Introduction
Lung cancer has continued to be the most common type
of cancer worldwide for several decades, with the highest incidence
and mortality rates (1). Only 13%
of patients with lung cancer survive for >5 years (2). As the second leading risk factor for
lung cancer, asbestos exposure is responsible for an estimated 5–7%
of all these cancers (3).
Asbestos-related lung carcinoma is considered to be one of the most
devastating occupational cancers (4). Although the use of asbestos has been
banned or severely restricted since the early 1970's in many
developed countries, asbestos-related lung cancer still poses a
great public health threat due to the long latency period from
asbestos exposure to the incidence of asbestos-induced cancer
(5).
It has been widely accepted that early detection and
precise diagnosis of cancer subtypes could greatly enhance the
efficacy of targeted therapies and improve disease outcome, and in
fact, markedly increase the patient survival rate (6). To date, many imaging and
cytology-based methods have been applied for early detection
(7–9). However, most techniques have limited
sensitivity to detect asbestos-related lung cancer, as the
histopathological subtypes of lung cancer patients with and without
asbestos exposure are quite similar (10).
Among asbestos-related lung cancer, non-small cell
lung cancer (NSCLC) accounts for at least 80% of these cases
(2). There are three primary
subtypes of NSCLC distinguishable by the appearance and chemical
makeup of the cells: adenocarcinoma (LC-AC), squamous cell
carcinoma (LC-SCC) and large-cell carcinoma. It has been shown that
gene expression profiles could be used to distinguish
asbestos-exposed from non-exposed lung cancer patients (11). Gene expression profiles in
asbestos-exposed epithelial and mesothelial lung cell lines have
revealed that the expression levels of genes such as nuclear
factor-κB (NF-κB) subunit 2 (NFKB2), IKBKB, thioredoxin (TXN),
thioredoxin reductase (TXNRD1), BCL2 interacting protein 3 like
(BNIP3L), protein kinase C (PKC)δ and adducin (ADD)3 were
significantly altered in response to asbestos exposure in all the
cell lines (12). Moreover,
accumulating in vivo and in vitro studies have
identified asbestos-related gene expression changes involved in
activation of the NF-κB pathway, p53 promoter activation, MAPK
signaling pathway and cell proliferation induced by tumor necrosis
factor-α (TNF-α) and TNF-β as well as PDGFA and PDGFB (13,14). The increase in available tumor
samples of patients diagnosed with different subtypes of a certain
tumor makes it possible to detect subtype-specific biomarkers.
Class comparison analysis of tumors of 36 patients with primary
LC-AC identified ADAM28 as a potential oncogene involved in
asbestos-related LC-AC, with expression verified in three
independent test sets (15).
Similarly, gene expression profiling of asbestos-related lung
squamous cell carcinomas (ARLC-SCCs) identified MS4A1 as a
potential candidate (10).
However, immunohistochemical staining showed that expression of
MS4A1 was primarily localized to stromal lymphocytes rather than
tumor cells. Thus, identification of candidate biomarkers in
ARLC-SCC is needed.
Using the same data from Wright et al
(10), we aimed to further
identify differentially expressed genes (DEGs) according to a
cut-off point of a log2 fold-change (FC) >1 or <-1
and P-value <0.05. Furthermore, the protein-protein interaction
(PPI) network was constructed and a novel, pathway-deviation-based
approach was proposed to detect potential ARLC-SCC-specific
biomarkers from the networks. Notably, a new scoring method was
developed to weight each DEG by integrating its expression
deviation score and network degree. In addition, a newly defined
parameter, pathscore, was used to measure the deviation in each
pathway enriched by DEGs. Finally, the molecular pathways involved
in the disease development of these two types of lung cancer
samples and their molecular heterogeneity, were systematically
analyzed by hierarchical clustering and pathway correlation
analysis. Several candidate marker genes and multi-effect targets
were identified, which may provide insight into the development of
novel diagnostic and therapeutic tools for ARLC-SCC. In addition,
as these genes are involved in multiple pathaws, they may thus
function as 'multi-effect targets', as it is possible, that drugs
targeting these genes may influence multiple pathways
simultaneously.
Materials and methods
Gene differential expression
analysis
The microarray data were retrieved from Gene
Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database under
accession no. GSE23822 (10).
Total RNAs from 56 human lung squamous cell carcinoma samples,
consisting of 26 ARLC-SCCs and 30 non-asbestos-related squamous
cell lung carcinomas (NARLC-SCCs), were hybridized to 48K Illumina
HumanHT-12 v3.0 Expression BeadChips, respectively. These cases
were classified as ARLC-SCC if there were >20 asbestos
bodies/gram wet weight (AB/g ww) in the non-tumor tissue, or
NARLC-SCC if no asbestos bodies were found. No statistically
significant differences in age, gender, smoking history or tumor
stage was noted between the two types of samples. After background
correction, the intensities of multiple probes for a gene were
averaged into one expression value which was then transformed as a
normalized expression value by Z-score. The differential expression
analysis between the two groups of samples was performed using the
R package known as linear models for microarray data (LIMMA)
(16). To minimize the potential
information loss caused by multi-hypothesis test and reserve more
inter-group DEGs, a P-value before false discovery rate (FDR)
adjustment was used to detect the significant DEGs and the detailed
criterion of DEGs was defined as P<0.05.
Protein-protein interaction (PPI) network
construction
PPI data were downloaded from the Biological General
Repository for Interaction Datasets (BioGRID; http://www.thebiogrid.org) interaction database
(17) and Human Protein Reference
Database (HPRD; http://www.hprd.org) (18), and merged into the background PPI
network. Then, DEGs were mapped to the background PPI network to
build the sub-network of DEGs which was extended with those
proteins (not included in DEGs) interacting with at least three
DEGs and trimmed by removing those orphan nodes. The PPI network
was visualized using Cytoscape (http://cytoscape.org) (19).
Network topological analysis
The network topological analysis was performed using
Network Analyzer (20) plug-in of
Cytoscape. Five topological parameters were used in this study,
including node degree, average shortest path length (ASPL),
closeness centrality (CC), eccentricity (EC) and topological
coefficient (TC).
Node degree
In the undirected network, the node degree of a node
x is the number of edges linked to x. And a node with
a high degree is referred to as hub.
Average shortest path length
(ASPL)
The length of a path between node x and
y is the number of edges forming it. The ASPL is the average
length of all the shortest paths between nodes in the network,
which indicates the expected distance between any two connected
nodes.
Closeness centrality (CC)
The CC of a node x is the reciprocal of the
ASPL from x to other nodes in the network. The CC of each
node is a number between 0 and 1. Closeness centrality is a measure
of how quickly information spreads from a given node to other
reachable nodes in the network.
Eccentricity (EC)
The EC of a node x is the maximum
non-infinite length of a shortest path between n and another node
in the network. Specially, the network diameter is the maximum node
EC.
Topological coefficient (TC)
The TC of a node x is a relative measure for
the extent to which a node shares neighbors with other nodes.
Weighting of DEGs based on deviation
score and node degree
For each gene, a reference interval (RI) was defined
as [min, max] in which min refers to the mean - SD (standard
deviation) based on the expression value vector of this gene in
NARLC-SCC patients and max refers to the mean + SD. Those genes
with an expression value within the RI were thought to resemble the
expression pattern of NARLC-SCC patients; otherwise an ARLC-SCC
pattern was preferred. Then for gene x, the difference
beyond the RI in each ARLC-SCC patient was summed up as its
deviation score of expression. A higher deviation score represents
a larger difference between the expression values of the gene in
ARLC-SCC patients and those in NARLC-SCC. Meanwhile, a higher node
degree of gene x in the PPI network indicates that it has
more neighbors and is of more biological importance. In this study,
these two measures were integrated as following to weigh the
importance of a gene to ARLC-SCC carcinogenesis. The importance
score W of gene x was calculated as below:
W=score×degree
in which di is the expression value of gene
x in ARLC-SCC sample i, and n is the amount of
ARLC-SCC samples. The value of d was set as max if
di was larger than max, otherwise it was
set as min. Each score in a sample was scaled into the range
of 0 and 10. The degree was log2 normalized. Finally all
genes were sorted based on the value of W, and the top 50%
of genes were selected as important genes which were used for
further analysis.
Pathway enrichment analysis
To further investigate the functional pathways
regulated by these important genes, the Kyoto Encyclopedia of Genes
and Genomes (KEGG, http://www.genome.jp/kegg) pathway (21) enrichment analysis was performed
with the important upregulated and downregulated genes,
respectively, using online tools from the Database for Annotation,
Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov) (22). At the significance cut-off
P<0.1 in Fisher exact test, 22 enriched pathways were obtained,
among which 12 were kept when the statistical significance was set
as P<0.05. To keep sufficient amount of pathways for statistical
analysis and subsequent hierarchical clustering, the cut-off of the
P-value was set as 0.1. The pathways enriched by either upregulated
or downregulated genes were combined together as important pathways
for further analysis.
Hierarchical clustering based on pathway
interference
For pathway p in important pathways, the
interference (defined as pathscore) in each tumor sample was
assessed as below:
Assuming there are m upregulated and n
downregulated genes in pathway p, di
refers to the mean of expression values of the i-th
upregulated gene in NARLC-SCC samples, while di
means that of the j-th downregulated gene. Each gene is
weighted with its degree in the network as coefficient w. A
positive pathscore means this pathway is upregulated in the tumor
patients compared with the average level in all NARLC-SCC patients,
while a negative pathscore means downregulated. Finally,
hierarchical clustering was performed on both samples and pathways
using the software Cluster 3.0 (http://bonsai.ims.u-tokyo.ac.jp/~mdehoon/software/cluster/)
(23). Before clustering, the
pathscore matrix was log-transformed and median-center scaled.
Then, the similarity matrix was calculated using the correlation
center. The clustering results were visualized via the Java plugin
TreeView (http://jtreeviewsourceforge.net/) (24).
Pathway correlation analysis
The correlation between two pathways was measured
with Spearman's rank correlation coefficient of pathscores across
all the samples via software SPSS 19.0 (IBM Corp., Armonk, NY,
USA). The pathway pairs with a larger absolute value of r
than 0.5 and a significance P<0.05 were assumed to be the
significantly correlated pathway pairs. Based on the distribution
of DEGs in each pathway, crosstalk genes among these correlated
pathways were obtained. The cross-talk genes may influence multiple
functional correlated pathways and could be used as critical
biomarkers and multi-effect drug targets. Identification of these
biomarkers is meaningful for both the understanding of the
mechanisms and the clinical diagnosis and therapy for ARLC-SCC.
Results
Differentially expressed gene
extraction
The gene expression difference between 26 ARLC-SCC
and 30 NARLC-SCC patients was detected using package LIMMA in
Bioconductor, and finally 1,333 DEGs were obtained, in which 391
genes were upregulated and 942 genes were downregulated in the
ARLC-SCC patients.
PPI network construction
The background PPI network was constructed based on
data from BioGrid and HPRD databases, including 14,553 proteins and
662,360 interactions. To obtain the interaction relationship among
these 1,333 DEGs, they were mapped to the background PPI network.
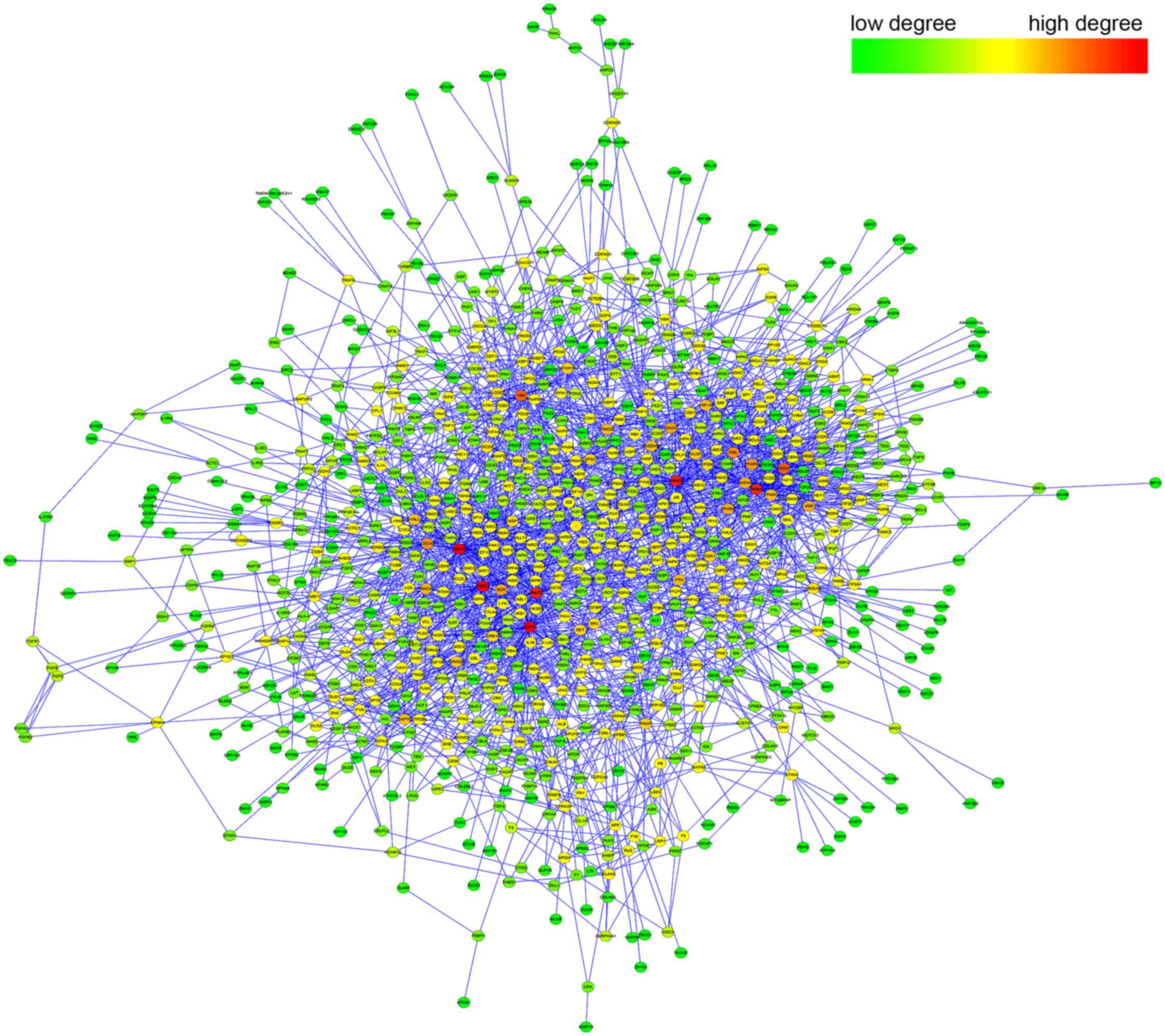
Then, the PPI sub-network of DEGs (Fig. 1) was constructed using Cytoscape,
in which 981 nodes and 2,568 interactions were kept.
A gene was represented as hub in the network
if it could interact with multiple DEGs, and thereby this gene
could regulate multiple biological processes. To highlight these
hub genes, topological analysis was performed on each node
in the network. As shown in Fig.
1, the high-degree nodes (red) were enriched at the center
region of the network, while the low-degree nodes (green) were
mostly distributed at the outer region.
Topological analysis of the PPI
network
To further analyze the specificity of the PPI
sub-network of DEGs, five topological parameters were measured for
the sub-network and background PPI network, respectively. As shown
in Table I, the sub-network of
DEGs showed a decreased average node degree (5.23) compared with
that (7.01) in the background PPI network. Meanwhile, the closeness
centrality (CC, 0.24 vs. 0.35) was also reduced in the sub-network.
On the contrary, the other three parameters derived from the
sub-network were increased compared to those derived from the
background network, specifically the average shortest path length
(ASPL, 4.17 vs. 2.97) and the eccentricity (EC, 8.52 vs. 6.51) were
greatly increased and the topological coefficient (TC, 0.24 vs.
0.17) with a minor increase. Taken together, these five parameters
consistently showed an obvious network efficiency reduction in the
sub-network of DEGs compared with that in the background network.
The lowered average degree may suggest a reduced contribution of
each node to the network, while the increase in ASPL, EC and TC
together with the decrease in CC may imply that both the tightness
of the network and the information spreading efficiency among the
genes were reduced.
 | Table IComparison of network topological
parameters in the ARLC-SCC-specific sub-network and the background
PPI network. |
Table I
Comparison of network topological
parameters in the ARLC-SCC-specific sub-network and the background
PPI network.
| Parameter | ARLC-SCC-specific
sub-network | Background PPI
network |
|---|
| Degree | 5.23 | 7.01 |
| EC | 8.52 | 6.51 |
| ASPL | 4.17 | 2.97 |
| CC | 0.24 | 0.35 |
| TC | 0.24 | 0.17 |
Detection of ARLC-SCC-related important
genes
To assess the importance of each DEG, an importance
score was assigned by integrating its deviation score of expression
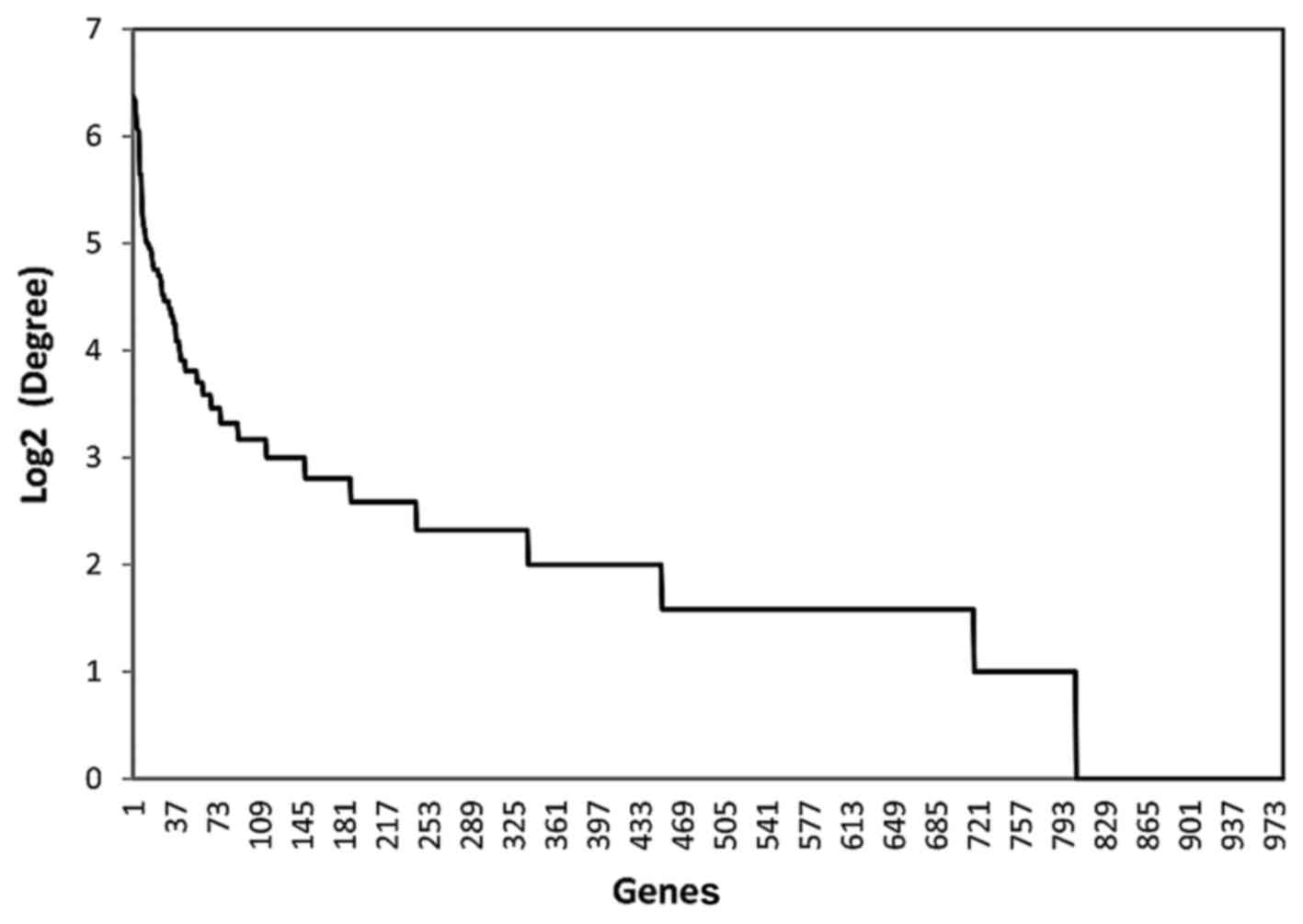
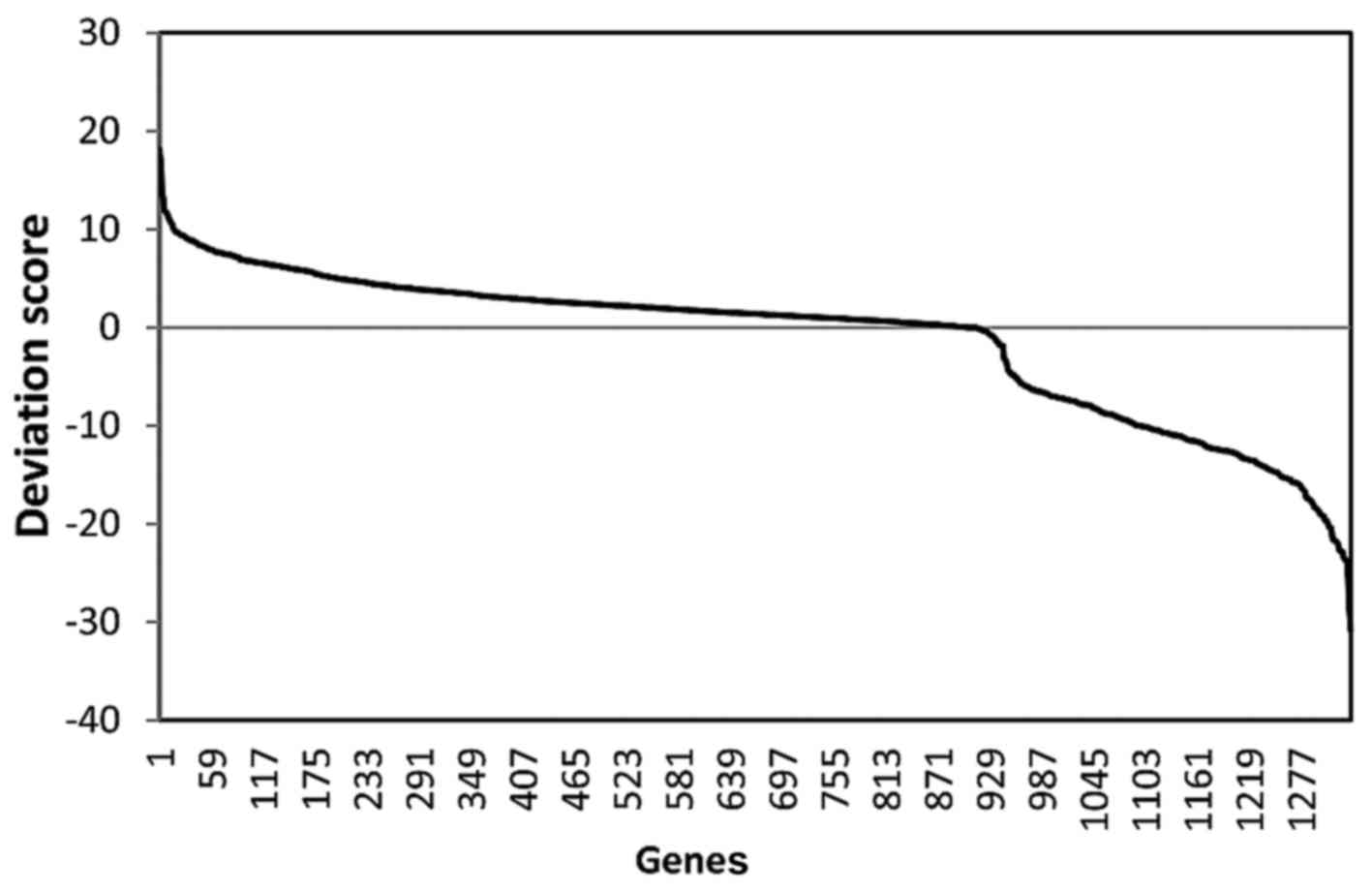
and node degree in the network. The distribution of these two
parameters among all the DEGs is presented as Figs. 2 and 3, respectively. Based on the descending
order of importance score, the top 50% of DEGs were kept as
ARLC-SCC-related important genes (524 genes in total). These
important genes showed large deviations from the RI and higher node
degrees. The higher node degrees indicated that they could interact
with multiple DEGs and play a role in multiple biological
processes. Thus, these genes are thought to be significantly
associated with ARLC-SCC carcinogenesis. Moreover, further studies
were carried out on these genes and the biological pathways in
which they are involved.
Biological pathways involved in ARLC-SCC
carcinogenesis
To get insight into ARLC-SCC-related pathways, the
524 important genes were uploaded to DAVID for KEGG pathway
enrichment analysis. Twenty-two KEGG pathways were obtained by
combining the enrichment results for the important upregulated and
downregulated genes. Specifically, these genes were significantly
enriched in specific pathways of cancer, proliferation-related
pathways regulating cell cycle and apoptosis, and
metastasis-related pathways involved in the regulation of cell
adhesion and the cytoskeleton. These results indicated that
cancer-related pathways may play an important role in ARLC-SCC
development, and the deregulation in cell cycle and apoptosis
process may be a major cause for SCC of the lung. Meanwhile, the
changes in metastasis-related pathways may account for the poor
prognosis and rapid spreading in ARLC-SCC patients.
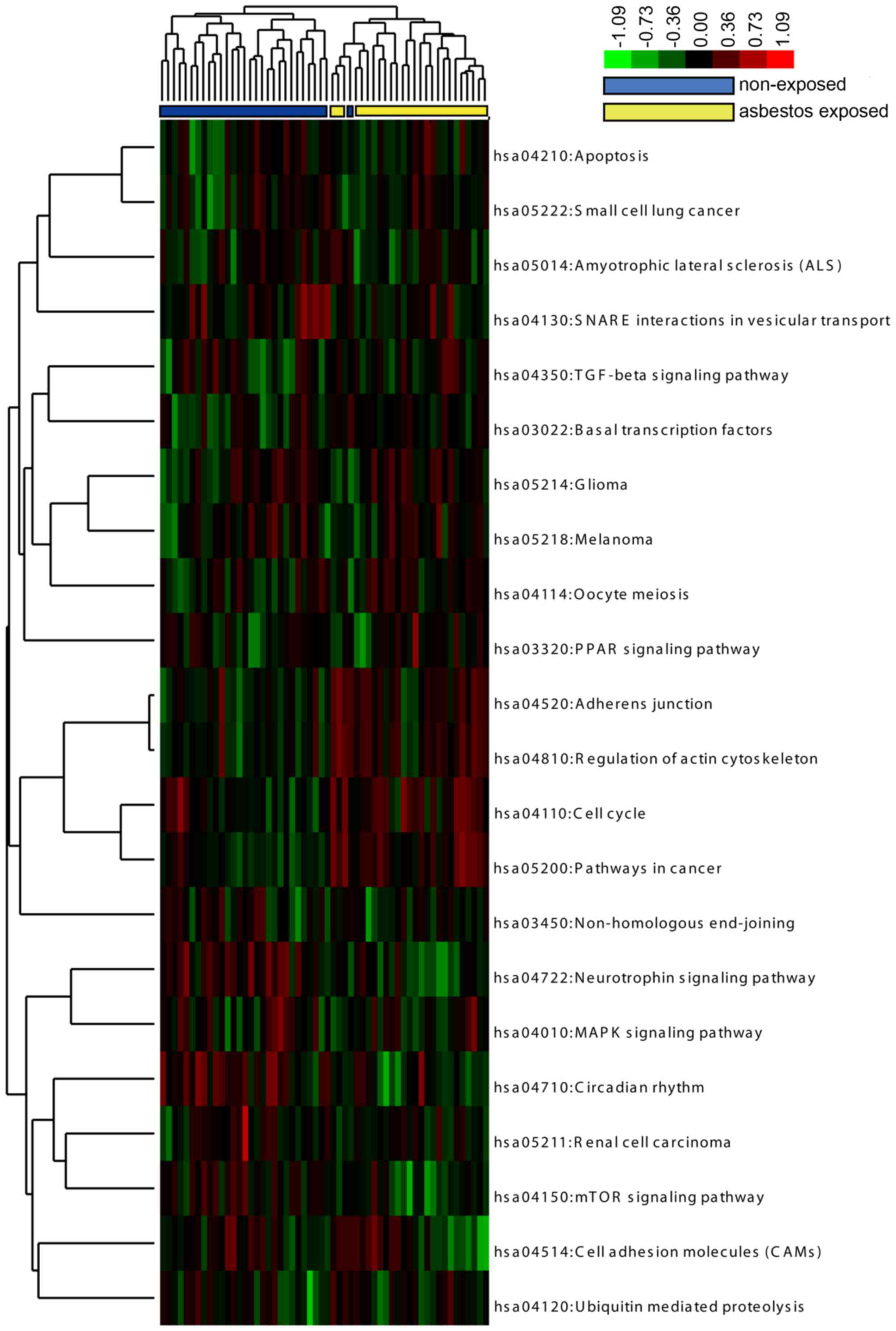
Hierarchical clustering based on pathway
interference path-score
To measure the extent to which a pathway was
interfered with in each tumor patient compared with the average
level in all NARLC-SCC patients, a pathscore was calculated for
each pathway integrating the deviation score and node degree of all
DEGs in the pathway. Then, the pathscore matrix for those 22
pathways in 56 patients was used for hierarchical clustering
analysis. The clustering dendrogram was visualized via TreeView and
is shown in Fig. 4. As shown in
the dendrogram, ARLC-SCC and NARLC-SCC patients were clearly
separated based on the pathscore of 22 KEGG pathways, except one
NARLC-SCC sample which was grouped into the ARLC-SCC cluster. The
clustering analysis in patients indicated a great difference in the
expression pattern of these pathways between these two types of SCC
samples. On the other side, it also suggests intrinsic associations
among these pathways by sharing similar variation trends under
different disease phenotypes. For example, the four KEGG pathways
such as adherens junction (hsa04520), regulation of actin
cytoskeleton (hsa04810), cell cycle (hsa04110) and pathway in
cancer (hsa05200) were consistently upregulated in the ARLC-SCC
patients compared with the average level of gene expression in all
the NARLC-SCC patients. Thus, identification of the correlated
pathways with the same or similar deviation pattern and the
cross-talk genes between them are critical for the identification
and functional analysis of ARLC-SCC-related biomarkers. For this
purpose, a correlation study was subsequently applied on the 22
important pathways.
Pathway correlation study and cross-talk
gene detection
The correlation between each two important pathways
was measured with Spearman's rank correlation coefficient r
based on their pathscore across the 56 lung cancer patients.
According to the correlation result, 13 significantly correlated
pathway pairs were obtained. For better visualization, a
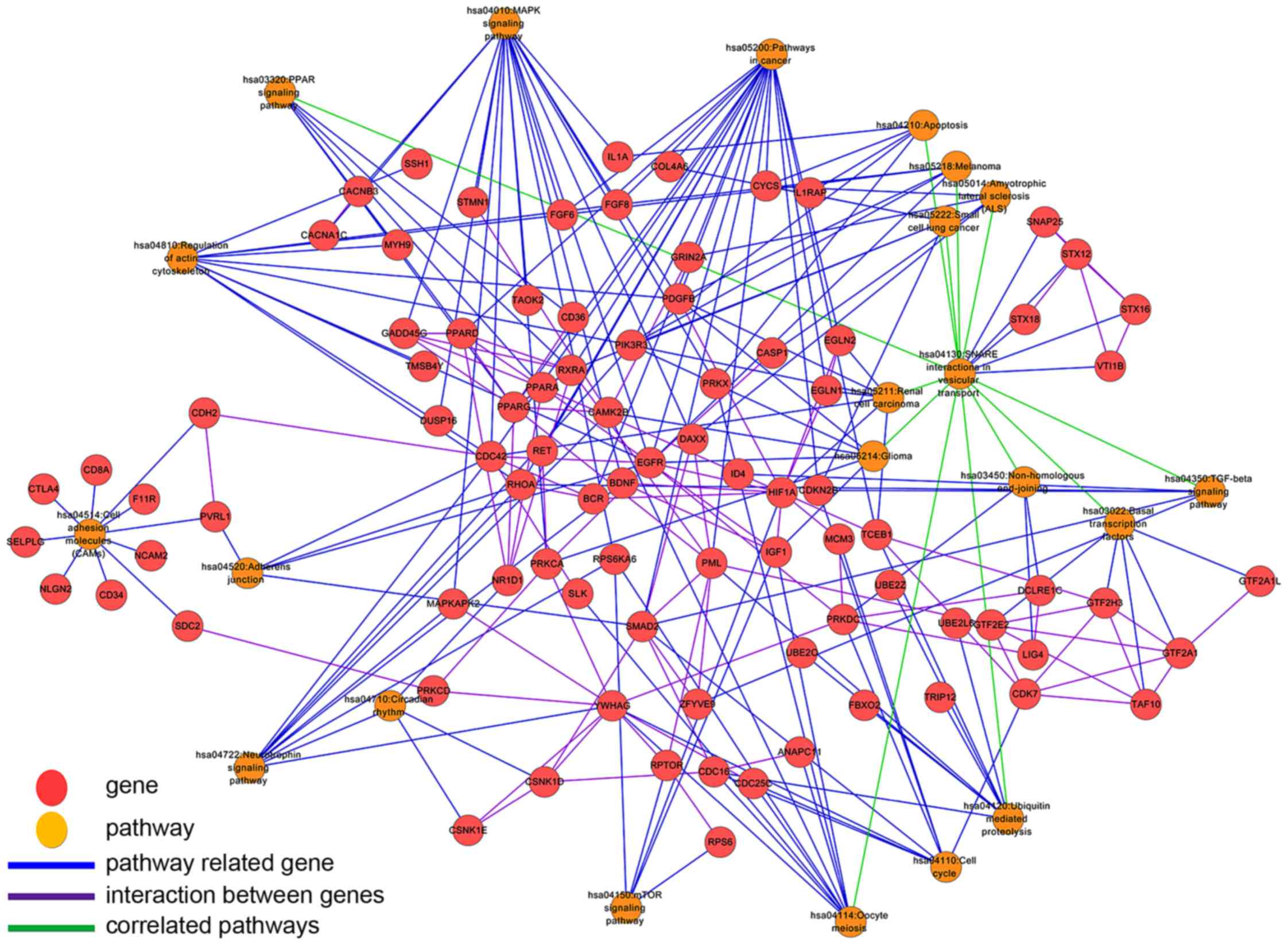
pathway-gene bipartite network (Fig.
5) was constructed based on the pathway-gene relationship,
gene-gene interaction and pathway-pathway correlation, which
included 109 nodes (22 pathways, 87 genes) and 263 edges. As shown
in Fig. 5, 12 pathways presented
a significant correlation with SNARE interactions in vesicular
transport pathway (hsa04130), which indicates that deregulation in
the synaptic vesicle transport process may play a central role in
the carcinogenesis of ARLC-SCC. Interactions were also found
between genes in the same pathway, indicating that these genes
interact with each other to regulate the same biological process.
In addition, many pathways were not independent but were connected
by sharing cross-talk genes. For example, 4 cross-talk genes (EGFR,
PRKX, PDGFB and PIK3R3) were shared by 4 pathways such as MAPK
signaling pathway, glioma, regulation of actin cytoskeleton and
apoptosis. In the same way, the cell cycle, circadian rhythm,
oocyte meiosis and neurotrophin signaling pathways were linked
together through cross-talk genes [SLK, insulin-like growth factor
(IGF1), CDC42 and PRKCA] among which IGF1 and CDC42 codes two
critical immunological proteins. This implies that the cross-talk
genes between different pathways may take part in the regulation of
specific biological functions. The abnormality in these genes may
lead to an inbalance of the pathways regulated by them, and finally
cause the onset of diseases. Thus, these cross-talk genes could be
used as ARLC-SCC-specific biomarker genes and even potential
multi-effect drug targets. It is possible, that drugs targeting
these genes may influence multiple pathways simultaneously
Discussion
To date, only one study has attempted to detect the
genetic heterogeneity between ARLC-SCC and NARLC-SCC patients. In
the study of Wright et al (10), 6 candidate genes (CARD18, MS4A1,
ABHD12, API5, ANKRD20A3 and LOC402117) were identified as being
differentially expressed between ARLC-SCC and NARLC-SCC cases,
while subsequent validations failed to support any of them as
markers of asbestos etiology. Compared to their research, a
relatively less stringent statistical significance (P<0.05) was
used to screen the DEGs in the present study. Hence, more candidate
genes were reserved for systematical network and pathway analysis,
and thereby provided a comprehensive landscape of ARLC-SCC
specificity at the gene expression level.
To detect the genetic specificity and biomarker
genes in ARLC-SCC patients, gene expression profiles from 26
ARLC-SCC patients and 30 NARLC-SCC patients were systematically
analyzed. As a result, 1,333 DEGs were found, for which 524 genes
were ARLC-SCC-specific weighted by a novel scoring method. Based on
pathway enrichment analysis, dramatic deviation in 22 KEGG pathways
was found between ARLC-SCC and NARLC-SCC patients. Subsequent
pathway correlation study showed that functional correlations may
exist between these 22 pathways by sharing similar expression
deviation patterns or differentially expressed cross-talk
genes.
Notably, based on the result of the pathway
correlation study, the pathway of SNARE interactions in vesicular
transport was found to be significantly correlated with the other
12 enriched pathways of the ARLC-SCC-specific genes, which
indicated that abnormal vesicular transport may play a latent role
in ARLC-SCC carcinogenesis and development. To date, several
transport mechanisms through which cells communicate with the
outside environment (within its microenvironment or even at distant
sites) have been identified. Among them, vesicular transport,
particularly exosome-mediated transport stands out (25). A considerable number of studies
over several decades have revealed that the exosome takes part in
the transport of multiple biological materials such as proteins,
RNAs (26), breakdown product of
signaling pathways and viruses (27), and plays a critical role in
disease development (28).
Furthermore, emerging evidence suggests that exosome-secreted
proteins can propel fibroblast growth (29), which may account for the fibrosis
of the lungs induced by asbestos exposure in ARLC-SCC cases. To the
best of our knowledge, this is the first study to elucidate the
potential association between exosome-mediated transport and
asbestosis, which may provide a novel direction of drug development
for ARLC-SCCs.
Based on the pathway correlation study, the pathways
with a similar deviation pattern often presented functional
correlation in the 56 SCC patients. These correlated pathways were
bridged by the DEGs between them. The shared DEGs constitute
cross-talk genes of which the deregulation in expression may
simultaneously disturb the function of two or more pathways.
Together with PDGFB, PIK3R3 and EGFR, CDC42 is involved in the
regulation of the actin cytoskeleton. Abnormal regulation or
functioning in cytoskeletal components is often a cause of many
diseases including cancers (30,31). Partial disassembly of cytoskeletal
actin may account for the respiratory barrier function of pulmonary
epithelium induced by ROS in response to asbestos exposure
(32). Overexpression of PDGFB
can stimulate increased collagen deposition and vascular smooth
muscle hyperplasia following asbestos inhalation (33). SLK may promote the cytoskeletal
rearrangements and disassembly of actin stress fibers, focal
adhesions and induced apoptosis (34), and therefore may contribute to the
development of ARLC-SCC. Growth factors such as IGF1 and
platelet-derived growth factor (PDGFB) are also known to promote
cell cycle re-entry through progression from G1- to S-phase after
asbestos exposure (35).
Activation of PRKCA is reported to have multiple effects on
peribronchiolar cell proliferation, pro-inflammatory and
pro-fibrotic cytokine expression, and immune cell profiles in the
lung, which may lead to asbestos-induced fibrogenesis (36,37). Taken together, the cross-talk
genes deregulated in ARLC-SCC patients may be used as biomarkers
and multi-effect targets for ARLC-SCC.
Due to the unavailability of these lung cancer
patient samples, experimental validations were beyond our reach.
Nonetheless, the present study provided a novel insight into the
genetic heterogeneity in ARLC-SCC cases. In particular, the finding
of the potential role of vesicular transport in ARLC-SCC
development provides a new opportunity for the development of a
novel therapy for ARLC-SCCs.
In the present study, a novel scoring approach was
proposed to detect potential ARLC-SCC-specific marker genes. Based
on network topological and pathway correlation analysis, obvious
heterogeneity was found between the ARLC-SCC and NARLC-SCC cases.
Notably, the undiscovered role of vesicular transport reported
herein may provide new insight into the targeted therapy of
ARLC-SCCs.
References
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality
Worldwide: IARC CancerBase No. 11 (Internet). International Agency
for Research on Cancer; Lyon, France: 2013, Available from:
http://globocan.iarc.fr.2014.
|
|
2
|
Balgkouranidou I, Liloglou T and Lianidou
ES: Lung cancer epigenetics: Emerging biomarkers. Biomarkers Med.
7:49–58. 2013. View Article : Google Scholar
|
|
3
|
LaDou J: The asbestos cancer epidemic.
Environ Health Perspect. 112:285–290. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nymark P, Guled M, Borze I, Faisal A,
Lahti L, Salmenkivi K, Kettunen E, Anttila S and Knuutila S:
Integrative analysis of microRNA, mRNA and aCGH data reveals
asbestos- and histology-related changes in lung cancer. Genes
Chromosomes Cancer. 50:585–597. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin RT, Takahashi K, Karjalainen A,
Hoshuyama T, Wilson D, Kameda T, Chan CC, Wen CP, Furuya S, Higashi
T, et al: Ecological association between asbestos-related diseases
and historical asbestos consumption: An international analysis.
Lancet. 369:844–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu X, Li X, Cheng J, Liu Z, Thrall M, Wang
X, Wang X and Wong S: Quantitative label-free multimodality
nonlinear optical imaging for in situ differentiation of cancerous
lesions. Proc SPIE 8565; March 8 2013; Photonic Therapeutics and
Diagnostics; IX:pp. 85653A2013, View Article : Google Scholar : http://dx.doi.org/10.1117/12.2002633.
|
|
7
|
Henschke CI; International Early Lung
Cancer Action Program Investigators: Survival of patients with
clinical stage I lung cancer diagnosed by computed tomography
screening for lung cancer. Clin Cancer Res. 13:4949–4950. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Henschke CI, Yankelevitz DF, Libby DM,
Pasmantier MW, Smith JP and Miettinen OS; International Early Lung
Cancer Action Program Investigators: Survival of patients with
stage I lung cancer detected on CT screening. N Engl J Med.
355:1763–1771. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schwartz AG, Prysak GM, Bock CH and Cote
ML: The molecular epidemiology of lung cancer. Carcinogenesis.
28:507–518. 2007. View Article : Google Scholar
|
|
10
|
Wright CM, Savarimuthu Francis SM, Tan ME,
Martins MU, Winterford C, Davidson MR, Duhig EE, Clarke BE, Hayward
NK, Yang IA, et al: MS4A1 dysregulation in asbestos-related lung
squamous cell carcinoma is due to CD20 stromal lymphocyte
expression. PLoS One. 7:e349432012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wikman H, Ruosaari S, Nymark P, Sarhadi
VK, Saharinen J, Vanhala E, Karjalainen A, Hollmén J, Knuutila S
and Anttila S: Gene expression and copy number profiling suggests
the importance of allelic imbalance in 19p in asbestos-associated
lung cancer. Oncogene. 26:4730–4737. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nymark P, Lindholm PM, Korpela MV, Lahti
L, Ruosaari S, Kaski S, Hollmén J, Anttila S, Kinnula VL and
Knuutila S: Gene expression profiles in asbestos-exposed epithelial
and mesothelial lung cell lines. BMC Genomics. 8:622007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nymark P, Wikman H, Hienonen-Kempas T and
Anttila S: Molecular and genetic changes in asbestos-related lung
cancer. Cancer Lett. 265:1–15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ruosaari S, Hienonen-Kempas T, Puustinen
A, Sarhadi VK, Hollmén J, Knuutila S, Saharinen J, Wikman H and
Anttila S: Pathways affected by asbestos exposure in normal and
tumour tissue of lung cancer patients. BMC Med Genomics. 1:552008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wright CM, Larsen JE, Hayward NK, Martins
MU, Tan ME, Davidson MR, Savarimuthu SM, McLachlan RE, Passmore LH,
Windsor MN, et al: ADAM28: A potential oncogene involved in
asbestos-related lung adenocarcinomas. Genes Chromosomes Cancer.
49:688–698. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smyth GK: Limma: Linear models for
microarray data. Bioinformatics and Computational Biology Solutions
Using R and Bioconductor. Springer; pp. 397–420. 2005, http://link.springer.com/chapter/10.1007%2F0-387-29362-0_23#page-1.
View Article : Google Scholar
|
|
17
|
Chatr-Aryamontri A, Breitkreutz B-J,
Heinicke S, Boucher L, Winter A, Stark C, Nixon J, Ramage L, Kolas
N, O'Donnell L, et al: The BioGRID interaction database: 2013
update. Nucleic Acids Res. 41:D816–D823. 2013. View Article : Google Scholar :
|
|
18
|
Keshava Prasad TS, Goel R, Kandasamy K,
Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R,
Shafreen B, Venugopal A, et al: Human protein reference
database-2009 update. Nucleic Acids Res. 37:D767–D772. 2009.
View Article : Google Scholar
|
|
19
|
Saito R, Smoot ME, Ono K, Ruscheinski J,
Wang PL, Lotia S, Pico AR, Bader GD and Ideker T: A travel guide to
Cytoscape plugins. Nat Methods. 9:1069–1076. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Doncheva NT, Assenov Y, Domingues FS and
Albrecht M: Topological analysis and interactive visualization of
biological networks and protein structures. Nat Protoc. 7:670–685.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kanehisa M, Goto S, Sato Y, Kawashima M,
Furumichi M and Tanabe M: Data, information, knowledge and
principle: Back to metabolism in KEGG. Nucleic Acids Res.
42:D199–D205. 2014. View Article : Google Scholar :
|
|
22
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
23
|
de Hoon MJ, Imoto S, Nolan J and Miyano S:
Open source clustering software. Bioinformatics. 20:1453–1454.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saldanha AJ: Java Treeview - extensible
visualization of microarray data. Bioinformatics. 20:3246–3248.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
EL Andaloussi S, Mäger I, Breakefield XO
and Wood MJ: Extracellular vesicles: Biology and emerging
therapeutic opportunities. Nat Rev Drug Discov. 12:347–357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vinciguerra P and Stutz F: mRNA export: An
assembly line from genes to nuclear pores. Curr Opin Cell Biol.
16:285–292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schorey JS and Bhatnagar S: Exosome
function: From tumor immunology to pathogen biology. Traffic.
9:871–881. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Staals RH and Pruijn GJ: The human exosome
and disease. RNA Exosome. Springer; pp. 132–142. 2010, http://link.springer.com/chapter/10.1007%2F978-1-4419-7841-7_11#page-1.
View Article : Google Scholar
|
|
29
|
Azmi AS, Bao B and Sarkar FH: Exosomes in
cancer development, metastasis, and drug resistance: A
comprehensive review. Cancer Metastasis Rev. 32:623–642. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reymond N, Im JH, Garg R, Vega FM, Borda
d'Agua B, Riou P, Cox S, Valderrama F, Muschel RJ and Ridley AJ:
Cdc42 promotes transendothelial migration of cancer cells through
β1 integrin. J Cell Biol. 199:653–668. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee SH and Dominguez R: Regulation of
actin cytoskeleton dynamics in cells. Mol Cells. 29:311–325. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Boardman KC, Aryal AM, Miller WM and
Waters CM: Actin re-distribution in response to hydrogen peroxide
in airway epithelial cells. J Cell Physiol. 199:57–66. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J, Poovey HG, Rodriguez JF, Brody A and
Hoyle GW: Effect of platelet-derived growth factor on the
development and persistence of asbestos-induced fibroproliferative
lung disease. J Environ Pathol Toxicol Oncol. 23:253–266. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sabourin LA, Tamai K, Seale P, Wagner J
and Rudnicki MA: Caspase 3 cleavage of the Ste20-related kinase SLK
releases and activates an apoptosis-inducing kinase domain and an
actin-disassembling region. Mol Cell Biol. 20:684–696. 2000.
View Article : Google Scholar
|
|
35
|
Brody AR: Asbestos-induced lung disease.
Environ Health Perspect. 100:21–30. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shukla A, Lounsbury KM, Barrett TF, Gell
J, Rincon M, Butnor KJ, Taatjes DJ, Davis GS, Vacek P, Nakayama KI,
et al: Asbestos-induced peribronchiolar cell proliferation and
cytokine production are attenuated in lungs of protein kinase C-δ
knockout mice. Am J Pathol. 170:140–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shukla A, Barrett TF, Nakayama KI,
Nakayama K, Mossman BT and Lounsbury KM: Transcriptional
upregulation of MMP12 and MMP13 by asbestos occurs via a
PKCdelta-dependent pathway in murine lung. FASEB J. 20:997–999.
2006. View Article : Google Scholar : PubMed/NCBI
|