Introduction
Atherosclerosis (AS) is the predominant pathological
basis of cardiovascular disease, and its prevalence worldwide has
reached epidemic proportions. Current evidence suggests that the
subendothelial retention of apoB100 containing lipoproteins (e.g.,
low-density lipoprotein, LDL) is the initial step of atherogenesis,
which is usually termed the 'response to retention' hypothesis in
the literature (1–4). Since the diameter of LDL particles
(20–30 nm) is much larger than the gap junctions (3–6 nm) between
vascular endothelial cells (ECs), the only pathway for LDL
particles to traffic across the intact endothelial barrier is
through a transporting process termed 'transcytosis', which was
postulated to describe the transport of macromolecular cargo from
one side of polar cells to the other within membrane-bound carriers
(5,6). Notably, many studies have pointed to
a possible link between LDL transcytosis across ECs and the
initiation of AS (2,6,7).
There is increasing evidence of the tight
interactions between AS development and renin-angiotensin system
(RAS) activation. A meta-analysis reported that Ang-converting
enzyme (ACE) inhibitors or Ang receptor blockers (ARBs) are
beneficial in normotensive (systolic pressure <130 mmHg)
atherosclerotic patients (8). As
we know, ACE plays a key role in RAS as it converts angiotensin I
(Ang I) to Ang II. Furthermore, ACE is abundant in vulnerable
lesions and is expressed in macrophage foam cells, the ECs of
neovessels, as well as smooth muscle cells (SMCs) (9,10).
Moreover, as the principal effector molecule of RAS,
Ang II is implicated in several important steps of AS, such as
vascular inflammation, vascular remodeling, thrombosis, and plaque
rupture. Ang II has been shown to accelerate AS by activating
various signaling pathways and augmenting oxidative stress
(11–14). In injured arteries, Ang II
provides a positive feedback loop for vascular inflammation by
recruiting inflammatory cells, which then generate more Ang II,
therefore perpetuating vascular inflammation (15). It has been suggested that Ang II
upregulates the levels of proinflammatory cytokines [interleukin
(IL)-6, monocyte chemoattractant protein-1 (MCP-1), vascular cell
adhesion molecule-1 (VCAM-1)] via type 1 Ang II receptor
(AT1R) and activates the nuclear factor-κB (NF-κB)
signaling pathway, which would deteriorate the atherosclerotic
inflammation response (16–19).
Ang II-induced reactive oxygen species
(ROS)-mediated DNA damage may also aggravate the progression of AS.
Indeed, evidence indicates that Ang II is able to induce the
production of superoxide anions and activate the NADH/NADPH
signaling pathway (20).
Meanwhile, data have revealed that AT1AR
deficiency-induced reduction of oxidative stress, apoptosis, and
matrix metalloproteinase expression in atherosclerotic lesions may
decrease plaque size (21).
Interestingly, Daugherty et al found that AT1AR
deficiency-induced reduction in AS was independent of systolic
blood pressure, oxidation and chemoattractants. They confirmed that
hypercholesterolemia-induced Ang peptide production provides a
basis for AT1AR deficiency-induced reductions in AS
(22).
In addition, AS is considered to be associated with
premature biological aging. Kunieda et al found that Ang II
promotes vascular inflammation and augments AS by indu cing
premature senescence of SMCs via a p21-dependent pathway (23). Wang et al suggested that
whole body receptor-associated protein (RAP) deficiency could
attenuate the incidence of AS in hypercholesterolemic mice infused
with Ang II (24). In addition,
evidence demonstrated that increased local RAS production leads to
the pathophysiological process of vascular remodeling, which is an
important event in the progression of AS (25,26). Some studies reported that Ang II
mediates the expression of focal adhesion kinases and integrins in
cardiac fibroblasts, the levels of c-fos, EGFR1, transforming
growth factor-β and extracellular matrix proteins in cardiac
myocytes as well as SMC proliferation and hypertrophy (27,28).
It should also be noted that the crosstalk between
dyslipidemia and RAS activation in atherogenesis has been
suggested. Previous research revealed that Ang II infusion
accelerated an increased severity of aortic atherosclerotic lesions
and aneurysms in apoE−/− mice (11). A recent study revealed that Ang II
infusion exaggerated AS development in apoE−/− mice by
enhancing the accumulation of dihydroethidium-positive mononuclear
cells in the intima and mRNA expression levels of Nox2 (29). Native or oxidized low-density
lipoprotein (ox-LDL) has previously been reported to enhance the
levels of ACE and AT1AR in human ECs and SMCs through
LDL receptors (LDLRs) or lectin-like ox-LDL receptor-1 (LOX-1),
respectively (30). In addition,
Ang II may facilitate LDL oxidation and uptake, as well as
thrombosis (31). It was shown
that Ang II infusion stimulated a significant increase in aortic
LDL receptor-related protein (LRP1) expression and lipid
infiltration in the arterial intima (32). Previously, evidence indicates that
LDL uptake by rat aorta is increased by Ang II, whereas this effect
may be independent of its pressor action (33). Moreover, Keidar et al
observed that the mechanism of Ang II atherogenicity is associated
with its effect on the uptake of ox-LDL by macrophage and foam cell
formation, a process mediated by IL-6 (34).
In our previous study, we demonstrated that
C-reactive protein (CRP) promotes AS by directly increasing the
transcytosis of LDL across ECs and accelerating LDL retention in
vascular walls, which is mediated by ROS (2). Moreover, tumor necrosis factor-α
(TNF-α) was also proven to promote early AS by upregulating LDL
transcytosis and LDL retention in vascular walls (3). However, whether or not Ang II is
able to directly exert a pro-atherogenic effect by promoting LDL
transcytosis across the endothelial barrier and LDL retention in
vascular walls, has not been defined.
In the present study, based on the in vitro
model to assay the transcytosis of LDL, we found that Ang II indeed
increased LDL transcytosis across ECs and accelerated LDL retention
in the subendothelial space of human umbilical venous walls.
Following this, we further explored the underlying molecular
mechanisms and revealed that the upregulation of
transcytosis-related proteins is involved in Ang II-induced LDL
transcytosis, which is associated with the production of ROS.
Materials and methods
The present study was approved by the Ethics
Committee of Tongji Medical College, Huazhong University of Science
and Technology (Wuhan, China) and conducted in accordance with the
Declaration of Helsinki and all applicable national and local
regulations. All subjects provided written informed consent prior
to the initiation of the study.
Reagents and chemicals
Endothelial cell medium (ECM) (cat. no. 1001) with
10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 U/ml
streptomycin and endothelial cell growth supplement (ECGS) were
purchased all from ScienCell Research Laboratories (Carlsbad, CA,
USA). Fluorescein isothiocyanate (FITC) (cat. no. FD150S) was
purchased from Sigma-Aldrich (St. Louis, MO, USA). LDL (cat. no.
YB-001) was obtained from Yiyuan Biotechnology Co., Ltd.
(Guangzhou, China). 2′,7′-Dichlorofluorescein (DCF-DA) was obtained
from Applygen Technologies, Inc. (Beijing, China). BCA assay kit
(cat. no. 23235) was obtained from Thermo Fisher Scientific, Inc.
(Rockford, IL, USA). Ang II and methyl-β-cyclodextrin (MβCD) were
supplied by Sigma-Aldrich. Dithiothreitol (DTT) was purchased from
MDBio, Inc. (Piscataway, NJ, USA). RIPA lysis buffer (cat. no.
AR0105) was from Beyotime Institute of Biotechnology (Shanghai,
China). Caveolin-1 (cat. no. 3238) was obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Cavin-1 (cat. no. 18892), LDLR
(cat. no. 10785), clathrin (cat. no. 10852) and β-actin (cat. no.
14395) were supplied by Proteintech Group, Inc. (Chicago, IL,
USA).
Isolation and culture of human umbilical
vein endothelial cells (HUVECs)
Isolation of HUVECs was performed as described in
our previous study (2). HUVECs
were cultured in ECM with 10% FBS, 100 U/ml penicillin, 100 U/ml
streptomycin and 30 µg/ml ECGS at 37°C with 5%
CO2. Cells were passaged when 80–90% confluent and were
used between passages 3 and 9.
LDL labeling
LDL was labeled using FITC as described in our
previous study (2). In brief, 2
mg LDL and 120 µg FITC were mixed and incubated in the dark
(37°C, 2 h). Unbound FITC was removed by dialysis against PBS in
the dark (4°C, 72 h). After the measurement of protein with the BCA
assay kit, FITC-LDL was then stored at 4°C in the dark for further
use.
Measurements of intracellular ROS
levels
For determining the intracellular levels of ROS,
HUVECs were incubated with 3 µmol/l DCF-DA at room
temperature in the dark for 30 min as previously described
(2), and then the medium with DCF
was removed. The cells were pretreated with 30 µmol/l DTT
for a further 30 min, followed by exposure to 10−9 M Ang
II for 30 min. Using a fluorescence spectrophotometer (Infinite
F200 PRO; Tecan, Männedorf, Switzerland) with excitation and
emission wavelengths of 490 and 520 nm, respectively, the
fluorescence intensity of the ROS-reactive dichlorofluorescin was
dynamically monitored.
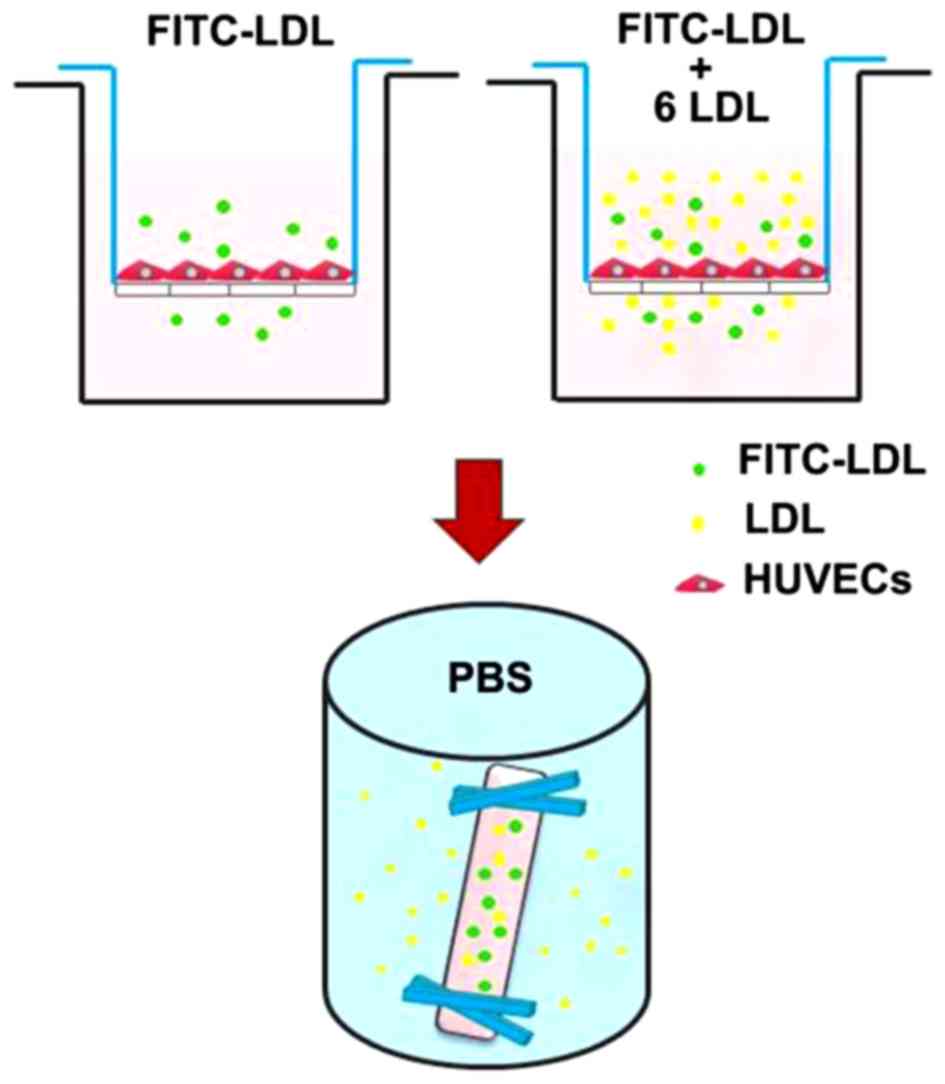
Analysis of the transcytosis of LDL
across HUVECs using an in vitro transcytosis model
LDL transcytosis across HUVECs was determined based
on the newly established transcytosis model established in our
laboratory. As shown in Fig. 1,
cells were seeded (~4×104 cells/insert) on a polyester
membrane of a Costar Transwell (6.5-mm diameter, 0.4-µm pore
size) (Corning Costar, Cambridge, MA, USA) to form an integrated
cell monolayer. As depicted in a previous study (2), two inserts of cell monolayers with
equal integrity were divided into the same group: the control
insert and the naive insert, respectively. The control insert was
stimulated with FITC-LDL to determine the total amount of LDL
transport. Paracellular transport was analyzed by treatment with
FITC-LDL and 6-fold excess of unlabeled LDL in the naive insert.
Thereafter, the amount of LDL transcytosis was calculated by
subtracting the paracellular transport (the competitive insert)
from the total transport (the non-competitive insert). In the
present study, following pretreatment with 3,000 µM MβCD or
30 µmol/l DTT for 30 min, HUVECs were then stimulated with
50 µg/ml FITC-LDL (and/or LDL) and 10−9 M Ang II
for 24 h. After 24 h, the samples were collected from the outer
chambers and further dialyzed against PBS to remove the free FITC.
The relative fluorescence was measured via a fluorescence
spectrophotometer with excitation and emission wavelengths of 490
and 520 nm, respectively. The amount of LDL transcytosis was
normalized to that obtained in the control group.
Confocal imaging analysis of the uptake
of LDL in HUVECs
Cells were seeded on gelatin-coated glass coverslips
in 24-well culture plates (37°C, 5% CO2). To determine
LDL uptake in the HUVECs, the cells were first incubated with 50
µg/ml FITC-LDL for 24 h and then treated with or without
10−9 M Ang II, 3,000 µM MβCD or 30 µmol/l
DTT for 24 h at 37°C. Images were obtained by confocal laser
scanning microscopy (Olympus FV500; Olympus, Center Valley, PA,
USA) (the excitation wavelength, 490 nm; the emission wavelength,
520 nm). The fluorescence images were analyzed using ImageJ
software. Each individual microscopic field was randomly selected
to include at least 15 cells, and the numbers of cells were
counted. The integrated fluorescence intensities were measured and
normalized to the number of cells.
Confocal imaging analysis of LDL
retention in the isolated human umbilical venous wall
The human umbilical venous rings were bubbled with a
mixed gas (95% O2, 5% CO2) and incubated with
50 µg/ml FITC-LDL, and/or 10−9 M Ang II, 3,000
µM MβCD or 30 µmol/l DTT for 3 h at 37°C. Then, all
tissues were frozen and consecutively cut into sections of
10-µm in thickness with a cryostat (Leica CM1900; Leica
Microsystems, Wetzlar, Germany), and further stained with DAPI. For
the fluorescence quantification, we used a weighted protocol
described previously (2). The
region above the basilar membrane was defined as the region of
interest (ROI). The fluorescent area and the area of ROI were
quantified using ImageJ software. As previously described (2), the weighted analysis was performed
by first determining the area of fluorescence within the ROI of
each optical section for three fluorescence intensity value ranges:
86 to 123, 124 to 161, and 162 to 200. These three area
measurements were then multiplied by 1, 2 or 3, respectively, to
give greater weight to areas of highest intensities. These weighted
values were then summed for each optical section and divided by the
area of the ROI.
Western blotting
HUVECs were seeded in 100-mm culture dishes.
Following pretreatment with 3,000 µM MβCD or 30
µmol/l DTT for 30 min, the HUVECs were then stimulated with
or without 10−9 M Ang II for 24 h. Cells were lysed with
the RIPA lysis buffer. Resuspended proteins were separated by
SDS-PAGE gel and transferred to a PVDF membrane. The membranes were
probed with primary antibodies against LDLR (1:700), caveolin-1
(1:8,000), cavin-1 (1:5,000), clathrin (1:1,000) and β-actin
(1:4,000). The immunoreactive bands were visualized by an ECL
western blot detection system. The expression of proteins was
normalized to the control group.
Statistical analysis
All data are expressed as the mean ± SEM from at
least three separate experiments. SPSS 13.0 software (SPSS, Inc.,
Chicago, IL, USA) was used for all statistical analysis. Student's
unpaired t-test was used to analyze individual group statistical
comparisons, and one-way ANOVA with post-hoc test was performed to
evaluate multiple group comparisons. Statistical significance is
defined as P<0.05.
Results
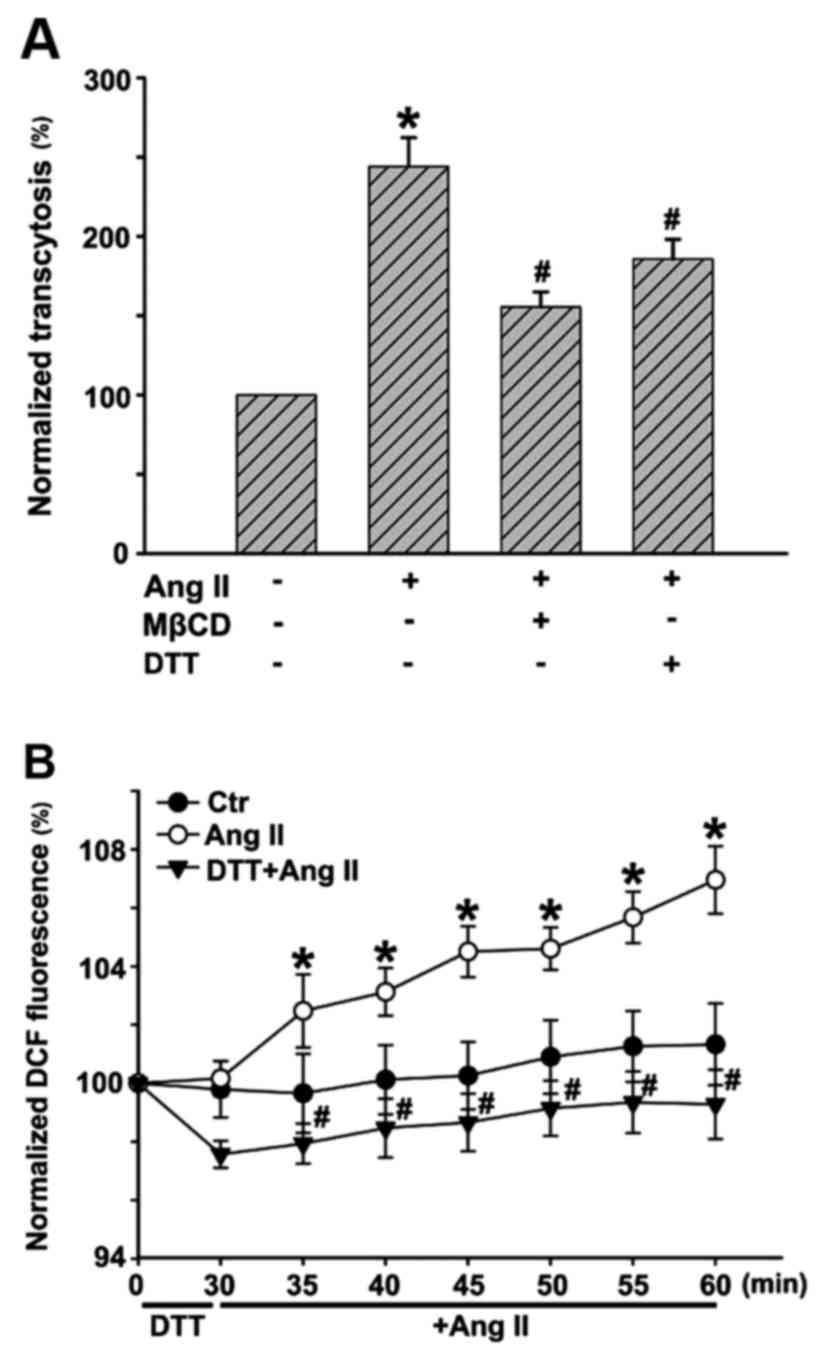
ROS are involved in Ang II-induced LDL
transcytosis across HUVECs
In this study, we ascertained whether Ang II could
promote LDL transcytosis across HUVECs based on the established
transcytosis model. The amount of LDL transcytosis was normalized
to that obtained in the control group. As shown in Fig. 2A, Ang II exposure for 24 h
significantly elevated the level of LDL transcytosis. Transcytosis
inhibitor, MβCD, highly attenuated Ang II-induced LDL transcytosis.
Ang II increased intracellular ROS levels, in agreement with
previous studies. ROS inhibitor, DTT, markedly decreased the level
of ROS induced by Ang II (Fig.
2B). In a previous study, we demonstrated that
H2O2 was able to increase LDL transcytosis
(2). In this study, ROS
inhibitor, DTT, significantly decreased LDL transcytosis induced by
Ang II, as depicted in Fig.
2A.
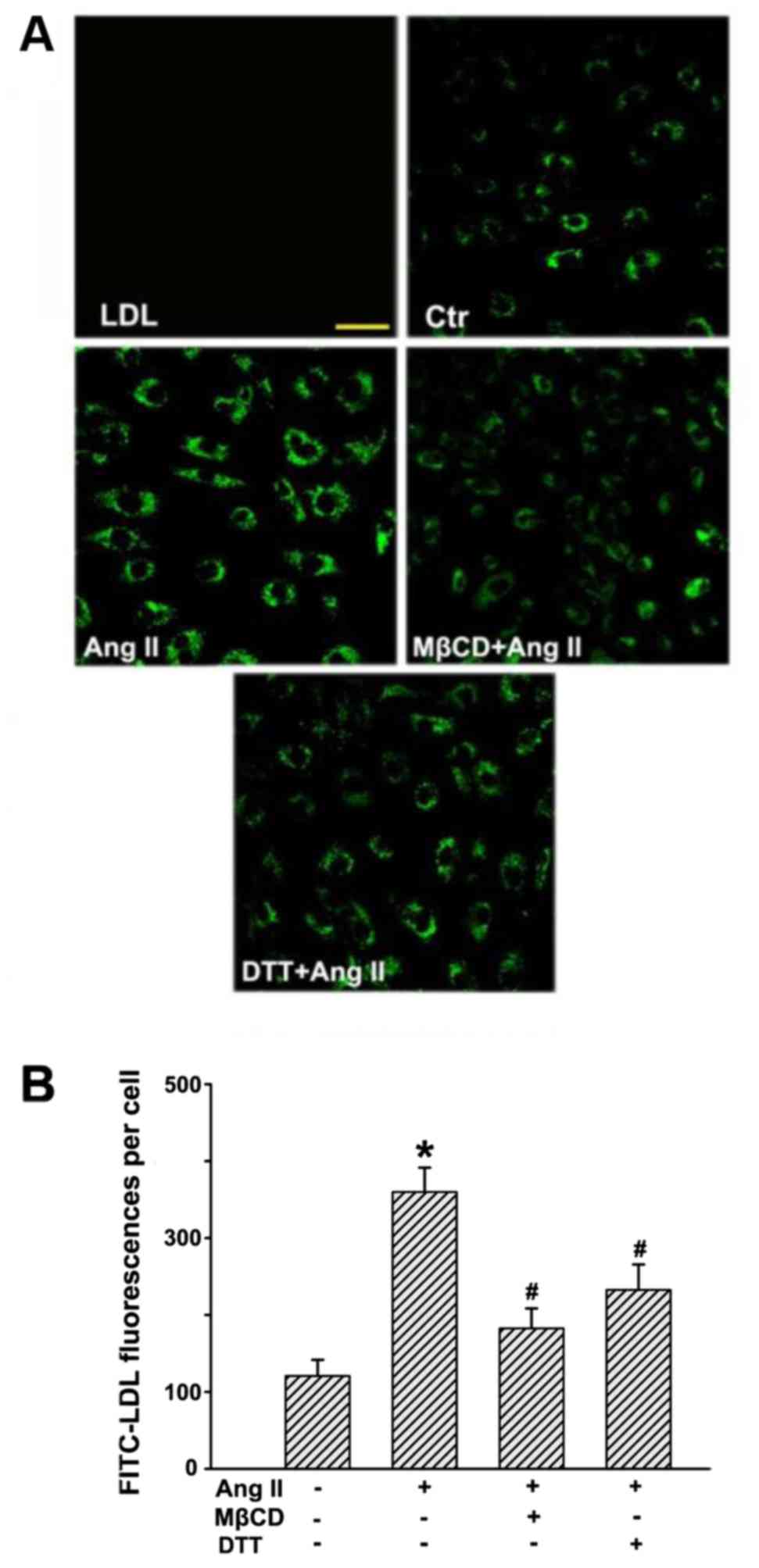
Ang II stimulation increases the uptake
of LDL in HUVECs
After incubation with FITC-LDL, the HUVECs were
found to be full of small, individual, discrete vesicles throughout
the cells. The fluorescent intensity in each individual cell
reflected the extent of LDL uptake. Since LDL uptake is an
intermediate phase of LDL transcytosis, it may also represent the
amount of LDL transcytosis to a certain degree. As shown in
Fig. 3, Ang II significantly
enhanced fluorescence intensities in the cells, which indicated an
increase in LDL uptake. In contrast, MβCD and DTT markedly
inhibited Ang II-stimulated LDL internalization
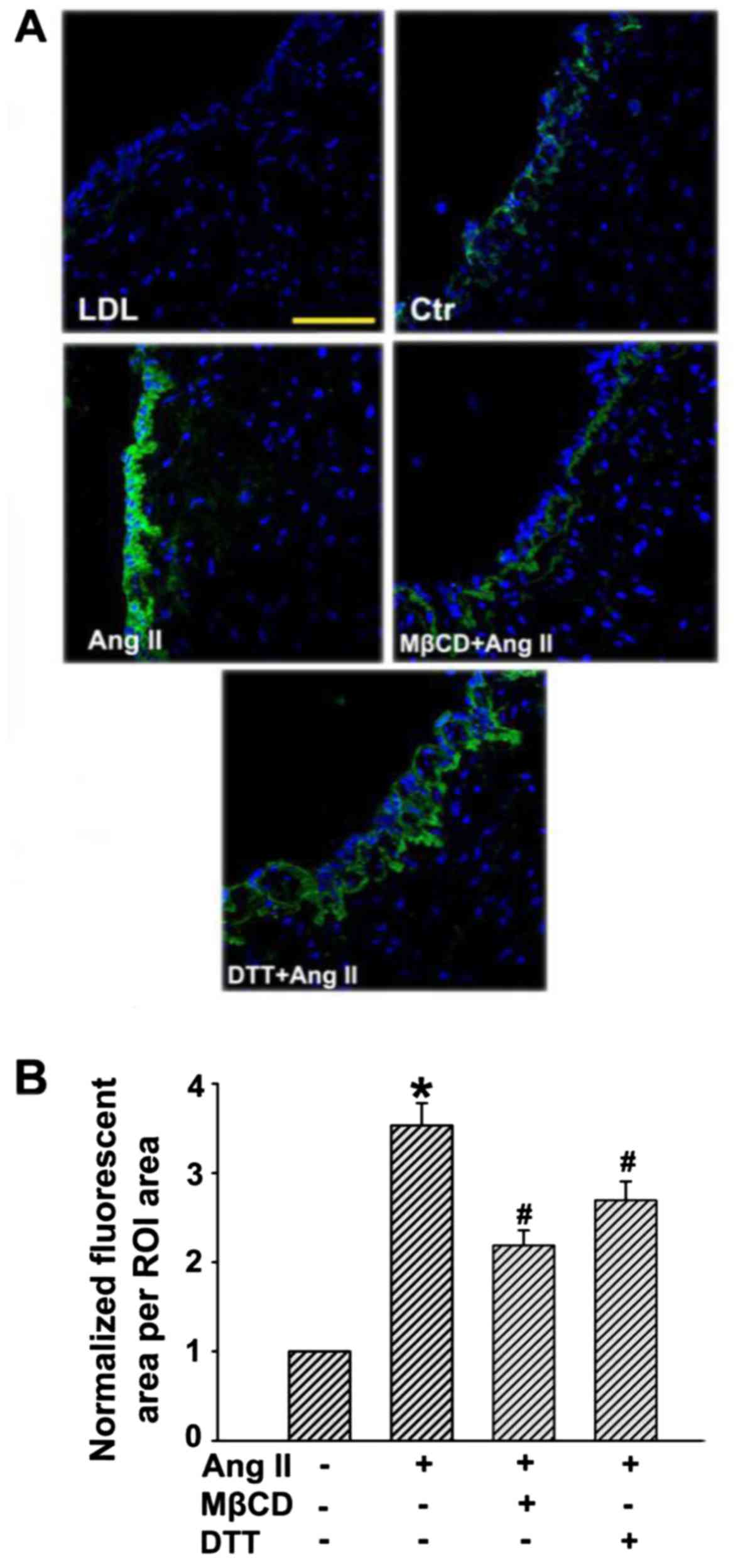
Ang II stimulation increases the
retention of LDL in human umbilical venous walls
Subendothelial retention of apoB-containing
lipoprotein particles (e.g., LDL) is important for the initiation
of AS. An experiment was conducted to demonstrate whether Ang II
could promote subendothelial retention of LDL in human umbilical
venous walls. As depicted in Fig.
4, only a small amount of LDL was determined in human umbilical
venous walls after incubation with FITC-LDL for 24 h, while much
more FITC-LDL accumulated in the region above the basilar membrane
after Ang II stimulation. However, the retention of FITC-LDL was
markedly inhibited in the presence of MβCD or DTT.
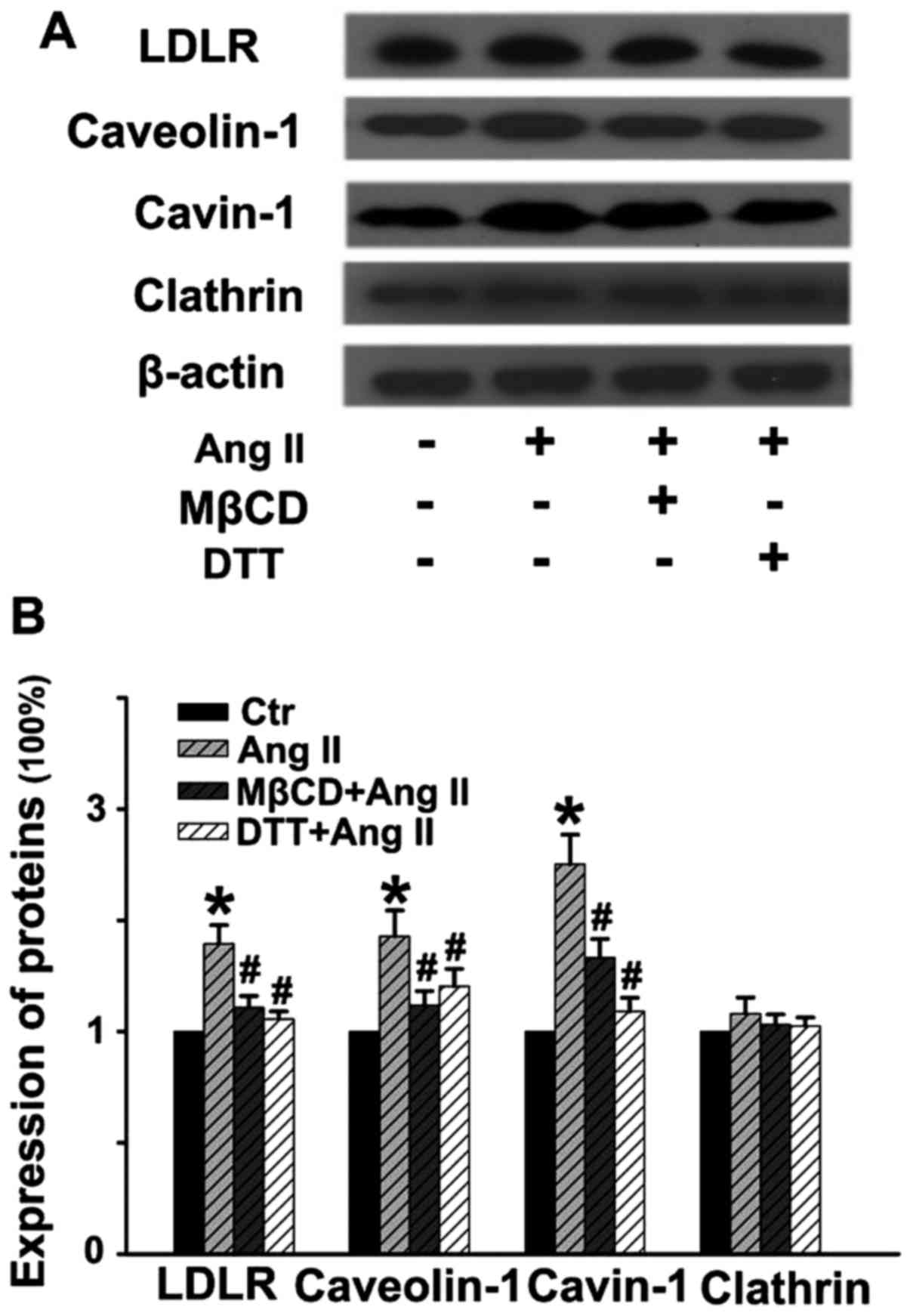
Ang II stimulates the expression of
molecules involved in LDL transcytosis
As shown in Fig.
5, Ang II stimulation for 24 h distinctly upregulated the
expression of LDLR, caveolin-1 and cavin-1, while it had no obvious
effect on the level of clathrin. Pretreatment with MβCD or DTT for
30 min significantly blocked Ang II-induced upregulation of these
proteins. Thus, Ang II-stimulated LDL transcytosis may be partly
due to the elevated expression of the proteins involved in
caveolae-mediated transcytosis. Of note, ROS are critical factors
in Ang II-induced LDL transcytosis.
Discussion
Currently, it is generally accepted that the
subendothelial retention of apoB-containing lipoprotein particles
(e.g., LDL) is the crucial initiating event in early AS. LDLs are
lipoprotein particles 20–30 nm in diameter and cannot pass through
the intact endothelium via a paracellular pathway, but rather via
the transporting process termed transcytosis. Importantly,
accumulating evidence has pointed to the possible link between LDL
transcytosis across ECs and the initiation of AS (2,3,7).
Meanwhile, there is increasing evidence of the tight
interactions between AS development and RAS activation. Of note,
the crosstalk between dyslipidemia and RAS activation in
atherogenesis has been emphasized. Ang II, as the principal
effector molecule of RAS, is shown to be implicated in several
important steps of AS development. However, whether or not Ang II
can directly exert a pro-atherogenic effect by promoting LDL
transcytosis across endothelial barriers, has not been defined.
Previously, based on our newly established in vitro
transcytosis model, our studies demonstrated that inflammatory
factors CRP (9) and TNF-α
(3) could increase
caveolae-mediated LDL transcytosis across ECs and therefore promote
LDL retention in human umbilical venous walls, as well as
atherosclerotic lesion formation. Nevertheless, endogenous ceramide
has also been shown to enhance the transcytosis of ox-LDL across
ECs and promote the initiating step of AS, the subendothelial
retention of lipids in the vascular wall (7). ROS have been emerging as essential
intracellular secondary messengers and are also important signaling
molecules known to increase endothelial permeability. Notably, we
revealed that CRP indeed promoted a significant increase in ROS
production in the ECs and DTT substantially decreased the
CRP-stimulated upregulation of LDL transcytosis. Moreover,
exogenous H2O2 was also found to cause an
increase in LDL transcytosis in vitro (9), further supporting the involvement of
ROS signaling in the CRP-stimulated increase in LDL transcytosis.
In the present study, we aimed to ascertain whether or not Ang II
is able to directly exert a pro-atherogenic effect by increasing
LDL transcytosis across the endothelial barrier and promoting LDL
retention in vascular walls.
With our in vitro transcytosis model, we
firstly investigated the effects of Ang II on the transcytosis of
LDL across ECs and found that Ang II significantly increased LDL
transcytosis. Transcytosis inhibitor, MβCD, markedly prevented Ang
II-stimulated LDL transcytosis. Of note, ROS inhibitor, DTT, also
decreased LDL transcytosis induced by Ang II. To the best of our
knowledge, this is the first study to ascertain that Ang II
directly enhances LDL transcytosis across ECs. Such an effect may
underlie the predictive value of Ang II in cardiovascular diseases.
In the process of LDL transcytosis, LDL particles must be
endocytosed into cells and then be transferred to the basolateral
side and thus be exocytosed to the subendothelial space. During
this process, an intermediate event of transcytosis occurs - LDL
particles are taken up into the cytosol, but are not yet released
(35). Therefore, determination
of the intermediate form of LDL particles and the subendothelial
retention of LDL can also reflect the activity of transcytosis. In
the present study, we revealed that both the uptake of LDL in cells
and the retention of LDL in human umbilical venous walls were
largely elevated after Ang II exposure, which were consistent with
the findings observed in the in vitro transcytosis model.
But how does Ang II affect LDL transcytosis aross ECs?
Transcytosis is a complex multi-step process which
includes endocytosis, intracellular trafficking and exocytosis. To
date, clathrin-mediated endocytosis (CME) is the most widely
studied endocytic pathway and plays critical roles in human health
and disease. This pathway encompasses the ubiquitous uptake of
nutrient-receptor complexes, adhesion molecules, growth factors,
membrane transporters, toxin and viruses, the recycling of synaptic
vesicles and activation of signaling pathways that regulate
development and immune responses (36). Cargos delivered via the CME
pathway are digested and degraded in the endolysosome (pH, ~4.5)
(37). Generally,
receptor-mediated endocytosis via CME is for internal use.
Specifically, researchers claim that LDLR binds LDL and is
endocytosed in clathrin-coated pits via clathrin-dependent
mechanisms. Acidification of early endocytic vesicles liberates LDL
from the receptor and allows the cargo to be transported into
lysosomes where LDL is degraded and cholesterol is salvaged for
cellular use (38,39).
Numerous studies have focused on transcytosis via
caveolae. Caveolae are a particular type of lipid raft and have
been described as 50–100 nm, flask-shaped, non-clathrin-coated
invaginations of the plasma membrane. A variety of receptors [e.g.,
LDLR, high-density lipoprotein receptor (HDL-R), albumin receptor
(Alb-R), transferrin receptor (Tf-R), insulin receptor (Ins-R)],
channels, and enzyme systems are located in caveolae microdomains
(40). Accumulating evidence
suggests that LDL transcytosis occurs mainly through a
receptor-dependent pathway (41).
Moreover, caveolae exhibit their highest frequency in ECs
(~10,000/cell). Since the population of clathrin-coated pits and
vesicles is relatively small in ECs (~3% of the total endothelial
vesicles), caveolae play a major role in the process of
transcytosis across the endothelium (41,42).
Caveolin-1, an integral membrane protein (20–22
kDa), is the major structural component of caveolae. Recently,
evidence suggests that cavins, identified as a class of caveolae
regulatory proteins, may also play an important role in caveolae
formation. Data indicate a correlation between the levels of
cavin-1 (polymerase transcript release factor, PTRF) and caveolin-1
and the abundance of caveolae. Notably, cavin-1 can be recruited by
caveolins to plasma membrane caveolar domains and is necessary for
caveolae formation (43,44). In the present study, by
determining the expression of these essential proteins involved in
LDL transcytosis, including LDLR, caveolin-1, cavin-1 and clathrin,
we observed that Ang II significantly upregulated the levels of
LDLR, caveolin-1 and cavin-1, but Ang II exhibited no obvious
effects on clathrin expression. These data provide compelling
evidence proving that Ang II-induced LDL transcytosis across ECs is
tightly associated with caveolae-mediated transcytosis. More
importantly, ROS inhibitor, DTT, significantly blocked the
expression of proteins involved in LDL transcytosis mediated by
caveolae.
Taken together, the present study for the first time
demonstrated that Ang II exerted its pro-atherogenic effects by
directly increasing LDL transcytosis across ECs and promoting LDL
retention in vascular walls. Ang II-stimulated LDL retention in
vascular walls appears to be a novel and key step in initiating AS.
Mechanistically, proteins involved in caveolae-mediated
transcytosis, including LDLR, caveolin-1 and cavin-1, were found to
be tightly associated with Ang II-induced LDL transcytosis across
ECs. However, this process was independent of clathrin in our
study. Of note, ROS are critical factors in Ang II-induced LDL
transcytosis. Hopefully, these findings will provide new insight
into the crosstalk between dyslipidemia and RAS activation in
atherogenesis, as well as novel strategies for the prevention or
treatment of diseases related to AS.
Acknowledgments
We are grateful to the National Natural Science
Founda tion of China (no. 81503072 and no. 81373413) and the Hubei
Province Health and Family Planning Scientific Research Project
(no. WJ2015Q037).
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bian F, Yang X, Zhou F, Wu PH, Xing S, Xu
G, Li W, Chi J, Ouyang C, Zhang Y, et al: C-reactive protein
promotes atherosclerosis by increasing LDL transcytosis across
endothelial cells. Br J Pharmacol. 171:2671–2684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Y, Yang X, Bian F, Wu P, Xing S, Xu
G, Li W, Chi J, Ouyang C, Zheng T, et al: TNF-α promotes early
atherosclerosis by increasing transcytosis of LDL across
endothelial cells: crosstalk between NF-κB and PPAR-γ. J Mol Cell
Cardiol. 72:85–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tabas I, Williams KJ and Borén J:
Subendothelial lipoprotein retention as the initiating process in
atherosclerosis: update and therapeutic implications. Circulation.
116:1832–1844. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tuma P and Hubbard AL: Transcytosis:
crossing cellular barriers. Physiol Rev. 83:871–932. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bian F, Xiong B, Yang X and Jin S: Lipid
rafts, ceramide and molecular transcytosis. Front Biosci (Landmark
Ed). 21:806–838. 2016. View
Article : Google Scholar
|
|
7
|
Li W, Yang X, Xing S, Bian F, Yao W, Bai
X, Zheng T, Wu G and Jin S: Endogenous ceramide contributes to the
transcytosis of oxLDL across endothelial cells and promotes its
subendothelial retention in vascular wall. Oxid Med Cell Longev.
2014:8230712014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McAlister FA; Renin Angiotension System
Modulator Meta-Analysis Investigators: Angiotensin-converting
enzyme inhibitors or angiotensin receptor blockers are beneficial
in normotensive atherosclerotic patients: a collaborative
meta-analysis of randomized trials. Eur Heart J. 33:505–514. 2012.
View Article : Google Scholar
|
|
9
|
Ohishi M, Dusting GJ, Fennessy PA,
Mendelsohn FA, Li XC and Zhuo JL: Increased expression and
co-localization of ACE, angiotensin II AT(1) receptors and
inducible nitric oxide synthase in atherosclerotic human coronary
arteries. Int J Physiol Pathophysiol Pharmacol. 2:111–124.
2010.PubMed/NCBI
|
|
10
|
Schieffer B, Schieffer E, Hilfiker-Kleiner
D, Hilfiker A, Kovanen PT, Kaartinen M, Nussberger J, Harringer W
and Drexler H: Expression of angiotensin II and interleukin 6 in
human coronary atherosclerotic plaques: potential implications for
inflammation and plaque instability. Circulation. 101:1372–1378.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Daugherty A, Manning MW and Cassis LA:
Angiotensin II promotes atherosclerotic lesions and aneurysms in
apolipo-protein E-deficient mice. J Clin Invest. 105:1605–1612.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weiss D, Kools JJ and Taylor WR:
Angiotensin II-induced hypertension accelerates the development of
atherosclerosis in apoE-deficient mice. Circulation. 103:448–454.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Griendling KK, Minieri CA, Ollerenshaw JD
and Alexander RW: Angiotensin II stimulates NADH and NADPH oxidase
activity in cultured vascular smooth muscle cells. Circ Res.
74:1141–1148. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Osterud B and Bjorklid E: Role of
monocytes in atherogenesis. Physiol Rev. 83:1069–1112. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pacurari M, Kafoury R, Tchounwou PB and
Ndebele K: The Renin-Angiotensin-aldosterone system in vascular
inflammation and remodeling. Int J Inflamm. 2014:6893602014.
View Article : Google Scholar
|
|
16
|
Ni W, Kitamoto S, Ishibashi M, Usui M,
Inoue S, Hiasa K, Zhao Q, Nishida K, Takeshita A and Egashira K:
Monocyte chemoattractant protein-1 is an essential inflammatory
mediator in angiotensin II-induced progression of established
atherosclerosis in hypercholesterolemic mice. Arterioscler Thromb
Vasc Biol. 24:534–539. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li XC and Zhuo JL: Nuclear factor-kappaB
as a hormonal intracellular signaling molecule: focus on
angiotensin II-induced cardiovascular and renal injury. Curr Opin
Nephrol Hypertens. 17:37–43. 2008. View Article : Google Scholar
|
|
18
|
Li W, Li Z, Chen Y, Li S, Lv Y, Zhou W,
Liao M, Zhu F, Zhou Z, Cheng X, et al: Autoantibodies targeting
AT1 receptor from patients with acute coronary syndrome
upregulate proinflammatory cytokines expression in endothelial
cells involving NF-κB pathway. J Immunol Res. 2014:3426932014.
|
|
19
|
Rius C, Abu-Taha M, Hermenegildo C,
Piqueras L, Cerda-Nicolas JM, Issekutz AC, Estañ L, Cortijo J,
Morcillo EJ, Orallo F, et al: Trans- but not cis-resveratrol
impairs angiotensin-II-mediated vascular inflammation through
inhibition of NF-κB activation and peroxisome
proliferator-activated receptor-gamma upregulation. J Immunol.
185:3718–3727. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rajagopalan S, Kurz S, Münzel T, Tarpey M,
Freeman BA, Griendling KK and Harrison DG: Angiotensin II-mediated
hypertension in the rat increases vascular superoxide production
via membrane NADH/NADPH oxidase activation. Contribution to
alterations of vasomotor tone. J Clin Invest. 97:1916–1923. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eto H, Miyata M, Shirasawa T, Akasaki Y,
Hamada N, Nagaki A, Orihara K, Biro S and Tei C: The long-term
effect of angiotensin II type 1a receptor deficiency on
hypercholesterolemia-induced atherosclerosis. Hypertens Res.
31:1631–1642. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Daugherty A, Rateri DL, Lu H, Inagami T
and Cassis LA: Hypercholesterolemia stimulates angiotensin peptide
synthesis and contributes to atherosclerosis through the AT1A
receptor. Circulation. 110:3849–3857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kunieda T, Minamino T, Nishi J, Tateno K,
Oyama T, Katsuno T, Miyauchi H, Orimo M, Okada S, Takamura M, et
al: Angiotensin II induces premature senescence of vascular smooth
muscle cells and accelerates the development of atherosclerosis via
a 21-dependent pathway. Circulation. 114:953–960. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang S, Subramanian V, Lu H, Howatt DA,
Moorleghen JJ, Charnigo R, Cassis LA and Daugherty A: Deficiency of
receptor-associated protein attenuates angiotensin II-induced
atherosclerosis in hypercholesterolemic mice without influencing
abdominal aortic aneurysms. Atherosclerosis. 220:375–380. 2012.
View Article : Google Scholar :
|
|
25
|
Wu SJ, Soulez M, Yang YH, Chu CS, Shih SC,
Hébert MJ, Kuo MC and Hsieh YJ: Local augmented angiotensinogen
secreted from apoptotic vascular endothelial cells is a vital
mediator of vascular remodelling. PLoS One. 10:e01325832015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Libby P: Inflammation in atherosclerosis.
Arterioscler Thromb Vasc Biol. 32:2045–2051. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schupp N, Kolkhof P, Queisser N, Gärtner
S, Schmid U, Kretschmer A, Hartmann E, Oli RG, Schäfer S and
Stopper H: Mineralocorticoid receptor-mediated DNA damage in
kidneys of DOCA-salt hypertensive rats. FASEB J. 25:968–978. 2011.
View Article : Google Scholar
|
|
28
|
Calhoun DA, White WB, Krum H, Guo W,
Bermann G, Trapani A, Lefkowitz MP and Ménard J: Effects of a novel
aldosterone synthase inhibitor for treatment of primary
hypertension: results of a randomized, double-blind, placebo- and
active-controlled phase 2 trial. Circulation. 124:1945–1955. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takata H, Yamada H, Kawahito H, Kishida S,
Irie D, Kato T, Wakana N, Miyagawa S, Fukui K and Matsubara H:
Vascular angiotensin II type 2 receptor attenuates atherosclerosis
via a kinin/NO-dependent mechanism. J Renin Angiotensin Aldosterone
Syst. 16:311–320. 2015. View Article : Google Scholar
|
|
30
|
Catar RA, Müller G, Heidler J, Schmitz G,
Bornstein SR and Morawietz H: Low-density lipoproteins induce the
renin-angiotensin system and their receptors in human endothelial
cells. Horm Metab Res. 39:801–805. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang X, Phillips MI and Mehta JL: LOX-1
and angiotensin receptors, and their interplay. Cardiovasc Drugs
Ther. 25:401–417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sendra J, Llorente-Cortés V, Costales P,
Huesca-Gómez C and Badimon L: Angiotensin II upregulates LDL
receptor-related protein (LRP1) expression in the vascular wall: a
new pro-atherogenic mechanism of hypertension. Cardiovasc Res.
78:581–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cardona-Sanclemente LE, Medina R and Born
GV: Effect of increasing doses of angiotensin II infused into
normal and hypertensive Wistar rats on low density lipoprotein and
fibrinogen uptake by aortic walls. Proc Natl Acad Sci USA.
91:3285–3288. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Keidar S, Heinrich R, Kaplan M, Hayek T
and Aviram M: Angiotensin II administration to atherosclerotic mice
increases macrophage uptake of oxidized ldl: a possible role for
interleukin-6. Arterioscler Thromb Vasc Biol. 21:1464–1469. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frank PG, Pavlides S and Lisanti MP:
Caveolae and transcytosis in endothelial cells: role in
atherosclerosis. Cell Tissue Res. 335:41–47. 2009. View Article : Google Scholar
|
|
36
|
Kirchhausen T, Owen D and Harrison SC:
Molecular structure, function, and dynamics of clathrin-mediated
membrane traffic. Cold Spring Harb Perspect Biol. 6:a0167252014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
El-Sayed A and Harashima H: Endocytosis of
gene delivery vectors: from clathrin-dependent to lipid
raft-mediated endocytosis. Mol Ther. 21:1118–1130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sorrentino V, Nelson JK, Maspero E,
Marques AR, Scheer L, Polo S and Zelcer N: The LXR-IDOL axis
defines a clathrin-, caveolae-, and dynamin-independent endocytic
route for LDLR internalization and lysosomal degradation. J Lipid
Res. 54:2174–2184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Howes MT, Mayor S and Parton RG:
Molecules, mechanisms, and cellular roles of clathrin-independent
endocytosis. Curr Opin Cell Biol. 22:519–527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Simionescu M, Popov D and Sima A:
Endothelial transcytosis in health and disease. Cell Tissue Res.
335:27–40. 2009. View Article : Google Scholar
|
|
41
|
Pavlides S, Gutierrez-Pajares JL,
Iturrieta J, Lisanti MP and Frank PG: Endothelial caveolin-1 plays
a major role in the development of atherosclerosis. Cell Tissue
Res. 356:147–157. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Predescu SA, Predescu DN and Malik AB:
Molecular determinants of endothelial transcytosis and their role
in endothelial permeability. Am J Physiol Lung Cell Mol Physiol.
293:L823–L842. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nassar ZD, Hill MM, Parton RG and Parat
MO: Caveola-forming proteins caveolin-1 and PTRF in prostate
cancer. Nat Rev Urol. 10:529–536. 2013.PubMed/NCBI
|
|
44
|
Regazzetti C, Dumas K, Lacas-Gervais S,
Pastor F, Peraldi P, Bonnafous S, Dugail I, Le Lay S, Valet P, Le
Marchand-Brustel Y, et al: Hypoxia inhibits cavin-1 and cavin-2
expression and down-regulates caveolae in adipocytes.
Endocrinology. 156:789–801. 2015. View Article : Google Scholar
|