Introduction
Nuclear factor (NF)-κB is a transcription factor
member of the Rel family. The Rel family includes five members,
RelA/p65, RelB, c-Rel, p50 and p52 (1–3),
which have sequence similarity to over ~300 amino acids in the
amino-terminal half of these proteins. These Rel family members can
form heterodimers or homodimers with a range of DNA-binding and
activation potentials. Even though all family members contain a Rel
homology domain that is required for DNA binding, dimerization, and
nuclear localization, only RelA/p65 (hereafter referred to as p65),
RelB and c-Rel, have a specific carboxy terminal transactivation
domain that can regulate NF-κB-dependent gene expression (3,4).
The most widely studied active form of NF-κB is a p65-p50
heterodimer, which is a main activator of gene transcription
(5,6).
Although this transcription factor was first known
as a regulator of inflammation and immune responses, recent studies
show that NF-κB also regulates cell proliferation (7,8).
The mechanism of proliferation regulation involves the stimulation
of cyclin E/CDK2 activity and c-Myc expression (9–11),
as well as other proliferation-associated genes, such as colony
stimulating factor, platelet-derived growth factor, and cyclin D1
(4,12–14).
Cyclin D1 is a member of the D-type cyclins. It
regulates the G1/S phase transition of the cell cycle. Cyclin D1 is
induced by mitogens and can dimerize with CDK4/CDK6 to
phosphorylate retinoblastoma (Rb) protein, which then derepresses
E2F transcriptional activity to allow entry into the S-phase
(15,16). Analysis of the cyclin D1 promoter
revealed the presence of two κB sites located at positions −840 and
−33, which have been deemed to be κB1 and κB2 (12), respectively. Mutational analysis
demonstrated that binding of the p65/p50 complex to the −33 κB2
site is important for cyclin D1 transcriptional regulation
during the proliferative response induced by growth factors or
serum or by stimulation via the Ras/Rac pathway (12,13,17).
Human T cell leukemia virus type 1 (HTLV-1) is a
complex retrovirus that can cause adult T cell leukemia (ATL)
(18,19). HTLV-1 encodes several unique
proteins that can participate in viral infectivity, replication,
persistence and transformation (20). Among these proteins, HTLV-1 basic
leucine zipper (bZIP) factor (HBZ) expression is conserved in all
ATL cells and is thought to be essential for leukemogenesis
(21). Additionally, the HBZ gene
can promote ATL cell proliferation (21). Recent studies have shown that HBZ
can inhibit Tax-mediated NF-κB activation and suppress the
transcription of various NF-κB target genes (22). These findings suggest that HBZ is
involved in the NF-κB signaling pathway, and also contributes to
the regulation of cyclin D1, an NF-κB target gene.
In a previous study, we showed that HBZ
downregulated cyclin D1 expression via interactions with
cAMP-response element binding protein CREB (23). To characterize other mechanisms of
cyclin D1 suppression via HBZ, we utilized 293T and Jurkat cells in
which HBZ was stably expressed and studied the effects of HBZ on
cyclin D1 expression. We found that HBZ mediated the suppression of
cyclin D1 by interacting with p65.
Materials and methods
Cell culture
Jurkat cells (purchased from the China Center for
Type Culture Collection, Wuhan, China) were cultured in RPMI-1640
medium (HyClone, Logan, UT, USA). 293T cells (purchased from the
China Center for Type Culture Collection) were cultured in
Dulbecco's modified Eagle's medium (DMEM) (HyClone). Media were
supplemented with 10% fetal bovine serum (FBS; HyClone), 2 mM
glutamine, 50 mg/ml streptomycin, and 100 U/ml penicillin. Cells
were synchronized by serum starvation for 18 h, followed by
stimulation in medium containing 10% FBS for 12 h.
Lentiviral vector construction and
transfection with the recombinant lentivirus
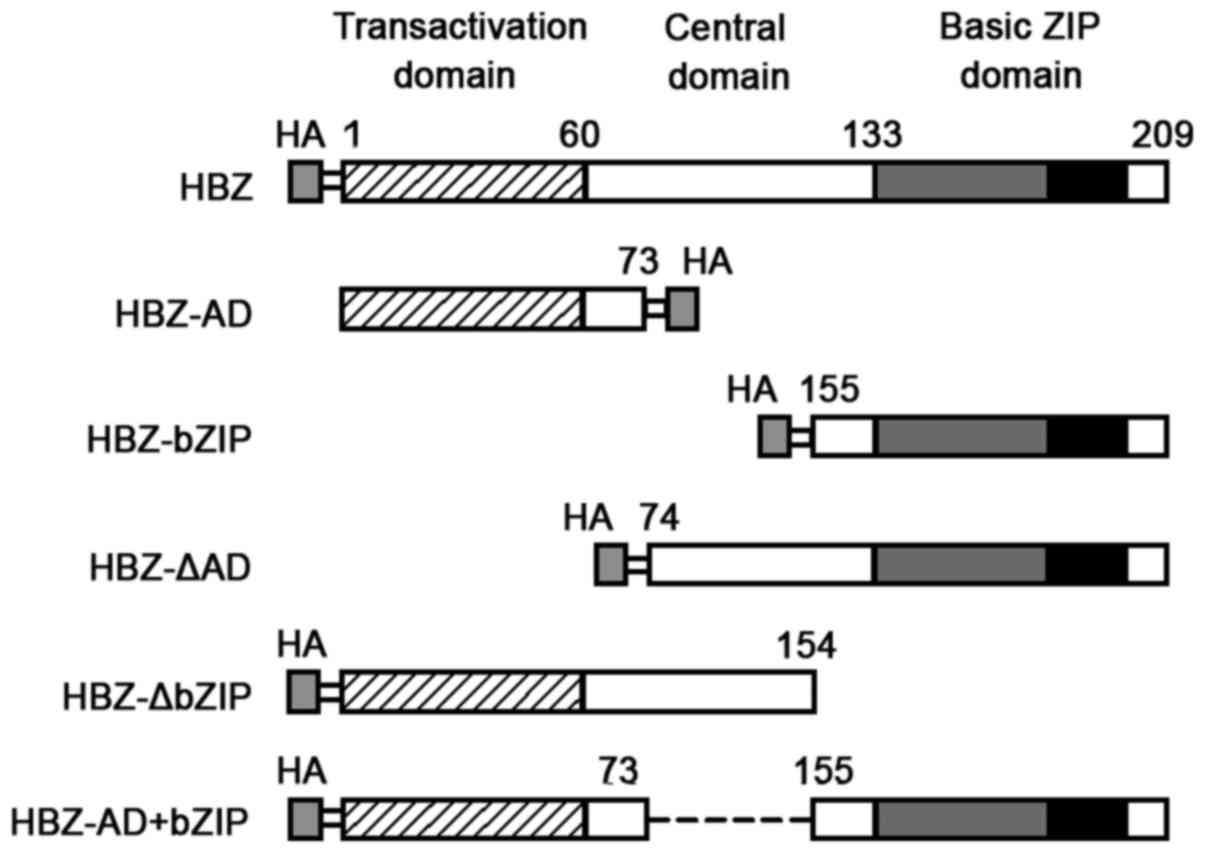
Full-length sHBZ (amino acids 1-209) and deletion
mutants (sHBZ-AD, amino acids 1-73; sHBZ-basic leucine zipper
domain (bZIP), amino acids 155-209; sHBZ-ΔAD, amino acids 74-209;
sHBZ-ΔbZIP, amino acids 1-154; and sHBZ-AD+bZIP, amino acids
(1-73)+(155-209) were generated by PCR from pET-29a-HBZ (provided
by Dr H.J. Zhi; Uniformed Services University of the Health
Sciences, Bethesda, MD, USA) with an HA epitope tagged at the C- or
N-terminus (Fig. 1). These
fragments were subcloned into a lentiviral vector (LV5) and were
transfected into 293T cells using Lipofectamine™ 2000 (Invitrogen,
Carlsbad, CA, USA) as reported previously (23). Infection or transduction with
lentiviral vectors was also performed as described previously
(23).
Synthesis of cDNA and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from the stable
lentiviral-infected 293T cells, Jurkat cells and uninfected control
cells using Total RNA kit I (R6834-01; Omega, Norcross, GA, USA)
according to the manufacturer's instructions. For cDNA synthesis,
2.5 µg total RNA was reverse transcribed using the
RevertAid™ First-Strand cDNA Synthesis kit (K1621; Fermentas,
Carlsbad, CA, USA). For RT-PCR, cDNA products were quantified with
Power SYBR-Green PCR Master Mix and an ABI 7500 Fast Real-Time PCR
system (Applied Biosystems, Carlsbad, CA, USA). Glyceraldehyde
3-phosphate dehydrogenase (GAPDH) mRNA transcript levels were
quantified to normalize the amount of cDNA that was loaded. The
customized primers used were: cyclin D1 forward,
5′-GACCATCCCCCTGACGGCCGAG-3′ and reverse,
5′-CGCACGTCGGTGGGTGTGC-3′; and GAPDH forward,
5′-AGAGGCAGGGATGATGTTCTG-3′ and reverse,
5′-GACTCATGACCACAGTCCATGC-3′. All PCR experiments were repeated at
least 3 times. A melting curve analysis was performed to ensure the
specificity of the products after amplification. The median in each
triplicate dataset was used to calculate the relative cyclin
D1 mRNA concentration (ΔCt = Ct median mRNA − Ct median
GAPDH), which was then converted to x-fold-changes
(2−ΔΔCt) (24).
Construction of cyclin D1 promoter
deletion mutants and luciferase assays
The deletion mutants of human cyclin D1
promoter that lacked AP1-, STAT-/SP1-, CREB- and NF-κB-binding
sites (CD1 to CD5) were generated by PCR and then cloned into the
vector pGL3-enhancer (Promega, Madison, WI, USA), as previously
reported (23). Luciferase assays
were performed using 293T and Jurkat cells, as described previously
(23). All reporter assays were
performed in triplicate and repeated in three independent
experiments. Firefly luciferase values were normalized by
Renilla luciferase values from pRL-TK (Promega).
Immunoprecipitation and
immunoblotting
To examine the protein-protein interactions in the
293T cells, subconfluent cells were transfected with various
combinations of expression vectors. Cells were lysed 48 h later in
RIPA buffer containing 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM
MgCl2, 1% Triton X-100, 0.5% Nonidet P-40 and protease
inhibitors. Lysates were precleared by incubation with 20 µl
suspension of 50% protein G-agarose (GE Healthcare Life Sciences,
Uppsala, Sweden) for 1 h at 4°C. Precleared lysates were incubated
with anti-HA (Y-11) or anti-NF-κB p65 (C-20) (both from Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) antibodies for 1 h at
4°C, and immune complexes were collected by incubation with protein
G-agarose at 4°C for 1 h. After extensive washing,
immunoprecipitated proteins were resolved by 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed
by western blotting with anti-p65 (SC-109), anti-p50 (H-119), or
anti-HA (F-7) (all from Santa Cruz Biotechnology, Inc.) antibodies.
Membranes were developed with ECL (GE Healthcare Life Sciences).
Other antibodies were as follows: anti-mouse IgG and anti-rabbit
IgG (GE Healthcare Life Sciences).
Statistical analysis
All data are presented as the means ± standard
deviation (SD) from at least 3 independent experiments. Statistical
analyses were performed with SPSS (version 19.0; SPSS Inc.,
Chicago, IL, USA). Statistical significance was assessed by one-way
analysis of variance (ANOVA) or the Student's t-test. Differences
were considered statistically significant when P-values were
<0.05.
Results
HBZ inhibits the expression of cyclin
D1
To analyze the effect of HBZ on cyclin D1
expression, we first transduced 293T cells with a lentiviral vector
carrying the HBZ gene and evaluated cyclin D1 levels by RT-qPCR and
western blot analysis. The expression of HBZ resulted in reduced
expression levels of cyclin D1 mRNA and protein (Fig. 2A and C). To further characterize
this effect in T cells, we transduced HBZ into Jurkat cells using a
lentiviral vector. As in 293T cells, HBZ expression resulted in
reduced cyclin D1 mRNA and protein levels in the Jurkat cells as
detected by RT-qPCR and western blot assays.
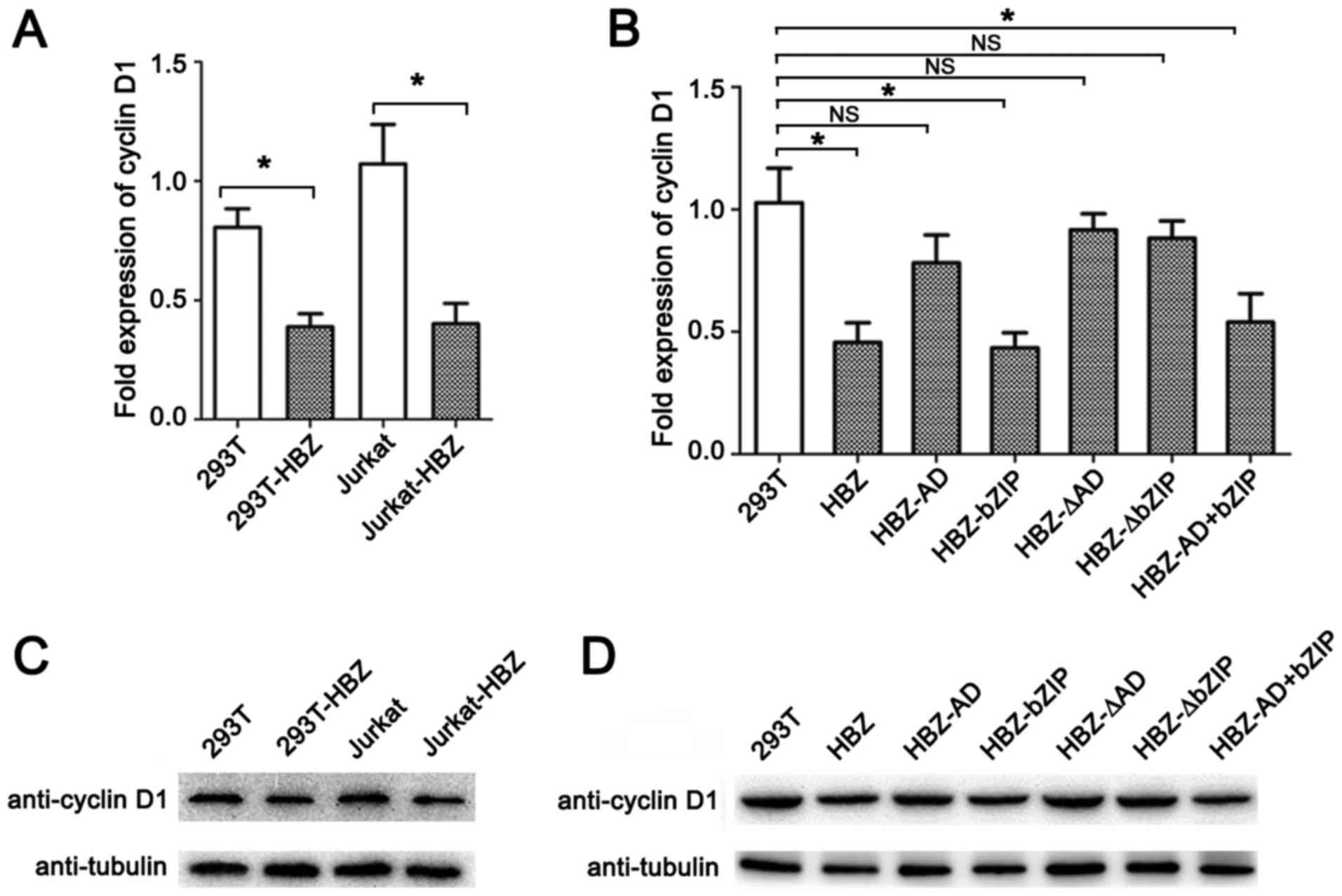 | Figure 2Repression of cyclin D1 gene
expression by HBZ. Cells that stably expressed HBZ or its mutants
were generated by lentiviral-mediated transfection of the HBZ genes
(HBZ, HBZ-AD, HBZ-bZIP, HBZ-ΔAD, HBZ-ΔbZIP or HBZ-AD+bZP). 293T
cells were transduced with either LV-HBZ, LV-HBZ-AD, LV-HBZ-bZIP,
HBZ-ΔAD, HBZ-ΔbZIP or HBZ-AD+bZP at an MOI of 5. Jurkat cells were
transduced with LV-HBZ at an MOI of 30. Cells were selected in
liquid media containing 1 mg/ml puromycin. (A) Levels of cyclin
D1 mRNA transcripts were reduced in the HBZ-expressing cells.
Real-time PCR was performed to analyze the expression of cyclin
D1 mRNA in untransduced (293T or Jurkat) cells and transduced
(293T-HBZ or Jurkat-HBZ) cells; error bars represent standard
deviation (SD). (B) Cyclin D1 mRNA transcript levels were
reduced by the bZIP and bZIP+AD domains of HBZ. Stable expression
of HBZ and its mutants in 293T cells and empty 293T cells was
confirmed by real-time PCR, as in (A); error bars represent SD. (C)
Cyclin D1 expression was reduced by HBZ. The blot shows that 50
µg total cell protein from each HBZ stably transfected cell
line was resolved using 15% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) and immunoblotted using cyclin D1,
HA and β-tubulin antibodies. (D) Cyclin D1 expression was reduced
by HBZ via its bZIP and bZIP+AD domains. Cell lysates were prepared
from 293T cells expressing stable HBZ and its mutants and
empty-vector transduced 293T cells; immunoblots were carried out as
in (C). *P<0.01; NS, no significant difference. |
HBZ has three domains, and each has one of the
following distinct functions (Fig.
1): an activation domain (AD), a central domain (CD), and a
bZIP domain (25–27). To identify the portion of HBZ that
is necessary to inhibit the expression of cyclin D1, we constructed
deletion mutants (Fig. 1). 293T
cells were transduced with the lentiviral vectors carrying each of
the aforementioned HBZ mutants, followed by RT-qPCR and western
blot assays. Two mutants (HBZ-bZIP and HBZ-AD+bZIP) repressed
cyclin D1 mRNA and protein levels (Fig. 2B and D). By contrast, three other
mutants (HBZ-AD, HBZ-ΔAD and HBZ-ΔbZIP) caused no significant
changes in cyclin D1 mRNA and protein levels. These findings
indicate that the bZIP domain, with or without an AD domain, is
needed to suppress levels of cyclin D1 mRNA and protein.
HBZ suppresses cyclin D1 promoter
activity through the CRE and NF-κB sites
Based on the ability of HBZ to downregulate
cyclin D1 gene transcription, we assessed whether cyclin
D1 promoter activity was under the control of HBZ. We
constructed a cyclin D1 promoter reporter plasmid and
co-transfected it into HBZ stably expressing Jurkat cells with a
control plasmid, pRL-TK. Our findings revealed that HBZ reduced
cyclin D1 promoter activity in Jurkat T cells (data not
shown).
The cyclin D1 promoter has multiple
transcription factor-binding sites, such as AP-1, E2F, NF-κB and
Oct-1 (28). Extracellular
signals act via signal transduction pathways to converge at binding
sites to activate or inhibit promoter activity and regulate cell
cycle progression. To identify cis-element(s) in the
cyclin D1 promoter region that are responsive to HBZ, we
transfected luciferase report constructs that contained cyclin
D1 promoters of various lengths into empty 293T cells and
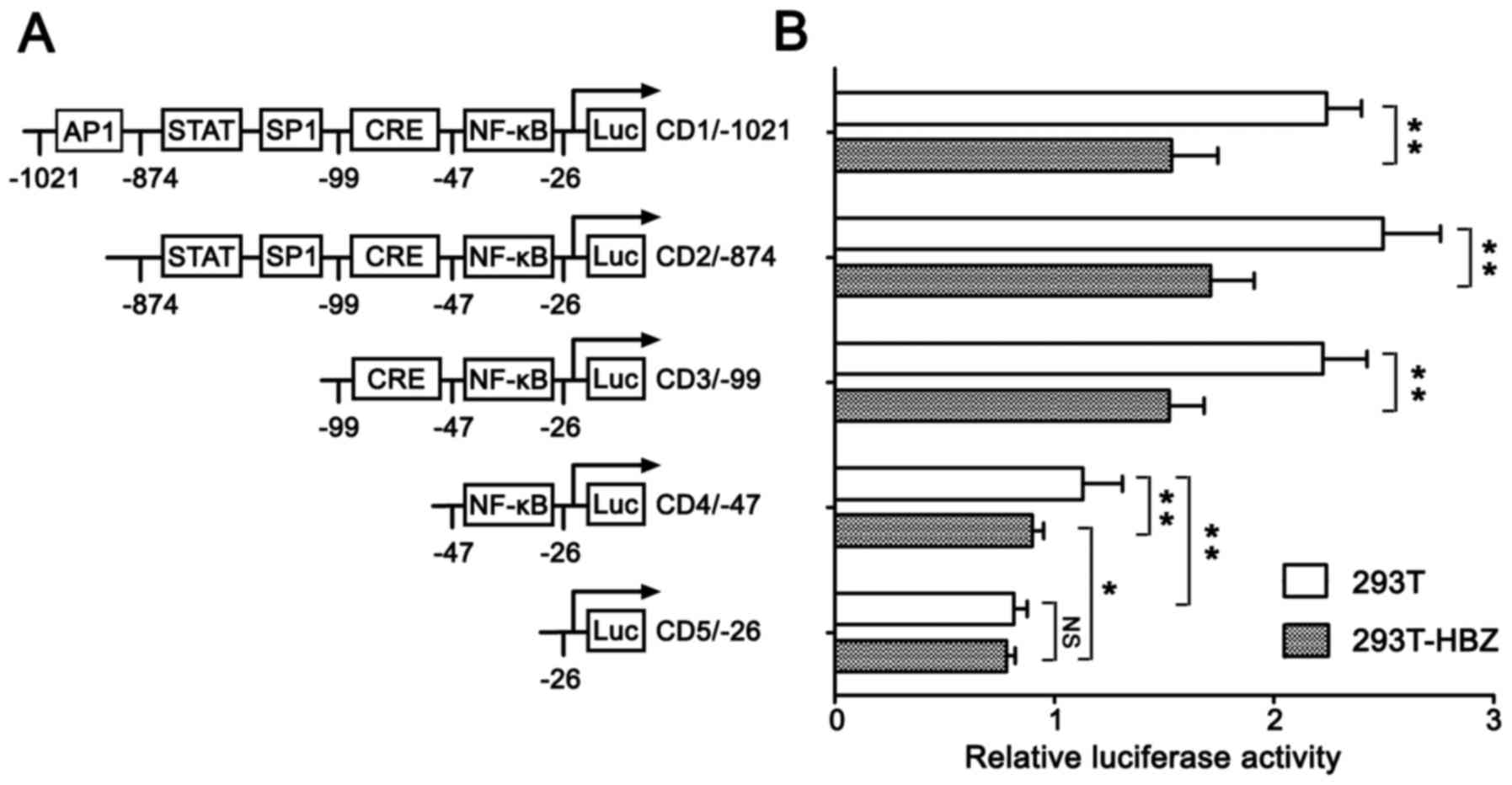
stable HBZ-expressing 293T cells. The promoter activity of
full-length pGL3-CD1/-1021 in 293T-HBZ cells was reduced by ~30%
compared with the activity noted in the empty 293T cells (Fig. 3). These findings are in accord
with the aforementioned studies carried out in the Jurkat human T
cell line. When the AP1 site was deleted (pGL3-CD2/-874), the
promoter activity was not reduced. Furthermore, deletion of the
STAT and SP1 (pGL3-CD3/-99) sites also did not reduce promoter
activity, and pGL3-CD3/-99, which contained CRE and NF-κB sites,
had promoter activity that was similar to pGL3-CD1/-1021, which
contained a full-length cyclin D1 promoter. However, when
the CRE site was deleted (pGL3-CD4/-47), promoter activity was
decreased to 50% of the pGL3-CD3/-99 activity in the empty 293T
cells, while in the 293T-HBZ cells it decreased to 40%. The
promoter activity of pGL3-CD4/-47 in the 293T-HBZ cells was reduced
20% compared with that noted in the empty 293T cells. Further
deletion of the NF-κB site (pGL3-CD5/-26) resulted in reduction of
the promoter activity to ~30% of pGL3-CD4/-47 in the empty 293T
cells. By contrast, there was no significant difference in
pGL3-CD5/-26 promoter activity between the 293T and 293T-HBZ cells.
Although we previously reported that the CRE sites were critical
for HBZ-mediated suppression of cyclin D1 promoter activity
(23), these findings indicate
that the NF-κB site is also responsible for HBZ-mediated
suppression of cyclin D1 promoter activity.
Domains of HBZ that are responsible for
NF-κB p65 inactivation
Previous studies suggest that NF-κB can activate
cyclin D1 promoter activity primarily by the proximal
binding site interacting with the classical p50/p65 complex
(12,13,29). We aimed to ascertain whether HBZ
contributes to the regulation of this signaling pathway.
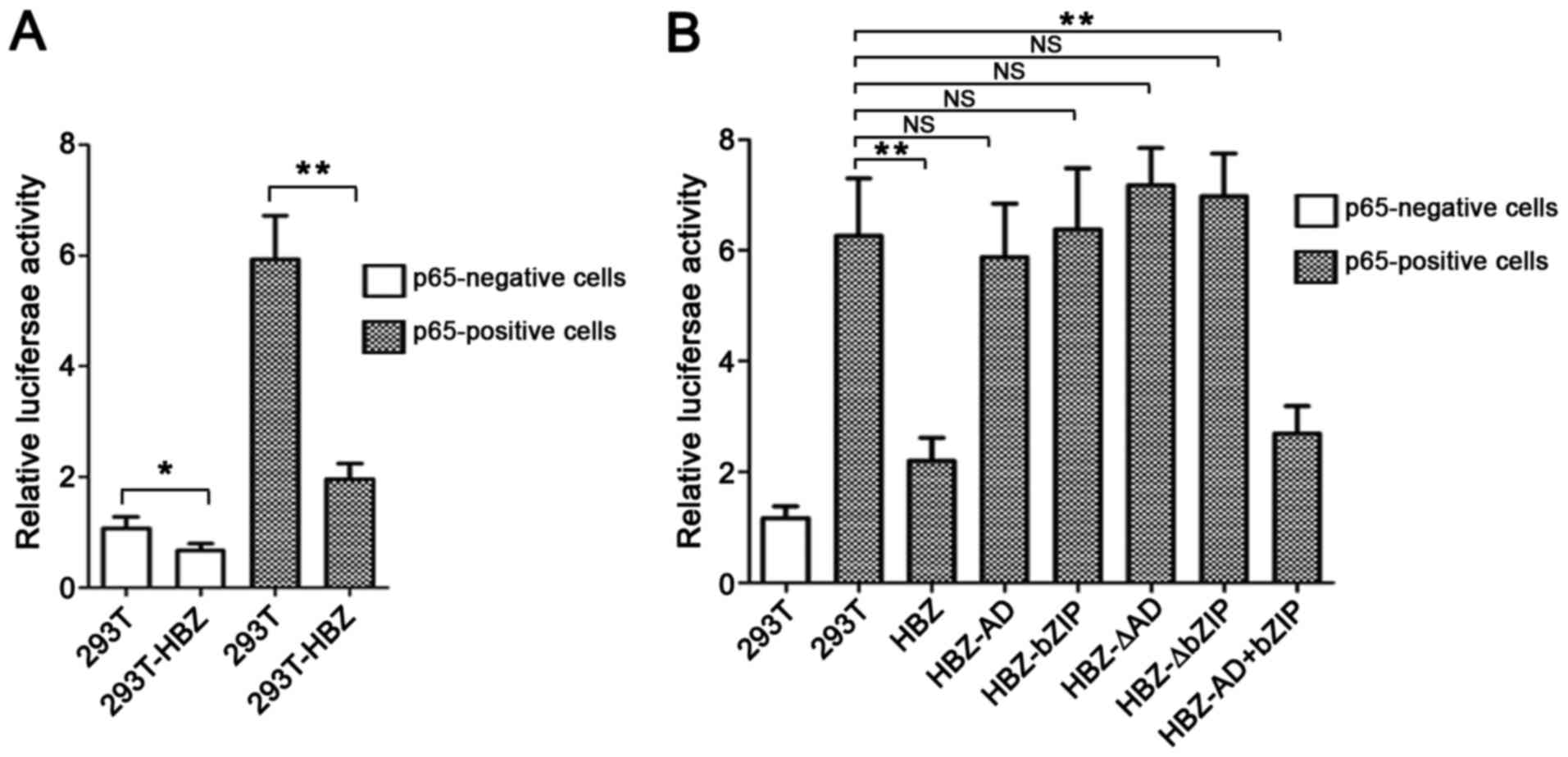
Accordingly, we first evaluated the effect of HBZ on the NF-κB
signaling pathway using a luciferase assay. We transfected 293T and
293T-HBZ cells with pGL3-CD4/-47 luciferase reporter plasmids with
or without p65-expressing vectors. HBZ expression suppressed
p65-mediated pGL3-CD4/-47 (NF-κB) activation (Fig. 4A). Next, we sought to identify the
region of HBZ responsible for repressing NF-κB activation. To this
end, we transfected pGL3-CD4/-47 luciferase reporter plasmids with
p65 vectors into HBZ-deletion mutants that were stably expressed in
293T cells and then performed a luciferase assy. Wild-type HBZ
markedly downregulated p65-mediated pGL3-CD4/-47 (NF-κB) activation
(Fig. 4B). Compared with other
mutants, only the HBZ-AD+bZIP mutant exhibited significant
suppressive activity.
HBZ represses NF-κB activity by binding
to p65
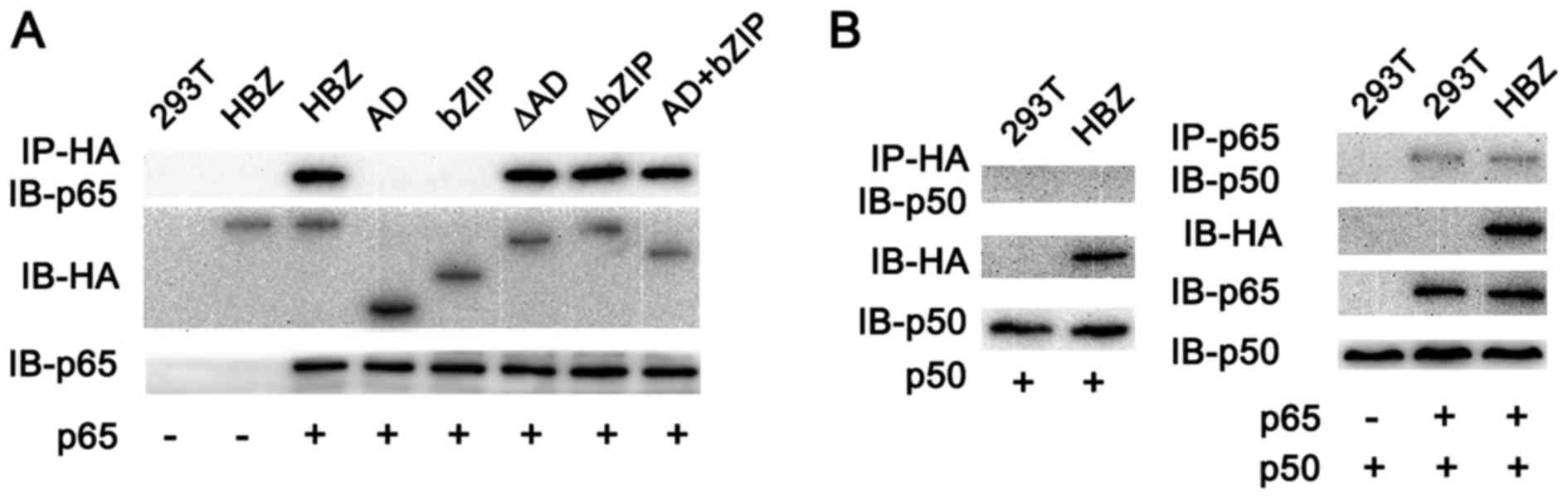
The data described above suggest that HBZ influences
cyclin D1 promoter activity via the NF-κB p65 signaling
pathway. To further investigate whether HBZ could physically
interact with NF-κB p65, and to identify whether the HBZ protein
was necessary for binding with the NF-κB p65 protein, we performed
a co-immunoprecipitation assay. We found that three mutants
(HBZ-ΔAD, HBZ-ΔbZIP and HBZ-AD+bZIP) could bind to p65 (Fig. 5A), demonstrating that at least two
of the three domains in HBZ were required for binding between HBZ
and p65. However, only HBZ-AD+bZIP suppressed NF-κB activation in
the luciferase assay (Fig. 4B),
indicating the significance of the AD and bZIP domains for p65
binding. In normal cells, p65 and p50 form heterodimers and bind to
the κB site in the cyclin D1 promoter. We next studied the
binding affinity of HBZ and p50 by immunoprecipitation. As shown in
Fig. 5B, HBZ did not bind to p50,
nor did it affect binding between p65 and p50.
Taken together, these observations demonstrate that
the HBZ-AD and bZIP domains directly physically associate with
NF-κB p65, and this interaction acts to inhibit cyclin D1
expression.
Discussion
Accumulating evidence indicates that the cell cycle
is regulated by oncogenes and tumor-suppressor genes, and that cell
cycle alterations can occur in response to various carcinogens
(30,31), implicating cell cycle regulation
in carcinogenesis (32). Normal
eukaryotic cells have a well-defined cell cycle that consists of
four distinct stages: G1, S, G2 and M. G1 and G2 phases represents
gaps between the M-S phase and S-M transitions, respectively. These
gaps allow for the repair of DNA damage and replication errors.
Moreover, G1 represents a period when many pathways can be engaged
to influence cell fate (33).
Extracellular changes, such as hypoxia, stress and diverse
metabolic responses, are integrated and interpreted during this
period. Aberrant control of G1, caused by the activation of many
oncogenes or the inactivation of tumor-suppressor genes, plays a
critical role in tumorigenesis (33). Cyclin D1 is a key regulator of
G1-S transition (34). As a
sensor that is activated in response to extracellular changes,
cyclin D1 can be induced by growth factors and stress in response
to the activation of various signaling pathways, including NF-κB
(35–37).
The transcription factor NF-κB belongs to the Rel
family. Mature dimeric NF-κB proteins can translocate to the
nucleus and activate genes involved in apoptosis, cell
proliferation and angiogenesis. It can be activated in response to
many viral infections, and is thought to be important in the host
protective response to viral pathogens. Thus, many viruses have
evolved distinct strategies to regulate the activation of NF-κB.
Furthermore, activation of the NF-κB signaling pathway has also
been reported in various types of cancer cells and is be thought to
play an important role in the development and progression of tumors
(38,39).
HTLV-1 was first reported in 1980 (18). After transmission, it increases
the viral copy number by driving the clonal proliferation of
infected cells, which results in the onset of ATL (40,41). In this strategy, Tax is known to
play a critical role in increasing the number of HTLV-1-infected
cells by promoting proliferation and inhibiting apoptosis (42,43). Tax can activate NF-κB by both
classical and alternative pathways via interactions with IKKγ and
p100 (44,45). However, Tax can often be
inactivated by genetic and epigenetic modifications in ATL
(40,46). HBZ, another viral gene
which is encoded by the minus strand of the HTLV-1 genome, is
expressed in all HTLV-1-infected and ATL cells, and promotes the
proliferation of these cells (21). Similar to other viral proteins of
oncogenic viruses, recent studies have demonstrated multiple
functions of HBZ. For example, HBZ can deregulate multiple cellular
signaling pathways, including the classical NF-κB, AP-1,
transforming growth factor-β (TGF-β), and the Wnt pathways, which
possibly contribute to viral persistence and the clonal expansion
of infected cells (22,47–49). These findings indicate that
HBZ is a critical viral gene involved in HTLV-1-mediated
oncogenesis.
The present study demonstrated a function of the
retroviral protein HBZ and provides some insights into the general
mechanism of the proliferation of viral-infected cells. Our data
indicate that HBZ can inhibit the expression of cyclin D1 via
repression of cyclin D1 promoter activity. Using cyclin
D1 promoter mutant reporter constructs, the luciferase assay
showed that HBZ downregulated cyclin D1 promoter activity
through interactions with the CREB and NF-κB binding sites
(Fig. 3), suggesting that HBZ
likely interacts with these transcription factors. Our previous
study showed that HBZ could directly bind to CREB via a bZIP domain
and inhibit CREB transcriptional activity at the CRE site, thereby
downregulating levels of cyclin D1 transcription (23). Therefore, in the present study, we
further explored the interaction between HBZ and NF-κB. We found
that HBZ suppressed activation of the cyclin D1 promoter,
which contained an NF-κB binding site, in a manner induced by p65.
This suppression was found to occur through the HBZ-AD and bZIP
domains (Fig. 4). This finding
suggests that HBZ exerts effects on the classical NF-κB signaling
pathway, although not all of the three HBZ domains participate in
this process, in accord with a previous study (22). Finally, our co-immunoprecipitation
findings confirmed that HBZ could bind to p65 via the HBZ-AD and/or
bZIP domains in vivo (Fig.
5A). However, HBZ was unable to bind to a single p50 molecule,
and binding between HBZ and p65 did not disturb the formation of
p65/p50 heterodimers (Fig.
5B).
As HBZ has been found to be an important viral
protein, it remains unclear how HTLV-1 induces transformation of T
cells into ATL. One viral protein, Tax, induces the expression of
many genes and causes the aberrant expression of many cellular
genes involved in the growth and survival of T cells (50). However, another viral protein,
HBZ, also interacts with many transcriptional factors and exerts an
effect that is opposite to Tax on the regulation of cellular
signaling pathways. As shown in the present study, HBZ functions as
a suppressor of cyclin D1. In accord with previous studies, we
identified a mechanism involved in the cell cycle regulation
mediated by HBZ. Although Tax has been found to activate cyclin D1
expression and the NF-κB pathway (45,51,52). we believe that HTLV-1 may take
advantage of this opposite function of Tax and HBZ in the
regulation of signaling pathways to allow for better survival and
regulation of the proliferation of HTLV-1-infected cells.
Acknowledgments
The present study was supported by the Foundation
for University Key Teacher by the Education Department Henan
Province (no. 2015GGJS-224) and the Research Fund for the Key
Program of Higher Education of Henan Province (no. 16A310026).
References
|
1
|
Verma IM, Stevenson JK, Schwarz EM, Van
Antwerp D and Miyamoto S: Rel/NF-kappa B/I kappa B family: Intimate
tales of association and dissociation. Genes Dev. 9:2723–2735.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baeuerle PA and Baltimore D: NF-kappa B:
Ten years after. Cell. 87:13–20. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baldwin AS Jr, Azizkhan JC, Jensen DE, Beg
AA and Coodly LR: Induction of NF-kappa B DNA-binding activity
during the G0-to-G1 transition in mouse fibroblasts. Mol Cell Biol.
11:4943–4951. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen FE and Ghosh G: Regulation of DNA
binding by Rel/NF-kappaB transcription factors: Structural views.
Oncogene. 18:6845–6852. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schmitz ML and Baeuerle PA: The p65
subunit is responsible for the strong transcription activating
potential of NF-kappa B. EMBO J. 10:3805–3817. 1991.PubMed/NCBI
|
|
6
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duyao MP, Buckler AJ and Sonenshein GE:
Interaction of an NF-kappa B-like factor with a site upstream of
the c-myc promoter. Proc Natl Acad Sci USA. 87:4727–4731. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kessler DJ, Spicer DB, La Rosa FA and
Sonenshein GE: A novel NF-kappa B element within exon 1 of the
murine c-myc gene. Oncogene. 7:2447–2453. 1992.PubMed/NCBI
|
|
11
|
Feng B, Cheng S, Hsia CY, King LB, Monroe
JG and Liou HC: NF-kappaB inducible genes BCL-X and cyclin E
promote immature B-cell proliferation and survival. Cell Immunol.
232:9–20. 2004. View Article : Google Scholar
|
|
12
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS Jr: NF-kappaB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hinz M, Krappmann D, Eichten A, Heder A,
Scheidereit C and Strauss M: NF-kappaB function in growth control:
Regulation of cyclin D1 expression and G0/G1-to-S-phase transition.
Mol Cell Biol. 19:2690–2698. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou A, Scoggin S, Gaynor RB and Williams
NS: Identification of NF-kappa B-regulated genes induced by
TNFalpha utilizing expression profiling and RNA interference.
Oncogene. 22:2054–2064. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sherr CJ: Mammalian G1 cyclins. Cell.
73:1059–1065. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beijersbergen RL and Bernards R: Cell
cycle regulation by the retinoblastoma family of growth inhibitory
proteins. Biochim Biophys Acta. 1287:103–120. 1996.PubMed/NCBI
|
|
17
|
Joyce D, Bouzahzah B, Fu M, Albanese C,
D'Amico M, Steer J, Klein JU, Lee RJ, Segall JE, Westwick JK, et
al: Integration of Rac-dependent regulation of cyclin D1
transcription through a nuclear factor-kappaB-dependent pathway. J
Biol Chem. 274:25245–25249. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn
PA, Minna JD and Gallo RC: Detection and isolation of type C
retrovirus particles from fresh and cultured lymphocytes of a
patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA.
77:7415–7419. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoshida M, Miyoshi I and Hinuma Y:
Isolation and characterization of retrovirus from cell lines of
human adult T-cell leukemia and its implication in the disease.
Proc Natl Acad Sci USA. 79:2031–2035. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kannian P and Green PL: Human T
lymphotropic virus type 1 (HTLV-1): Molecular biology and
oncogenesis. Viruses. 2:2037–2077. 2010. View Article : Google Scholar
|
|
21
|
Satou Y, Yasunaga J, Yoshida M and
Matsuoka M: HTLV-I basic leucine zipper factor gene mRNA supports
proliferation of adult T cell leukemia cells. Proc Natl Acad Sci
USA. 103:720–725. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao T, Yasunaga J, Satou Y, Nakao M,
Takahashi M, Fujii M and Matsuoka M: Human T-cell leukemia virus
type 1 bZIP factor selectively suppresses the classical pathway of
NF-kappaB. Blood. 113:2755–2764. 2009. View Article : Google Scholar
|
|
23
|
Ma Y, Zheng S, Wang Y, Zang W, Li M, Wang
N, Li P, Jin J, Dong Z and Zhao G: The HTLV-1 HBZ protein inhibits
cyclin D1 expression through interacting with the cellular
transcription factor CREB. Mol Biol Rep. 40:5967–5975. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gaudray G, Gachon F, Basbous J,
Biard-Piechaczyk M, Devaux C and Mesnard JM: The complementary
strand of the human T-cell leukemia virus type 1 RNA genome encodes
a bZIP transcription factor that downregulates viral transcription.
J Virol. 76:12813–12822. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hivin P, Frédéric M, Arpin-André C,
Basbous J, Gay B, Thébault S and Mesnard JM: Nuclear localization
of HTLV-I bZIP factor (HBZ) is mediated by three distinct motifs. J
Cell Sci. 118:1355–1362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao T and Matsuoka M: HBZ and its roles
in HTLV-1 oncogenesis. Front Microbiol. 3:2472012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo ZY, Hao XH, Tan FF, Pei X, Shang LM,
Jiang XL and Yang F: The elements of human cyclin D1 promoter and
regulation involved. Clin Epigenetics. 2:63–76. 2011. View Article : Google Scholar
|
|
29
|
Dahlman JM, Wang J, Bakkar N and Guttridge
DC: The RelA/p65 subunit of NF-kappaB specifically regulates cyclin
D1 protein stability: Implications for cell cycle withdrawal and
skeletal myogenesis. J Cell Biochem. 106:42–51. 2009. View Article : Google Scholar
|
|
30
|
Sgambato A, Han EK, Zhang YJ, Moon RC,
Santella RM and Weinstein IB: Deregulated expression of cyclin D1
and other cell cycle-related genes in carcinogen-induced rat
mammary tumors. Carcinogenesis. 16:2193–2198. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee CC, Yamamoto S, Wanibuchi H, Wada S,
Sugimura K, Kishimoto T and Fukushima S: Cyclin D1 overexpression
in rat two-stage bladder carcinogenesis and its relationship with
oncogenes, tumor suppressor genes, and cell proliferation. Cancer
Res. 57:4765–4776. 1997.PubMed/NCBI
|
|
32
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Semczuk A and Jakowicki JA: Alterations of
pRb1-cyclin D1-cdk4/6-p16(INK4A) pathway in endometrial
carcinogenesis. Cancer Lett. 203:1–12. 2004. View Article : Google Scholar
|
|
35
|
Watanabe G, Albanese C, Lee RJ, Reutens A,
Vairo G, Henglein B and Pestell RG: Inhibition of cyclin D1 kinase
activity is associated with E2F-mediated inhibition of cyclin D1
promoter activity through E2F and Sp1. Mol Cell Biol. 18:3212–3222.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee RJ, Albanese C, Stenger RJ, Watanabe
G, Inghirami G, Haines GK III, Webster M, Muller WJ, Brugge JS,
Davis RJ, et al: pp60(v-src) induction of cyclin D1 requires
collaborative interactions between the extracellular
signal-regulated kinase, p38, and Jun kinase pathways. A role for
cAMP response element-binding protein and activating transcription
factor-2 in pp60(v-src) signaling in breast cancer cells. J Biol
Chem. 274:7341–7350. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR,
Pfeffer LM and Donner DB: NF-kappaB activation by tumour necrosis
factor requires the Akt serine-threonine kinase. Nature. 401:82–85.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pikarsky E, Porat RM, Stein I, Abramovitch
R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E and
Ben-Neriah Y: NF-kappaB functions as a tumour promoter in
inflammation-associated cancer. Nature. 431:461–466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Matsuoka M and Jeang KT: Human T-cell
leukaemia virus type 1 (HTLV-1) infectivity and cellular
transformation. Nat Rev Cancer. 7:270–280. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matsuoka M: Human T-cell leukemia virus
type I (HTLV-I) infection and the onset of adult T-cell leukemia
(ATL). Retrovirology. 2:272005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mulloy JC, Kislyakova T, Cereseto A,
Casareto L, LoMonico A, Fullen J, Lorenzi MV, Cara A, Nicot C, Giam
C, et al: Human T-cell lymphotropic/leukemia virus type 1 Tax
abrogates p53-induced cell cycle arrest and apoptosis through its
CREB/ATF functional domain. J Virol. 72:8852–8860. 1998.PubMed/NCBI
|
|
43
|
Yoshida M: Multiple viral strategies of
HTLV-1 for dysregulation of cell growth control. Annu Rev Immunol.
19:475–496. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chu ZL, Shin YA, Yang JM, DiDonato JA and
Ballard DW: IKKgamma mediates the interaction of cellular IkappaB
kinases with the tax transforming protein of human T-cell leukemia
virus type 1. J Biol Chem. 274:15297–15300. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiao G, Cvijic ME, Fong A, Harhaj EW,
Uhlik MT, Waterfield M and Sun SC: Retroviral oncoprotein Tax
induces processing of NF-kappaB2/p100 in T-cells: Evidence for the
involvement of IKKalpha. EMBO J. 20:6805–6815. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kannagi M, Harada S, Maruyama I, Inoko H,
Igarashi H, Kuwashima G, Sato S, Morita M, Kidokoro M, Sugimoto M,
et al: Predominant recognition of human T-cell leukemia virus type
I (HTLV-I) pX gene products by human CD8+ cytotoxic T
cells directed against HTLV-I-infected cells. Int Immunol.
3:761–767. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Matsumoto J, Ohshima T, Isono O and
Shimotohno K: HTLV-1 HBZ suppresses AP-1 activity by impairing both
the DNA-binding ability and the stability of c-Jun protein.
Oncogene. 24:1001–1010. 2005. View Article : Google Scholar
|
|
48
|
Zhao T, Satou Y, Sugata K, Miyazato P,
Green PL, Imamura T and Matsuoka M: HTLV-1 bZIP factor enhances
TGF-β signaling through p300 coactivator. Blood. 118:1865–1876.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ma G, Yasunaga J, Fan J, Yanagawa S and
Matsuoka M: HTLV-1 bZIP factor dysregulates the Wnt pathways to
support proliferation and migration of adult T-cell leukemia cells.
Oncogene. 32:4222–4230. 2013. View Article : Google Scholar
|
|
50
|
Ressler S, Connor LM and Marriott SJ:
Cellular transformation by human T-cell leukemia virus type I. FEMS
Microbiol Lett. 140:99–109. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim YM, Geiger TR, Egan DI, Sharma N and
Nyborg JK: The HTLV-1 tax protein cooperates with phosphorylated
CREB, TORC2 and p300 to activate CRE-dependent cyclin D1
transcription. Oncogene. 29:2142–2152. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Harhaj NS, Sun SC and Harhaj EW:
Activation of NF-kappa B by the human T cell leukemia virus type I
Tax oncoprotein is associated with ubiquitin-dependent
relocalization of I kappa B kinase. J Biol Chem. 282:4185–4192.
2007. View Article : Google Scholar
|