Introduction
Ischemia/reperfusion (I/R) injury to the myocardium,
kidney and lungs results in fibrosis, which is mainly mediated by
inflammation, the transdifferentiation of fibroblasts to
fibroblasts and collagen deposition (1–3).
One of the molecular pathways involved is the epithelial to
mesenchymal transition (EMT) (4).
High-mobility group box 1 (HMGB1), a non-chromosomal
nuclear protein that regulates gene transcription and maintains the
nucleosome structure (5), has
been identified as an inflammatory factor that regulates the EMT
process (6). Additionally, HMGB1
has been proved to be associated with I/R injury. A previous study
has shown that cardiac HMGB1 expression is upregulated following
I/R injury of the heart, and the inhibition of HMGB1 significantly
reduced tissue inflammation, injury and fibrosis in addition to
improving cardiac performance (7). Taken together, these findings
suggest that HMGB1 is a novel target for suppressing the
inflammatory process and reducing I/R injury.
MicroRNAs (miRNAs or miRs), a type of small,
non-coding RNA that regulate protein expression (8), are also thought to play crucial
roles in the development of fibrosis. For example, miR-21
simultaneously regulated ERK1 signaling in hepatic stellate cell
(HSC) activation and hepatocyte EMT in hepatic fibrosis (9). miR-21 promoted fibrogenic EMT of
epicardial mesothelial cells which involved programmed cell death 4
and sprouty-1 (10). Furthermore,
miRNAs have been implicated in the regulation of HMGB1. For
example, miR-129-2 directly targeted HMGB1 and inhibited its
expression in glioma cells (11).
Lu et al demonstrated that miR-181b inhibited the expression
of HMGB1 and Mcl-1 by directly binding to their 3′-untranslated
regions (3′-UTRs) (12).
Furthermore, overexpressed miR-21 targeted the 3′-UTR of HMGB1 and
inhibited HMGB1-induced autophagy (13).
miR-25 is a member of the miR-106b~25 cluster that
is associated with EMT and potentially exerts effects on kidney
fibrosis (14). Divakaran et
al demonstrated that miR-25 reduced collagen deposition during
cardiac fibrosis (15). However,
whether miR-25 plays a role in hypoxia/reoxygenation (H/R)-induced
fibrosis and apoptosis, and whether HMGB1 is a target of miR-25,
remains largely unknown.
In this study, we examined the effects of miR-25 on
H/R-induced fibrosis and the apoptosis of cardiomyocyte H9c2 cells
as well as the precise mechanisms responsible for these effects.
Firstly, we detected the expression of miR-25 and HMGB1 in
H/R-exposed H9c2 cells. Cell cycle analysis as well as measurement
of the apoptotic rate was performed in the H/R-exposed cells.
Furthermore, we established miR-25-overexpressing and
miR-25-silenced cell lines in order to examine the effect of miR-25
on fibrosis and apoptosis. We then confirmed that miR-25 directly
targeted HMGB1 and downregulated HMGB1 expression. For signaling
pathway analysis, transforming growth factor-β1 (TGF-β1)/Smad3 and
ERK1/2 were analyzed following HMGB1 knockdown. Moreover, an
inhibitor of the TGF-β1/Smad3 pathway was used to evaluate the
effects of miR-25. Taken together, the findings of the present
study may provide novel insights into the pathogenesis of fibrosis,
particularly with regard to H/R-related heart failure.
Materials and methods
Cell culture
H9c2 cells were purchased from Auragene (Changsha,
China). This cell line was recently authenticated and tested for
contamination. The cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; HyClone, Logan, UT, USA) supplemented with
10% fetal calf serum (Gibco, Gaithersburg, MD, USA). For H/R
exposure, H9c2 cells were cultured under hypoxic conditions for 6,
12, 24 or 36 h, followed by reoxygenation for 1 h. The cells were
then harvested for use in subsequent experiments.
Flow cytometric analysis
An Annexin V apoptosis detection kit (Life
Technologies, Grand Island, NY, USA) was used for apoptosis
detection. Following exposure to H/R, H9c2 cells were collected,
and washed twice with cold phosphate-buffered saline (PBS) and 500
µl binding buffer was used to resuspend the cells. Annexin
V-FITC (5 µl) and propidium iodide (PI) (5 µl) were
added to the solution and mixed well. Following a 15-min incubation
period at room temperature in the dark, the cells were analyzed
using a flow cytometer (BD Biosciences, San Jose, CA, USA).
For cell cycle analysis, following exposure to H/R,
the H9c2 cells were collected, and washed twice with cold PBS and
500 µl PBS was used to resuspend the cells. The cells were
fixed in 70% ethanol overnight, centrifuged and washed with PBS
three times. Finally, 0.2 mg RNase A and 1 ml PI/Triton X-100
solution were used to resuspend the cells for 15 min. The cells
were analyzed using a flow cytometer (BD Biosciences).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from H9c2 cells using TRIzol
reagent (Life Technologies, Shanghai, China) according to the
manufacturer's instructions. cDNA was synthesized using the TaqMan
MicroRNA Reverse Transcription kit (Applied Biosystems, Foster
City, CA, USA). qPCR was performed using a quantitative
fluorescence PCR instrument (ABI 7500 Thermocycler; Life
Technologies) and SYBR-Green Universal PCR Master Mix (Bio-Rad,
Hercules, CA, USA). The following oligonucleotide sequences of the
primer sets were used: miR-25 (HmiR0109); U6 (HmiRQP9001) (both
from Fulengen, Guangzhou, China); HMGB1 sense, GGA GAG TAA TGT TAC
AGA GCG G and antisense, AGG ATC TCC TTT GCC CAT GT; collagen I
sense, ATC AGC CCA AAC CCC AAG GAG A and antisense, CGC AGG AAG GTC
AGC TGG ATA G; collagen III sense, TGA TGG GAT CCA ATG AGG GAG A
and antisense, GAG TCT CAT GGC CTT GCG TGT TT; matrix
metallopeptidase 2 (MMP2) sense, TGA TGG CAT CGC TCA GAT CC and
antisense, GGC CTC GTA TAC CGC ATC AA; TIMP metallopeptidase
inhibitor 2 (TIMP2) sense, TGT GAC TTC ATC GTG CCC TG and
antisense, ATG TAG CAC GGG ATC ATG GG; β-actin sense, AGG GGC CGG
ACT CGT CAT ACT and antisense, GGC GGC ACC ACC ATG TAC CCT). qPCR
was performed in a total volume of 20 µl, including 10
µl of 2X SYBR-Green qPCR mix, 1 µl of each forward
and reverse primer (10 µmol/l), 1 µl of each cDNA
sample and 7 µl H2O. Amplifications were
performed in triplicate in 96-well microtiter plates. The following
thermal cycling conditions were used: 95°C for 3 min, 35 cycles of
95°C for 10 sec, and 58°C for 30 sec, finally followed by 95°C for
12 sec, and 58°C for 50 sec.
Western blot analysis
Whole-cell lysates were harvested and washed with
PBS, and then lysed in a buffer containing 150 mM NaCl, 1 mM PMSF,
NaVO4, aprotinin and leupeptin as protease inhibitors,
in 50 mM Tris-HCl pH 8.0, 0.2% sodium dodecyl sulfate (SDS) and 1%
NP-40. Thirty micrograms of protein per sample was resolved on an
SDS-polyacrylamide gel with subsequent transfer blotting. The
membranes were incubated at 4°C with a primary antibody overnight
[HMGB1, (ab79823; Abcam, Cambridge, UK); TIMP2 (sc-6835) and MMP2
(sc-10736) (both from Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA); collagen I (ab34710) and collagen III (ab7778) (both from
Abcam); TGF-β1 (YT4632, Immunoway, Newark, DE, USA); ERK1/2
(ab17942; Abcam); phosphorylated (p-)ERK1/2 (sc-16982; Santa Cruz
Biotechnology, Inc.); Smad3 (ab40854; Abcam); p-Smad3 (sc-130218;
Santa Cruz Biotechnology, Inc.); Bcl-2 (ab117115) and cleaved
caspase-3, ab2302 (both from Abcam)]. After washing, the membranes
were incubated with the corresponding secondary antibody
[111-035-003; 111-035-008 (Jackson ImmunoResearch Laboratories,
West Grove, PA, USA)] for 1 h at room temperature, followed by
chemiluminescence for visualization. For the control group, the
membrane was stripped and reprobed using an actin antibody after
probing each membrane with the primary antibody.
Transfection
H9c2 cells were cultured as described above prior to
transfection. To knockdown the endogenous expression of miR-25, an
miR-25 inhibitor (a recombinant lentivirus of Lv-anti-miR-25) was
used and to upregulate miR-25, a recombinant lentivirus of
Lv-miR-25 (both from GeneChem, Shanghai, China) was used. As a
negative control, an miR inhibitor or overexpression control
(lentivirus of Lv-con and Lv-anti-con; GeneChem) was transfected
into the H9c2 cells. Then, miR-25 gain/loss models and their
controls with the neomycin resistance gene were successfully
isolated using G418 (10131-027; Invitrogen Life Technologies,
Carlsbad, CA, USA). The stably transfected cells were subjected to
hypoxic conditions for 24 h, followed by reoxygenation for 1 h. The
cells were then harvested for evaluation by western blot analysis,
RT-qPCR and flow cytometry.
HMGB1-shRNA and shRNA control were used to regulate
the expression of HMGB1. All the plasmids were purchased from
Auragene. Transfection was performed using Lipofectamine™ 2000
(Invitrogen) according to the manufacturer's instructions. Briefly,
100 pmol of mimics was diluted in 250 µl DMEM, which was
then mixed with 10 µl Lipofectamine 2000 (diluted in 250
µl serum-free medium). Following incubation at 37°C for 4 h,
the medium was replaced with complete medium and incubated for a
further 24 h. The cells were then collected for H/R exposure.
miR-25-overexpressing cells were incubated with or
without SB431542 (10 µM) (S1067; Selleck Chemicals, Houston,
TX, USA) for 24 h, and then subjected to H/R.
Prediction of miRNA-25 targets
The miRNA databases and target prediction tool,
microRNA.org-Targets and Expression, was used to identify potential
miR-25 targets.
Dual-luciferase reporter gene assay
A Dual-Luciferase Reporter Gene assay kit (E1910;
Promega, Madison, WI, USA) was used in the study. For the
luciferase reporter experiments, a 3′-UTR segment of the HMGB1 gene
(accession no. NM_002128.5) was amplified by PCR from human genomic
DNA using primers that included an XhoI and NotI
tails on the 5′ and 3′ strands, respectively. PCR products were
recombined with psi-CHECK2. The H9c2 cells were then transfected
with the firefly luciferase HMGB1-3′UTR-psi-CHECK2, combined with
miR-25 mimics (HmiR-AN0351), miR-25 inhibitor (HmiR-AN0351-SN-10),
miRNA NC mimics (CmiR-AN0001), and miRNA NC inhibitor
(CmiR-AN0001-SN) (all from Fulengen), respectively. Twenty-four
hours after transfection, the cells were lysed with a 1X passive
lysis buffer and the activity of both Renilla and firefly
luciferase was assayed using the dual-luciferase reporter assay
system (Promega) according to the manufacturer's instructions.
Statistical analysis
All experiments were repeated three times. All data
are presented as the means ± SD. Comparisons between groups were
performed by one-way analysis of variance (ANOVA) using SPSS 17.0
software (SPSS Inc., Chicago, IL, USA) and Prism 5.0 software
(GraphPad Software Inc., San Diego, CA, USA). A P-value <0.05
was considered to indicate a statistically significant
difference.
Results
H/R upregulates HMGB1 expression and
downregulates miR-25 expression as well as increasing fibrosis and
enhancin the apoptosis of H9c2 cells
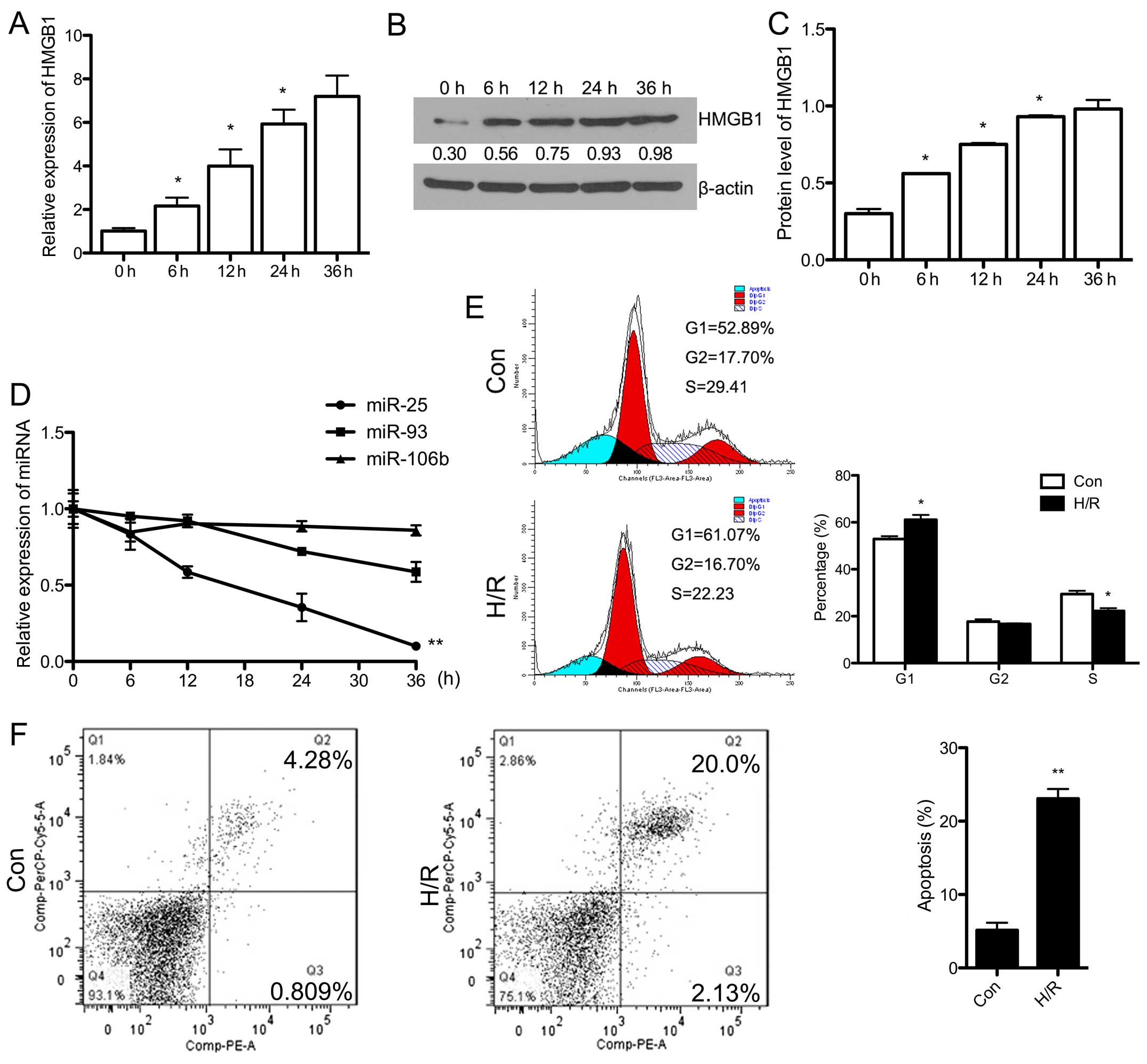
Firstly, the H9c2 cells were exposed to hypoxic
conditions for 6, 12, 24 and 36 h, respectively, followed by 1 h of
reoxygenation. HMGB1 expression was evaluated by RT-qPCR and
western blot analysis at different time points. The mRNA expression
of HMGB1 increased in a time-dependent manner (Fig. 1A). The protein expression of HMGB1
increased over time, although there was no significant difference
in expression between 24 and 36 h (Fig. 1B and C). Accordingly, we selected
24 h as the time point for use in subsequent experiments.
Additionally, we detected the expression of the miR-106b~25 cluster
that is associated with EMT. As shown in Fig. 1D, the downregulation of miR-25 and
miR-93 was observed in the H/R-exposed H9c2 cells over time.
Howerer, there were no significant changes in the expression of
miR-106b.
In order to examine the apoptosis of H/R-exposed
cells, flow cytometry and western blot analysis were performed,
respectively. As shown in Fig.
1F, the apoptosis rate was significantly upregulated following
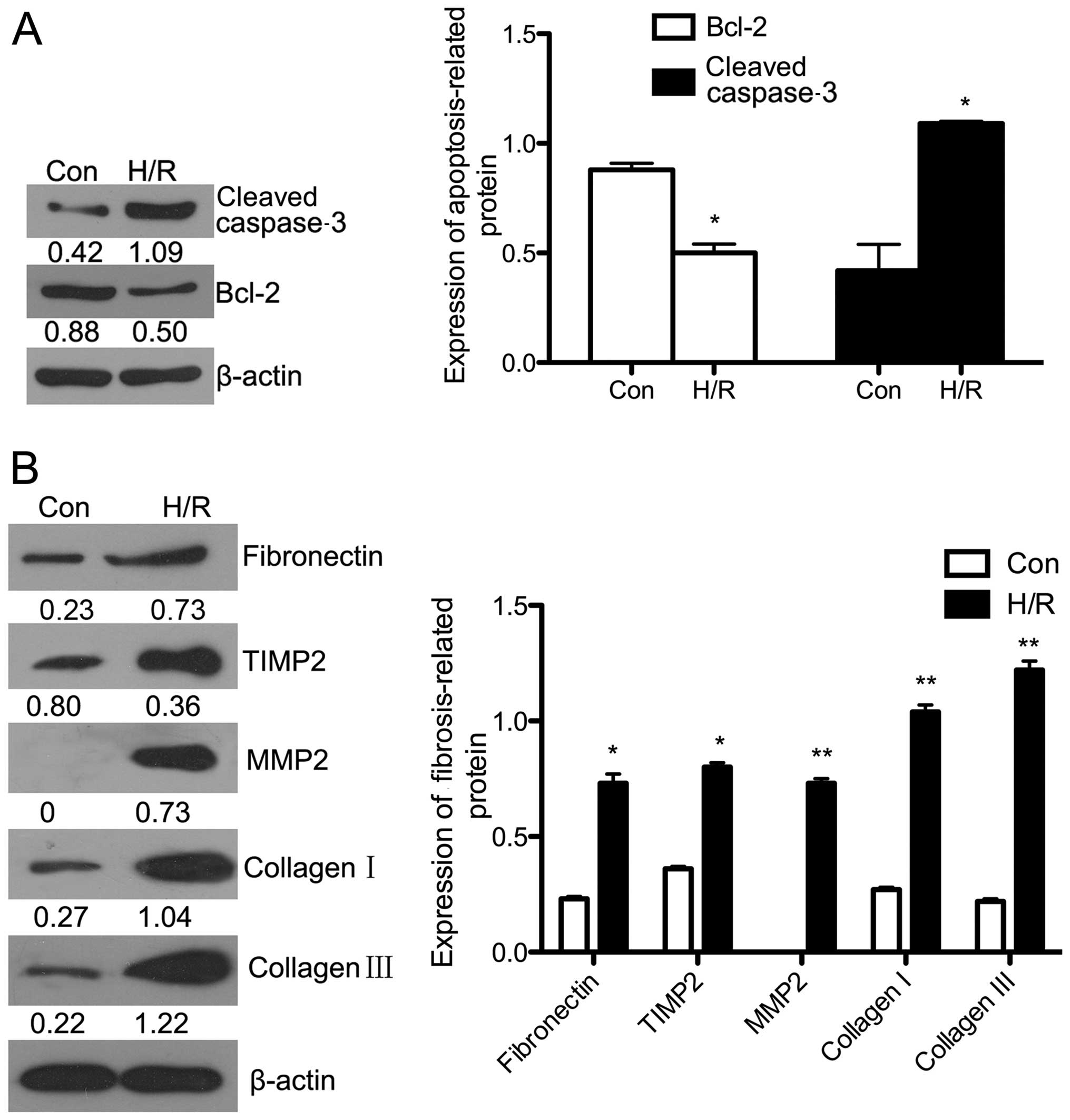
exposure to H/R (P<0.01). The protein expression of Bcl-2, which
inhibits cell apoptosis, was downregulated (P<0.05), and cleaved
caspase-3 expression was significantly upregulated in the
H/R-exposed cells (P<0.05) (Fig.
2A). Additionally, cell cycle analysis by flow cytometry
revealed that the number of cells in the G1 phase significantly
increased following exposure to H/R (P<0.05), whereas the number
of cells in the S phase decreased (P<0.05) (Fig. 1E). This indicated that the
H/R-induced apoptosis of H9c2 cells may involve blocking of the
cell cycle in the G1 phase. Finally, we evaluated the expression of
fibrosis-related proteins using western blot analysis (Fig. 2B). The protein expression of
fibronectin (P<0.05), TIMP2 (P<0.05), MMP2 (P<0.01),
collagen I (P<0.01) and collagen III (P<0.01) were all
significantly increased following exposure to H/R compared with
that in the control groups.
Collectively, these findings demonstrate that the
H/R-exposed cells showed increased levels of apoptosis and
fibrosis, upregulation of HMGB1 expression, downregulation of
miR-25 expression and blocking of the cell cycle in the G1
phase.
Overexpression of miR-25 reverses the
H/R-induced fibrosis and apoptosis of H9c2 cells
To examine the effects of miR-25, we used a
gain-of-function approach based on stable lentiviral transfection.
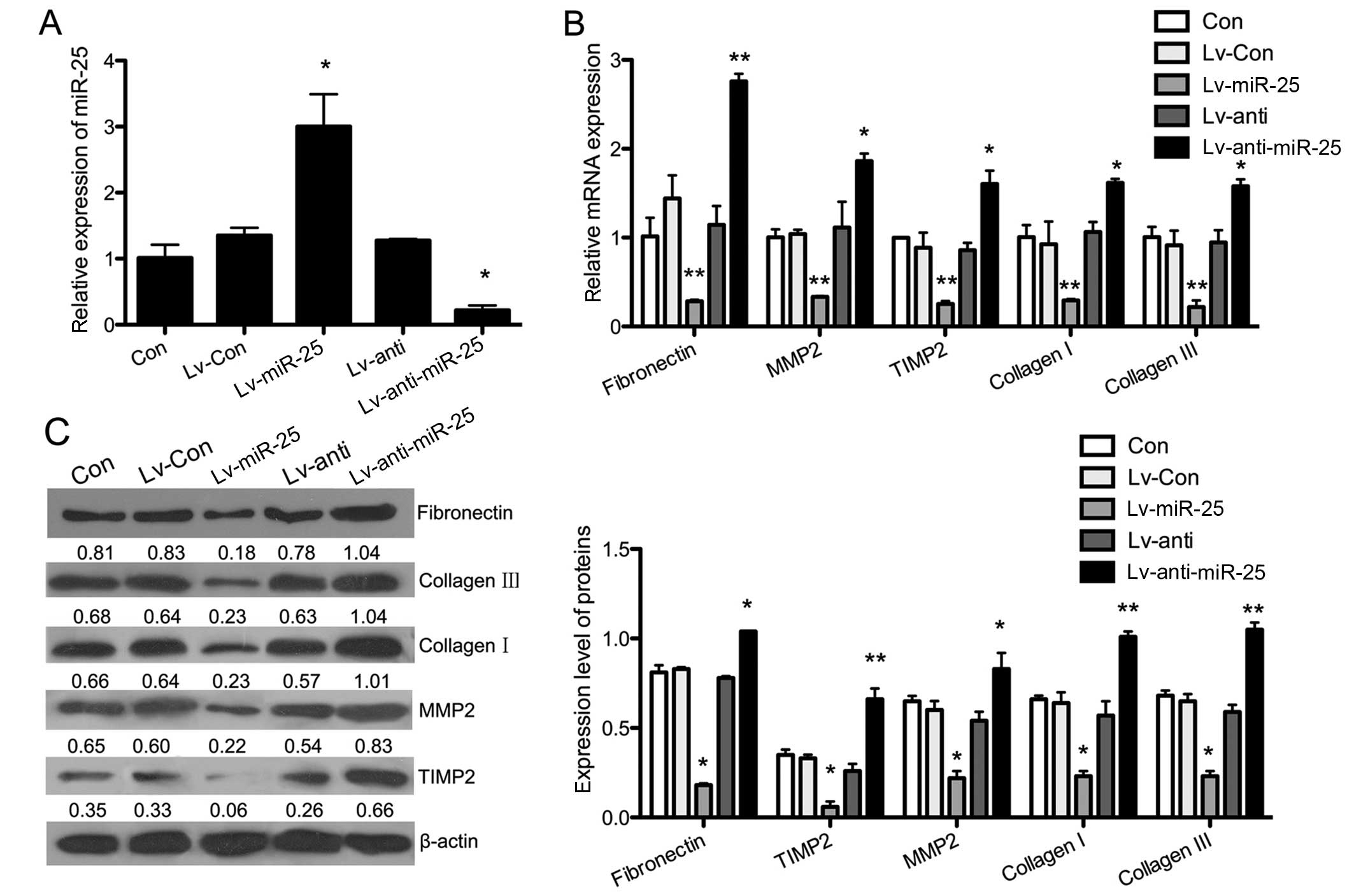
The expression of miR-25 was significantly upregulated in the
Lv-miR-25 group compared with the Con group (P<0.05), and it was
downregulated in the Lv-anti-miR-25 group (P<0.05). There were
no changes in miR-25 expression in the Lv-Con and Lv-anti groups
(Fig. 3A). The mRNA and protein
expression of fibrosis-related proteins was evaluated by RT-qPCR
and western blot analysis, respectively, in gain-of and
lost-of-function models of miR-25 cell lines subjected to H/R. As
shown in Fig. 3B, the
overexpression of miR-25 significantly reversed the H/R-induced
expression of fibronectin (P<0.01), collagen I (P<0.01),
collagen III (P<0.01), MMP2 (P<0.01) and TIMP2 (P<0.01).
The H/R-induced expression of fibronectin (P<0.01), collagen I
(P<0.05), collagen III (P<0.05), MMP2 (P<0.05) and TIMP2
(P<0.05) was further increased when the miR-25-silenced cells
were subjected to H/R. These findings suggesed that the
overexpression of miR-25 inhibited fibrosis, which was induced by
H/R in H9c2 cells. The results of western blot analysis showed that
miR-25 overexpression led to similar effects on the expression of
fibrosis-related genes (Fig.
3C).
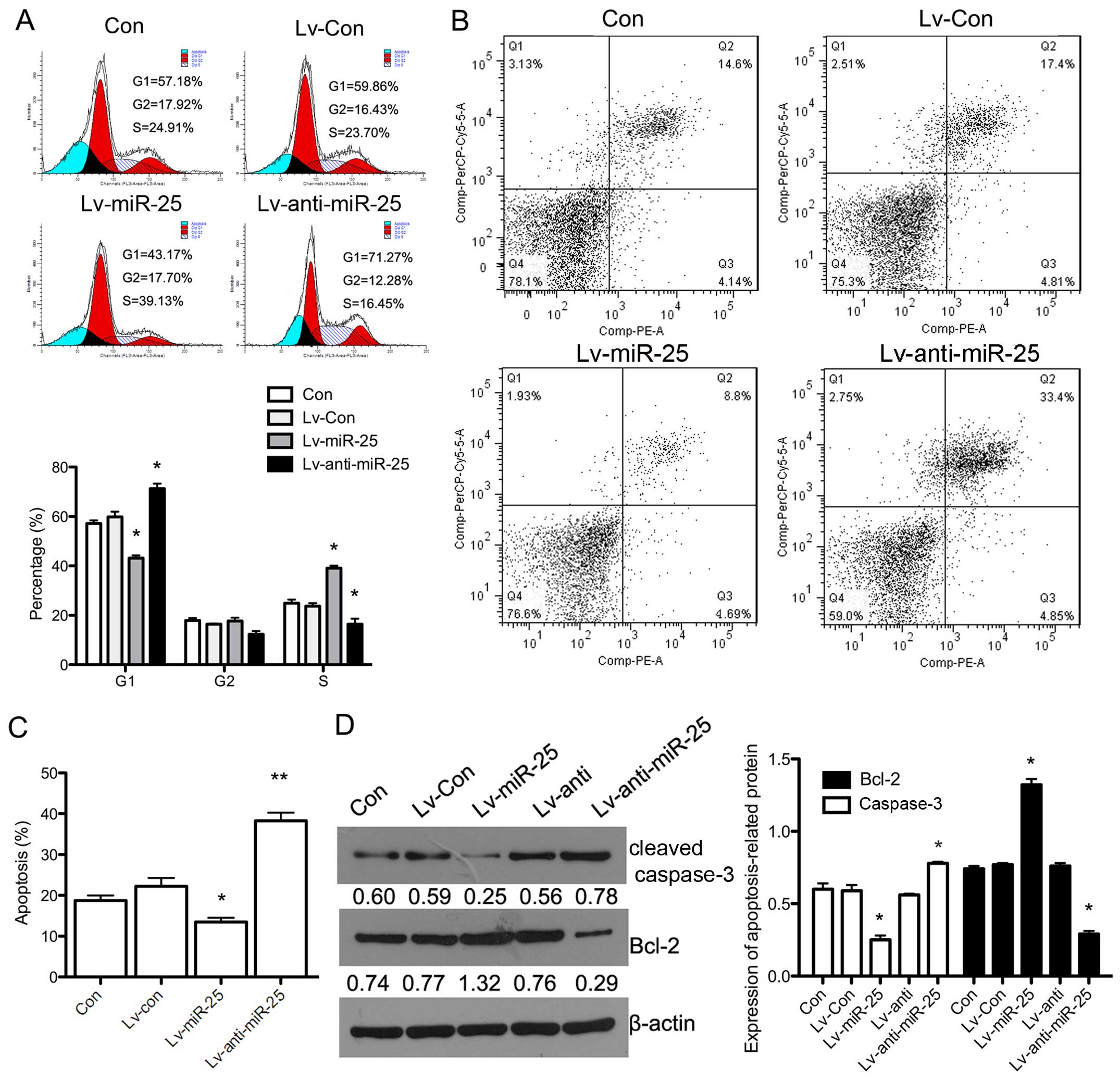
Additionally, we examined the cell cycle in H9c2
cells exhibiting stable overexpression and silencing of miR-25
following exposure to H/R. The overexpression of miR-25 led to a
significant decrease in the number of cells in the G1 phase
(P<0.05), whereas the number of cells in the S phase was
increased in miR-25-overexpressing cells exposed to H/R (P<0.05)
compared with the group of H9c2 cells exposed to H/R only (Con).
Silencing miR-25 expression in combination with H/R resulted in
increased numbers of cells in the G1 phase and decreased numbers of
cells in the S phase (Fig. 4A).
Furthermore, apoptosis was evaluated by flow cytometry and western
blot analysis. Fig. 4B and C show
that the apoptotic rate in miR-25-overexpressing cells exposed to
H/R was decreased compared with the control group (H9c2 cells
exposed only to H/R) (P<0.05). Reduced miR-25 expression with
H/R led to a significant increase in the apoptotic rate
(P<0.01). The protein expression of Bcl-2 was upregulated in
miR-25-overexpressing cells exposed to H/R (P<0.05), and cleaved
caspase-3 expression was significantly downregulated (P<0.05)
(Fig. 4D). This indicated that
the overexpression of miR-25 inhibited the H/R-induced apoptosis of
H9c2 cells through regulation of the cell cycle.
These findings demonstrated that the overexpression
of miR-25 reversed the enhanced expression of fibrosis- and
apoptosis-related genes induced by H/R.
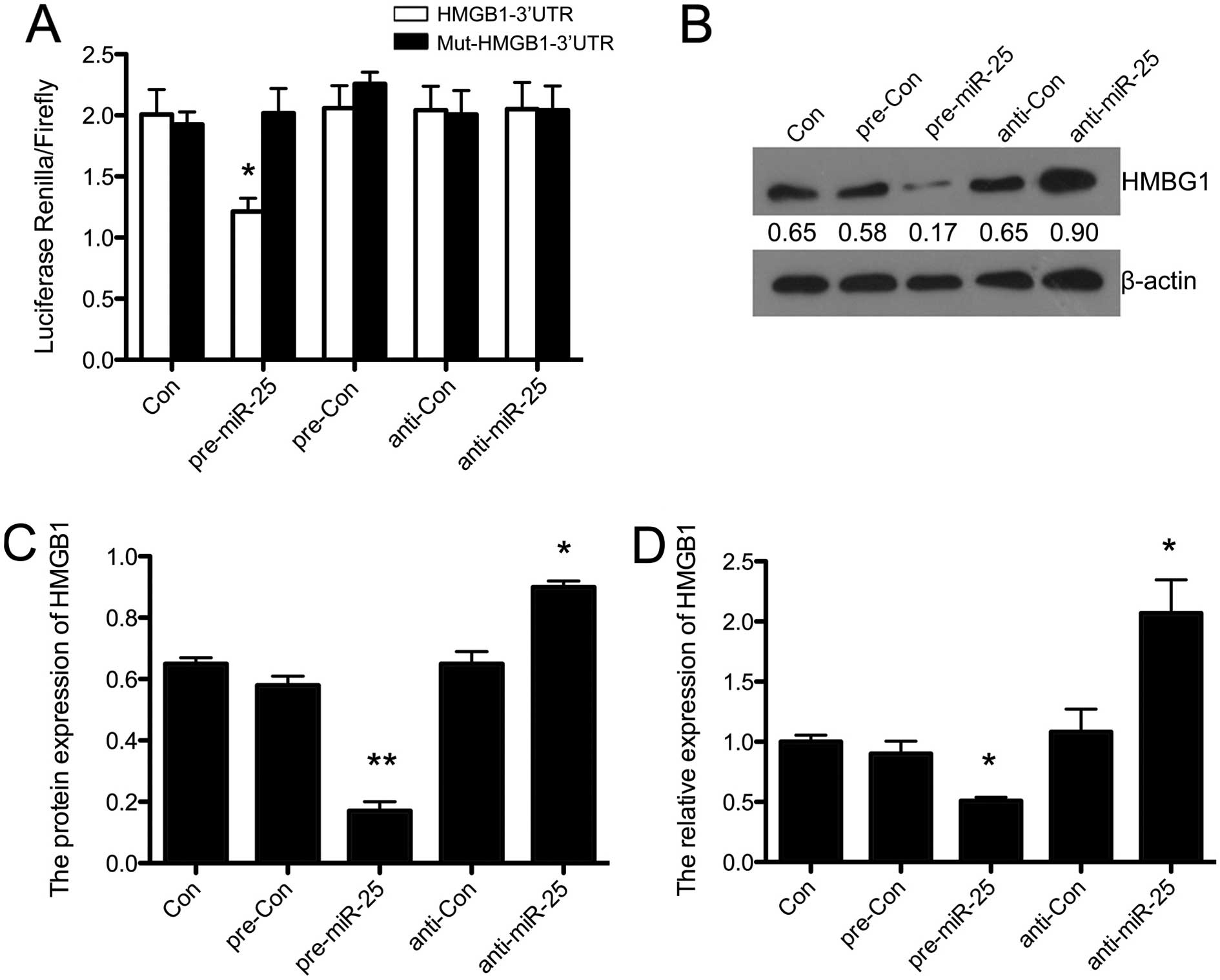
miR-25 directly targets HMGB1 and
inhibits HMGB1 expression in H9c2 cells
The bioinformatics tool, microRNA.org-Targets and
Expression, was used to predict the target genes for miR-25. HMGB1
was found to be a potential candidate target of miR-25. In order to
confirm whether HMGB1 was indeed functionally targeted by miR-25, a
dual-luciferase reporter gene assay was performed. Fig. 5A shows that the overexpression of
miR-25 inhibited luciferase activity in the construct with the
HMGB1-3′-UTR segment in H9c2 cells (P<0.05). There was no change
in luciferase reporter activity when the cells were transfected
with the miR-25-inhibitor or negative controls. This demonstrated
that HMGB1 was directly targeted by miR-25 in H9c2 cells. In
addition, we performed western blot analysis in
miR-25-overexpressing H9c2 cells (Fig. 5B). As shown in Fig. 5C, the overexpression of miR-25
suppressed the protein expression of HMGB1 compared with the
control (P<0.01), and silencing miR-25 upregulated the
expression of HMGB1 (P<0.05), with no changes observed in the
pre-Con and anti-Con groups. Furthermore, the results of RT-qPCR
showed similar effects of miR-25 on the mRNA expression of HMGB1
(P<0.05) (Fig. 5D). This
indicated that HMGB1 was directly targeted and inhibited by
miR-25.
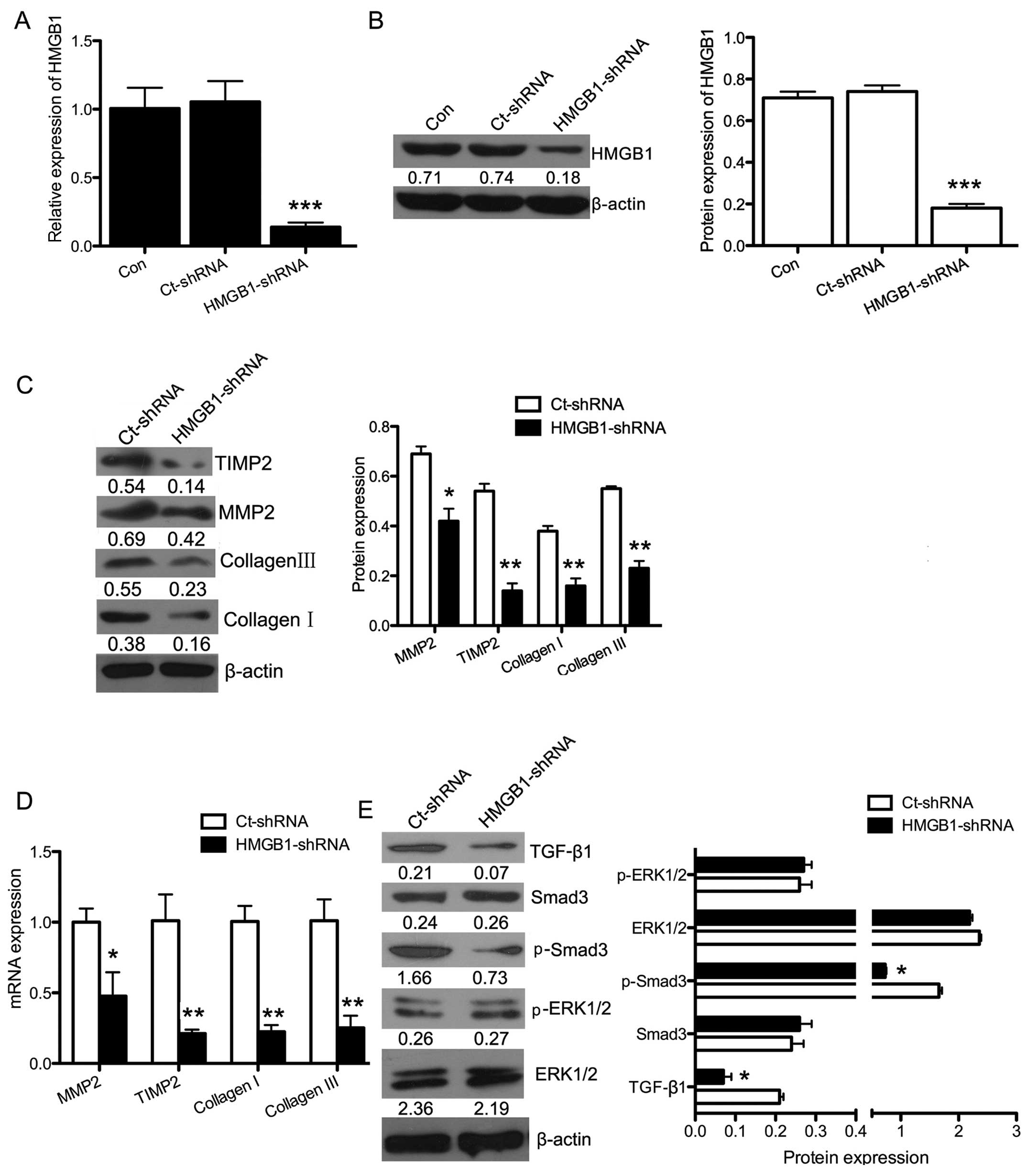
Downregulation of HMGB1 reduces fibrosis
via the TGF-β1/Smad3 pathway in H9c2 cells
To determine the effects of silencing HMGB1,
HMGB1-shRNA or Ct-shRNA were transfected into H9c2 cells. As shown
in Fig. 6A and B, the mRNA
(P<0.001) and protein (P<0.001) expression of HMGB1 was
significantly downregulated in the HMGB1-shRNA group.
To further examine the role of HMGB1 in fibrosis,
HMGB1-shRNA or Ct-shRNA was transfected into H9c2 cells exposed to
H/R. Then, the cells were analyzed by western blot analysis and
RT-qPCR for the expression of fibrosis-related genes. The mRNA and
protein expression levels of collagen I (P<0.01), collagen III
(P<0.01), MMP2 (P<0.05) and TIMP2 (P<0.01) were
significantly decreased after silencing HMGB1 (Fig. 6C and D) in the H/R-exposed cells.
This suggested that the downregulation of HMGB1 reduced the extent
of fibrosis induced by H/R. Furthermore, the HMGB1-related
signaling pathways were then evaluated by western blot analysis
(Fig. 6E). The protein expression
of TGF-β1 (P<0.05) and p-Smad3 (P<0.05) in the H/R-exposed
cells was significantly downregulated following HMGB1 silencing,
whereas no difference was observed in the expression of p-ERK1/2
after cell transfection. Taken together, these findings indicate
that HMGB1 may enhance fibrosis through the TGF-β1/Smad3 signaling
pathway in H9c2 cells exposed to H/R.
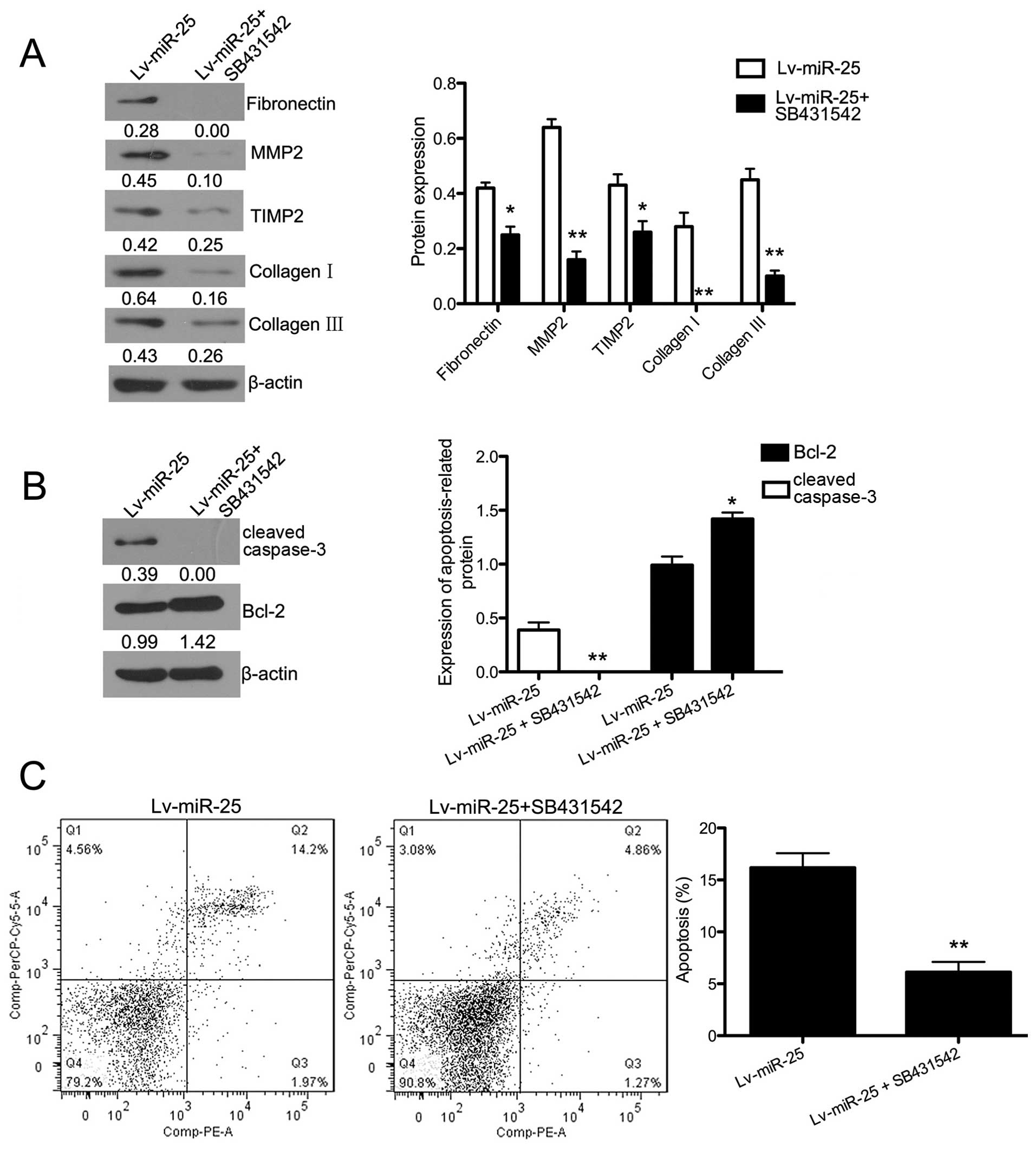
An inhibitor of the TGF-β1/Smad3
signaling pathway enhances the inhibitory effect of miR-25 on
fibrosis in H9c2 cells
A signaling pathway inhibitor of TGF-β1/Smad3,
SB431542, was used in miR-25-overexpressing cells exposed to H/R.
The expression of fibronectin, collagen I, collagen III, MMP2 and
TIMP2 was evaluated by western blot analysis (Fig. 7A). The expression of all the
fibrosis-related proteins in miR-25-overexpressing cells exposed to
H/R was significantly suppressed by SB431542 (P<0.05 for
fibronectin and TIMP2, P<0.01 for MMP2, collagen I and collagen
III). The protein expression of Bcl-2 was upregulated after
inhibitor treatment in the miR-25-overexpressing cells exposed to
H/R (P<0.01), and cleaved caspase-3 expression was significantly
downregulated (P<0.05) (Fig.
7B). Fig. 7C shows that the
apoptotic rate in inhibitor-treated cells exposed to H/R was
significantly lower (P<0.01). Collectively, the inhibitor of the
TGF-β1/Smad3 signaling pathway enhanced the inhibitory effect of
miR-25-overexpression on fibrosis in H/R-exposed cells.
Discussion
Myocardial I/R injury often induces myocardial cell
death and fibrosis (16), and
miRNAs maybe involved in reducing the risk of myocardial apoptosis
and fibrosis (17,18). This study examined the role of
miR-25 in fibrosis as well as in the apoptosis of H9c2 cells by
observing miR-25 expression in H/R and by identifying the signaling
pathway of apoptosis and fibrosis involving miR-25.
The expression of the miR-25 family of
inflammation-associated miRNAs in the plasma of patients with
unstable angina was downregulated in comparison with those who
received no statins (19). The
inhibition of miR-25 sensitized the murine myocardium to heart
failure in a Hand2-dependent manner (20). miR-25 has also been proved to
decrease collagen deposition (15). Moreover, we found that miR-25 was
the most downregulated miR in the miR-25-106b family in H9c2 cells
following exposure to H/R. Exposure to H/R induced the upregulation
of fibrosis-related proteins, namely fibronectin, collagen I,
collagen III, MMP2 and TIMP2, increased cell apoptosis and cell
cycle blocking in the G1 phase. These effects indicate that the
expression of miR-25 is associated with H/R-induced apoptosis and
fibrosis. Moreover, in the miR-25-overexpressing cells exposed to
H/R conditions, fibrosis was inhibited, cell apoptosis was
prevented, and cell cycle blocking in the G1 phase was reversed.
These results demonstrated that the overexpression of miR-25
inhibited H/R-induced apoptosis and fibrosis.
HMGB1, a chromatin-associated non-histone nuclear
protein and extracellular damage-associated molecular pattern
(DAMP), is a critical regulator of cell death and survival
(21). Accumulating evidence
indicates that some non-immune cells, such as endothelial cells,
hepatocytes (22) and
cardiomyocytes (23) also
actively secrete HMGB1. HMGB1 has been identified as a novel
pro-inflammatory cytokine in several cardiovascular diseases
(7,24–26). Additionally, HMGB1 has been found
to function as an early pro-inflammatory mediator during myocardial
I/R, and HMGB1 A box peptide (a specific HMGB1 antagonist) may
protect against apoptosis and fibrosis induced by myocardial I/R
injury (27,28). These results suggested that HMGB1
plays an important role in I/R injury, and is potentially a novel
target for reducing I/R injury. In our study, we found that HMGB1
was upregulated in the H9c2 cells following exposure to H/R.
Simultaneously, the expression of miR-25 was downregulated.
Moreover, we confirmed that HMGB1 may be a potential target of
miR-25 using a bioinformatics tool. A dual-luciferase reporter gene
assay confirmed that HMGB1 was a direct target of miR-25.
Additionally, the downregulation of miR-25 induced the upregulation
of HMGB1. Thus, miR-25 inhibited the apoptosis and fibrosis induced
by H/R by targeting HMGB1.
Su et al demonstrated that HMGB1 induced
cardiac collagen deposition through the PKCβ/ERK1/2 signaling
pathway (29). Additionally,
TGF-β expression correlates with fibrous tissue deposition by
inducing extracellular matrix protein synthesis following
myocardial infarction (30).
HMGB1 also prevents postinfarction adverse myocardial remodeling
through the TGF-β/Smad signaling pathway. It has been shown that
the overexpression of the miR-106b-25 cluster in gastrointestinal
cancer cells, and suggested that the miR-106b~25 cluster is a key
modulator of TGF-β signaling (31). Furthermore, Smad3 signaling was
proved to play roles in modulating myocardial fibrosis, possibly
through a pathway that entails the accumulation of miRNAs that
decrease collagen gene expression (15). Therefore, we examined whether the
PKCβ/ERK1/2 signaling pathway or the TGF-β1/Smad3 signaling pathway
was involved in the regulatory effects of HMGB1 in fibrosis. We
found that the downregulation of HMGB1 reduced fibrosis in the
H/R-exposed H9c2 cells through the TGF-β1/Smad3 pathway, rather
than the PKCβ/ERK1/2 signaling pathway. Moreover, an inhibitor of
the TGF-β1/Smad3 signaling pathway (SB431542) enhanced the effect
of miR-25.
In conclusion, the upregulation of miR-25 inhibited
apoptosis and fibrosis in H/R-exposed cells by directly targeting
HMGB1 through the TGF-β1/Smad3 pathway. These findings may provide
novel insight into the pathogenesis of fibrosis, particularly with
respect to H/R-related heart failure.
References
|
1
|
Lee SW, Won JY, Kim WJ, Lee J, Kim KH,
Youn SW, Kim JY, Lee EJ, Kim YJ, Kim KW and Kim HS: Snail as a
potential target molecule in cardiac fibrosis: paracrine action of
endothelial cells on fibroblasts through snail and CTGF axis. Mol
Ther. 21:1767–1777. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dhooghe B, Noël S, Huaux F and Leal T:
Lung inflammation in cystic fibrosis: pathogenesis and novel
therapies. Clin Biochem. 47:539–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Srivastava SP, Koya D and Kanasaki K:
MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT
and EndMT. BioMed Res Int. 2013:1254692013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
López-Novoa JM and Nieto MA: Inflammation
and EMT: an alliance towards organ fibrosis and cancer progression.
EMBO Mol Med. 1:303–314. 2009. View Article : Google Scholar
|
|
5
|
Andersson U and Tracey KJ: HMGB1 is a
therapeutic target for sterile inflammation and infection. Annual
Rev Immunol. 29:139–162. 2011. View Article : Google Scholar
|
|
6
|
Lynch J, Nolan S, Slattery C, Feighery R,
Ryan MP and McMorrow T: High-mobility group box protein 1: a novel
mediator of inflammatory-induced renal epithelial-mesenchymal
transition. Am J Nephrol. 32:590–602. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Andrassy M, Volz HC, Igwe JC, Funke B,
Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK,
et al: High-mobility group box-1 in ischemia-reperfusion injury of
the heart. Circulation. 117:3216–3226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roush S and Slack FJ: The let-7 family of
microRNAs. Trends Cell Biol. 18:505–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao J, Tang N, Wu K, Dai W, Ye C, Shi J,
Zhang J, Ning B, Zeng X and Lin Y: MiR-21 simultaneously regulates
ERK1 signaling in HSC activation and hepatocyte EMT in hepatic
fibrosis. PLoS One. 9:e1080052014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brønnum H, Andersen DC, Schneider M,
Sandberg MB, Eskildsen T, Nielsen SB, Kalluri R and Sheikh SP:
miR-21 promotes fibrogenic epithelial-to-mesenchymal transition of
epicardial mesothelial cells involving Programmed Cell Death 4 and
Sprouty-1. PLoS One. 8:e562802013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang Y, Huang JQ, Zhang X and Shen LF:
MiR-129-2 functions as a tumor suppressor in glioma cells by
targeting HMGB1 and is down-regulated by DNA methylation. Mol Cell
Biochem. 404:229–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu F, Zhang J, Ji M, Li P, Du Y, Wang H,
Zang S, Ma D, Sun X and Ji C: miR-181b increases drug sensitivity
in acute myeloid leukemia via targeting HMGB1 and Mcl-1. Int J
Oncol. 45:383–392. 2014.PubMed/NCBI
|
|
13
|
Li X, Wang S, Chen Y, Liu G and Yang X:
miR-22 targets the 3′UTR of HMGB1 and inhibits the HMGB1-associated
autophagy in osteosarcoma cells during chemotherapy. Tumour Biol.
35:6021–6028. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang Q, Zhong H, Xie F, Xie J, Chen H and
Yao G: Expression of miR-106b-25 induced by salvianolic acid B
inhibits epithelial- to-mesenchymal transition in HK-2 cells. Eur J
Pharmacol. 741:97–103. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Divakaran V, Adrogue J, Ishiyama M, Entman
ML, Haudek S, Sivasubramanian N and Mann DL: Adaptive and
maladptive effects of SMAD3 signaling in the adult heart after
hemodynamic pressure overloading. Circ Heart Fail. 2:633–642. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barallobre-Barreiro J, Didangelos A,
Schoendube FA, Drozdov I, Yin X, Fernández-Caggiano M, Willeit P,
Puntmann VO, Aldama-López G, Shah AM, et al: Proteomics analysis of
cardiac extracellular matrix remodeling in a porcine model of
ischemia/reperfusion injury. Circulation. 125:789–802. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Z, Hu Z, Lu Z, Cai S, Gu X, Zhuang H,
Ruan Z, Xia Z, Irwin MG, Feng D and Zhang L: Differential microRNA
profiling in a cellular hypoxia reoxygenation model upon
posthypoxic propofol treatment reveals alterations in autophagy
signaling network. Oxid Med Cell Longev. 2013:3784842013.
View Article : Google Scholar
|
|
18
|
Di Y, Lei Y, Yu F, Changfeng F, Song W and
Xuming M: MicroRNA expression and function in cerebral ischemia
reperfusion injury. J Mol Neurosci. 53:242–250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Ren JY, Chen H and Han GP:
Statins decrease expression of five inflammation-associated
microRNAs in the plasma of patients with unstable angina. Beijing
Da. Xue Xue Bao. 47:761–768. 2015.In Chinese.
|
|
20
|
Dirkx E, Gladka MM, Philippen LE, Armand
AS, Kinet V, Leptidis S, El Azzouzi H, Salic K, Bourajjaj M, da
Silva GJ, et al: Nfat and miR-25 cooperate to reactivate the
transcription factor Hand2 in heart failure. Nat Cell Biol.
15:1282–1293. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klune JR, Dhupar R, Cardinal J, Billiar TR
and Tsung A: HMGB1: endogenous danger signaling. Mol Med.
14:476–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsung A, Klune JR, Zhang X, Jeyabalan G,
Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR and Billiar TR:
HMGB1 release induced by liver ischemia involves Toll-like receptor
4-dependent reactive oxygen species production and calcium-mediated
signaling. J Exp Med. 204:2913–2923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu H, Su Z, Wu J, Yang M, Penninger JM,
Martin CM, Kvietys PR and Rui T: The alarmin cytokine, high
mobility group box 1, is produced by viable cardiomyocytes and
mediates the lipopolysaccharide-induced myocardial dysfunction via
a TLR4/phosphatidylinositol 3-kinase gamma pathway. J Immunol.
184:1492–1498. 2010. View Article : Google Scholar
|
|
24
|
Hu X, Jiang H, Bai Q, Zhou X, Xu C, Lu Z,
Cui B and Wen H: Increased serum HMGB1 is related to the severity
of coronary artery stenosis. Clin Chim Acta. 406:139–142. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Andrassy M, Volz HC, Riedle N, Gitsioudis
G, Seidel C, Laohachewin D, Zankl AR, Kaya Z, Bierhaus A,
Giannitsis E, et al: HMGB1 as a predictor of infarct transmurality
and functional recovery in patients with myocardial infarction. J
Intern Med. 270:245–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao D, Wang Y and Xu Y: Decreased serum
endogenous secretory receptor for advanced glycation endproducts
and increased cleaved receptor for advanced glycation endproducts
levels in patients with atrial fibrillation. Int J Cardiol.
158:471–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frangogiannis NG, Smith CW and Entman ML:
The inflammatory response in myocardial infarction. Cardiovasc Res.
53:31–47. 2002. View Article : Google Scholar
|
|
28
|
Hu X, Fu W and Jiang H: HMGB1: A potential
therapeutic target for myocardial ischemia and reperfusion injury.
Int J Cardiol. 155:4892012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Su Z, Yin J, Wang T, Sun Y, Ni P, Ma R,
Zhu H, Zheng D, Shen H, Xu W and Xu H: Up-regulated HMGB1 in EAM
directly led to collagen deposition by a PKCβ/Erk1/2-dependent
pathway: cardiac fibroblast/myofibroblast may be another source of
HMGB1. J Cell Mol Med. 18:1740–1751. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saltis J, Agrotis A and Bobik A:
Regulation and interactions of transforming growth factor-beta with
cardiovascular cells: implications for development and disease.
Clin Exp Pharmacol Physiol. 23:193–200. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Petrocca F, Vecchione A and Croce CM:
Emerging role of miR-106b-25/miR-17-92 clusters in the control of
transforming growth factor beta signaling. Cancer Res.
68:8191–8194. 2008. View Article : Google Scholar : PubMed/NCBI
|