Introduction
Proprotein convertase subtilisin/kexin type 9
(PCSK9) is a liver-derived plasma protein that regulates plasma
low-density lipoprotein-cholesterol (LDL-C) levels by diverting the
cell surface LDL receptor (LDLR) of hepatocytes to lysosomes for
degradation (1,2). Thus, PCSK9 plasma levels directly
influence the level of circulating LDL-C (3,4).
Recent clinical studies have shown that a reduction in circulating
PCSK9 using neutralizing anti-PCSK9 antibodies can lower serum
LDL-C levels in dyslipidemic and hypercholesterolemic patients
(5), which provide strong
validation to support the notion that lowering circulating PCSK9
levels to upregulate hepatic LDLR is beneficial for reducing the
risk of cardiovascular disease in humans.
In liver tissue, PCSK9 synthesis is largely
controlled at the gene transcriptional level by two transcription
factor families, sterol regulatory element-binding proteins
(SREBPs) (6,7) and hepatocyte nuclear factor 1 (HNF1)
(8). PCSK9 gene expression is
positively regulated by SREBP through an SRE motif of the proximal
promoter in response to depletion of intracellular levels of
sterols (9,10). Statins, a class of cholesterol
lowering drugs, increase the PCSK9 expression level by activating
the SREBP pathway and thus diminish their beneficial effects
(11). Our laboratory has
previously identified a highly conserved HNF1 binding site on the
PCSK9 promoter region as another critical regulatory sequence motif
of PCSK9 transcription (8). DNA
binding studies conducted in HepG2 cells identified HNF1α as the
primary transactivator that binds to the HNF1 site of PCSK9
promoter. We have shown that siRNA-mediated knockdown of HNF1α
reduced PCSK9 protein levels in HepG2 cells. In vivo, an
increased binding of HNF1α to the PCSK9 promoter was detected in
the liver of rosuvastatin (RSV)-treated hamsters, which was thought
to contribute to the strong induction of PCSK9 gene expression by
RSV in that animal model (12).
HNF1α and HNF1β, the two members of the HNF1 family,
are homeobox proteins that form homodimers or heterodimers to bind
HNF1 sites on the promoter of their target genes (13,14). The importance of HNF1α in PCSK9
expression has been clearly demonstrated in cell culture studies
and in mice where adenovirus-mediated overexpression of HNF1α led
to increased PCSK9 and reduced liver LDLR protein (15). Relative to HNF1α, studies to
address the role of HNF1β in PCSK9 transcription are scarce. One
earlier study reported that while HNF1β is normally expressed at
low levels in adult mouse hepatocytes, its expression is induced in
HNF1α−/− livers (16).
In vivo DNA binding experiments indicated that loss of HNF1α
triggers higher HNF1β binding at several HNF1α target loci,
including the PCSK9 promoter region, implicating that HNF1β could
also modulate PCSK9 gene expression. Previously, utilizing mouse
models, our laboratory attempted to examine the functional roles of
HNF1α and HNF1β in PCSK9 expression in liver tissue by applying
adenovirus-mediated delivery of shRNAs targeted to the HNF1 family
members in mice. We found that in mice adenovirus-mediated
liver-specific knockdown of HNF1α markedly decreased serum PCSK9
and hepatic PCSK9 expression whereas depletion of HNF1β by the same
approach had no effects (17).
Thus, our studies provided direct evidence to support the notion
that in mouse species HNF1α but not HNF1β regulates liver PCSK9
expression.
Evidence exists that the expression of endogenous
PCSK9 and its response to pharmacological and nutritional factors
vary among different species including mice, hamsters and non-human
primates (18–20). Since RSV has been shown to
strongly induce PCSK9 expression in hamsters by activating the
SREBP pathway and by increasing HNF1α protein levels (12), in this study, we thus examined the
effects of transient knockdown of HNF1 isoforms HNF1α and HNF1β
individually in hamster livers on secreted PCSK9 and hepatic LDLR
expression under statin treatment conditions. Our results for the
first time demonstrated that in hamster species, both HNF1α and
HNF1β are functionally involved in hepatic PCSK9 transcription and
that transient, liver-specific knockdown of either HNF1α or HNF1β
could antagonize the statin-induced elevation of serum PCSK9 and
lower circulating cholesterol levels.
Materials and methods
Cloning of hamster HNF1α complete coding
sequence
When this project was started, sequences for the
golden Syrian hamster HNF1α gene were not available. By applying a
homology based primer selection approach we amplified the whole
coding sequence of HNF1α hamster orthologue from a cDNA pool
generated from male hamster liver samples and cloned the PCR
product into pCR2.1-Topo vector (Invitrogen Life Technologies,
Carlsbad, CA, USA). After sequencing the entire coding sequence, we
cloned the hamster HNF1α coding region into pcDNA4.0-HisMax-TOPO
vector (Invitrogen Life Technologies) and transfected the plasmid
into 293 cells (American Type Culture Collection, Manassas, VA,
USA) to validate the correct expression of hamster HNF1α protein.
Recently, the genomic sequence of a female golden Syrian hamster
(Mesocricetus auratus) became available on NCBI. The NCBI
reference sequence (HNF1α transcript X2, XM_005078961.1) predicted
the HNF1α nucleotide coding sequence and protein sequence that are
100% identical to our hamster HNF1α cDNA and protein sequences.
shRNA design and adenoviral vector
productions
Hamster hepatic HNF1α and HNF1β were transiently
knocked down using adenoviral vectors expressing shRNA targeting to
HNF1α or HNF1β. Briefly, a sequence spanning hamster HNF1α mRNA
coding sequence from 786 to 806 that is identical to human, mouse,
and rat sequences was selected as the target for RNAi. For HNF1β,
we cloned a partial coding region of hamster HNF1β and sequenced.
The sequence of the partial clone was identical to the
corresponding sequence of the NCBI-predicted HNF1β transcript X1.
Thus, a sequence spanning nucleotides 1464–1484 of the hamster
HNF1β mRNA transcript X1 was targeted for shRNA-mediated knockdown.
This sequence is identical among all isoforms of the hamster, human
and mouse HNF1β sequence. To construct Ad-shHNF1α/Ad-shHNF1β
adenoviral vectors, a U6 promoter-based shuttle vector (pSH-HNF1α
or pSH-HNF1β) that expresses HNF1α or HNF1β shRNA was generated
using BLOCK-iT™ U6 RNAi entry vector kit (Invitrogen Life
Technologies) following the manufacturer's instructions as
previously described (17).
Ad-shLacZ encoding an shRNA for the LacZ gene was used as control
in this study.
Animals and adenoviral injection
Male Syrian golden hamsters were purchased from
Harlan Laboratories, Inc. (Indianapolis, IN, USA) at 8–10 weeks of
age and weighed 100–110 g. Animals were housed 2 per cage in the VA
Palo Alto Health Care System (VAPAHCS) Veterinary Medical Unit
under a 12 h light and dark cycle (6:00 a.m.–6:00 p.m.). Hamsters
were fed a regular rodent chow diet and water ad libitum.
Hamsters had a 7-day acclimation period before the experiments. All
experimental analyses were conducted in accordance with animal use
protocols approved by the Institutional Animal Care and Use
Committee at the VAPAHCS. Hamster body weight and food intake were
recorded every two days throughout the duration of the experiment.
Animals were administered with 5×1011 viral particles
under isoflurane anesthesia via the orbital venous plexus. Three
days post injection, animals were orally administered with RSV at a
daily dose of 15 mg/kg for seven days. The control group
(Ad-shLacZ) received an equal volume of the vehicle (sterilized
water).
Blood samples were collected by administering
isoflurane and bleeding from the orbital venous plexus in lithium
heparinized capillary tubes before the study and at the end of the
study. At the termination, animals were dosed with RSV and the food
was removed. Blood and liver were harvested after a 4-h fast.
Livers were collected, weighed, frozen in liquid nitrogen, and
stored at −80°C until processing. RSV was purchased from AK
Scientific, Inc. (Union City, CA, USA). Serum levels of total
cholesterol and HDL cholesterol were measured using respective kits
from Stanbio Laboratory, (Boerne, TX, USA) according to the
manufacturer's instructions. Non-HDL-C was calculated as total
cholesterol minus HDL-C. Serum ALT activity was measured using the
ALT/SGPT Liqui-UV kit (Stanbio Laboratory) following the
manufacturer's instructions.
Detection of hamster PCSK9 in serum
Levels of hamster serum PCSK9 were measured using
the mouse PCSK9 quantikine ELISA kit (R&D Systems, Inc.,
Minneapolis, MN, USA) (21).
Briefly, serum samples were diluted 1:10 in calibrator diluent and
allowed to bind for 2 h onto microplate wells that were precoated
with the capture antibody. Samples were then sequentially incubated
with PCSK9 conjugate followed by the PCSK9 substrate solution with
extensive intermittent washes between each step. The amount of
PCSK9 in serum was estimated colorimetrically using a standard
microplate reader (MDS Analytical Technologies, Silicon Valley, CA,
USA). Additionally, PCSK9 in hamster serum samples was detected by
immunoprecipitation, followed by western blot analysis using
anti-hamster PCSK9 antibody as we described previously (21,22).
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from 20 mg of individual
flash frozen mouse liver tissue samples using the RNeasy PLUS Mini
kit (Qiagen, Inc., Valencia, CA, USA). After RNA integrity was
confirmed by gel electrophoresis, 1.5 µg of total RNA was
reverse transcribed by random priming using the High-Capacity cDNA
reverse transcription kit (Invitrogen Life Technologies) according
to the manufacturer's guidelines. Quantitative PCR (qPCR) was then
performed in an ABI 7900HT sequence detection system using
SYBR-Green PCR Master Mix (Invitrogen Life Technologies) and PCR
primers specific for each gene being amplified (Table I). With duplicate measurements
from each cDNA sample, the data were analyzed using the ΔΔCT method
and the relative expression of target mRNA level was normalized to
that of GAPDH in each sample.
 | Table IList of hamster primers for
RT-qPCR. |
Table I
List of hamster primers for
RT-qPCR.
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|
| GAPDH |
AACTTTGGCATTGTGGAAGG |
GGATGCAGGGATGATGTTCT |
| HMGCR |
GACGGTGACACTTACCATCTGT |
GATGCACCGTGTTATGGTGA |
| HNF1α |
GAGGTGGCTCAGCAATTCAC |
CACTCCTCCACCAAGGTCTC |
| HNF1β |
CAGCCAGTCGGTTTTACAGC |
ATTCGTCAAGGTGCTGACGG |
| LDLR |
TTGGGTTGATTCCAAACTCC |
GATTGGCACTGAAAATGGCT |
| PCSK9 |
TGCTCCAGAGGTCATCACAG |
GTCCCACTCTGTGACATGAAG |
| SREBP1 |
GCACTTTTTGACACGTTTCTTC |
CTGTACAGGCTCTCCTGTGG |
Western blot analysis
Total protein was extracted from flash-frozen mouse
liver samples by homogenizing 50 mg of tissue in RIPA buffer
supplemented with 1 mM PMSF and protease inhibitor cocktail (Roche,
Mannheim, Germany). Protein content was quantified using BCA
protein assay reagent (Pierce) and 50 µg of protein from
individual samples was resolved by SDS-PAGE. Following transfer
onto nitrocellulose membranes, LDLR, PCSK9, HNF1α and β-actin
proteins were detected by immunoblotting using a rabbit anti-LDLR
(BioVision, Inc., Milpitas, CA, USA), anti-PCSK9 (8), goat anti-HNF1α (sc-6547; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and monoclonal anti-β-actin
(Clone AC-15; Sigma-Aldrich, St. Louis, MO, USA) antibodies.
Immunoreactive bands of predicted molecular mass were visualized
using SuperSignal West Femto chemiluminescent substrate (Pierce
Chemical Co., Rockford, IL, USA) and FluorChem E western blot
analysis imaging system (Protein Simple, San Jose, CA, USA).
Background subtracted band densities were quantified using Alpha
View SA imaging software (Protein Simple). Signal intensities for
β-actin were used to normalize differences in protein loading among
individual samples.
Statistical analysis
All values are expressed as mean ± SEM. One way
ANOVA with Dunnett's post-test was performed using Prism 5 software
(GraphPad Software Inc., La Jolla, CA, USA). Statistical
significance is displayed as P<0.05, P<0.01 or
P<0.001.
Results
Cloning of hamster HNF1α coding region
and adenovirus-mediated gene knockdown
To assess the role of HNF1α in hepatic PCSK9
transcription in hamster species, we first cloned the entire coding
region of hamster HNF1α orthologue from cDNAs generated from
hamster liver tissue. Hamster HNF1α protein sequence is highly
homologous to human, mouse and rat species with amino acid sequence
identity in the range of 94–97%. Based upon the cDNA sequence, we
constructed several plasmids that express shRNAs targeting
different coding regions of hamster HNF1α. One of the shRNAs
targets hamster HNF1α mRNA coding sequence from 786–806 that is
identical to human, mouse and rat sequences (Fig. 1A).
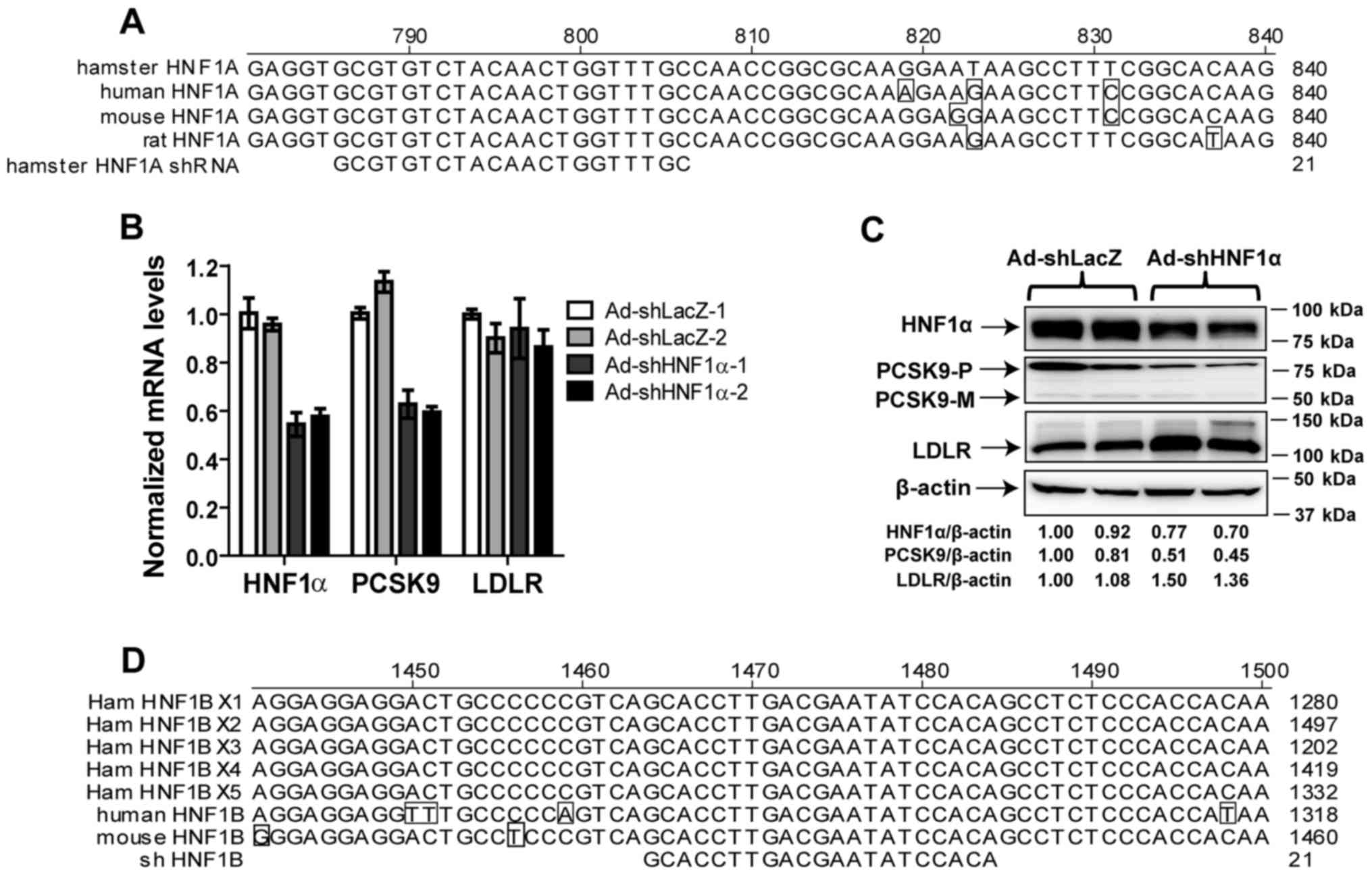 | Figure 1shRNA sequence alignment and
validation of shHNF1α-targeted knockdown in hamsters. (A) The
sequence of HNF1α shRNA is aligned with its target sequences in the
HNF1α-coding sequences of hamster, human, mouse and rat species.
Nucleotides of human, mouse and rat HNF1α that differ from those of
the hamster sequence are boxed. (B) Two male Syrian golden hamsters
were injected with Ad-shLacZ or Ad-shHNF1α (5×1011 virus
particles per hamster) adenoviral particles. Ten days after the
viral injection, all animals were euthanized and liver total RNA
was isolated. Individual levels of HNF1α mRNA, LDLR mRNA and PCSK9
mRNA were assessed by quantitative real-time PCR using
hamster-specific PCR primers as described in the Materials and
methods section. After normalization with GAPDH mRNA levels, the
relative expression levels of mRNAs of interest are presented. The
results are expressed as the means ± SEM of triplicate measurements
of each cDNA sample of individual hamsters. (C) Individual liver
protein extracts were prepared and protein concentrations were
determined. A total of 50 µg of homogenate proteins from
individual liver samples was analyzed by western blot analysis and
the expression levels of LDLR, PCSK9 and HNF1α were quantified by
Alpha View software with normalization to signals of β-actin. (D)
The sequence of HNF1β shRNA is aligned with its target sequences in
the HNF1β-coding sequences of 5 predicted isoforms of hamster,
human and mouse HNF1β. Nucleotides of human and mouse HNF1β that
differ from those of the hamster sequence are boxed. HNF1,
hepatocyte nuclear factor 1; PCSK9, proprotein convertase
subtilisin/kexin type 9. |
First, to demonstrate an effective knockdown of
HNF1α by this shRNA in hamsters without RSV treatment, we infected
two hamsters with Ad-shHNF1α and another two hamsters with control
virus Ad-shLacZ for 10 days. Hepatic gene expression analysis by
qPCR demonstrated that the mRNA levels of HNF1α and PCSK9 were both
reduced by ~40–50% in hamsters injected with Ad-shHNF1α as compared
to hamsters injected with Ad-shLacZ. By contrast, the mRNA
expression of LDLR was not affected by HNF1α knockdown (Fig. 1B). Reductions of HNF1α and PCSK9
mRNA expression by the shRNA expression were further corroborated
by western blot analysis that showed reduced liver HNF1α and PCSK9
protein levels and increased LDLR protein abundance in both
hamsters infected with Ad-shHNF1α (Fig. 1C). Taken together, these results
validated the success of this shRNA in knockdown of HNF1α
expression and demonstrated its impact on PCSK9 expression in
hamsters without statin treatment.
To examine the potential role of HNF1β in PCSK9
transcription in hamster species, from a hamster liver cDNA pool,
we cloned a partial coding region of hamster HNF1β and sequenced.
The sequence of the partial clone was identical to the
corresponding sequence of the NCBI-predicted HNF1β transcript X1. A
sequence spanning nucleotides 1464–1484 of the hamster HNF1β
transcript X1 was targeted for shRNA-mediated knockdown. Of note,
this shRNA is also capable of targeting other predicted hamster
HNF1β transcripts as well as mouse and human transcripts (Fig. 1D). The specificity of this HNF1β
shRNA has been validated in cultured human and mouse hepatic cells
and in mouse liver tissue (17).
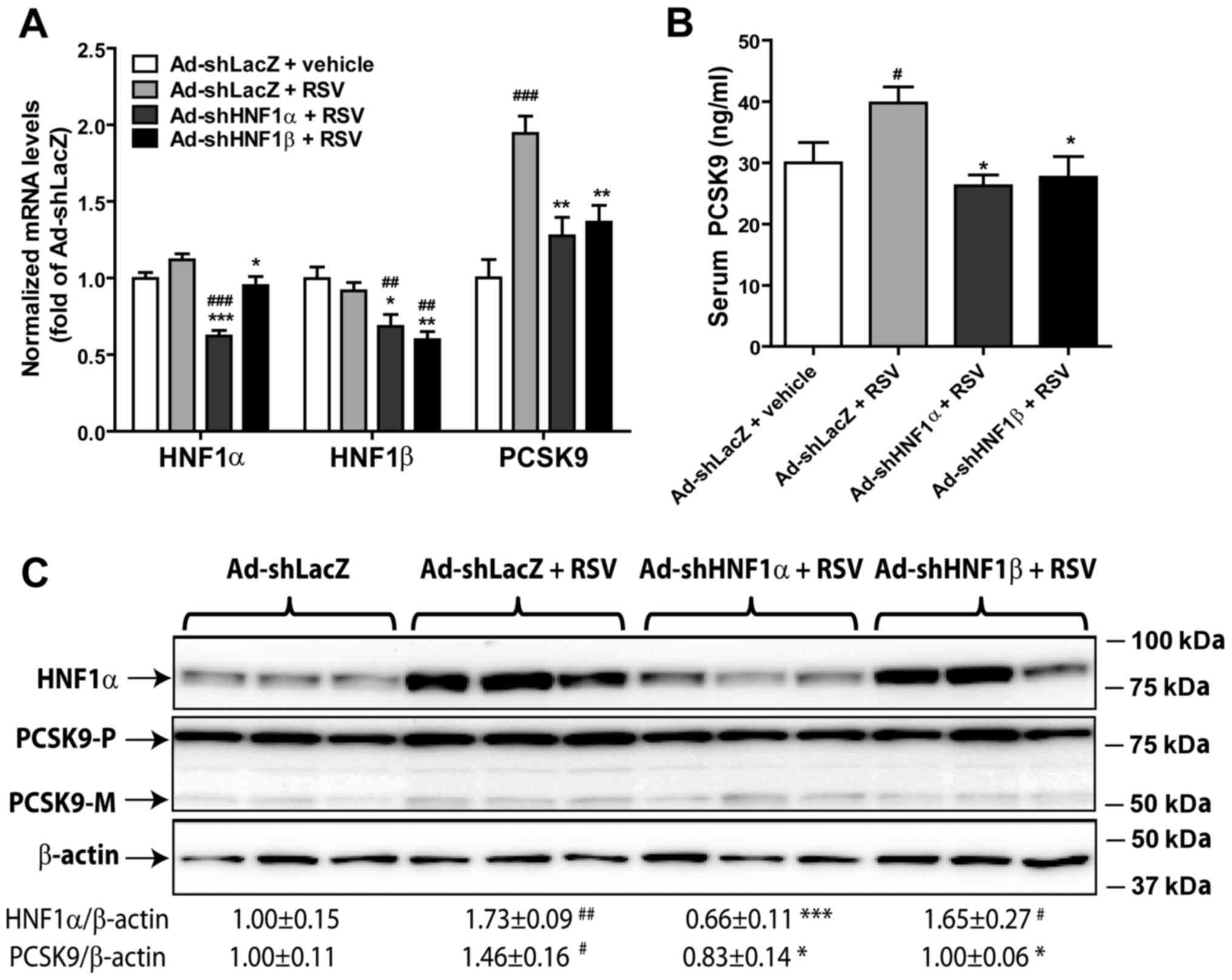
Knockdown of HNF1α or HNF1β in hamsters
abolishes the RSV-induced elevation of serum PCSK9 levels
Since our previous study showed that RSV treatment
significantly increased circulating PCSK9 levels in dyslipidemic
hamsters (12), we wanted to know
whether knockdown of HNF1α or HNF1β in hamsters could antagonize
the RSV-induced elevation of serum PCSK9 levels. Hamsters were
injected with Ad-shHNF1α, Ad-shHNF1β or a control virus
(Ad-shLacZ). Three days after viral infection, the animals were
orally administered RSV (15 mg/kg/day) or vehicle for 7 days. The
results of RT-qPCR confirmed the successful knockdown of both HNF1α
and HNF1β by the hepatic expression of their specific shRNAs
(Fig. 2A). The mRNA level of
HNF1α was not changed by RSV treatment, but it was reduced to 60%
of the control by injection of Ad-shHNF1α into hamsters. Likewise,
the expression levels of HNF1β mRNA were unaffected by the RSV
treatment but they were reduced to 60% of the control by Ad-shHNF1β
injection and to 70% of the control by Ad-shHNF1α injection. The
inhibition of HNF1β mRNA expression by Ad-shHNF1α was also observed
in our previous study in mice (17). RSV treatment significantly
increased the PCSK9 mRNA level by 94% (P<0.01) in the control
hamsters but not in hamsters injected with Ad-shHNF1α or Ad-shHNF1β
(Fig. 2A). Furthermore, RSV
treatment elevated serum PCSK9 levels by ~32.5% (P<0.05) in the
wild-type hamsters (Ad-shLacZ). Expression of HNF1α shRNA or HNF1β
shRNA in the liver of hamsters both prevented the RSV-induced
elevation of PCSK9 serum levels (Fig.
2B).
Examination of HNF1α protein content by western blot
analysis showed that the liver HNF1α protein level was
significantly induced by RSV treatment (Fig. 2C), that confirmed our previous
finding made in dyslipidemic hamsters (12). Injection of Ad-shHNF1α, but not
Ad-shHNF1β, significantly reduced liver HNF1α protein levels,
further demonstrating the specificity of the shRNA. Because an
antibody against hamster HNF1β is not available, we were unable to
measure liver HNF1β protein levels in this study. Importantly,
western blot analysis showed that RSV treatment increased the
hepatic PCSK9 protein amount. This effect was again abolished by
adenovirus-directed expressions of HNF1 shRNAs (Fig. 2C).
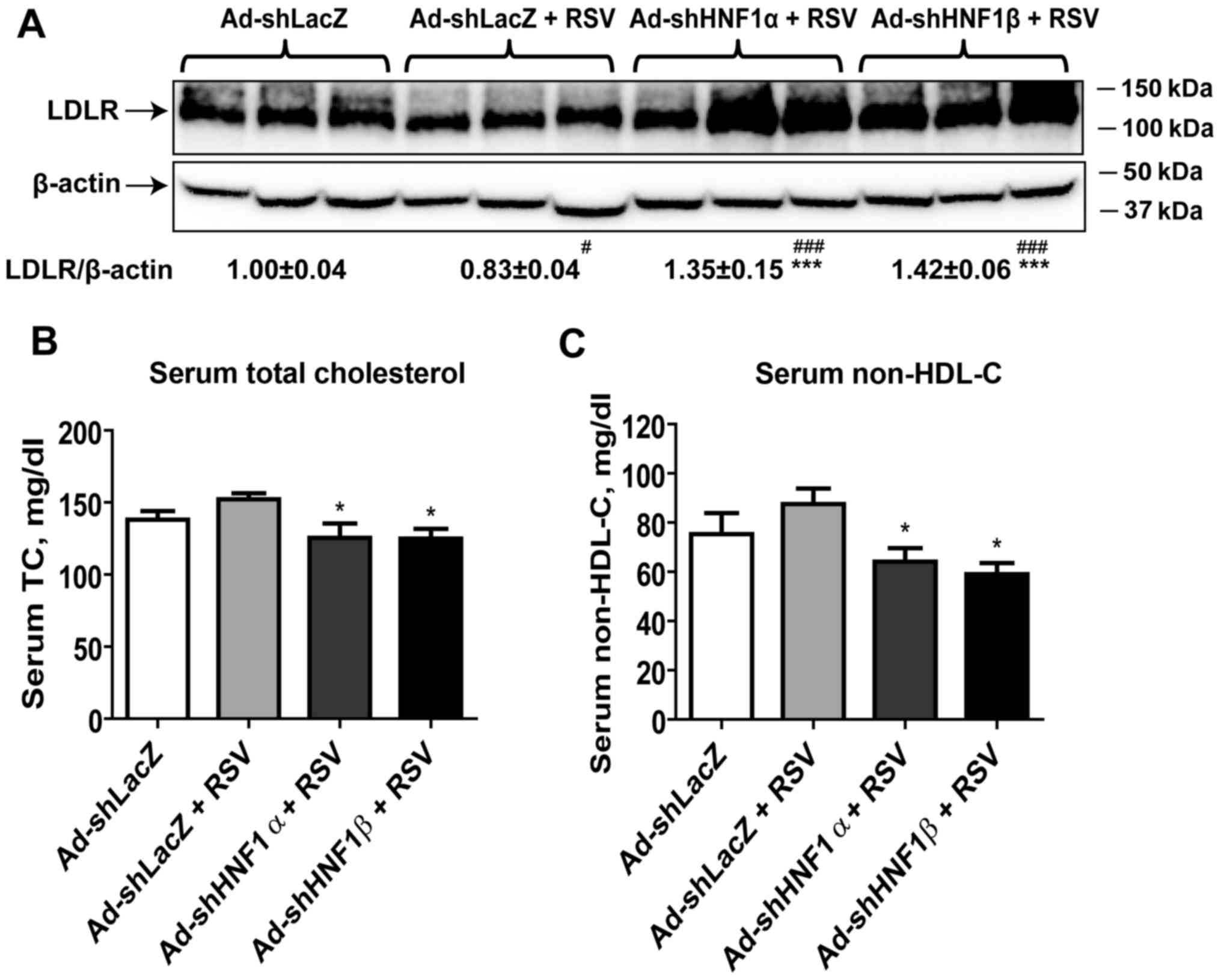
Depletion of HNF1 isoforms increases
liver LDLR protein content and reduces serum cholesterol
levels
Our previous study on dyslipidemic hamsters detected
a reduced hepatic LDLR content in RSV-treated hamsters (12). This finding was observed again in
the normolipidemic hamsters treated with RSV in the wild-type
animals (Ad-shLacZ) (Fig. 3A).
However, knockdown of HNF1α as well as HNF1β both prevented the
negative effect of RSV and further increased LDLR protein levels to
135% (P<0.001) and 142% of the control (P<0.001),
respectively.
In these normolipidemic hamsters, RSV treatment
showed a trend in the elevation of serum TC and non-HDL cholesterol
levels in the control hamsters (Fig.
3B and 3C). However,
injection of Ad-shHNF1α or Ad-shHNF1β into hamsters under RSV
treatment reduced serum cholesterol levels. Serum TC and non-high
density lipoprotein cholesterol (HDL-C) levels were 18% (P<0.05)
and 27% (P<0.05) lower in the Ad-shHNF1α + RSV group as compared
with the Ad-shLacZ + RSV group, respectively. Likewise, serum TC
was reduced by 18% (P<0.05) and non-HDL-C was lowered by 32%
(P<0.05) in the Ad-shHNF1β + RSV group compared to the control.
Liver cholesterol levels were similar among all groups.
Furthermore, serum levels of liver transaminase ALT were not
elevated by adenoviral injection or RSV treatment (data not shown),
suggesting that the normal function of the liver was not disrupted
in these animals.
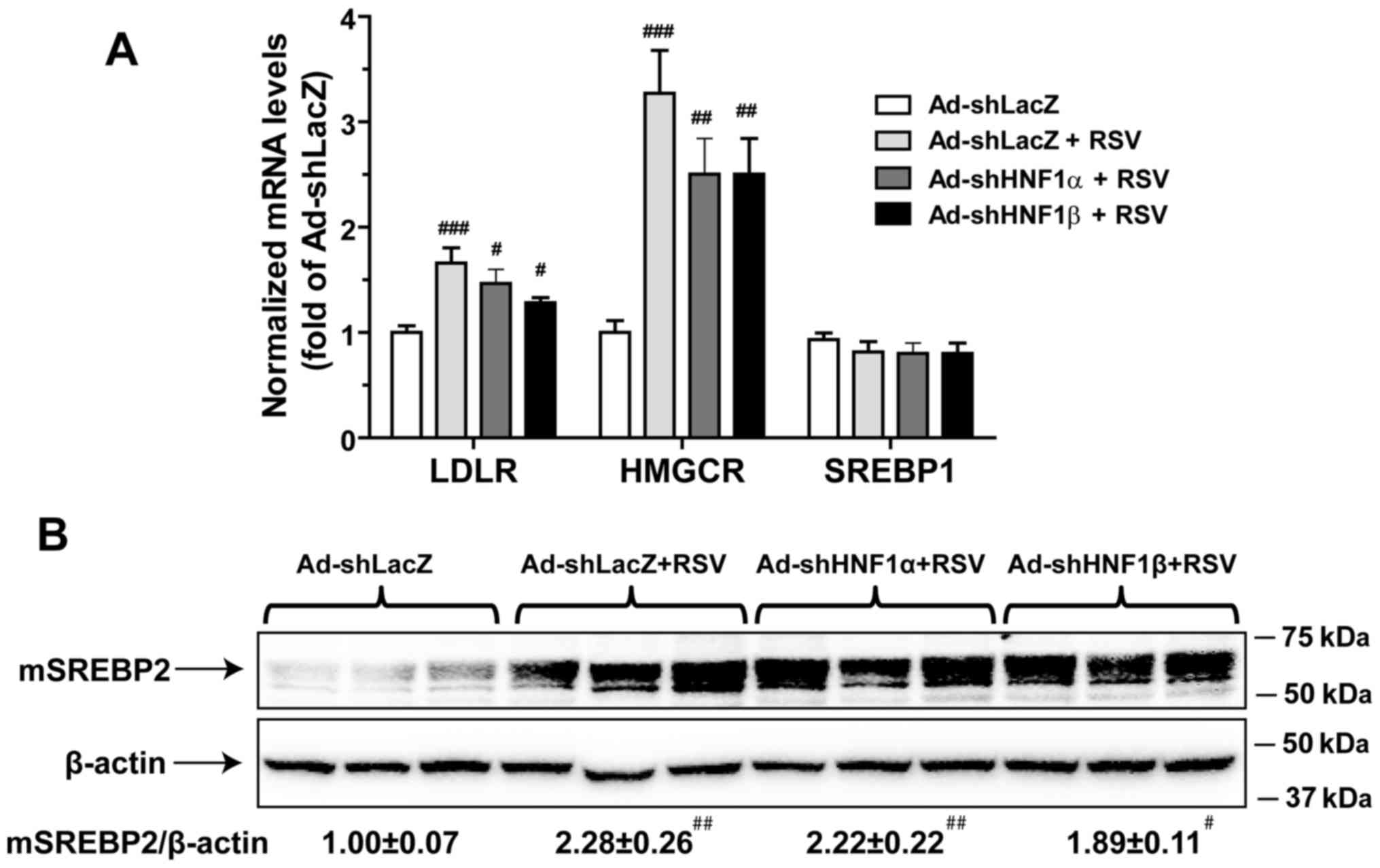
Hepatic depletion of HNF1 isoforms does
not affect the SREBP pathway
The transcription of PCSK9 is controlled by both
SRE-1 and HNF1 sites on its promoter region. To examine the
involvement of SRE-1, we measured mRNA levels of LDLR and HMG-CoA
reductase (HMGCR) in all groups. Fig.
4A showed that RSV treatment increased the mRNA levels of LDLR
and HMGCR. However, in contrast to PCSK9, injection of Ad-shHNF1α
or Ad-shHNF1β did not inhibit RSV-induced mRNA expression of these
SRE target genes. We further examined the liver contents of mature
SREBR2 in different liver samples by western blot analysis and
demonstrated that the induction of mSREBP2 by RSV treatment was
unaffected by the knockdown of either HNF1α or HNF1β (Fig. 4B). This data excluded the
involvement of the SREBP2 pathway in the reduction of PCSK9
transcription in liver tissues with specific knockdown of HNF1α and
HNF1β.
Discussion
PCSK9 has become an important new therapeutic target
for the management of hyperlipidemia and reduction in
cardiovascular disease. With the rapid development of anti-PCSK9
antibody as new therapy (23–25), many patients with high circulating
LDL-C levels could be treated with PCSK9 inhibitors to drastically
reduce serum PCSK9 levels to protect liver LDLR from degradation.
On the other hand, our current understanding of the physiological
regulation of PCSK9 expression at the transcriptional level as well
as the translational level remains incomplete.
In the present study, we applied the approach of
adenovirus-mediated gene knockdown to investigate the individual
roles of HNF1α and HNF1β in circulating PCSK9 levels in the
hamster, a species that shares more characteristics in lipid
metabolism with humans than any other rodents including mice and
rats (26–30). This study led to several important
new findings.
First, we demonstrated that liver-specific depletion
of HNF1α in hamsters without statin treatment reduced PCSK9 mRNA
levels by 40% and increased hepatic LDLR levels substantially.
These results are highly consistent with the effect of HNF1α
knocking down in normolipidemic mice, and further underscore the
physiological role of HNF1α as a transactivator for PCSK9 gene
expression in liver tissue of different animal models.
Secondly, our previous study in dyslipidemic
hamsters showed that in addition to an increase in SREBP2
expression, RSV treatment increased the liver abundance of HNF1α
protein (12). Our study
suggested that the inducing effect of RSV on HNF1α is likely an
underlying mechanism accounting for the higher induction of PCSK9
than LDLR due to the utilization of two transactivators (HNF1α and
SREBP2) in PCSK9 transcription versus one (SREBP2) in LDLR
transcription. From this perspective, HNF1α shRNA could act as an
antagonist to directly inhibit RSV-induced PCSK9 expression. Thus,
in this investigation, we focused on the examination of the effects
of hepatic depletion of HNF1α in hamsters under RSV treatment.
Indeed, injection of Ad-shHNF1α into hamsters totally abolished
RSV-induced elevation in serum PCSK9 and hepatic PCSK9 mRNA and
protein levels. This led to a marked increase in liver LDLR and
significant reductions in serum cholesterol levels. Interestingly,
we again observed the elevated HNF1α protein levels in the liver of
RSV-treated hamsters. The precise mechanism by which RSV affects
HNF1α protein abundance is presently unclear.
The third important outcome of our study is the
demonstration of the nonexpendable role of HNF1β in PCSK9
transcription in hamster liver tissue. We demonstrated that
injection of Ad-shHNF1β into hamsters substantially lowered serum
PCSK9 and hepatic PCSK9 to levels nearly identical to the effects
generated by depleting HNF1α. The fact that expression levels of
HNF1α protein and mRNA were not reduced in hamster livers with
HNF1β knockdown excluded the possibility of the cross-reactivity of
HNF1β shRNA. The strong inhibitory effects of HNF1β shRNA on PCSK9
expression in the hamsters were strikingly different from the
results following the knockdown of HNF1β in mouse liver. In our
previous study conducted in mice, we did not observe any inhibition
of PCSK9 expression levels in both liver and serum by
Ad-shHNF1β-directed expression of HNF1β shRNA. The lack of effect
on PCSK9 expression was not due to insufficient depletion of HNF1β
because we showed that the HNF1β mRNA abundance and the mRNA
expression of three HNF1β target genes (ApoC3, HNF4α and FABP5)
were all greatly reduced in the liver of the Ad-shHNF1β-infected
mice.
The different roles of HNF1β in PCSK9 transcription
between the hamster and mouse could be caused by variations in
species-specific expression of mature transcripts. By quantitative
PCR measurement, it was reported that in the mouse adult liver,
HNF1α is a dominant isoform with mRNA levels more than 7-fold the
levels of HNF1β (31). We
compared the relative mRNA levels of HNF1α and HNF1β in hamster
liver and showed that HNF1β mRNA was expressed at a level of 31% of
that of HNF1α mRNA, which is substantially higher than the HNF1β
mRNA amount detected in mouse liver. We attempted to detect HNF1β
protein levels in liver tissue using several commercial anti-HNF1β
antibodies. However, none of these produced specific signals on
western blot analyses. Thus, it is unclear whether RSV treatment
also increased HNF1β protein levels. Another possible explanation
for the effectiveness of HNF1β shRNA on RSV-induced PCSK9
expression is that in hamster liver, HNF1α and HNF1β might bind to
the HNF1 site of the PCSK9 promoter as an obligated heterodimer.
Thus, reducing either one would attenuate the HNF1-mediated
transactivation of the PCSK9 gene.
In conclusion, we demonstrated that liver-specific
knockdown of HNF1α or HNF1β in hamsters produced antagonistic
effects on the RSV-induced elevation of serum PCSK9 and cholesterol
levels. Thus, both HNF1α and HNF1β play key roles in the control of
PCSK9 gene transcription in liver tissue of the hamster species.
Combined with the difference observed in Ad-shHNF1β-mediated
knockdown between hamsters and mice and the strong induction of
HNF1α protein by RSV treatment in hamster liver tissue, our study
results further support the notion that the regulatory mechanism
governing PCSK9 expression is species-specific, which could be a
contributing factor for different efficacies of statins and other
lipid-modulating drugs observed in various animal studies. The
question of whether statins regulate hepatic HNF1α in other species
including humans has clinical relevance and requires further
investigation.
Acknowledgments
The present study was supported by the Department of
Veterans Affairs (Office of Research and Development, Medical
Research Service) and by grants (no. 1R01AT006336-01A1) from the
National Center of Complementary and Alternative Medicine.
References
|
1
|
Zhang DW, Lagace TA, Garuti R, Zhao Z,
McDonald M, Horton JD, Cohen JC and Hobbs HH: Binding of proprotein
convertase subtilisin/kexin type 9 to epidermal growth factor-like
repeat A of low density lipoprotein receptor decreases receptor
recycling and increases degradation. J Biol Chem. 282:18602–18612.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lagace TA, Curtis DE, Garuti R, McNutt MC,
Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE and Horton JD:
Secreted PCSK9 decreases the number of LDL receptors in hepatocytes
and in livers of parabiotic mice. J Clin Invest. 116:2995–3005.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lambert G, Ancellin N, Charlton F, Comas
D, Pilot J, Keech A, Patel S, Sullivan DR, Cohn JS, Rye KA and
Barter PJ: Plasma PCSK9 concentrations correlate with LDL and total
cholesterol in diabetic patients and are decreased by fenofibrate
treatment. Clin Chem. 54:1038–1045. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lakoski SG, Lagace TA, Cohen JC, Horton JD
and Hobbs HH: Genetic and metabolic determinants of plasma PCSK9
levels. J Clin Endocrinol Metab. 94:2537–2543. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ling H, Burns TL and Hilleman DE: An
update on the clinical development of proprotein convertase
subtilisin kexin 9 inhibitors, novel therapeutic agents for
lowering low-density lipoprotein cholesterol. Cardiovasc Ther.
32:82–88. 2014. View Article : Google Scholar
|
|
6
|
Horton JD, Shah NA, Warrington JA,
Anderson NN, Park SW, Brown MS and Goldstein JL: Combined analysis
of oligonucleotide microarray data from transgenic and knockout
mice identifies direct SREBP target genes. Proc Natl Acad Sci USA.
100:12027–12032. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maxwell KN, Soccio RE, Duncan EM, Sehayek
E and Breslow JL: Novel putative SREBP and LXR target genes
identified by microarray analysis in liver of cholesterol-fed mice.
J ipid Res. 44:2109–2119. 2003.
|
|
8
|
Li H, Dong B, Park SW, Lee HS, Chen W and
Liu J: Hepatocyte nuclear factor 1alpha plays a critical role in
PCSK9 gene transcription and regulation by the natural
hypocholesterolemic compound berberine. J Biol Chem.
284:28885–28895. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dubuc G, Chamberland A, Wassef H, Davignon
J, Seidah NG, Bernier L and Prat A: Statins upregulate PCSK9, the
gene encoding the proprotein convertase neural apoptosis-regulated
convertase-1 implicated in familial hypercholesterolemia.
Arterioscler Thromb Vasc Biol. 24:1454–1459. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D
and Park SW: Sterol-dependent regulation of proprotein convertase
subtilisin/kexin type 9 expression by sterol-regulatory element
binding protein-2. J Lipid Res. 49:399–409. 2008. View Article : Google Scholar
|
|
11
|
Raal F, Panz V, Immelman A and Pilcher G:
Elevated PCSK9 levels in untreated patients with heterozygous or
homozygous familial hypercholesterolemia and the response to
high-dose statin therapy. J Am Heart Assoc. 2:e0000282013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dong B, Wu M, Li H, Kraemer FB, Adeli K,
Seidah NG, Park SW and Liu J: Strong induction of PCSK9 gene
expression through HNF1alpha and SREBP2: Mechanism for the
resistance to LDL-cholesterol lowering effect of statins in
dyslipidemic hamsters. J Lipid Res. 51:1486–1495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Costa RH, Kalinichenko VV, Holterman AX
and Wang X: Transcription factors in liver development,
differentiation, and regeneration. Hepatology. 38:1331–1347. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mendel DB, Hansen LP, Graves MK, Conley PB
and Crabtree R: HNF-1α and HNF-1β (vHNF-1) share dimerization and
homeo domains, but not activation domains, and form heterodimers in
vitro. Genes Dev. 5:1042–1056. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ai D, Chen C, Han S, Ganda A, Murphy AJ,
Haeusler R, Thorp E, Accili D, Horton JD and Tall AR: Regulation of
hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest.
122:1262–1270. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Servitja J-M, Pignatelli M, Maestro MA,
Cardalda C, Boj SF, Lozano J, Blanco E, Lafuente A, McCarthy MI,
Sumoy L, et al: Hnf1α (MODY3) controls tissue-specific
transcriptional programs and exerts opposed effects on cell growth
in pancreatic islets and liver. Mol Cell Biol. 29:2945–2959. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shende VR, Wu M, Singh AB, Dong B, Kan CF
and Liu J: Reduction of circulating PCSK9 and LDL-C levels by
liver-specific knockdown of HNF1α in normolipidemic mice. J Lipid
Res. 56:801–809. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong B, Singh AB, Azhar S, Seidah NG and
Liu J: High-fructose feeding promotes accelerated degradation of
hepatic LDL receptor and hypercholesterolemia in hamsters via
elevated circulating PCSK9 levels. Atherosclerosis. 239:364–374.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miao J, Manthena PV, Haas ME, Ling AV,
Shin DJ, Graham MJ, Crooke RM, Liu J and Biddinger SB: Role of
insulin in the regulation of proprotein convertase subtilisin/kexin
type 9. Arterioscler Thromb Vasc Biol. 35:1589–1596. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hentze H, Jensen KK, Chia SM, Johns DG,
Shaw RJ, Davis HR Jr, Shih SJ and Wong KK: Inverse relationship
between LDL cholesterol and PCSK9 plasma levels in dyslipidemic
cynomolgus monkeys: Effects of LDL lowering by ezetimibe in the
absence of statins. Atherosclerosis. 231:84–90. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dong B, Li H, Singh AB, Cao A and Liu J:
Inhibition of PCSK9 transcription by berberine involves
down-regulation of hepatic HNF1α protein expression through the
ubiquitin-proteasome degradation pathway. J Biol Chem.
290:4047–4058. 2015. View Article : Google Scholar
|
|
22
|
Cao A, Wu M, Li H and Liu J: Janus kinase
activation by cytokine oncostatin M decreases PCSK9 expression in
liver cells. J Lipid Res. 52:518–530. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu M, Dong B, Cao A, Li H and Liu J:
Delineation of molecular pathways that regulate hepatic PCSK9 and
LDL receptor expression during fasting in normolipidemic hamsters.
Atherosclerosis. 224:401–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Verbeek R, Stoekenbroek RM and Hovingh GK;
CSK9 inhibitors: Novel therapeutic agents for the treatment of
hypercholesterolemia. Eur J Pharmacol. 763:38–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huynh K: Dyslipidaemia. Assessing the
efficacy and safety of evolocumab and alirocumab. Nat Rev Cardiol.
12:2612015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Desai NR and Sabatine MS: PCSK9 inhibition
in patients with hypercholesterolemia. Trends Cardiovasc Med.
25:567–574. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yin W, Carballo-Jane E, McLaren DG,
Mendoza VH, Gagen K, Geoghagen NS, McNamara LA, Gorski JN, Eiermann
GJ, Petrov A, et al: Plasma lipid profiling across species for the
identification of optimal animal models of human dyslipidemia. J
Lipid Res. 53:51–65. 2012. View Article : Google Scholar :
|
|
28
|
Dietschy JM, Turley SD and Spady DK: Role
of liver in the maintenance of cholesterol and low density
lipoprotein homeostasis in different animal species, including
humans. J Lipid Res. 34:1637–1659. 1993.PubMed/NCBI
|
|
29
|
Quig DW, Arbeeny CM and Zilversmit DB:
Effects of hyperlipidemias in hamsters on lipid transfer protein
activity and unidirectional cholesteryl ester transfer in plasma.
Biochim Biophys Acta. 1083:257–264. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Taghibiglou C, Carpentier A, Van Iderstine
SC, Chen B, Rudy D, Aiton A, Lewis GF and Adeli K: Mechanisms of
hepatic very low density lipoprotein overproduction in insulin
resistance. Evidence for enhanced lipoprotein assembly, reduced
intracellular ApoB degradation, and increased microsomal
triglyceride transfer protein in a fructose-fed hamster model. J
Biol Chem. 275:8416–8425. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harries LW, Brown JE and Gloyn AL:
Species-specific differences in the expression of the HNF1A, HNF1B
and HNF4A genes. PLoS One. 4:e78552009. View Article : Google Scholar : PubMed/NCBI
|