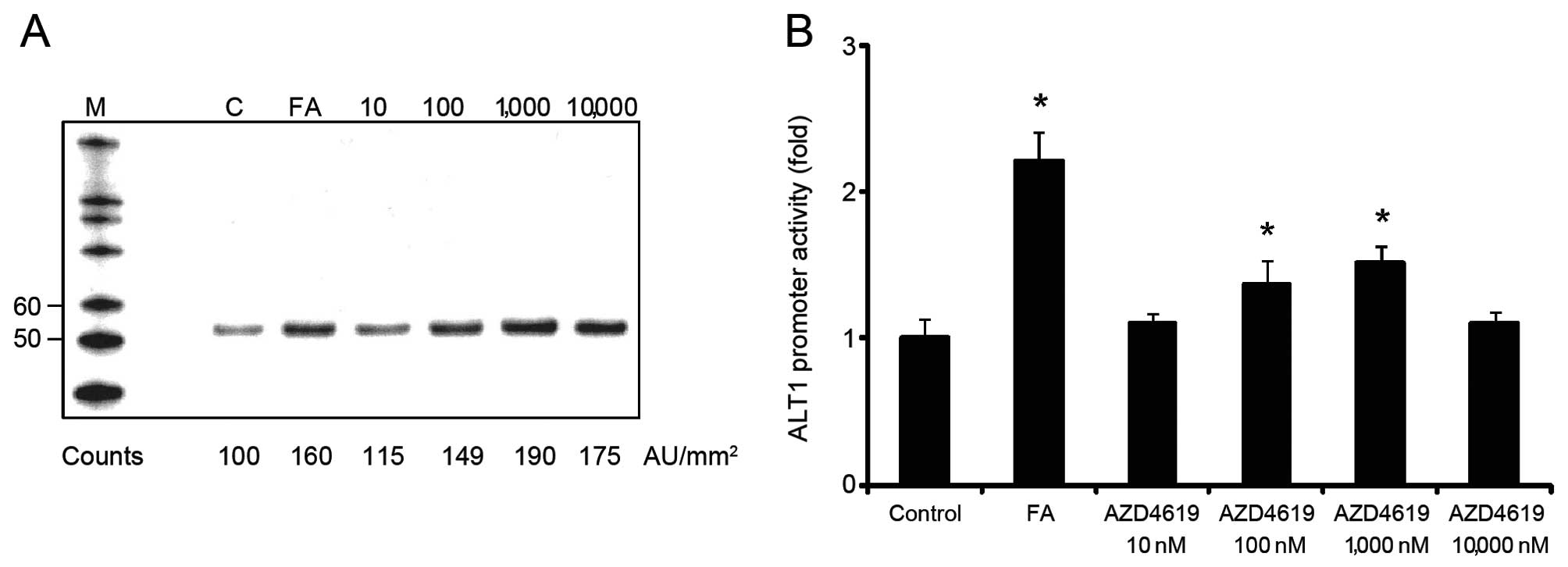
compared to the cells treated with the vehicle (Fig. 1A). To study the species-specific
responses of AZD4619 on the ALT1 gene, promoter constructs of
approximately 2 kb of either the human or the rat gene promoter
were generated and subsequently transfected into human (HuH-7) or
rat hepatoma cells (MH1C1), respectively. Treatment of the human
cells which were transfected with the human ALT1 construct with
AZD4619 resulted in a statistically significant increase in
reporter gene activity at 100–1000 nM (Fig. 1B). By contrast, as shown by
western blot analysis, there were no changes in rat ALT1 protein
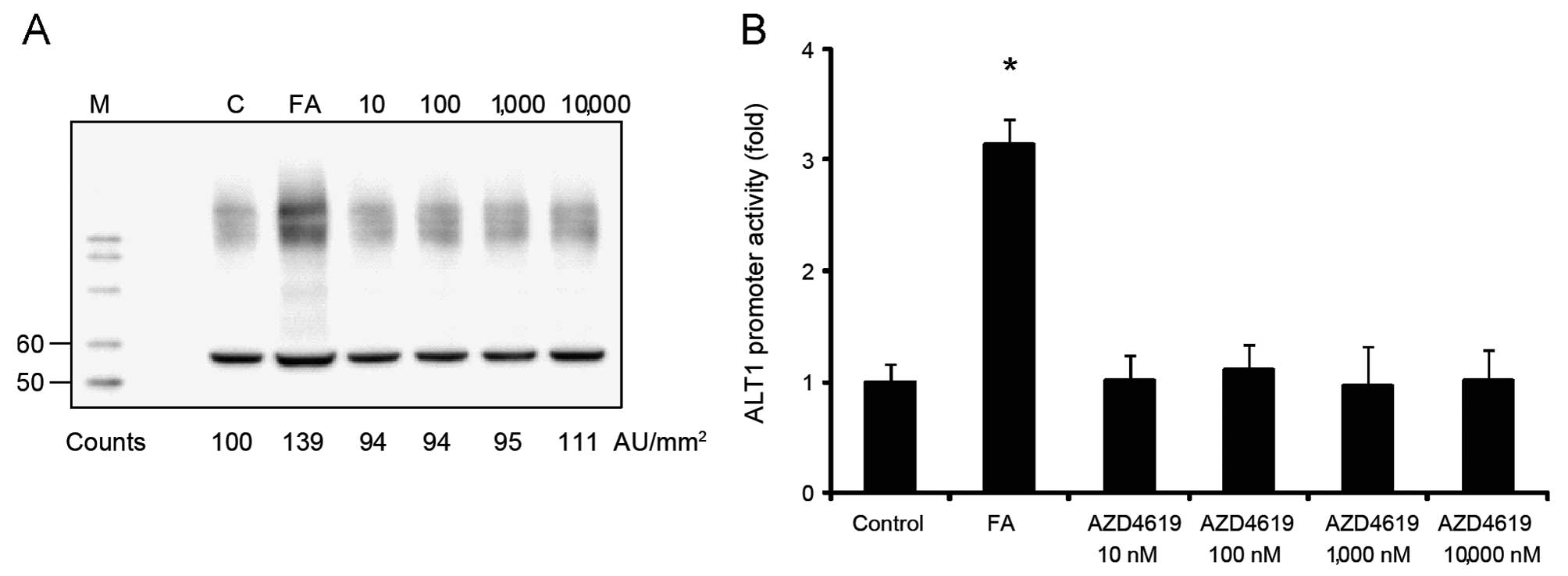
expression at equivalent concentrations of AZD4619 (Fig. 2A). This was also true for the rat
MH1C1 cells transfected with the rat ALT1 promoter construct.
AZD4619 did not have the capacity to provoke any change in rat ALT1
promoter activity in the concentrations tested (10–10,000 nM)
(Fig. 2B). The PPARα agonist,
fenofibric acid, was used as a positive control in both protein and
promoter assays, which also demonstrated the expected responses in
the systems.
AZD4619 displays specificity for human
PPARα in reporter gene assays
To elucidate the mechanisms responsible for the
induction of human ALT1 and not rat ALT1 levels by AZD4619 in the
concentration range tested, we investigated receptor selectivity by
the compound. AZD4619 was found to be a highly specific and potent
agonist of human PPARα with a calculated EC50 value of
0.10 µM in reporter gene assays. However, the potency of
AZD4619 on rat PPARα was >100-fold lower, with an
EC50 value of 10.3 µM (Table III). The evaluation of
fenofibric acid in the same assay resulted in an EC50
value of 13.1 and 17.0 µM for human and rat PPARα,
respectively (data not shown). The amino acid compositions for
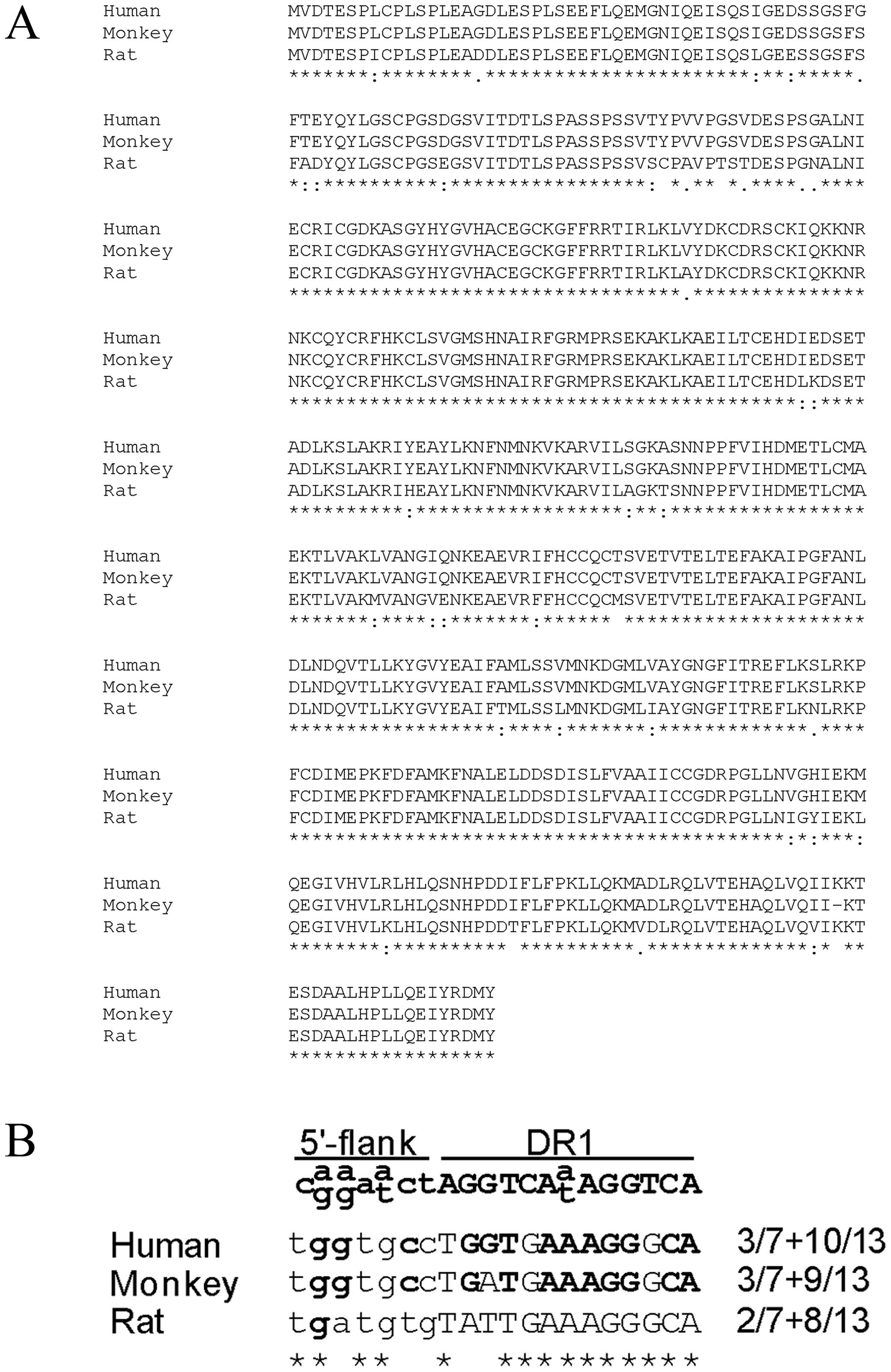
human and rat PPARα were compared in a multiple sequence alignment
and several amino acids deviated between the human and rat
ligand-binding domains (Fig.
3A).
| Table IIIEC50 values (µM)
for AZD4619 in PPARα-GAL4 reporter gene assay. |
Table III
EC50 values (µM)
for AZD4619 in PPARα-GAL4 reporter gene assay.
| PPARα |
EC50 |
|---|
| Human | 0.10 |
| Monkey | 0.47 |
| Rat | 10.3 |
| Mouse | 6.2 |
Functional PPRE in the human ALT1
promoter is not conserved in the rat
To further investigate the reason for the
differential responses by AZD4619 on the human and rat ALT1 genes,
approximately 2 kb of the human and rat promoters were compared
using pairwise sequence alignment. The functional PPRE at −574 in
the human promoter was found within a 400 bp evolutionary conserved
region where the nucleotide sequences matched by 67.5% between the
two species. However, the core sequence of the human PPRE at −574
was not identical in the rat promoter (Fig. 3B).
Glucocorticoids also show the capacity to
induce the activation of the ALT1 promoter
To determine whether another compound known to
induce moderate serum ALT activity in rats and humans would cause
the activation of the ALT1 promoter, transient transfections with
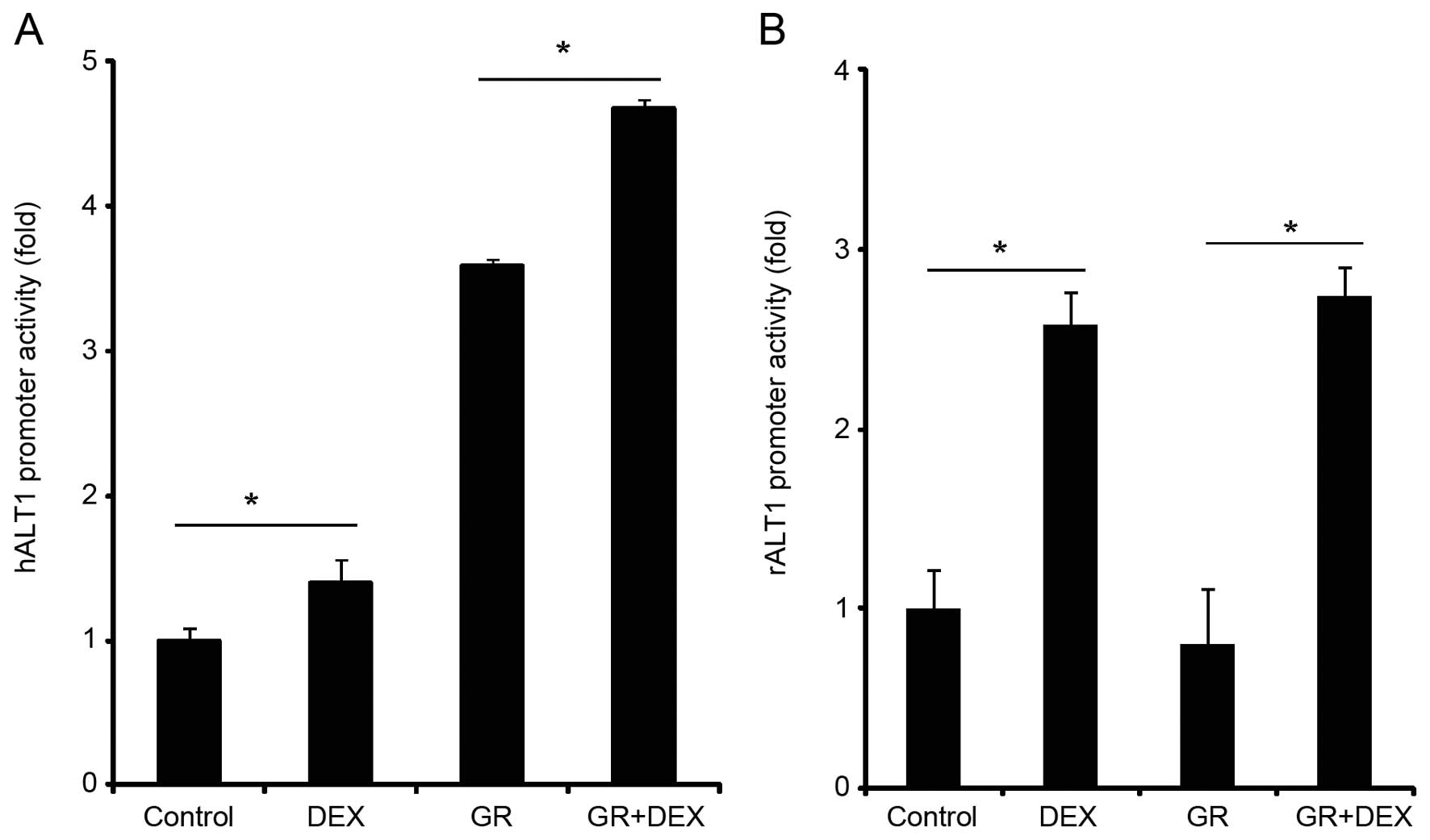
ALT1 promoter constructs and treatment with dexamethasone was
performed. The human hepatoma cell line, HuH-7, and the rat MH1C1
cells were transfected with the promoter constructs for human or
rat ALT1, respectively, and dexamethasone treatment increased the
promoter activity in both species (Fig. 4A and B). Co-transfection of the
HuH-7 cells with a vector expressing the human GR (hGR) increased
the ALT1 promoter more than dexamethasone treatment alone, and the
combination of hGR and dexamethasone in the HuH-7 cells gave rise
to an additional effect on the promoter (Fig. 4A). In the rat cells, transfection
with the rat GR (rGR) did not affect the ALT1 expression levels,
neither alone nor in combination with dexamethasone (Fig. 4B).
Discussion
A novel PPAR agonist (AZD4619) did not increase
serum ALT levels in a one month rat toxicological study, but
unexpectedly did so in the first human clinical trial (13). To elucidate the mechanisms
responsible for this discrepancy, we investigated the effects of
AZD4619 on ALT1 gene and protein expression and compared the
response between human and rat species. AZD4619 was tested in
vitro at concentrations ranging from its human receptor potency
up to >100-fold higher concentrations (0.01 to 10 µM).
Still, there was no increase in rat ALT1 levels, neither in ALT1
reporter gene assay nor in ALT1 protein expression assessed by
western blot analysis in primary rat hepatocytes. Treatment with
AZD4619 at the same concentrations (0.01 to 10 µM) in human
hepatocytes gave rise to statistically significant elevations, both
in ALT1 promoter gene activity and protein expression. In the human
subjects in which plasma ALT1 levels were elevated, the plasma
concentrations of AZD4619 were in the same concentration range
(Cmax, 0.40 µM) as where the in vitro assays detected
increased ALT1 promoter activity and protein expression (0.1–1
µM). In the in vitro assays, compounds dissolved in
cell medium (containing FBS, about 23 mg/l albumin) are protein
bound to albumin (99.97%), as in in vivo conditions. In the
rat in vivo study, the concentration of AZD4619 in rat
plasma ranged from 0.64 up to 742 µM between the dose
groups. Hence, the plasma levels of AZD4619 in the high-dose group
were >6,000-fold higher than the human EC50 value in
the PPAR binding assay, without any effects on serum ALT-levels
in vivo. Furthermore, the lowest plasma concentration where
a pharmacological effect of AZD4619 was observed (i.e., decrease in
circulating triglycerides) in humans, occurred already in the 0.5
mg dose group (Cmax, 0.05 µM) (Table I). On the other hand, in rats,
decreases in circulating triglycerides occurred first in the
high-dose group, 517 mg/kg (Cmax >536 µM). These data
point to a clear species-related difference in the ability of
AZD4619 to activate PPARα.
To mechanistically understand the difference between
human and rat species in response to AZD4619, PPARα-GAL4 assays
were performed. The PPAR reporter gene assays demonstrated that
AZD4619 is a very human-specific PPARα ligand activating human PPAR
at a 100-fold lower concentration compared to rat PPARα, which
might fully or partly explain the lack of plasma ALT elevations in
the rat. The ligand-binding domains (LBDs) of human and rat PPARα
are 93% identical, key amino acids yet to be confirmed are likely
contributing to this species selective activation. Furthermore,
analysis of the genomic region in the rat corresponding to the
functional human −574 PPRE denoted a difference in the core PPRE
between human and rat, which might be sufficient to make the rat
PPRE substantially less functional. In a comparison of the two
sites with the PPRE consensus described by Juge-Aubry et al
(17), the human site matched by
3/7+10/13, whereas the rat site only corresponded by 2/7+8/13.
However, fenofibric acid induced both the human and rat ALT1
promoter genes and increased ALT1 protein expression in
vitro. The EC50 for fenofibric acid in the
PPARα-GAL4 assay was similar for both species (13 and 17 µM)
(data not shown). These results indicate that the PPRE in the rat
is functional and that the species-selective activation of the
human vs. the rat ALT1 gene by AZD4619 can be explained by selected
amino acid differences in the respective LBD. The support for
species-selective PPARα activation comes from studies with another
PPAR agonist, Wy14643, shown to be at least 38-fold more rodent
selective compared to humans in a time-resolved fluoresence
resonance energy transfer (TR-FRET) binding assay (18). Even though the PPRE in the ALT1
promoter was not identical in the human and rat, the surrounding
region was evolutionary conserved between the two species, implying
its importance. Another explanation for the induction of fenofibric
acid on the rat ALT1 gene might be that the effect is mediated
through an unidentified unique rat PPRE distinct from the −574 PPRE
in the ALT1 promoter (13).
It has been reported in the literature that
fenofibrate induces the rat ALT gene even though no exact mechanism
has been established (10). Male
rats treated with fenofibrate at 180 and 1,000 mg/kg for 13 weeks
were shown to have increased ALT levels in serum, whereas AST
levels increased already at 30 mg/kg fenofibrate dose (10). Both AST and ALT have been reported
to increase mildly in some patients who receive fenofibrate
(11,19) and in vitro studies have
shown that human hepatocytes respond to fenofibrate by a
PPARα-dependent increase in aminotransferases (12,20). In humans receiving the dose of 5
mg/kg fenofibrate, approximately 9% develop mild serum
aminotransferase increases without any reports on hepatic
pathologies (11). The transient
and small elevation of aminotransferases in clinical trials and the
historical absence of liver injury using fenofibrates, have been
suggested to be due to the induction of the ALT and AST genes,
rather than damage to hepatic cells (12). Knowledge gained from the
application of genome-wide approaches and 'omics' technologies has
given way to a more complex and interconnected view of the
importance of liver transaminases in the regulation of systemic
metabolic function (21).
Dexamethasone is another drug that is capable of
inducing ALT expression in the livers of rats and contributes to
increased serum ALT levels without involving hepatic cell death
(8,22,23). We previously found a consensus
binding site for glucocorticoids (GRE, −1253), highly conserved
between the human and rat ALT1 promoter, which may explain the
promoting effect of glucocorticoids on ALT1 expression in the liver
and serum (13). In the present
study, we confirmed that dexamethasone increased the transcription
of both the rat and the human ALT1 reporter genes (Fig. 4).
Although not shown in this study, preclinical
toxicological testing of AZD4619 in cynomolgus monkeys treated for
28 days with the same doses as the rats (0, 3, 15, 93 and 517
mg/kg) resulted in a weak serum ALT increase (2-fold) in females
and a trend towards increased serum ALT levels in males in the mid-
and high-dose groups. Of note, no histopathological changes were
detected in any organs with known ALT expression such as the liver,
skeletal muscle, heart, pancreas and kidneys (data not shown). The
magnitudes of the observed transcriptional changes for ALT1 by
AZD4619 in cynomolgus monkeys are very much in line with the
changes in human hepatocytes reported herein. In the PPARα-GAL4
reporter assay comparing different species, human and cynomolgus
monkey PPARα exhibited EC50 values similar to each other
compared to human and rat/mouse PPARα following treatment with
AZD4619 (Table III).
Furthermore, an in silico comparison of the cynomolgus PPARα
LBD with the human PPAR LBD revealed that they are identical, apart
from one amino acid deletion in the monkey compared to the human
LBD (Fig. 3A). In addition, the
PPRE in the monkey ALT1 gene only deviates one nucleotide from the
human PPRE (Fig. 3B), which would
explain an induction by PPARα agonists in this species.
To summarise, AZD4619 induces the human ALT1 gene
and protein expression in hepatocytes, but has no effect on rat
ALT1 gene and protein expression in the concentrations tested. The
most likely reason for this difference is the high species
(>100-fold) selectivity of AZD4619 for the human PPARα. In the
in vivo study, serum ALT was increased in the human clinical
trial, an effect not observed in the preceding rat toxicity study.
Discrepancies of ALT1 induction between species might result in
problematic drug development, where drugs designed for humans, give
rise to findings not detected in pre-clinical toxicity testing:
mechanistic data to provide context to these findings is therefore
key in considering the clinical risk assessment. These data
highlight the importance of translational assessment for
hepatoxicity testing and assessment of candidate compounds in
humanised model systems is recommended; such systems could include
assay of ALT1 gene induction to reveal if drug candidates result in
species specific effects.
Acknowledgments
We would like to thank Mrs. Ingalill Rafter for
performing western blot analysis in the present study.
Abbreviations:
|
ALT
|
alanine aminotransferase
|
|
AST
|
aspartate aminotransferase
|
|
Dex
|
dexamethasone
|
|
FA
|
fenofibric acid
|
|
LBD
|
ligand-binding domain
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
PPRE
|
PPAR response element
|
References
|
1
|
Karmen A, Wroblewski F and Ladue JS:
Transaminase activity in human blood. J Clin Invest. 34:126–131.
1955. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ladue JS and Wroblewski F: The
significance of the serum glutamic oxalacetic transaminase activity
following acute myocardial infarction. Circulation. 11:871–877.
1955. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lindena J, Sommerfeld U, Höpfel C and
Trautschold I: Catalytic enzyme activity concentration in tissues
of man, dog, rabbit, guinea pig, rat and mouse. Approach to a
quantitative diagnostic enzymology, III. Communication. J Clin Chem
Clin Biochem. 24:35–47. 1986.PubMed/NCBI
|
|
4
|
Yang RZ, Blaileanu G, Hansen BC, Shuldiner
AR and Gong DW: cDNA cloning, genomic structure, chromosomal
mapping, and functional expression of a novel human alanine
aminotransferase. Genomics. 79:445–450. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sohocki MM, Sullivan LS, Harrison WR,
Sodergren EJ, Elder FF, Weinstock G, Tanase S and Daiger SP: Human
glutamate pyruvate transaminase (GPT): Localization to 8q24.3, cDNA
and genomic sequences, and polymorphic sites. Genomics. 40:247–252.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lindblom P, Rafter I, Copley C, Andersson
U, Hedberg JJ, Berg AL, Samuelsson A, Hellmold H, Cotgreave I and
Glinghammar B: Isoforms of alanine aminotransferases in human
tissues and serum–differential tissue expression using novel
antibodies. Arch Biochem Biophys. 466:66–77. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miyazaki M, Rosenblum JS, Kasahara Y,
Nakagawa I and Patricelli MP: Determination of enzymatic source of
alanine aminotransferase activity in serum from dogs with liver
injury. J Pharmacol Toxicol Methods. 60:307–315. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Brien PJ, Slaughter MR, Polley SR and
Kramer K: Advantages of glutamate dehydrogenase as a blood
biomarker of acute hepatic injury in rats. Lab Anim. 36:313–321.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reagan WJ, Yang RZ, Park S, Goldstein R,
Brees D and Gong DW: Metabolic adaptive ALT isoenzyme response in
livers of C57/BL6 mice treated with dexamethasone. Toxicol Pathol.
40:1117–1127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kobayashi A, Suzuki Y, Kuno H, Sugai S,
Sakakibara H and Shimoi K: Effects of fenofibrate on plasma and
hepatic transaminase activities and hepatic transaminase gene
expression in rats. J Toxicol Sci. 34:377–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blane GF: Comparative toxicity and safety
profile of fenofibrate and other fibric acid derivatives. Am J Med.
83:26–36. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Edgar AD, Tomkiewicz C, Costet P, Legendre
C, Aggerbeck M, Bouguet J, Staels B, Guyomard C, Pineau T and
Barouki R: Fenofibrate modifies transaminase gene expression via a
peroxisome proliferator activated receptor alpha-dependent pathway.
Toxicol Lett. 98:13–23. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thulin P, Rafter I, Stockling K,
Tomkiewicz C, Norjavaara E, Aggerbeck M, Hellmold H, Ehrenborg E,
Andersson U, Cotgreave I and Glinghammar B: PPARalpha regulates the
hepatotoxic biomarker alanine aminotransferase (ALT1) gene
expression in human hepatocytes. Toxicol Appl Pharmacol. 231:1–9.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ljung B, Bamberg K, Dahllof B, Kjellstedt
A, Oakes ND, Ostling J, Svensson L and Camejo G: AZ 242, a novel
PPARalpha/gamma agonist with beneficial effects on insulin
resistance and carbohydrate and lipid metabolism in ob/ob mice and
obese Zucker rats. J Lipid Res. 43:1855–1863. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ovcharenko I, Nobrega MA, Loots GG and
Stubbs L: ECR Browser: a tool for visualizing and accessing data
from comparisons of multiple vertebrate genomes. Nucleic Acids Res.
32:W280–W286. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ovcharenko I, Loots GG, Hardison RC,
Miller W and Stubbs L: zPicture: Dynamic alignment and
visualization tool for analyzing conservation profiles. Genome Res.
14:472–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Juge-Aubry C, Pernin A, Favez T, Burger
AG, Wahli W, Meier CA and Desvergne B: DNA binding properties of
peroxisome proliferator-activated receptor subtypes on various
natural peroxisome proliferator response elements. Importance of
the 5′-flanking region. J Biol Chem. 272:25252–25259. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Caplan S, Yu J, Chen P, Sabio M and
Boettcher B: Comparison of ligand-binding affinities in the human,
mouse and hamster PPAR[alpha] ligand-binding domains with
PPAR[alpha] and PPAR[alpha]/[gamma] dual agonists. 64th Scientific
Sessions(Category: Clinical Therapeutics/New Technology -
Pharmacologic Treatment of Diabetes or its Complications). American
Diabetes Association; pp. p5092004
|
|
19
|
Balfour JA, McTavish D and Heel RC:
Fenofibrate. A review of its pharmacodynamic and pharmacokinetic
properties and therapeutic use in dyslipidaemia. Drugs. 40:260–290.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tomkiewicz C, Muzeau F, Edgar AD, Barouki
R and Aggerbeck M: Opposite regulation of the rat and human
cytosolic aspartate aminotransferase genes by fibrates. Biochem
Pharmacol. 67:213–225. 2004. View Article : Google Scholar
|
|
21
|
Sookoian S and Pirola CJ: Liver enzymes,
metabolomics and genome-wide association studies: From systems
biology to the personalized medicine. World J Gastroenterol.
21:711–725. 2015.PubMed/NCBI
|
|
22
|
Jackson ER, Kilroy C, Joslin DL, Schomaker
SJ, Pruimboom-Brees I and Amacher DE: The early effects of
short-term dexamethasone administration on hepatic and serum
alanine aminotransferase in the rat. Drug Chem Toxicol. 31:427–445.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu R, Pan X and Whitington PF: Increased
hepatic expression is a major determinant of serum alanine
aminotransferase elevation in mice with nonalcoholic
steatohepatitis. Liver Int. 29:337–343. 2009. View Article : Google Scholar
|