Introduction
Congenital heart defects (CHDs), a series of
developmental anomalies in the structures of the heart and the
great endothoracic blood vessels, encompassing ventricular septal
defect (VSD), atrial septal defect, tetralogy of Fallot, double
outlet right ventricle (DORV), transposition of the great arteries,
pulmonary atresia and persistent truncus ateriosus, are the most
common form of birth defects in humans, with an estimated
prevalence of 1% in live births worldwide (1). It was reported that in 2013, there
were >34 million individuals living with CHDs worldwide
(2). Severe CHDs may result in
reduced exercise performance and quality of life (3–6),
fetal brain injury and neurodevelopmental delay (7,8),
pulmonary hypertension and Eisenmenger syndrome (9), cardiac enlargement and heart failure
(10,11), cardiac arrhythmias and sudden
cardiac death (12–14). In fact, CHDs are responsible for
approximately 30% of all birth defect-related deaths (1). Globally in 2010, CHDs led to
approximately 223,000 deaths (15). Although great advances in cardiac
surgical techniques and intensive care have allowed the
overwhelming majority of children with CHDs to survive into
adulthood, unfortunately, the late morbidity and mortality rates
are still high in the survivors (16–18). Therefore, CHDs have laid a heavy
economic burden on patients and healthcare systems, and this burden
is anticipated to be even heavier in the future due to an
increasing number of CHDs in adults (16–18). Despite the important clinical
significance, the causes of CHDs among most patients remain largely
unclear.
It has been previously reported that non-inherited
modifiable factors, including maternal illnesses, nutritional
deficiencies, and exposure to drugs, toxicants or polluted air
during the first trimester of pregnancy may confer an increased
vulnerability to CHDs (19).
However, a growing body of evidence strongly suggests that genetic
defects are the predominant cause of CHDs, and mutations in a great
number of genes, particularly those coding for transcription
factors essential for cardiovascular morphogenesis, including NK2
homeobox (NKX2)-5, NKX2-6, GATA binding protein (GATA)4, GATA5,
GATA6, T-Bo (TBX)1, TBX5, TBX20, paired like homeodomain 2 (PITX2)
and heart and neural crest derivatives expressed transcript
(HAND)2, have been associated with various CHDs (20–57). Nevertheless, CHDs are a
genetically heterogeneous disorder, and the genetic basis
underlying CHDs in an overwhelming majority of cases remains to be
elucidated.
The HAND subset of basic helix-loop-helix (bHLH)
transcription factors is composed of two members, HAND1 and HAND2,
which are required for the normal cardiovascular development in
fish, chicks, rodents and humans (58). The HAND1 protein has a
functionally important structural domain termed bHLH, which
consists of a short stretch of basic amino acids followed by an
amphipathic α helix, a loop and an additional α helix, and is
required for binding to target gene DNA and protein-protein
combinatorial interactions (59).
A previous study demonstrated that HAND1 can directly activate the
cardiac ANF promoter, alone or in synergy with
transcriptionally cooperative partners, including GATA4, myocyte
enhancer factor 2 (MEF2) and HAND2 (60). In chicks, HAND1 and HAND2 are both
expressed in the bilateral heart primordia and subsequently
throughout the primitive tubular heart, as well as its derivatives
during embryonic genesis, and the treatment of chick embryos with
HAND1 and HAND2 antisense oligonucleotides has
revealed that either oligonucleotide alone has no effect on
embryonic development, whereas together they arrest development at
the looping heart tube stage (61). In mice, HAND1 is highly expressed
in distinct regions of the linear heart tube during embryogenesis
and, after looping, becomes localized to both primary heart fields,
specifically in the outer curvature of the presumptive left
ventricle and the developing outflow tract, and also at a lower
level in the outer curvature of the right ventricle (62). Mice lacking Hand1 suffer
from defective cardiac looping, failed chamber septation, anomalous
ventricular myocardial differentiation and early embryonic
lethality resulting from cardiac failure (63,64). Mouse embryos homozygous for the
cardiac-specific Hand1-null allele present diverse cardiac
deformations, including membranous VSD, overriding aorta and
hyperplastic atrioventricular valves, and DORV (65). In humans, HAND1 is expressed in
cardiac tissues within both ventricles (66), and mutations in HAND1 have
been causally linked to hypoplastic hearts and cardiac septal
defects (67–69). However, the prevalence and
spectrum of HAND1 mutations in other cohorts of patients
with various CHDs remain to be investigated.
Thus, in this study, the coding exons and flanking
introns of the HAND1 gene, which encodes a basic
helix-loop-helix transcription factor crucial for cardiovascular
development, were sequenced in 158 unrelated patients with CHDs. We
identified a de novo heterozygous mutation, p.K132X in a
patient with DORV. Thus, this mutation may be associated with an
enhanced susceptibility to DORV. Our findings may provide new
insight into the pathogenesis of DORV and CHDs at the genetic
level.
Materials and methods
Ethics
This study was conducted in conformity with the
ethical principles for medical research outlined in the Declaration
of Helsinki. The study protocol was approved by the local
institutional Ethics Committee of Tongji Hospital, Tongji
University, Shanghai, China [approval no. LL(H)-09-07], and written
informed consent was obtained from the patients or their guardians
prior to the study.
Study population
A cohort of 158 unrelated patients (87 males and 71
females, with an average age of 3.3 years) with non-syndromic CHDs
was recruited from the Chinese Han population. The available
relatives of the CHD cases were also enlisted. All patients
underwent a comprehensive clinical evaluation, including medical
history, complete physical examination, 12-lead electrocardiogram
and two-dimensional transthoracic echocardiography with color flow
Doppler. Cardiac catheterization, angiography and cardiac magnetic
resonance imaging were carried out only when indicated. The cardiac
phenotype was confirmed by echocardiography in all patients with
CHDs. Patients with known chromosomal abnormalities or other
recognized syndromic CHDs were excluded from the study. A total of
300 healthy subjects (162 males and 138 females, with an average
age of 3.2 years), who were matched to the patietns with CHD in
age, gender and ethnicity, were enrolled as controls. All the
control individuals underwent transthoracic echocardiography and
their cardiac morphologic structures were shown to be normal.
Mutational analysis of HAND1
Peripheral venous blood samples were drawn from the
study participants and the genomic DNA was isolated from leukocytes
using the Wizard Genomic DNA Purification Kit (Promega Corp.,
Madison, WI, USA) according to the manual of procedure. The
referential genomic DNA sequence of HAND1 was derived from
GenBank (GenBank ID: NC_000005.10), an online nucleotide database
at the National Center for Biotechnology Information (NCBI;
http://www.ncbi.nlm.nih.gov/nucleotide/). With the aid
of the online Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?ORGANISM=9606&INPUT_SEQUENCE=NC_000005.10&LINK_LOC=nuccore&PRIMER5_START=154474972&PRIMER3_END=154478264),
the primers used to amplify the coding exons and splicing junction
sites of HAND1 by polymerase chain reaction (PCR) were
designed as shown in Table I. PCR
was carried out using HotStar Taq DNA Polymerase (Qiagen, Hilden,
Germany) on a Veriti Thermal Cycler (Applied Biosystems, Foster,
CA, USA). The amplicons were fractionated by electrophoresis on a
2% agarose gel and then purified with the QIAquick Gel Extraction
kit (Qiagen, Hilden, Germany). The purified amplicons were
PCR-sequenced under an ABI PRISM 3130 XL DNA Analyzer (Applied
Biosystems) with BigDye® Terminator v3.1 Cycle
Sequencing kits (Applied Biosystems). The DNA sequences were
analyzed with the DNA Sequencing Analysis Software v5.1 (Applied
Biosystems). For an identified sequence variance, the Human Gene
Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/), the 1000 Genomes Project
(1000GP; http://www.1000genomes.org/data) database, the NCBI's
single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/snp) database and PubMed
(http://www.ncbi.nlm.nih.gov/pubmed)
database were consulted to confirm its novelty.
 | Table IPrimers used for the amplification of
the coding regions and splicing junction sites of the HAND1
gene. |
Table I
Primers used for the amplification of
the coding regions and splicing junction sites of the HAND1
gene.
| Coding exon | Forward primer
(5′→3′) | Reverse primer
(5′→3′) | Product size
(bp) |
|---|
| 1 |
GAGCGGCGTTAATAGGGCTG |
TTCGACTACCTGCATGGCCT | 666 |
| 2 |
GGAACTCCGCGCATAAAGGC |
CGTGCGATCCAAGTGTGTGG | 478 |
Alignment of multiple HAND1 protein
sequences among species
The amino acid sequences of the HAND1 protein in
humans were aligned with those in the chimpanzee, monkey, cattle,
mouse, rat, fowl, fruitfly and frog using MUSCLE, an online program
(http://www.ncbi.nlm.nih.gov/homologene), in order to
show the evolutionary conservation for an altered amino acid.
Expression plasmids and site-directed
mutagenesis
The recombinant expression plasmid, HAND1-pcDNA3.1,
which contains the full-length cDNA of human HAND1, was
constructed as previously described (69). The identified mutation was
introduced into the wild-type HAND1-pcDNA3.1 plasmid by
site-directed mutagenesis using a QuickChange II XL Site-Directed
Mutagenesis kit (Stratagene, La Jolla, CA, USA) and a complementary
pair of primers, and was verified by sequencing. The expression
plasmid GATA4-pSSRa and the ANF-luciferase reporter (ANF-luc)
plasmid, which contains the 2600-bp 5′-flanking region of the
ANF gene and expresses the Firefly luciferase, were kind
gifts from Dr Ichiro Shiojima at Chiba University School of
Medicine (Chiba, Japan).
Cell culture, DNA transfection and
luciferase assays
HeLa and NIH3T3 cells were cultured in Dulbecco's
modified Eagle's medium supplemented with 10% fetal bovine serum,
and plated at a density of 1×105 cells per well on
24-well plates 24 h prior to transfection. Transfection was
performed using Lipofectamine® 2000 reagent (Invitrogen
Life Technologies, Carlsbad, CA, USA) following the manufacturer's
instructions. The internal control vector pGL4.75 (Promega Corp.),
which expresses the Renilla luciferase, was co-transfected
in transfection assays to normalize transfection efficiency. For
the transfection of HeLa cells, 1.0 µg of empty pcDNA3.1
vector, 1.0 µg of wild-type HAND1-pcDNA3.1, 1.0 µg of
mutant HAND1-pcDNA3.1, 0.5 µg of wild-type HAND1-pcDNA3.1,
or 0.5 µg of wild-type HAND1-pcDNA3.1 plus 0.5 µg of
mutant HAND1-pcDNA3.1 were used in combination with 1.0 µg
of ANF-luc and 0.04 µg of pGL4.75. For the transfection of
NIH3T3 cells, the same amount (0.5 µg) of plasmid DNA (empty
pcDNA3.1 vector, wild-type HAND1-pcDNA3.1, GATA4-pSSRa or mutant
HAND1-pcDNA3.1) was used alone or in combination, in the presence
of 1.0 µg of ANF-luc and 0.04 µg of pGL4.75. The
transfected cells were incubated for 48 h at 37°C with 5%
CO2, then washed and lysed using 1X passive lysis buffer
provided by the Dual-Glo luciferase reporter assay kit (Promega
Corp.). The Firefly and Renilla luciferase activities were
measured using the Dual-Glo luciferase reporter assay kit (Promega
Corp.) according to the manufacture's instructions using a
GloMax® 96 Luminometer (Promega Corp.). The activity of
the ANF promoter was expressed as the fold activation of the
Firefly luciferase value relative to the Renilla luciferase
value. At least 3 independent transfection experiments, all of
which were conducted in triplicate, were performed to calculate
average values and standard deviations.
Statistical analysis
Statistical analyses were performed using the SPSS
version 17.0 software package (SPSS, Inc., Chicago, IL, USA). Data
are expressed as the means and standard deviation, unless otherwise
indicated. The numeric variables were compared between 2 groups
using the Student's unpaired t-test. A comparison of the
categorical variables between 2 groups was made using Pearson's
χ2 test or Fisher's exact test where appropriate. A
two-tailed value of P<0.05 was considered to indicate a
statistically significant difference.
Results
Baseline clinical features of the study
subjects
In this study, 158 unrelated patients with isolated
CHDs (87 males and 71 females, with an average age of 3.3 years)
were clinically evaluated in contrast with 300 ethnically-matched,
unrelated healthy individuals (162 males and 138 females, with an
average age of 3.2 years). All the patients had
echocardiographically documented CHDs. Of the 158 patients with
CHD, 11 (approximately 7%) had a positive family history of CHDs.
The control individuals were physically and mentally healthy with
no structural cardiac defects confirmed by echocardiogram, and they
had a negative family history of CHDs. There were no significant
differences between the patient and control groups as regards
demographic characteristics, including age, gender and ethnicity.
The baseline clinical characteristics of the 158 unrelated patients
with CHDs are presented in Table
II.
| Table IIClinical characteristics of the
patients with congenital heart defects. |
Table II
Clinical characteristics of the
patients with congenital heart defects.
| Parameters | No. or mean | % or range |
|---|
| Male/female | 87/71 | 55/45 |
| Age, years | 3 | 0–12 |
| Positive family
history of CHDs | 11 | 7 |
| Distribution of
distinct forms of CHDs | | |
| Isolated CHDs | 95 | 60 |
| VSD | 31 | 20 |
| ASD | 19 | 12 |
| PDA | 13 | 8 |
| PS | 10 | 6 |
| DORV | 5 | 3 |
| TGA | 4 | 3 |
| PTA | 3 | 2 |
| HLV | 3 | 2 |
| PA | 2 | 1 |
| TAPVC | 2 | 1 |
| CoA | 2 | 1 |
| CAC | 1 | 1 |
| Complex CHDs | 63 | 40 |
| TOF | 25 | 16 |
| VSD + ASD | 15 | 9 |
| DORV + VSD | 13 | 8 |
| VSD + PDA | 8 | 5 |
| PTA + VSD | 1 | 1 |
| TGA + VSD | 1 | 1 |
| Incidence of
arrhythmias | | |
| Atrioventricular
block | 8 | 5 |
| Atrial
fibrillation | 5 | 3 |
| Treatment | | |
| Surgical
repair | 92 | 58 |
| Percutaneous
closure | 32 | 22 |
| Follow-up | 31 | 20 |
Discovery of a de novo HAND1
mutation
By PCR sequencing, a heterozygous mutation in
HAND1 was identified in one of the 158 unrelated patients
with isolated CHDs, with a mutational prevalence of approximately
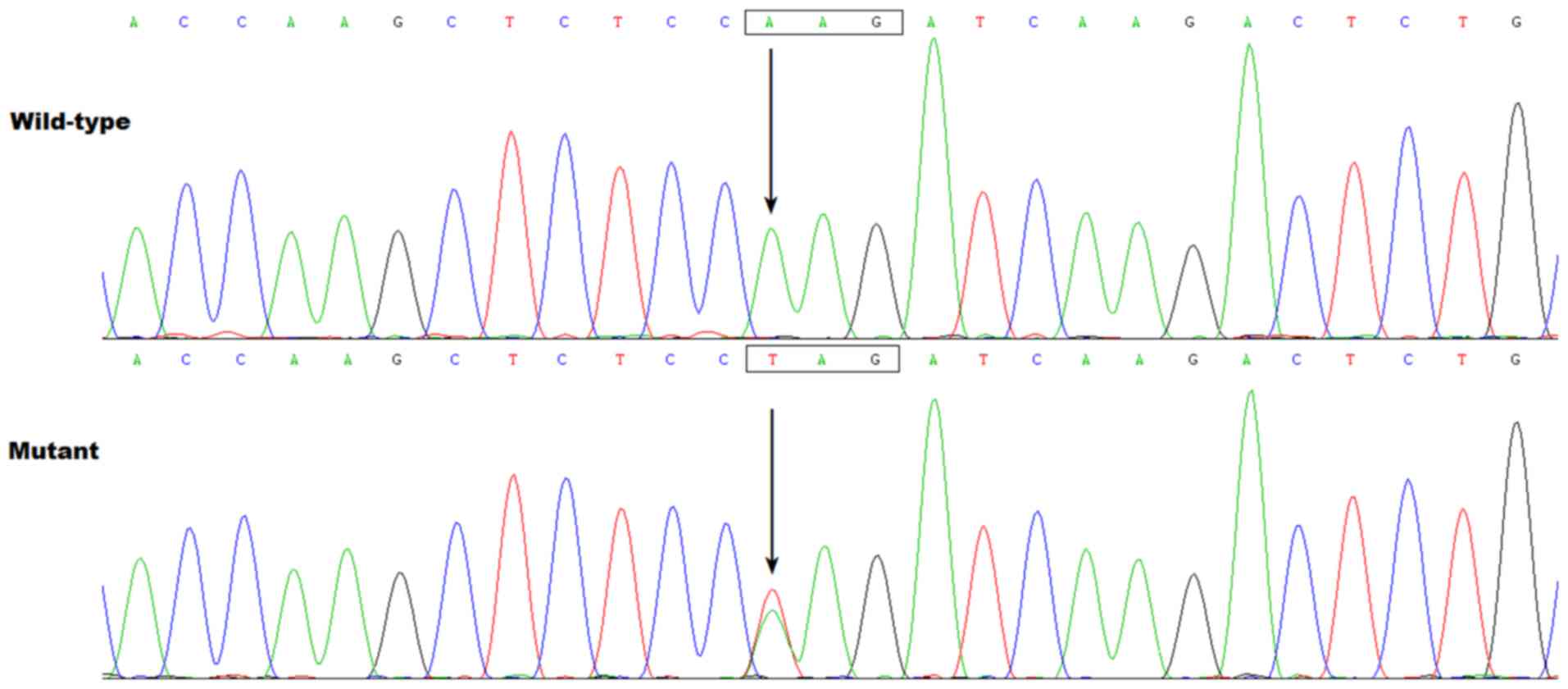
0.63%. Specifically, a substitution of thymine (T) for adenine (A)
in the first nucleotide of codon 132 (c.394A>T), predicting the
conversion of the codon coding for lysine (K) into a stop codon (X)
at amino acid position 132 (p.K132X), was discovered in a
5-month-old boy affected with congenital DORV, as well as VSD, who
had no family history of CHDs. The nonsense mutation was neither
found in the mutation carrier's healthy parents, nor detected in
the 300 unrelated control individuals, indicating it is a de
novo mutation. The sequence chromatograms showing the
heterozygous HAND1 mutation of c.394A>T, as well as its
control sequence are shown in Fig.
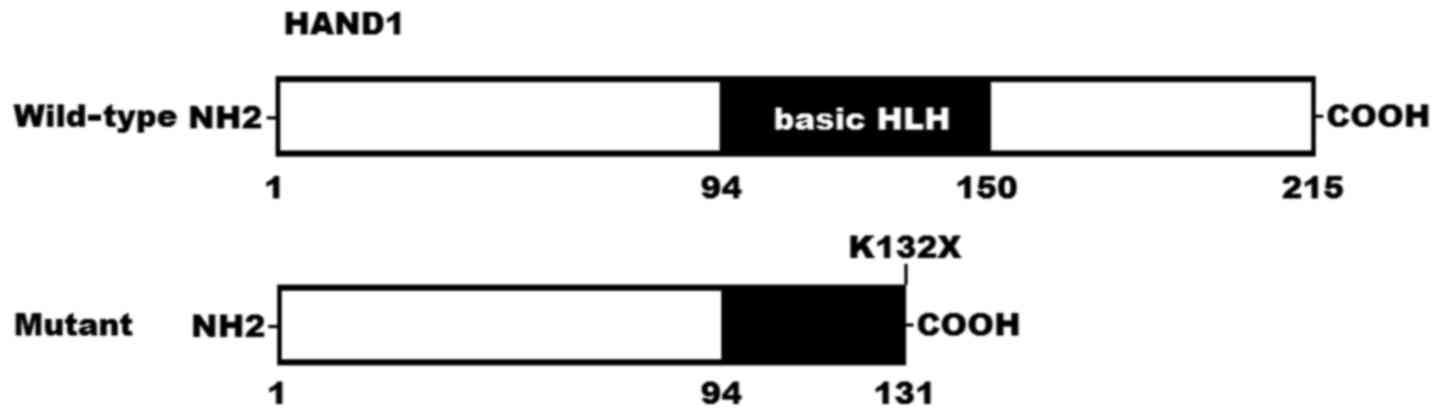
1. A schematic diagram of HAND1 showing the bHLH structural
domain and the location of the identified mutation is shown in
Fig. 2. The identified
HAND1 mutation c.394A>T has not been reported in the
HGMD, 1000GP, SNP and PubMed databases (accessed on September 12,
2016), suggesting that it is a novel mutation.
Alignment of multiple HAND1 protein
sequences across species
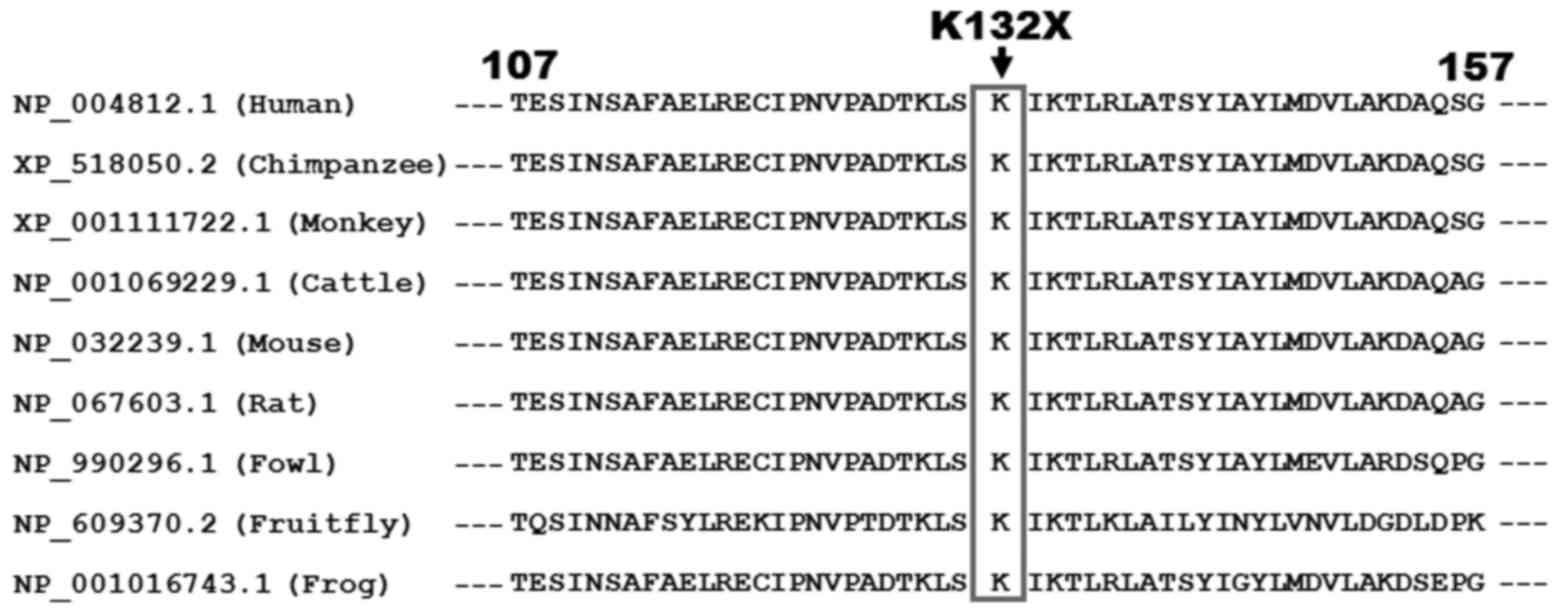
Multiple alignments of the HAND1 protein sequences
among various species displayed that the altered lysine at amino
acid position 132 (p.K132) was completely conserved evolutionarily
(Fig. 3).
No transcriptional activity of the mutant
HAND1 protein
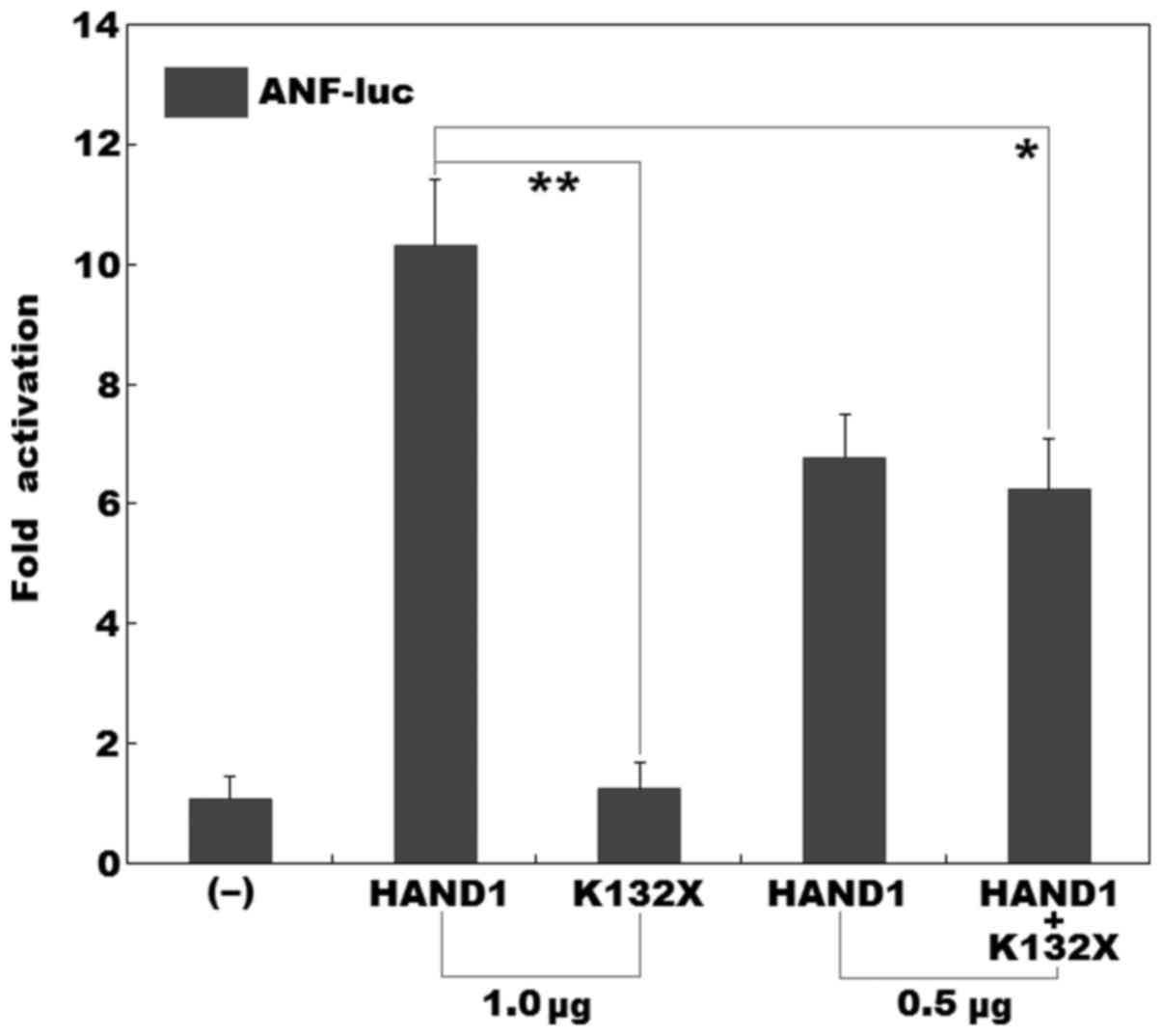
As shown in Fig.
4, dual-luciferase assays in the cultured HeLa cells revealed
that the same amount (1.0 µg) of wild-type and K132X-mutant
HAND1-pcDNA3.1 plasmids transcriptionally activated the ANF
promoter by approximately 10-fold and approximately 1-fold,
respectively. When 0.5 µg of wild-type HAND1-pcDNA3.1 was
used together with the same amount (0.5 µg) of 132X-mutant
HAND1-pcDNA3.1, the induced activation of the ANF promoter
was approximately 6-fold.
Synergistic transactivational failure
caused by the mutation
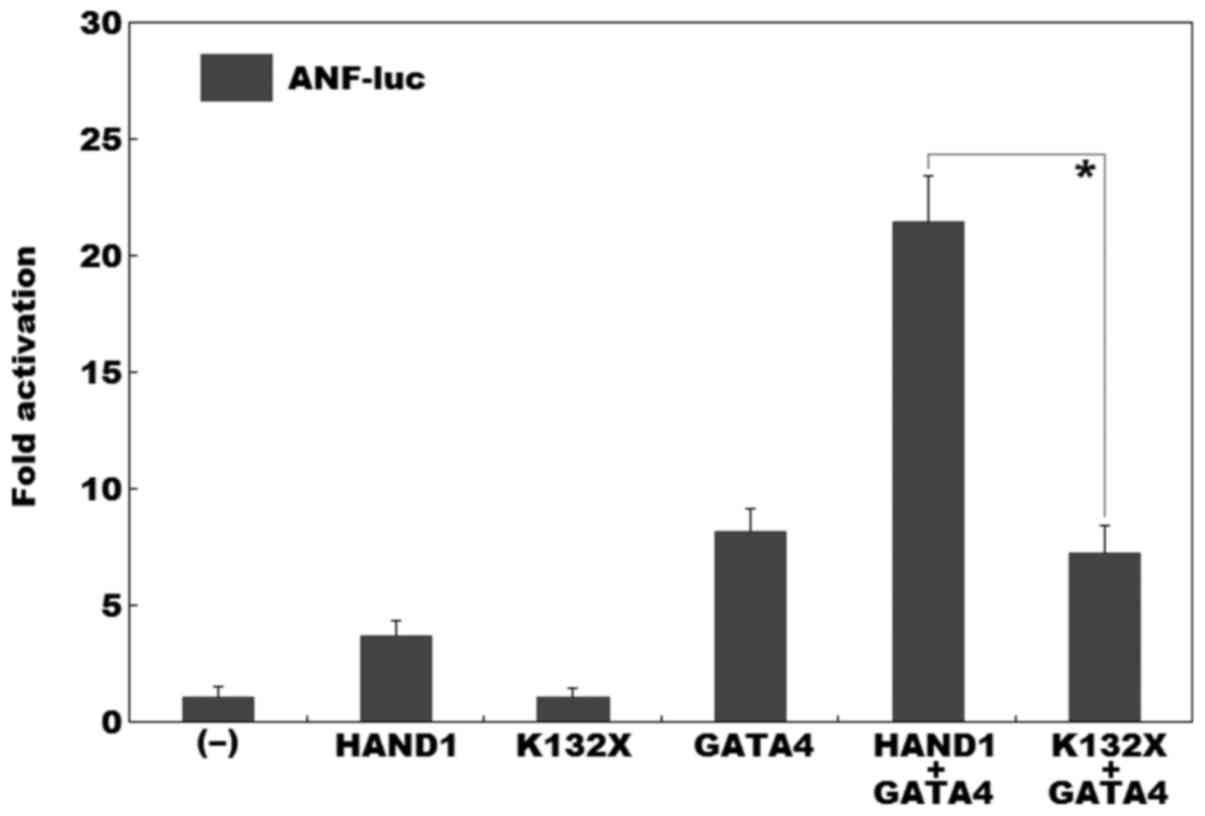
As shown in Fig.
5, dual-luciferase assays in the cultured NIH3T3 cells revealed
that the same amount (0.5 µg) of wild-type and K132X-mutant
HAND1 activated the ANF promoter by approximately 4-fold and
approximately 1-fold, respectively; while in the presence of 0.5
µg of wild-type GATA4, the same amount (0.5
µg) of wild-type and K132X-mutant HAND1 activated the
ANF promoter by approximately 22-fold and approximately
7-fold, respectively.
Discussion
In the current study, a novel heterozygous mutation,
p.K132X, was identified in a patient with isolated DORV, as well as
VSD. The nonsense mutation, which was absent in the 600 reference
chromosomes, altered the amino acid that was completely conserved
evolutionarily across species, and was predicted to generate a
truncated protein with partial bHLH domain left. Functional tests
revealed that the K132-mutant HAND1 protein had no transcriptional
activation of the ANF promoter. Furthermore, the mutation
abrogated the synergistic activation of the ANF promoter
between HAND1 and GATA4. Therefore, it is likely that the
identified HAND1 mutation predisposes to DORV, as well as
VSD.
In humans, the HAND1 gene, as the
eHAND gene, is located at chromosome 5q33, coding for a
transcription factor protein of 215 amino acids. In this study, the
HAND1 mutation identified in a patient with CHD was
predicted to generate a truncated protein losing the partial bHLH
domain; thus, it is reasonably anticipated to disable HAND1.
Functional analyses substantiated that the mutant HAND1 lost
the transcriptional activation of the ANF promoter.
Furthermore, the mutation disrupted the synergistic activation
between HAND1 and GATA4, another cardiac core transcription factor
previously associated with CHDs in humans (70). These findings strongly suggest
that haploinsufficiency caused by the HAND1 mutation is
probably an alternative pathological mechanism of CHDs.
Somatic or germline mutations in HAND1 have
been previously associated with various CHDs in humans (67–69). By direct PCR sequencing of the
HAND1 gene in human heart tissues derived from 31 unrelated
patients with hypoplastic hearts, Reamon-Buettner et al
(67) identified a common
frameshift mutation (p.A126fsX12) in 24 of 31 hypoplastic left or
right ventricles. Luciferase assays revealed that the resulting
mutant protein was unable to modulate the transcription of reporter
genes, suggesting that functionally impaired HAND1 leads to
hypoplastic human hearts. Subsequently, Reamon-Buettner et
al (67) sequenced
HAND1 in a cohort of 68 malformed hearts affected primarily
by septation defects, and detected 32 different nonsynonymous
mutations, of which 12 are in the bHLH domain of HAND1. Functional
analyses using yeast and mammalian cells have revealed that the
transcriptional activity of HAND1 is reduced or abolished by
certain mutations, suggesting that genetically compromised
HAND1 may also be responsible for septation defects of the
human hearts. Chen et al (68) screened the coding regions of
HAND1 in 498 unrelated individuals affected with
non-syndromic CHDs, and found 2 novel non-synonymous mutations of
p.G73S and p.K152N in 2 patients suffering from VSD, respectively.
Yeast two-hybrid and liquid β-galactosidase assays indicated that
both mutations increased the transcriptional activity of HAND1,
probably by enhancing the capability of HAND1 to form homodimers.
In this study, a de novo HAND1 mutation of p.K132X was
discovered in a patient with DORV and VSD, thus expanding the
clinical phenotypic spectrum linked to HAND1 mutations.
Taken collectively, these findings highlight the exquisite
sensitivity of the developing cardiovascular system to the function
of HAND1, suggesting a key role of HAND1 in human
heart development and CHDs.
In conclusion, this study firstly associates
HAND1 loss-of-function mutation with enhanced susceptibility
to DORV and VSD, which adds significant insight to the molecular
pathogenesis underpinning CHDs, suggesting potential implications
for precise diagnosis and genetic counseling of patients with
CHD.
Acknowledgments
We are really grateful to the study participants for
their dedication to the study. This study was supported by grants
from the National Basic Research Program of China (no.
2012CB9668003), the National Natural Science Fund of China (nos.
81470372, 81270231, 81270161 and 31170791), the Key Program for
Basic Research of Shanghai, China (no. 14JC1405500), the Natural
Science Fund of Shanghai, China (no. 16ZR1432500), and the
Fundamental Research Funds for the Central Universities (to Li
Li).
References
|
1
|
Mozaffarian D, Benjamin EJ, Go AS, Arnett
DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ,
Howard VJ, et al American Heart Association Statistics Committee
and Stroke Statistics Subcommittee: Heart disease and stroke
statistics - 2015 update: A report from the American Heart
Association. Circulation. 131:e29–e322. 2015. View Article : Google Scholar
|
|
2
|
Global Burden of Disease Study 2013
Collaborators: Global, regional, and national incidence,
prevalence, and years lived with disability for 301 acute and
chronic diseases and injuries in 188 countries, 1990–2013. A
systematic analysis for the Global Burden of Disease Study 2013.
Lancet. 386:743–800. 2015. View Article : Google Scholar
|
|
3
|
Feltez G, Coronel CC, Pellanda LC and
Lukrafka JL: Exercise capacity in children and adolescents with
corrected congenital heart disease. Pediatr Cardiol. 36:1075–1082.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosenblum O, Katz U, Reuveny R, Williams
CA and Dubnov-Raz G: Exercise performance in children and young
adults after complete and incomplete repair of congenital heart
disease. Pediatr Cardiol. 36:1573–1581. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kahr PC, Radke RM, Orwat S, Baumgartner H
and Diller GP: Analysis of associations between congenital heart
defect complexity and health-related quality of life using a
meta-analytic strategy. Int J Cardiol. 199:197–203. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Diller GP, Bräutigam A, Kempny A, Uebing
A, Alonso-Gonzalez R, Swan L, Babu-Narayan SV, Baumgartner H,
Dimopoulos K and Gatzoulis MA: Depression requiring anti-depressant
drug therapy in adult congenital heart disease: Prevalence, risk
factors, and prognostic value. Eur Heart J. 37:771–782. 2016.
View Article : Google Scholar
|
|
7
|
Sun L, Macgowan CK, Sled JG, Yoo SJ,
Manlhiot C, Porayette P, Grosse-Wortmann L, Jaeggi E, McCrindle BW,
Kingdom J, et al: Reduced fetal cerebral oxygen consumption is
associated with smaller brain size in fetuses with congenital heart
disease. Circulation. 131:1313–1323. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marelli A, Miller SP, Marino BS, Jefferson
AL and Newburger JW: Brain in congenital heart disease across the
lifespan: The cumulative burden of injury. Circulation.
133:1951–1962. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Galiè N, Humbert M, Vachiery JL, Gibbs S,
Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A,
Beghetti M, et al: 2015 ESC/ERS Guidelines for the diagnosis and
treatment of pulmonary hypertension: The Joint Task Force for the
Diagnosis and Treatment of Pulmonary Hypertension of the European
Society of Cardiology (ESC) and the European Respiratory Society
(ERS): Endorsed by: Association for European Paediatric and
Congenital Cardiology (AEPC), International Society for Heart and
Lung Transplantation (ISHLT). Eur Heart J. 37:67–119. 2016.
View Article : Google Scholar
|
|
10
|
Stout KK, Broberg CS, Book WM, Cecchin F,
Chen JM, Dimopoulos K, Everitt MD, Gatzoulis M, Harris L, Hsu DT,
et al American Heart Association Council on Clinical Cardiology,
Council on Functional Genomics and Translational Biology, and
Council on Cardiovascular Radiology and Imaging: Chronic Heart
Failure in Congenital Heart Disease: A Scientific Statement From
the American Heart Association. Circulation. 133:770–801.
2016.PubMed/NCBI
|
|
11
|
Wald RM, Valente AM and Marelli A: Heart
failure in adult congenital heart disease: Emerging concepts with a
focus on tetralogy of Fallot. Trends Cardiovasc Med. 25:422–432.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McLeod CJ and Warnes C: Recognition and
management of arrhythmias in adult congenital heart disease. Curr
Opin Cardiol. 31:117–123. 2016. View Article : Google Scholar
|
|
13
|
Priori SG, Blomström-Lundqvist C, Mazzanti
A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R,
Hindricks G, et al: 2015 ESC Guidelines for the management of
patients with ventricular arrhythmias and the prevention of sudden
cardiac death: The Task Force for the Management of Patients with
Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death
of the European Society of Cardiology (ESC). Endorsed by:
Association for European Paediatric and Congenital Cardiology
(AEPC). Eur Heart J. 36:2793–2867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Diller GP and Baumgartner H: Sudden
cardiac death during exercise in patients with congenital heart
disease: The exercise paradox and the challenge of appropriate
counselling. Eur Heart J. 37:627–629. 2016. View Article : Google Scholar
|
|
15
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the Global
Burden of Disease Study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van der Bom T, Zomer AC, Zwinderman AH,
Meijboom FJ, Bouma BJ and Mulder BJ: The changing epidemiology of
congenital heart disease. Nat Rev Cardiol. 8:50–60. 2011.
View Article : Google Scholar
|
|
17
|
Tutarel O, Kempny A, Alonso-Gonzalez R,
Jabbour R, Li W, Uebing A, Dimopoulos K, Swan L, Gatzoulis MA and
Diller GP: Congenital heart disease beyond the age of 60: Emergence
of a new population with high resource utilization, high morbidity,
and high mortality. Eur Heart J. 35:725–732. 2014. View Article : Google Scholar
|
|
18
|
Niwa K: Adults with congenital heart
disease transition. Curr Opin Pediatr. 27:576–580. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jenkins KJ, Correa A, Feinstein JA, Botto
L, Britt AE, Daniels SR, Elixson M, Warnes CA and Webb CL; American
Heart Association Council on Cardiovascular Disease in the Young:
Noninherited risk factors and congenital cardiovascular defects:
current knowledge: a scientific statement from the American Heart
Association Council on Cardiovascular Disease in the Young:
endorsed by the American Academy of Pediatrics. Circulation.
115:2995–3014. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Andersen TA, Troelsen KL and Larsen LA: Of
mice and men: Molecular genetics of congenital heart disease. Cell
Mol Life Sci. 71:1327–1352. 2014. View Article : Google Scholar :
|
|
21
|
Homsy J, Zaidi S, Shen Y, Ware JS, Samocha
KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et
al: De novo mutations in congenital heart disease with
neurodevelopmental and other congenital anomalies. Science.
350:1262–1266. 2015. View Article : Google Scholar
|
|
22
|
Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ,
Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, et al: Global
genetic analysis in mice unveils central role for cilia in
congenital heart disease. Nature. 521:520–524. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guimier A, Gabriel GC, Bajolle F, Tsang M,
Liu H, Noll A, Schwartz M, El Malti R, Smith LD, Klena NT, et al:
MMP21 is mutated in human heterotaxy and is required for normal
left-right asymmetry in vertebrates. Nat Genet. 47:1260–1263. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Racedo SE, McDonald-McGinn DM, Chung JH,
Goldmuntz E, Zackai E, Emanuel BS, Zhou B, Funke B and Morrow BE:
Mouse and human CRKL is dosage sensitive for cardiac outflow tract
formation. Am J Hum Genet. 96:235–244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mlynarski EE, Sheridan MB, Xie M, Guo T,
Racedo SE, McDonald-McGinn DM, Gai X, Chow EW, Vorstman J, Swillen
A, et al International Chromosome 22q11.2 Consortium: International
Chromosome 22q11.2 Consortium: Copy-number variation of the glucose
transporter gene SLC2A3 and congenital heart defects in the 22q11.2
deletion syndrome. Am J Hum Genet. 96:753–764. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo T, Chung JH, Wang T, McDonald-McGinn
DM, Kates WR, Hawuła W, Coleman K, Zackai E, Emanuel BS and Morrow
BE: Histone modifier genes alter conotruncal heart phenotypes in
22q11.2 deletion syndrome. Am J Hum Genet. 97:869–877. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Quintero-Rivera F, Xi QJ, Keppler-Noreuil
KM, Lee JH, Higgins AW, Anchan RM, Roberts AE, Seong IS, Fan X,
Lage K, et al: MATR3 disruption in human and mouse associated with
bicuspid aortic valve, aortic coarctation and patent ductus
arteriosus. Hum Mol Genet. 24:2375–2389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sanchez-Castro M, Pichon O, Briand A,
Poulain D, Gournay V, David A and Le Caignec C: Disruption of the
SEMA3D gene in a patient with congenital heart defects. Hum Mutat.
36:30–33. 2015. View Article : Google Scholar
|
|
29
|
Theis JL, Zimmermann MT, Evans JM, Eckloff
BW, Wieben ED, Qureshi MY, O'Leary PW and Olson TM: Recessive MYH6
mutations in hypoplastic left heart with reduced ejection fraction.
Circ Cardiovasc Genet. 8:564–571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kassab K, Hariri H, Gharibeh L, Fahed AC,
Zein M, El-Rassy I, Nemer M, El-Rassi I, Bitar F and Nemer G: GATA5
mutation homozygosity linked to a double outlet right ventricle
phenotype in a Lebanese patient. Mol Genet Genomic Med. 4:160–171.
2015. View Article : Google Scholar
|
|
31
|
Perrot A, Schmitt KR, Roth EM, Stiller B,
Posch MG, Browne EN, Timmann C, Horstmann RD, Berger F and Özcelik
C: CCN1 mutation is associated with atrial septal defect. Pediatr
Cardiol. 36:295–299. 2015. View Article : Google Scholar
|
|
32
|
Wang J, Mao JH, Ding KK, Xu WJ, Liu XY,
Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, et al: A novel NKX2.6
mutation associated with congenital ventricular septal defect.
Pediatr Cardiol. 36:646–656. 2015. View Article : Google Scholar
|
|
33
|
Zheng J, Li F, Liu J, Xu Z, Zhang H, Fu Q,
Wang J and Sun K: Investigation of somatic NKX2-5 mutations in
Chinese children with congenital heart disease. Int J Med Sci.
12:538–543. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pan Y, Wang ZG, Liu XY, Zhao H, Zhou N,
Zheng GF, Qiu XB, Li RG, Yuan F, Shi HY, et al: A novel TBX1
loss-of-function mutation associated with congenital heart disease.
Pediatr Cardiol. 36:1400–1410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao CM, Peng LY, Li L, Liu XY, Wang J,
Zhang XL, Yuan F, Li RG, Qiu XB and Yang YQ: PITX2 loss-of-function
mutation contributes to congenital endocardial cushion defect and
Axenfeld-Rieger syndrome. PLoS One. 10:e01244092015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Deng X, Pan H, Wang J, Wang B, Cheng Z,
Cheng L, Zhao L, Li H and Ma X: Functional analysis of two novel
mutations in TWIST1 protein motifs found in ventricular septal
defect patients. Pediatr Cardiol. 36:1602–1609. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pan Y, Geng R, Zhou N, Zheng GF, Zhao H,
Wang J, Zhao CM, Qiu XB, Yang YQ and Liu XY: TBX20 loss-of-function
mutation contributes to double outlet right ventricle. Int J Mol
Med. 35:1058–1066. 2015.PubMed/NCBI
|
|
38
|
Yang J, Zhu M, Wang Y, Hou X, Wu H, Wang
D, Shen H, Hu Z and Zou J: Whole-exome sequencing identify a new
mutation of MYH7 in a Chinese family with left ventricular
noncompaction. Gene. 558:138–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sifrim A, Hitz MP, Wilsdon A, Breckpot J,
Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan
GJ, et al INTERVAL Study; UK10K Consortium; Deciphering
Developmental Disorders Study: Distinct genetic architectures for
syndromic and nonsyndromic congenital heart defects identified by
exome sequencing. Nat Genet. 48:1060–1065. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Boyle L, Wamelink MM, Salomons GS, Roos B,
Pop A, Dauber A, Hwa V, Andrew M, Douglas J, Feingold M, et al:
Mutations in TKT are the cause of a syndrome including short
stature, developmental delay, and congenital heart defects. Am J
Hum Genet. 98:1235–1242. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li N, Subrahmanyan L, Smith E, Yu X, Zaidi
S, Choi M, Mane S, Nelson-Williams C, Bahjati M, Kazemi M, et al:
Mutations in the histone modifier PRDM6 are associated with
isolated nonsyndromic patent ductus arteriosus. Am J Hum Genet.
98:1082–1091. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fregeau B, Kim BJ, Hernández-García A,
Jordan VK, Cho MT, Schnur RE, Monaghan KG, Juusola J, Rosenfeld JA,
Bhoj E, et al: De novo mutations of RERE cause a genetic syndrome
with features that overlap those associated with proximal 1p36
deletions. Am J Hum Genet. 98:963–970. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Priest JR, Osoegawa K, Mohammed N, Nanda
V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott
D, et al: De novo and rare variants at multiple loci support the
oligogenic origins of atrioventricular septal heart defects. PLoS
Genet. 12:e10059632016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Werner P, Latney B, Deardorff MA and
Goldmuntz E: MESP1 mutations in patients with congenital heart
defects. Hum Mutat. 37:308–314. 2016. View Article : Google Scholar :
|
|
45
|
LaHaye S, Corsmeier D, Basu M, Bowman JL,
Fitzgerald-Butt S, Zender G, Bosse K, McBride KL, White P and Garg
V: Utilization of whole exome sequencing to identify causative
mutations in familial congenital heart disease. Circ Cardiovasc
Genet. 9:320–329. 2016.PubMed/NCBI
|
|
46
|
Liu D, Liu QQ, Guan LH, Jiang X, Zhou DX,
Beghetti M, Qu JM and Jing ZC: BMPR2 mutation is a potential
predisposing genetic risk factor for congenital heart disease
associated pulmonary vascular disease. Int J Cardiol. 211:132–136.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu
YJ, Qu XK, Shi HY, Hou XM, Huang RT, et al: A HAND2
loss-of-function mutation causes familial ventricular septal defect
and pulmonary stenosis. G3 (Bethesda). 6:987–992. 2016. View Article : Google Scholar
|
|
48
|
Yoshida A, Morisaki H, Nakaji M, Kitano M,
Kim KS, Sagawa K, Ishikawa S, Satokata I, Mitani Y, Kato H, et al:
Genetic mutation analysis in Japanese patients with non-syndromic
congenital heart disease. J Hum Genet. 61:157–162. 2016. View Article : Google Scholar
|
|
49
|
Lu CX, Gong HR, Liu XY, Wang J, Zhao CM,
Huang RT, Xue S and Yang YQ: A novel HAND2 loss-of-function
mutation responsible for tetralogy of Fallot. Int J Mol Med.
37:445–451. 2016.
|
|
50
|
Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H,
Meng M, Wang W and Jiang L: Genetic variations of NKX2-5 in
sporadic atrial septal defect and ventricular septal defect in
Chinese Yunnan population. Gene. 575:29–33. 2016. View Article : Google Scholar
|
|
51
|
Chen J, Qi B, Zhao J, Liu W, Duan R and
Zhang M: A novel mutation of GATA4 (K300T) associated with familial
atrial septal defect. Gene. 575:473–477. 2016. View Article : Google Scholar
|
|
52
|
Tong YF: Mutations of NKX2.5 and GATA4
genes in the development of congenital heart disease. Gene.
588:86–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang X, Wang J, Wang B, Chen S, Fu Q and
Sun K: A novel missense mutation of GATA4 in a Chinese family with
congenital heart disease. PLoS One. 11:e01589042016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li
RG, Qu XK, Huang RT, Xue S and Yang YQ: ITX2 loss-of-function
mutation contributes to tetralogy of Fallot. Gene. 577:258–264.
2016. View Article : Google Scholar
|
|
55
|
Zhou YM, Dai XY, Huang RT, Xue S, Xu YJ,
Qiu XB and Yang YQ: A novel TBX20 loss of function mutation
contributes to adult onset dilated cardiomyopathy or congenital
atrial septal defect. Mol Med Rep. 14:3307–3314. 2016.PubMed/NCBI
|
|
56
|
Li FF, Deng X, Zhou J, Yan P, Zhao EY and
Liu SL: Characterization of human bone morphogenetic protein gene
variants for possible roles in congenital heart disease. Mol Med
Rep. 14:1459–1464. 2016.PubMed/NCBI
|
|
57
|
Kelle AM, Bentley SJ, Rohena LO, Cabalka
AK and Olson TM: Ebstein anomaly, left ventricular non-compaction,
and early onset heart failure associated with a de novo
α-tropomyosin gene mutation. Am J Med Genet A. 170:2186–2190. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Thattaliyath BD, Livi CB, Steinhelper ME,
Toney GM and Firulli AB: HAND1 and HAND2 are expressed in the
adult-rodent heart and are modulated during cardiac hypertrophy.
Biochem Biophys Res Commun. 297:870–875. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Reamon-Buettner SM, Ciribilli Y, Inga A
and Borlak J: A loss-of-function mutation in the binding domain of
HAND1 predicts hypoplasia of the human hearts. Hum Mol Genet.
17:1397–1405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Morin S, Pozzulo G, Robitaille L, Cross J
and Nemer M: MEF2-dependent recruitment of the HAND1 transcription
factor results in synergistic activation of target promoters. J
Biol Chem. 280:32272–32278. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Srivastava D, Cserjesi P and Olson EN: A
subclass of bHLH proteins required for cardiac morphogenesis.
Science. 270:1995–1999. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Risebro CA, Smart N, Dupays L,
Breckenridge R, Mohun TJ and Riley PR: Hand1 regulates
cardiomyocyte proliferation versus differentiation in the
developing heart. Development. 133:4595–4606. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Firulli AB, McFadden DG, Lin Q, Srivastava
D and Olson EN: Heart and extra-embryonic mesodermal defects in
mouse embryos lacking the bHLH transcription factor Hand1. Nat
Genet. 18:266–270. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Riley P, Anson-Cartwright L and Cross JC:
The Hand1 bHLH transcription factor is essential for placentation
and cardiac morphogenesis. Nat Genet. 18:271–275. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
McFadden DG, Barbosa AC, Richardson JA,
Schneider MD, Srivastava D and Olson EN: The Hand1 and Hand2
transcription factors regulate expansion of the embryonic cardiac
ventricles in a gene dosage-dependent manner. Development.
132:189–201. 2005. View Article : Google Scholar
|
|
66
|
Natarajan A, Yamagishi H, Ahmad F, Li D,
Roberts R, Matsuoka R, Hill S and Srivastava D: Human eHAND, but
not dHAND, is down-regulated in cardiomyopathies. J Mol Cell
Cardiol. 33:1607–1614. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Reamon-Buettner SM, Ciribilli Y, Traverso
I, Kuhls B, Inga A and Borlak J: A functional genetic study
identifies HAND1 mutations in septation defects of the human heart.
Hum Mol Genet. 18:3567–3578. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Cheng Z, Lib L, Li Z, Liu M, Yan J, Wang B
and Ma X: Two novel HAND1 mutations in Chinese patients with
ventricular septal defect. Clin Chim Acta. 413:675–677. 2012.
View Article : Google Scholar
|
|
69
|
Zhou YM, Dai XY, Qiu XB, Yuan F, Li RG, Xu
YJ, Qu XK, Huang RT, Xue S and Yang YQ: HAND1 loss-of-function
mutation associated with familial dilated cardiomyopathy. Clin Chem
Lab Med. 54:1161–1167. 2016. View Article : Google Scholar
|
|
70
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|