Introduction
Williams-Beuren syndrome (WBS, no. OMIM 194050) is a
multisystem genetic disorder resulting from the hemizygous deletion
of a region spanning 1.5–1.8 Mb on chromosome 7q11.23 which
contains 28 coding genes and 2 miRNAs. This syndrome is
characterized by cardiovascular malformations (mostly supravalvular
aortic stenosis), mental retardation and a specific facial
dysmorphism (1). We recently
reported the occurrence of several cases of B-cell non-Hodg-kin's
lymphoma (B-NHL) in children with WBS as well as the existence of
somatic deletion of the WBS critical region (WBSCR) in sporadic
B-NHL. We also characterized the WBSCR using CGH-array and
high-throughput sequencing in both normal and tumor samples from
WBS patients and we found no second hits on the remaining allele.
Thus, we suggested that the haploinsufficiency of the WBSCR genes
might be associated with a predisposition to cancer, particularly
B-NHL (2,3).
Genomic instability is a hallmark of B-NHL. The
mechanisms by which B cells somatically engineer their genomes to
generate the vast diversity of antibodies induce genomic
instability. These mechanisms are highly regulated but, in some
rare cases, abnormal B-cells might escape from controls and evolve
toward malignancy. A number of congenital disorders associated with
a DNA damage response and/or repair defect such as
ataxia-telangiectasia, Bloom syndrome or Nijmegen breakage syndrome
have been linked to a predisposition to B-NHL (4–7).
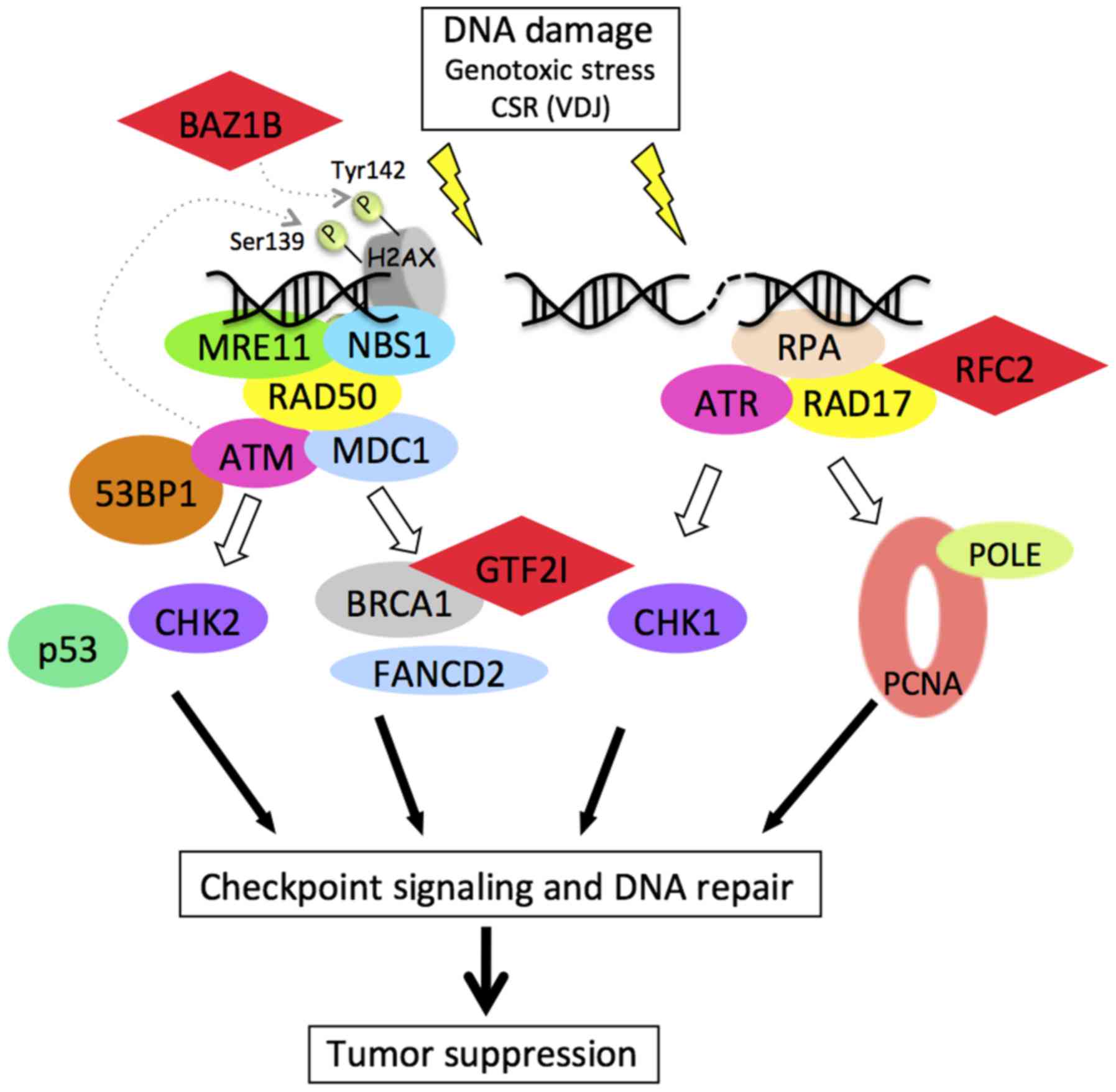
In addition, three genes that are hemizygously
deleted in WBS encode proteins that are involved in DNA damage
response and/or repair signaling pathways (Fig. 1).
First, RFC2 is one of the five subunits of the
replication factor C that loads the proliferating cell nuclear
antigen (PCNA) onto chromatin during DNA replication thus
facilitating DNA polymerase action. RFC2 forms a multiprotein
complex with Rad17 that plays a major role in ATR signaling.
Notably, it has been shown that mutant cells of Saccharomyces
cerevisiae having a thermosensitive RFC2 mutation exhibit
temperature-sensitive growth, sensitivity to hydroxyurea (HU) and
UV light and an increased rate of mitotic recombination DNA
replication and cell cycle checkpoint (8).
Second, GTF2I (also called TFII-I) is a
multifunctional transcription factor that activates
growth-promoting target genes upon tyrosine phosphorylation in
response to mitogenic signaling. In 2005, Desgranges et al
demonstrated that GTF2I is ubiquitinated and targeted to
proteasomal degradation in response to genotoxic stress in a p53-
and ATM-dependent manner resulting in cell cycle arrest (9). More recently, Tanikawa et al
showed that GTF2I plays an important role in regulating
BRCA1-mediated homologous recombination (HR) mechanisms. After
irradiation, GTF2I forms foci with γ-H2A.X and the depletion of
endogenous GTF2I using siRNA knockdown results in the inhibition of
HR efficiency (10). GTF2I is
also involved in translesion synthesis (TLS) mechanisms. Notably,
it has been shown that GTF2I bridges PCNA and Pol ζ to promote TLS
(11).
Finally, BAZ1B (also called WSTF) is a component of
the WSTF-ISWI chromatin-remodeling complex (WICH) that is involved
in maintaining chromatin organization (12,13). Recently, BAZ1B has also been
linked to the DNA damage response pathway and was shown to possess
tyrosine kinase activity phosphorylating Tyr142 of H2A.X. In
response to genotoxic stress, the balance of the phosphorylation
state of both Tyr142 and Ser139 of H2A.X appears to determine the
cell fate between DNA repair and apoptosis (14,15).
However, it is unclear whether the
haploinsufficiency of RFC2, GTF2I and BAZ1B is involved in DNA
damage response and/or repair deficiency in WBS patients.
The aim of our study was to investigate the
sensitivity to genotoxic stress and the DNA damage response in both
primary cell lines derived from WBS patients and in WBS
gene-specific siRNA knockdown cells.
Materials and methods
Ethical approval
Participation in this study by patients and their
relatives along with skin biopsy donations and informed consent
procedures were approved by the ethics committees of the Genomic
and Genetic Disorder Biobank (Casa Sollievo della Sofferenza, San
Giovanni Rotondo, Italy) and the University of Franche-Comté (UFC;
Besançon, France).
Cell lines, cell culture and
transfections
Primary fibroblast cell lines from WBS patients
(GDB306FIBRO, GDB863FIBRO, GDB728FIBRO) and from healthy donors
(GDB380-2FIBRO, GDB809-1FIBRO, GDB819-1FIBRO) were provided by Dr
Giuseppe Merla from the Genomic and Genetic Disorders Biobank
(GGDB, Network of Telethon Genetic Biobanks, project no. GTB07001G)
in San Giovanni Rotondo, Italy. The 293T cell line (no. CRL-3216)
was purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). Cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Lonza, Verviers, Belgium) supplemented with
10% heat-inactivated endotoxin-free fetal bovine serum (FBS) and 1%
penicillin-streptomycin (Invitrogen Life Technologies, Carlsbad,
CA, USA).
293T cells were stably transfected individually with
1 µg of each of the 4 unique gene-specific 29mer shRNA constructs
(BAZ1B, no. TG306439; GTF2I, no. TG304176; RFC2, no. TG309864) or
no. TR30013 scrambled negative control non-effective shRNA cassette
in pGFP-V-RS (OriGene Technologies, Inc., Rockville, MD, USA) using
the Effectene transfection kit (Qiagen, Hilden, Germany) according
to the manufacturer's instructions. Transfected cells were selected
with 2 µg/m of puromycin (Invitrogen Life Technologies), 48 h
following transfection.
Drug treatments and cell proliferation
assay
Cells were treated with 0.05, 0.5 and 5 mM of HU
(no. H8627, 500 mM stock solution in ddH2O; Sigma, St.
Louis, MO, USA) or 1, 5 and 25 µM of etoposide (ETP, no. 2200, 50
mM stock solution in DMSO; Cell Signaling Technology, Inc.,
Danvers, MA, USA) for the indicated times.
For the cell proliferation assay, 10 μl of
Premix WST-1 cell proliferation assay reagent (no. MK400; Takara
Bio, Dalian, China) was added to the cultured cells in triplicate
in a 96-well plate at the indicated times and conditions with 100
μl of culture media and then the cells were incubated for 2
h before measuring the optical density (OD) following the
manufacturer's recommendations.
Cell cycle and γ-H2A.X analyses by flow
cytometry
For flow cytometry analyses a minimum of
105 cells from each condition was washed with PBS and
then fixed in pre-cooled 70% absolute ethanol and incubated at
−20°C for 60 min. Fixed cells were washed 3 times with cold PBS and
then stained with Alexa Fluor 647 anti-H2A.X-phosphorylated
(Ser139) antibody (no. 613408; BioLegend, Inc., San Diego, CA,
USA). Finally, the cells were washed once with PBS and resuspended
in 50 μl of propidium iodide (PI)/RNase staining solution
(no. 4087; Cell Signaling Technology, Inc.) 15 min before
fluorescence acquisition with BD FACS Canto II cytometer (BD
Biosciences, Franklin Lakes, NJ, USA).
Western blotting
For western blot analysis, whole-cell lysates were
separated by SDS-PAGE and transferred onto PVDF membranes using
Criterion TGX gels and Trans-Blot Turbo Midi PVDF Transfer Packs
(Bio-Rad, Berkeley, CA, USA). The blots were then incubated with
specific primary antibodies as follows: anti-CHK1 (no. 2360),
anti-CHK2 (no. 6334), anti-phospho-CHK1 Ser317 (no. 12302),
anti-phospho-CHK2 Thr68 (no. 2197), anti-BRCA1 (no. 9010),
anti-phospho-BRCA1 Ser1524 (no. 9009), anti-TFII-I (no. 4562) and
anti-WSTF (no. 2152) all purchased from Cell Signaling Technology,
Inc. The primary anti-RCF2 antibody (no. ab174271) was purchased
from Abcam (Cambridge, UK).
Results
A link between WBS and cancer has been suggested by
us and other authors. In the absence of epidemiological data on
this topic, we carried out a functional study in order to
investigate the DNA damage response in cells derived from WBS
patients.
Cell proliferation and sensitivity to
genotoxic stress
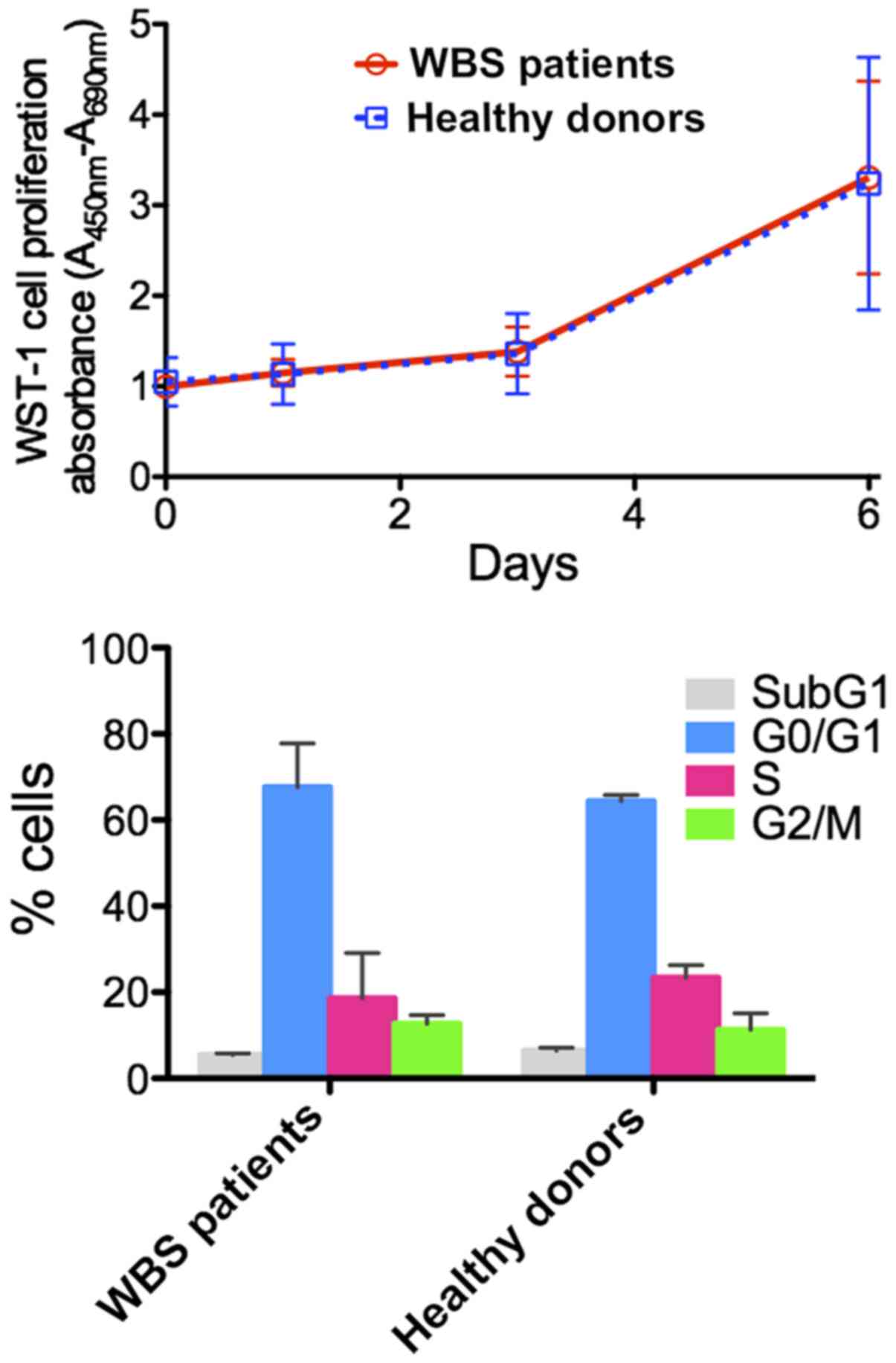
The proliferation of primary skin fibroblasts
derived from 3 WBS patients was studied on day 1, 3 and 6 and was
compared with the proliferation of fibroblasts derived from 3
healthy controls. The analysis of proliferation curves obtained
using the WST-1 proliferation assay showed no difference in cell
proliferation between the WBS patient and control fibroblasts.
Moreover, cell cycle analysis by flow cytometry revealed a similar
distribution of WBS cells in the G0/G1, S and G2/M phases relative
to that noted in the control cells (Fig. 2).
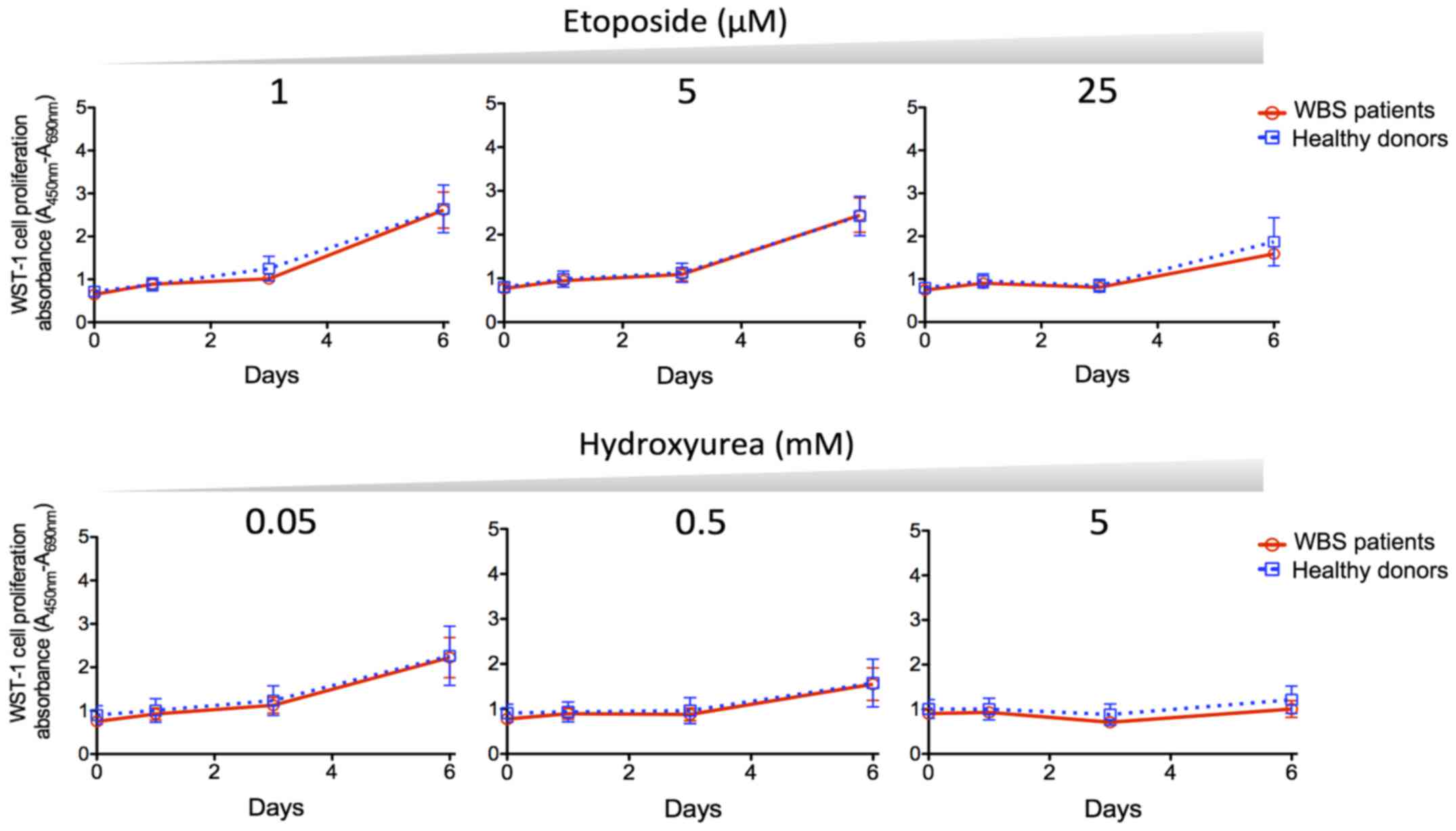
DNA damage response and repair defects are usually
associated with increased cellular sensitivity to genotoxic agents.
We used two different genotoxic agents to investigate the
sensitivity of WBS cells. ETP is a topoisomerase II inhibitor
commonly used as an antitumor agent that induces DNA double-strand
breaks. HU is a replication inhibitor that can induce DNA
double-strand breaks by causing replication fork arrest upon
nucleotide pool depletion. WST-1 cell proliferation assay of the
treated cells showed a dose-dependent effect of ETP and HU but a
difference in sensitivity to genotoxic agents between WBS patient
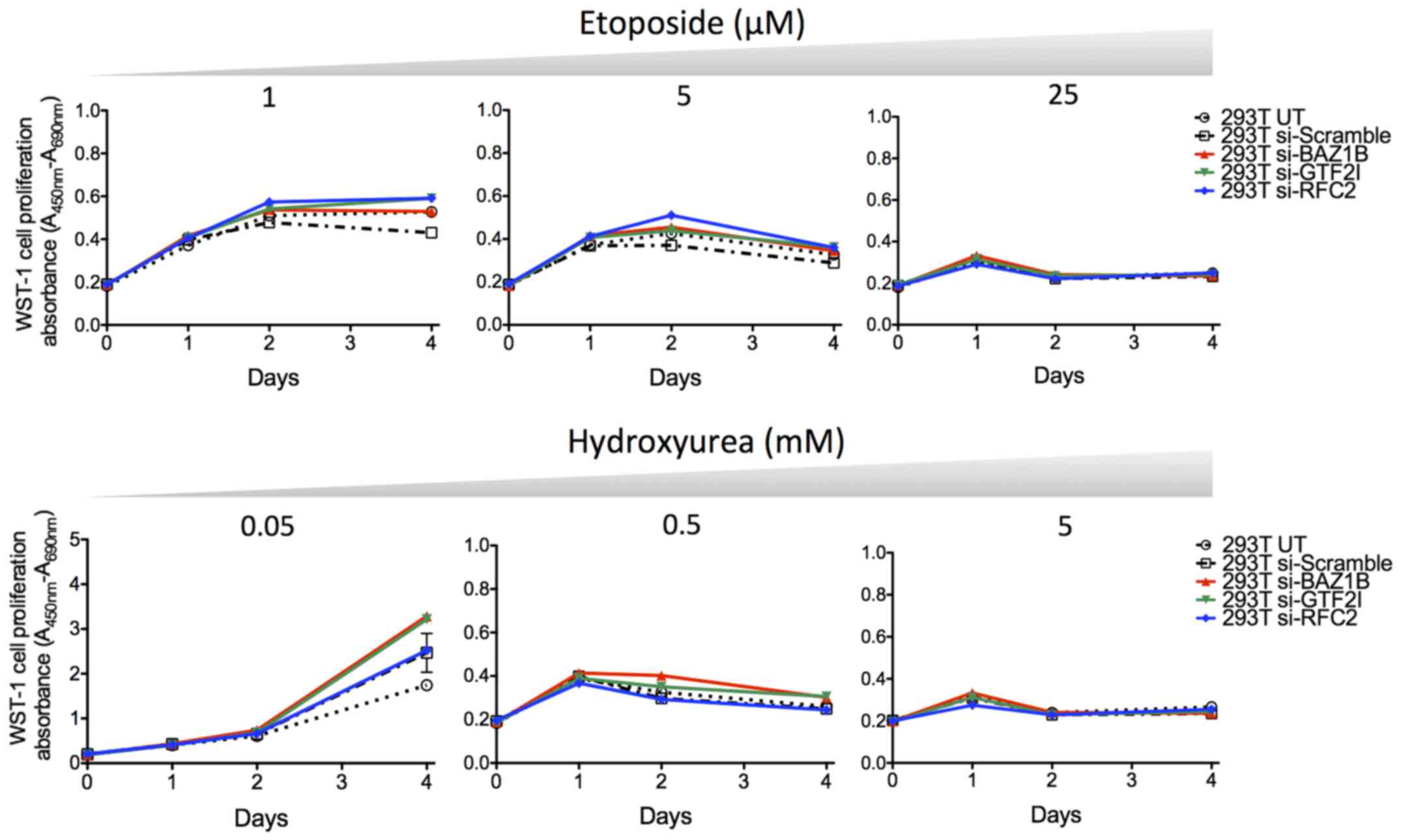
and healthy control fibroblasts was not observed (Fig. 3).
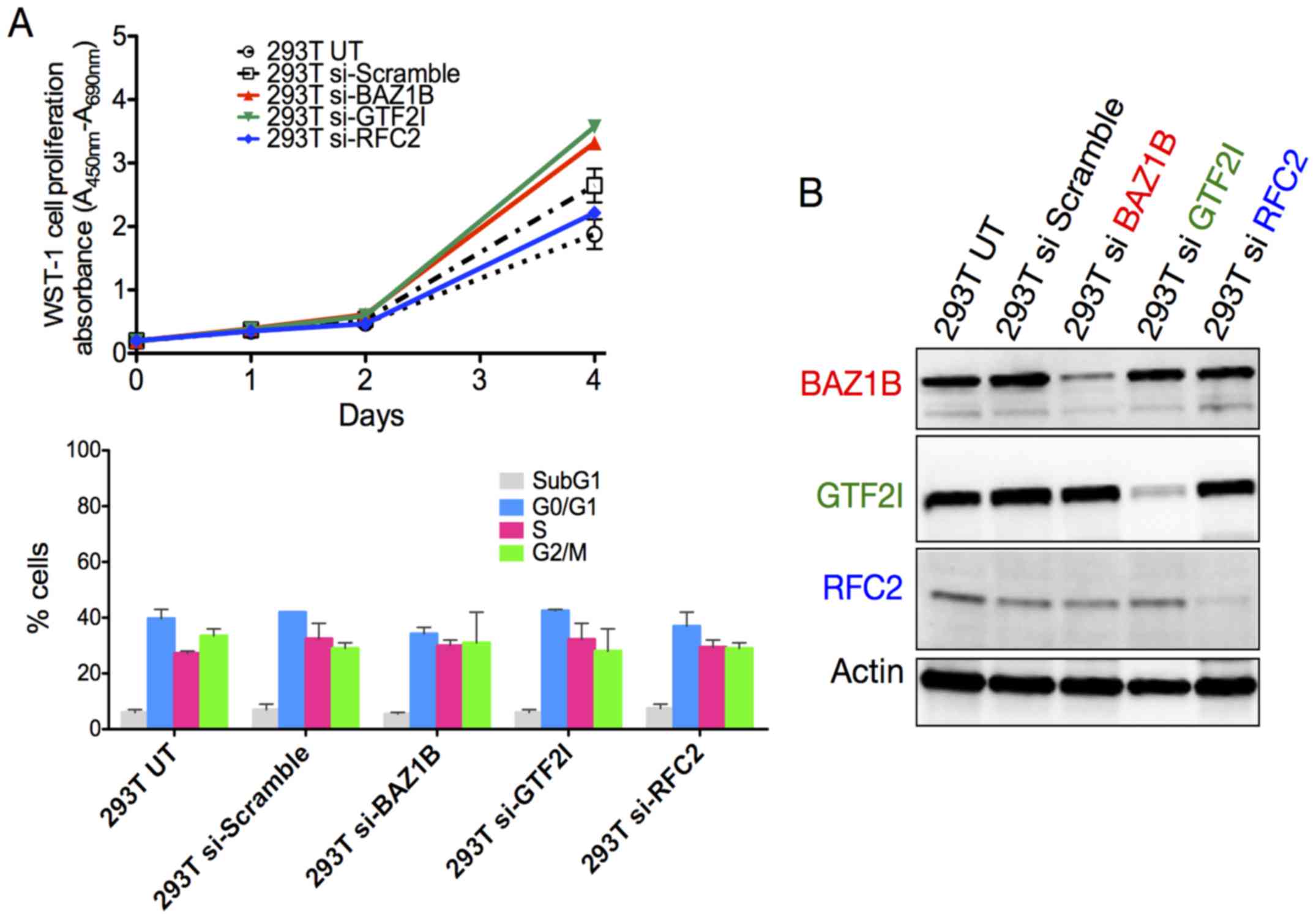
In order to further understand the individual
functions of WBS genes in DNA damage response and repair, we
generated RFC2, GTF2I and BAZ1B knockdown in 293T cells using
specific siRNA expression vectors. The 293T cells were stably
transfected and the expression of siRNA targets was validated by
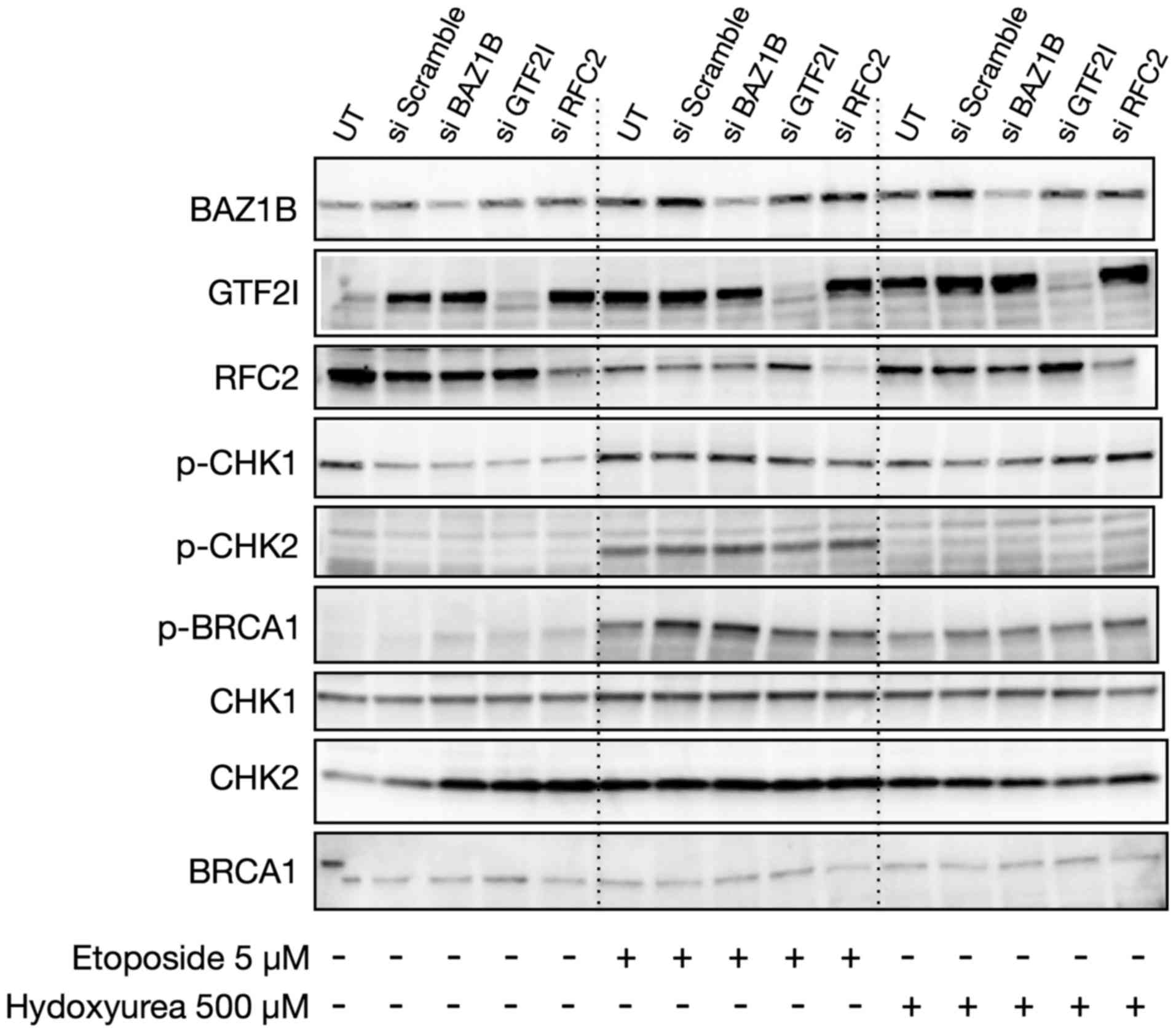
western blotting (Fig. 4B).
The 293T cells were subsequently used for
proliferation and cell cycle assays (Fig. 4A). In the absence of treatment,
293T cells depleted in BAZ1B and GTF2I showed a slightly increased
proliferation rate on day 4 compared with the untransfected cells
(UT) and cells transfected with the scramble siRNA. However, these
differences were not correlated with the data of the cell cycle
analysis that showed almost no differences between the different
transfected cells and controls. The sensitivity to HU and ETP was
also examined in the siRNA-transfected 293T cells. No difference
was observed relative to the UT and scramble controls (Fig. 5).
These results suggest that the haploinsufficiency of
WBS genes, including BAZ1B, GTF2I, and RFC2, is not associated with
a hypersensitivity to genotoxic agents.
γ-H2A.X induction and expression of DNA
damage response and repair proteins
During the DNA double-strand break response,
chromatin undergoes reorganization marked by H2A.X Ser 139
phosphorylation (γ-H2A.X). In the early phase of DNA damage
response, γ-H2A.X forms foci, which are platforms for recruiting
molecules involved in DNA damage repair and signaling. It has been
demonstrated that γ-H2A.X induction is reduced in cells derived
from patients with genetic syndromes associated with impaired DNA
damage (16). BAZ1B has a kinase
function that is responsible for H2A.X Tyr142 phosphorylation. A
crosstalk between Tyr142 and Ser139 of H2A.X has been demonstrated
recently and we aimed to ascertain whether the haploinsufficiency
of BAZ1B may be associated with abnormal γ-H2A.X induction. We,
therefore, analyzed γ-H2A.X induction in WBS fibroblasts, primary
cell lines and siRNA-transfected 293T cells.
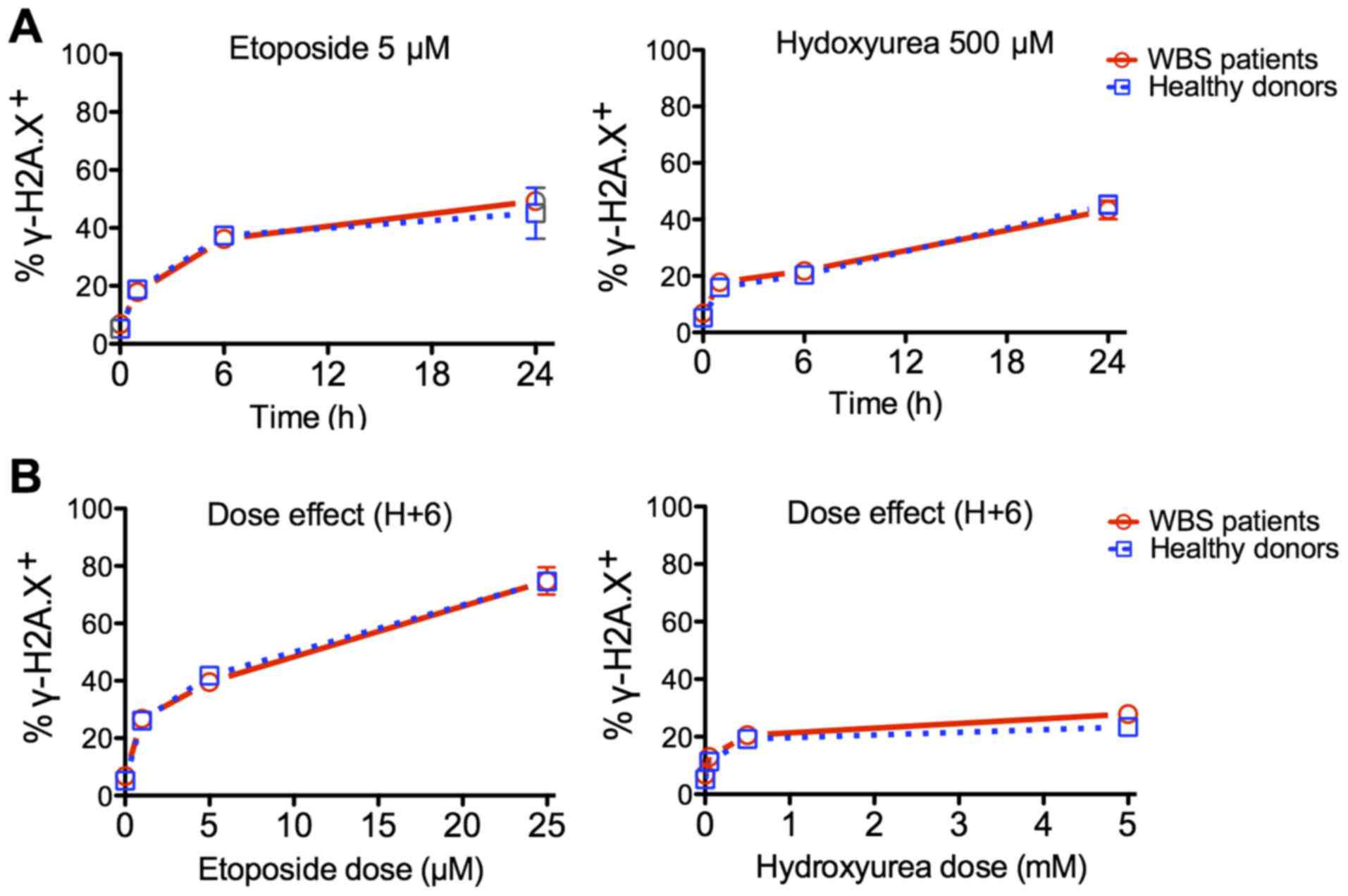
The dose-effects and kinetics of γ-H2A.X induction
following exposure to ETP and HU was investigated using
intracellular staining and flow cytometry. WBS patient and healthy
control cells displayed a similar proportion of γ-H2A.X-positive
cells under each condition (Fig.
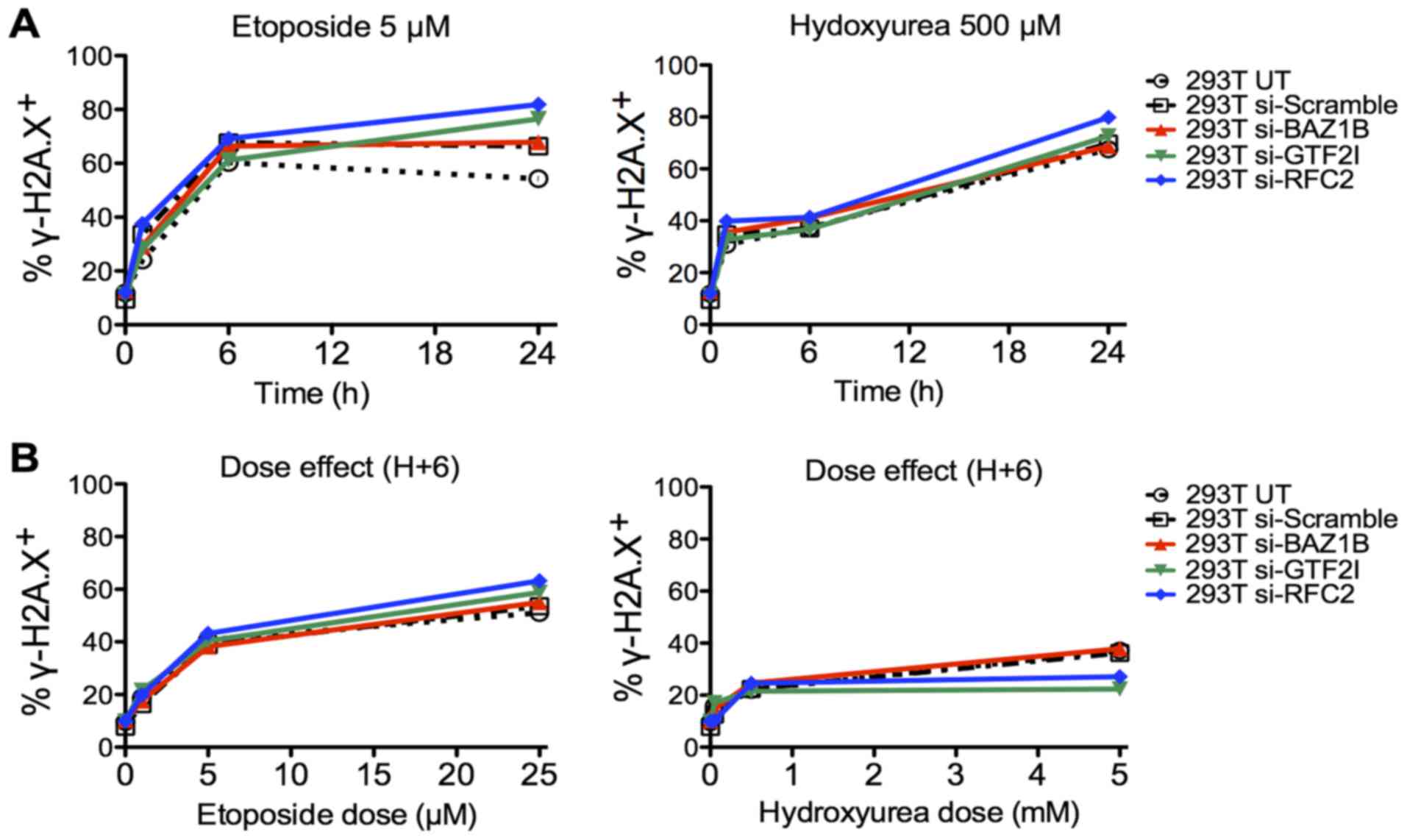
6). In the 293T cells, no significant difference was observed
between the siRNA-transfected cells and the controls (Fig. 7).
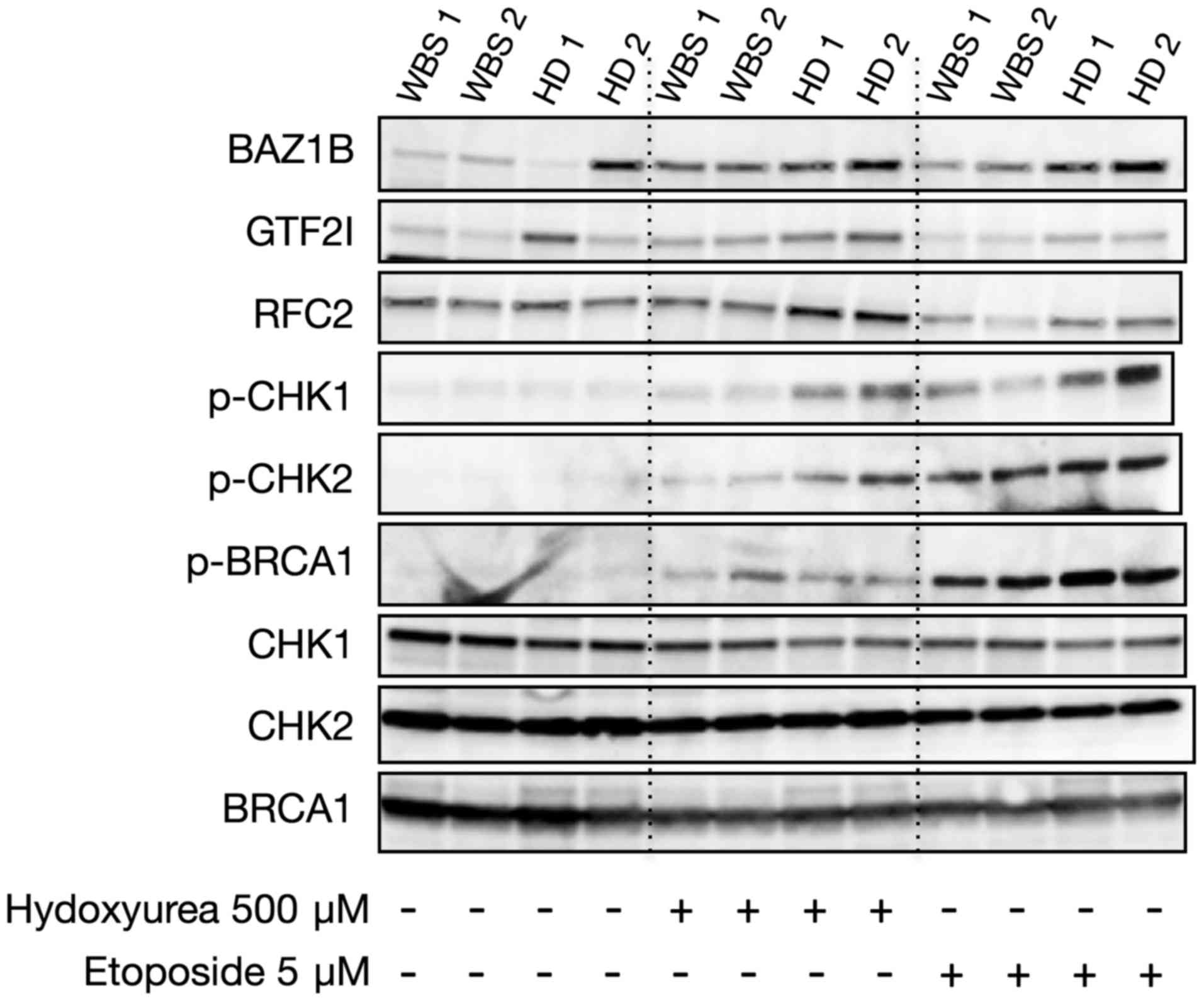
The expression and phosphorylation of DNA damage
response and repair proteins known to interact with RFC2 and GTF2I
were then studied by western blotting at the baseline (without any
treatment) as well as following genotoxic damage using ETP and HU
treatments.
As expected, the baseline expression of BAZ1B and
GTF2I in primary fibroblasts from WBS patients appeared to be
slightly lower compared with these levels noted in the healthy
donors. This difference in expression increased upon treatments.
Notably, the expression of RFC2 was similar in the WBS and control
cells. Nonetheless, upon treatments with ETP and HU, a lower
expression of RFC2 was observed in the WBS cell lines.
Interestingly, phosphorylation of CHK1 at Ser317 and CHK2 at Thr68
appeared to be lower in the WBS cells after HU exposure and, to a
lesser extent, after ETP exposure. No difference was found in the
phosphorylation of BRCA1 in the primary fibroblasts from WBS
patients and healthy donors (Fig.
8).
These results are concordant with previous research
(16) and demonstrate that an
impaired HU-induced phospho-CHK1 and phospho-CHK2 is associated
with WBS.
In 293T cells transfected with siRNAs, immunoblot
analysis demonstrated effective knockdown of the targets. However,
no effect on the expression or phosphorylation of CHK1, CHK2 and
BRCA1 was demonstrated between the transfected 293T cells and the
controls (Fig. 9).
Discussion
In the present study, we demonstrated that
ATR-dependent CHK1 and CHK2 phosphorylation upon DNA damage is
impaired in primary fibroblasts from WBS patients. However, these
results were not reproduced following silencing of each of the
genes RFC2, GTF2I and BAZ1B that are involved
in DNA damage responses. Moreover, exposure to HU or ETP did not
impair the cell cycle and proliferation in fibroblasts from WBS
patients as compared to normal cells.
Several previous studies on WBS patient-derived
cells demonstrated a DNA damage response defect in WBS. In 2011,
Savina et al demonstrated experimentally a relationship
between an abnormal DNA damage response and the 7q11.23 hemizygous
microdeletion using a comet assay in lymphocytes isolated from WBS
patients (17).
In 2007, O'Driscol et al found an impaired
ATR-dependent DNA damage response in WBS lymphoblastoid cell lines
(LBL) which they linked with the haploinsufficiency of RFC2, a
coding gene localized within the WBSCR (16).
Our results are consistent with the observations of
O'Driscoll et al (16) who
demonstrated a link between WBS and impaired ATR-dependent DNA
damage response. However, our results did not demonstrate a role of
RFC2 in the abnormal DNA damage response observed in WBS patients.
This may be due to the cellular model that we used. In the study of
O'Driscoll et al (16),
LBL cell lines were complemented after transfection of RFC2 cDNA
and might be more appropriate to show the role of RFC2 since
depletion of RFC2 alone in the 293T cells did not reproduce the
phenotype of WBS cells.
Additionally, the DNA damage response defect
observed in WBS patients was not associated with a hypersensitivity
to DNA damaging agents. Thus, our results suggest that the WBS gene
plays important roles in DNA damage response but are also
dispensable for WBS cell viability when these cells undergo a
genotoxic stress.
WBS is a contiguous gene syndrome and the DNA damage
response defect in WBS cells is more likely to be associated with
the depletion of a combination of genes.
Further studies are needed to elucidate the role of
each WBS gene and the combination of these genes in DNA damage
response and to understand their links with the susceptibility to
LNH-B in WBS patients.
Acknowledgments
We would like to acknowledge the 'Ligue Contre le
Cancer, Conférence de Coordination du Grand-Est (CCIR-GE)',
'Canceropôle du Grand-Est' and 'Cent pour Sang la Vie' for the
financial support of this study. We thank The Genomic and Genetic
Disorders Biobank, a member of the Telethon Network of Genetic
Biobanks funded by Telethon Italy (project no. GTB12001G) for the
banking of biospecimens.
References
|
1
|
Pober BR: Williams-Beuren syndrome. N Engl
J Med. 362:239–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guenat D, Quentin S, Rizzari C, Lundin C,
Coliva T, Edery P, Fryssira H, Bermont L, Ferrand C, Soulier J, et
al: Constitutional and somatic deletions of the Williams-Beuren
syndrome critical region in non-Hodgkin lymphoma. J Hematol Oncol.
7:822014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guenat D, Rizzari C, Lundin C, Borg C,
Soulier J and Rohrlich PS: Predisposition to Burkitt lymphoma in
Williams-Beuren syndrome. Blood. 124:21822014.
|
|
4
|
Chrzanowska KH, Gregorek H,
Dembowska-Bagińska B, Kalina MA and Digweed M: Nijmegen breakage
syndrome (NBS). Orphanet J Rare Dis. 7:132012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arora H, Chacon AH, Choudhary S, McLeod
MP, Meshkov L, Nouri K and Izakovic J: Bloom syndrome. Int J
Dermatol. 53:798–802. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Suarez F, Mahlaoui N, Canioni D,
Andriamanga C, Dubois d'Enghien C, Brousse N, Jais JP, Fischer A,
Hermine O and Stoppa-Lyonnet D: Incidence, presentation, and
prognosis of malignancies in ataxia-telangiectasia: A report from
the French National Registry of Primary Immune Deficiencies. J Clin
Oncol. 33:202–208. 2015. View Article : Google Scholar
|
|
7
|
Attarbaschi A, Carraro E, Abla O,
Barzilai-Birenboim S, Bomken S, Brugieres L, Bubanska E, Burkhardt
B, Chiang AK, Csoka M, et al: Non-Hodgkin's lymphoma and
pre-existing conditions: spectrum, clinical characteristics and
outcome in 213 children and adolescents. Haematologica. Aug
11–2016.Epub ahead of print. View Article : Google Scholar
|
|
8
|
Noskov VN, Araki H and Sugino A: The RFC2
gene, encoding the third-largest subunit of the replication factor
C complex, is required for an S-phase checkpoint in Saccharomyces
cerevisiae. Mol Cell Biol. 18:4914–4923. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Desgranges ZP, Ahn J, Lazebnik MB,
Ashworth T, Lee C, Pestell RC, Rosenberg N, Prives C and Roy AL:
Inhibition of TFII-I-dependent cell cycle regulation by 53. Mol
Cell Biol. 25:10940–10952. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tanikawa M, Wada-Hiraike O, Nakagawa S,
Shirane A, Hiraike H, Koyama S, Miyamoto Y, Sone K, Tsuruga T,
Nagasaka K, et al: Multifunctional transcription factor TFII-I is
an activator of BRCA1 function. Br J Cancer. 104:1349–1355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fattah FJ, Hara K, Fattah KR, Yang C, Wu
N, Warrington R, Chen DJ, Zhou P, Boothman DA and Yu H: The
transcription factor TFII-I promotes DNA translesion synthesis and
genomic stability. PLoS Genet. 10:e10044192014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poot RA, Bozhenok L, van denBerg DLC,
Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J and
Varga-Weisz PD: The Williams syndrome transcription factor
interacts with PCNA to target chromatin remodelling by ISWI to
replication foci. Nat Cell Biol. 6:1236–1244. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kitagawa H, Fujiki R, Yoshimura K, Oya H
and Kato S: Williams syndrome is an epigenome-regulator disease.
Endocr J. 58:77–85. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao A, Li H, Shechter D, Ahn SH, Fabrizio
LA, Erdjument- Bromage H, Ishibe-Murakami S, Wang B, Tempst P,
Hofmann K, et al: WSTF regulates the H2A.X DNA damage response via
a novel tyrosine kinase activity. Nature. 457:57–62. 2009.
View Article : Google Scholar
|
|
15
|
Singh N, Basnet H, Wiltshire TD, Mohammad
DH, Thompson JR, Héroux A, Botuyan MV, Yaffe MB, Couch FJ,
Rosenfeld MG, et al: Dual recognition of phosphoserine and
phosphotyrosine in histone variant H2A.X by DNA damage response
protein MCPH1. Proc Natl Acad Sci USA. 109:14381–14386. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
O'Driscoll M, Dobyns WB, van Hagen JM and
Jeggo PA: Cellular and clinical impact of haploinsufficiency for
genes involved in ATR signaling. Am J Hum Genet. 81:77–86. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Savina NV, Smal MP, Kuzhir TD, Egorova TM,
Khurs OM, Polityko AD and Goncharova RI: Chromosomal instability at
the 7q11.23 region impacts on DNA-damage response in lymphocytes
from Williams-Beuren syndrome patients. Mutat Res. 724:46–51. 2011.
View Article : Google Scholar : PubMed/NCBI
|