Introduction
Cardiac fibrosis, strongly associated with various
cardiovascular pathological processes, leads to many cardiac
diseases such as heart failure and types of arrhythmias especially
atrial fibrillation as well as sudden cardiac death by causing
ischemic and hypoxic changes in the myocardium, increasing
myocardium stiffness, reducing systolic heart ejection and blocking
electrical conduction (1). It is
characterized by excessive production and deposition of
extracellular matrix (ECM) proteins, including various types of
collagen protein, which are mainly produced by cardiac fibroblasts
(CFs) and contractile, hypersecretory, productive myofibroblasts
derived from CFs suffering a myriad of pathological stimuli
(2,3). CFs are the most abundant cell type
in the heart tissue, accounting for ~75% of all cardiac cells and
predominantly maintain the functional and structural integrity of
the entire heart. For a long time, on account of their small size,
comprising only ~10–15% of the heart volume and having no
excitability and contractibility, the pathological role of CFs was
overlooked by most investigators. However, in recent years,
research has shown that CFs play a vital role in the cardiac
fibrogenesis cascade. Regardless of the initial stimuli, CFs are
the final, decisive factor of the cardiac fibrosis process
(4,5). Therefore, restraining the activation
and function of CFs has been identified as a promising therapeutic
strategy for cardiac fibrosis-related diseases.
Ca2+ signaling is crucial for numerous
cellular functions including gene expression, cell growth,
proliferation, differentiation and death (6). Previous studies have shown that
Ca2+ signaling is closely related to the initiation and
development of fibrosis and is indispensable for the pro-fibrogenic
effect of CFs (7–9). Runnels et al (10) identified a new type of
Ca2+-permeable ion channel named transient receptor
potential melastatin 7 (TRPM7) in mammals. Our team and other
investigators have elucidated that TRPM7 channels are highly
expressed on the membrane of CFs, and may be the only calcium
permeable cation channel on the CF membrane, bearing the
Ca2+-influx signal and closely associated with the
cardiac fibrosis process (4,11).
However, whether TRPM7 channels contribute to cardiac fibrogenesis
by regulating functional changes of CFs remains elusive. Therefore,
by stimulating CFs with angiotensin II (Ang II) in vitro, we
demonstrated that TRPM7 channels modulate the proliferation and
differentiation capacity of CFs by mediating Ca2+
signaling on the membrane, and then increase collagen synthesis
finally resulting in myocardial interstitial fibrosis.
Materials and methods
Animals, CFs and reagents
Sprague-Dawley (SD) rats with an SPF status (1–3
days old) were purchased from the Animal Experimental Center of
Wuhan University and the Center for Disease Control and Prevention
of Hubei Province. Dulbecco's modified Eagle's medium (DMEM)-F12
was purchased from HyClone (Logan, UT, USA). Phosphate-buffered
saline (PBS), penicillin/streptomycin and trypsin were purchased
from Jinuo Co. (Hangzhou, China). Fetal bovine serum (FBS) was
obtained from Invitrogen and Gibco Life Technologies (Carlsbad, CA,
USA). The sources of the various primary and secondary antibodies
are provided in the text. Ang II (A9525) and collagenase type II
were acquired both from Sigma-Aldrich (St. Louis, MO, USA). CFs
were divided into five groups: i) blank control (BC), CFs with no
intervention; ii) negative control (NC), CFs transfected with
adenoviral vectors carrying green fluorescent protein (GFP) only
without the TRPM7-small interfering RNA (siRNA) sequence; iii)
NC+Ang II, CFs transfected with adenoviral vectors carrying GFP
only without the TRPM7-siRNA sequence and then treated with 1
µM/l Ang II for 24 h; iv) TRPM7-siRNA, CFs transfected with
adenoviral vectors carrying GFP and the TRPM7-siRNA sequence; v)
TRPM7-siRNA+Ang II, CFs transfected with adenoviral vectors
carrying GFP and the TRPM7-siRNA sequence and then treated with 1
µM/l Ang II for 24 h.
Primary CF isolation and culture
Our experimental protocol concerning the acquisition
and use of animal heart tissues was approved by the Ethics
Committee of Renmin Hospital of Wuhan University. Primary CFs were
obtained from 1- to 3-day-old SD rat pups applying the method of
enzymatic digestion and differential attachment as described in our
previous study with a few adjustments (4). Briefly, shredded heart tissues were
first enzymatically digested with 0.125% trypsin for 10 min and the
supernatant was discarding. Then, 0.125% trypsin was supplemented
with 0.08% collagenase type II for 5 min 8 times and the
supernatant was collected. After centrifugation, the cells were
resuspended in DMEM-F12 (HyClone) containing 10% FBS (Gibco) and 1%
penicillin/streptomycin for differential attachment. After 1.5 h,
the cell culture solution was completely replaced with fresh
culture medium and the dish was placed in a constant temperature
incubator at 37°C with 5% CO2. Every 2–3 days, the cells
were washed with PBS for 3 times and the complete medium was
exchanged. When the cell density reached 90%, passaging was
performed by digesting the cells using 0.25% trypsin plus 0.5 mM
EDTA. In general, the cells were passaged to 3–5 generations for
subsequent experimental use.
Western blot analysis
Western blot analysis was carried out to assess the
protein expression levels of TRPM7, α-smooth muscle actin (α-SMA),
Ki-67, PCNA, collagen I and III in the CFs following the different
treatments. The cell total protein was extracted by cracking CFs in
RIPA lysis buffer (AS1004; Aspen Biological, Wuhan, China)
including proteinase inhibitor cocktails. Protein concentrations
were probed with the BCA protein assay kit (AS1086; Aspen
Biological) for equal-izing the protein content for each group.
Equivalent amounts of proteins (40 µg/lane) were separated
by using distinct sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) (10% for GAPDH, α-SMA and Ki-67; 8% for
TRPM7, collagen I and III; 12% for PCNA). Subsequently, the
separated proteins were transferred onto PVDF membranes, blocked
with 5% skim milk, and dissolved in TBST for 1 h at room
temperature (RT). The blots were respectively incubated with
various primary antibodies against α-SMA (TDY210, 1:5,000), Ki-67
(bs-2130R, 1:500), PCNA (ab92552, 1:1,500), collagen I (ab6308,
1:1,000), collagen III (ab7778, 1:500), TRPM7 (ab109438, 1:1,000)
and GAPDH (ab37168, 1:10,000) (all from Abcam, Cambridge, MA, USA)
at 4°C overnight. After being washed with TBST for 3 times (5 min
each time), the membranes were treated with either HRP-conjugated
goat anti-rabbit (074-1506, 1:10,000) or goat anti-mouse (074-1806,
1:10,000) (both from Kirkegaard and Perry Laboratories, Inc.,
Gaithersburg, MD, USA) secondary antibodies for 1 h at RT. The
chemiluminescence of the blots was determined with an ECL kit
(AS1059; Aspen Biological), and AlphaEaseFC software (Alpha
Innotech, San Leandro, CA, USA) was employed to analyze the optical
density. GAPDH was used as the internal control.
RT-qPCR
Total RNA of the CFs grown on 6-well plates was
extracted with TRIzol reagent (15596-026; Invitrogen™) and reverse
transcribed to cDNA according to the manufacturer's instructions of
Toyobo First Strand cDNA synthesis kit (ReverTra Ace-α-, FSK-100;
Toyobo, Osaka, Japan). Quantitative PCR was carried out in three
stages: first, 95°C for 1 min for pre-denaturation; second, a
40-cycle reaction at 95°C for 15 sec, 58°C for 20 sec and 72°C for
45 sec; third, assessing the temperature from 60°C to 95°C (1°C
every 20 sec) for obtaining the melting curve using
SYBR® Premix Ex Taq™ kit (RR420A; Takara, Otsu, Japan)
on StepOne™ Real-Time PCR apparatus (Life Technologies, Carlsbad,
CA, USA). All the primers used in this study were designed by
Invitrogen Biotechnology Co., Ltd. (Shanghai, China) and are as
follows: TRPM7, 5′-GTACCTGGTCAGAGCACGATGT-3′ and
5′-TGGTATGGATTTGGGTTTCATC-3′; collagen I,
5′-AACTGGTACATCAGCCCAAACC-3′ and 5′-ATCGGAACCTTCGCTTCCAT-3′;
collagen III, 5′-ATGTGTCTGCGACTCGGGAT-3′ and
5′-ACAGGAGCAGGTGTAGAAGGC-3′; GAPDH, 5′-CGCTAACATCAAATGGGGTG-3′ and
5′-TTGCTGACAATCTTGAGGGAG-3′; actin, 5′-CGTTGACATCCGTAAAGACCTC-3′
and 5′-TAGGAGCCAGGGCAGTAATCT-3′. GADPH and actin gene were employed
as the internal control.
RNA interference
Adenovirus vectors carrying small interfering RNA
(siRNA) targeting rat TRPM7 and green fluorescent protein gene
(PHBAd-U6-Scramble-CMV-GFP) were designed and synthesized by
Hanheng Biological Co. (Shanghai, China). The silencing
oligonucleotide sequences are as follows: siRNA1,
5′-GATGTCAGATTTGTCAGCAACTTGT-3′; siRNA2,
5′-GCTCAGAATCTTATTGATGAT-3′. In addition, the adenovirus vectors
with GFP only, without the knockdown sequence were applied as the
negative control group (NC). According to the preliminary
experimental data, we defined the multiplicity of infection of the
adenovirus infecting CFs as 50 (MOI=50). CFs inoculated in 6-well
plates were treated with the viruses for 2 h with DMEM-free/F12.
After 24 h, the infected cells were examined by fluorescence
microscopy. After 72 h, RT-qPCR and western blot analysis were
conducted to monitor the silencing efficiency of TRPM7 at the mRNA
and protein levels, respectively.
Recording of the TRPM7 currents
1The whole-cell patch-clamp experiments were
performed as previously reported (4,12)
for recording the TRPM7 currents on the CF membrane under various
conditions. The internal solution perfused in the glass electrode
contained: 145 mmol/l Cs-mesilate, 8 mmol/l NaCl, 10 mmol/l EGTA
and 10 mmol/l HEPES, PH value adjusted to 7.2 with CsOH. The
standard extracellular Tyrode solution contained: 140 mmol/l NaCl,
5 mmol/l KCl, 2 mmol/l CaCl2, 20 mmol/l HEPES, 10 mmol/l
glucose adjusting PH value to 7.4 with NaOH. Borosilicate glass
electrodes were pulled using a microelectrode puller (PB-7;
Narishige, Tokyo, Japan) holding the resistance at 3–5 MΩ when
filled with pipette solution. The whole cell configuration was set
up so the impedance was rapidly increased and a slight negative
pressure was applied leading to the rupture of the membranes. For
acquiring the current-voltage relationships (I-V curves), voltage
step protocol was applied with a stimulus voltage ranging from −120
to +100 mV lasting for 500 msec. TRPM7 currents were recorded by
utilizing an Axopatch 200B Amplifier (Molecular Devices, LLC,
Sunnyvale, CA, USA) and Pulse+Pulsefit 8.8 software program (HEKA
Elektronik, Lambrecht, Germany). All the data were digitized at 5
kHz, filtered at 2 kHz and normalized according to cell size
(Cslows) as pA/pF.
Cell Counting Kit-8 (CCK-8) assay
CCK-8 assay was applied to assess the activity and
proliferation ability of the CFs. Cells were inoculated in 96-well
culture plates at a density of 2×104 cells/ml (200
µl, ~4,000 cells/well). After treatment with the viruses or
Ang II for 24 h, cells in each well were washed with PBS for 2
times and fresh complete medium containing 20 µl CCK-8
reagent was added. The plates then were incubated in a constant
temperature incubator with 5% CO2 at 37°C for 4 h.
Finally, the absorbance at 450 nm was measured with a microplate
reader (Tecan infinite M200; Tecan, Durham, NC, USA).
Immunofluorescence assay
CFs growing on rinsed aseptic coverslips in 6-well
plate were washed with PBS for 2 times, fixed with 4%
paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100
for 10 min and then blocked with 5% BSA for 1 h. After incubation
with the primary antibody against α-SMA (ab3280; Abcam) overnight
at 4°C, the cells were then incubated with a red
fluorescence-marked secondary antibody (AS1109; Aspen Biological)
for 1 h at RT. The coverslips were mounted on glass slides with
fluoromount-G resistance to fluorescence quenching containing
4′,6-diamidino-2-phenylindole (DAPI) (AS105; Aspen Biological). The
images were observed and saved using fluorescence microscopy with
200× objective.
Statistical analysis
SPSS 17.0 (SPSS, Inc., Chicago, IL, USA) was used
for data analysis. All data are presented as mean ± SD. Differences
among groups were assessed with one-way analysis of variance
(ANOVA) followed by least significant difference-t (LSD-t) test and
statistically significant differences were defined as having
P-values <0.05.
Results
Ang II induces time-dependent TRPM7
expression in CFs
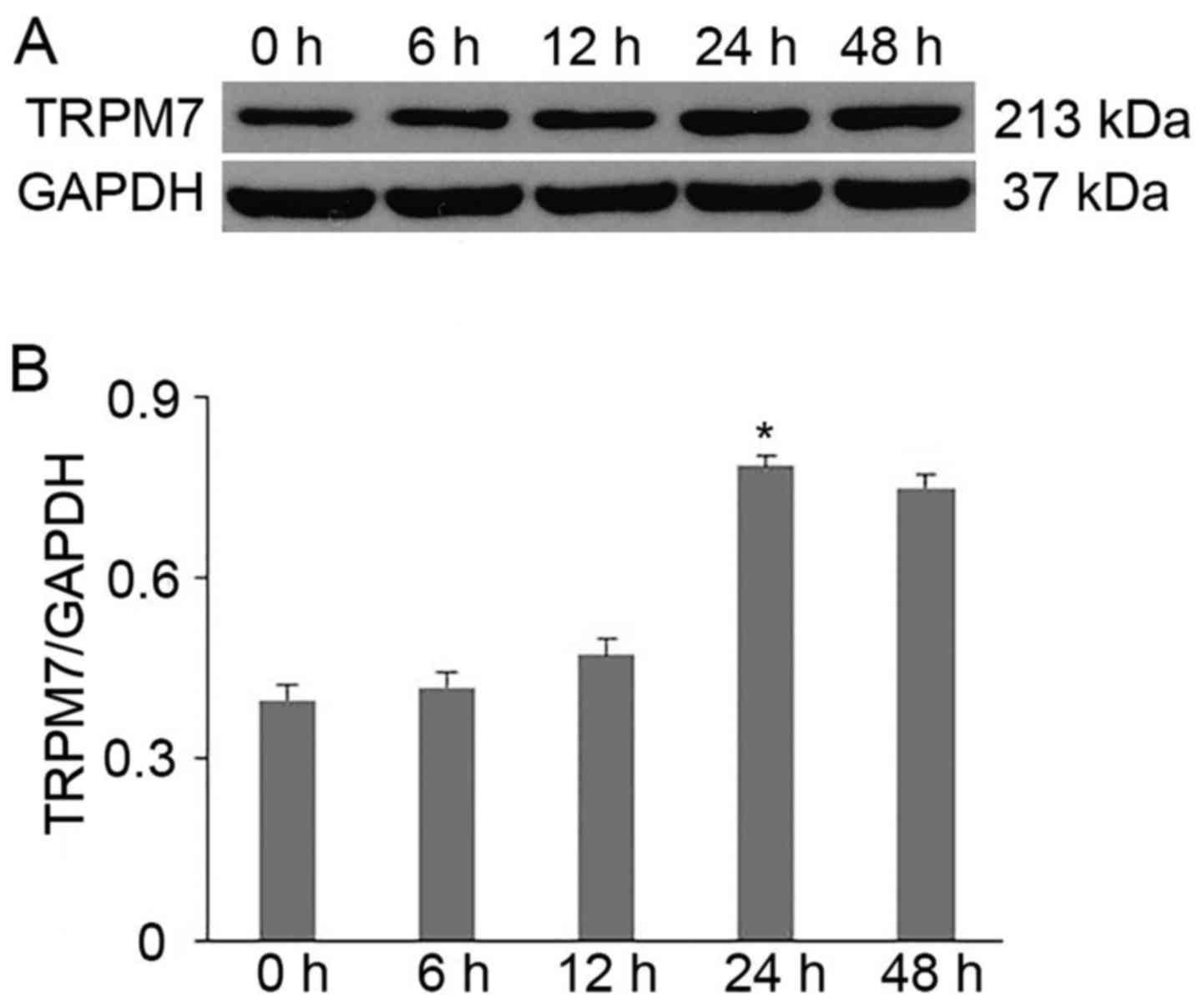
To determine the optimal time-point of 1 µM/l
Ang II for generating cardiac fibrosis, western blot technology was
applied to measure the TRPM7 channel protein expression levels in
Ang II-intervened primary cultured CFs at five different
time-points (0, 6, 12, 24 and 48 h). As shown in Fig. 1, our data indicated that TRPM7
channels were expressed in the primary cultured CFs (0 h). In
addition, the TRPM7 channel protein was time-dependently increased
after Ang II intervention and reached the highest level at 24 h
compared with the other time-points (p<0.01 vs. 0, 6 and 12 h)
(Fig. 1B); a slight decrease was
noted at 48 h, but with no statistical significance (p>0.05, 48
vs. 24 h) (Fig. 1B). Therefore,
we selected treatment with Ang II for 24 h as the optimum
time-point for the rest of our experiments.
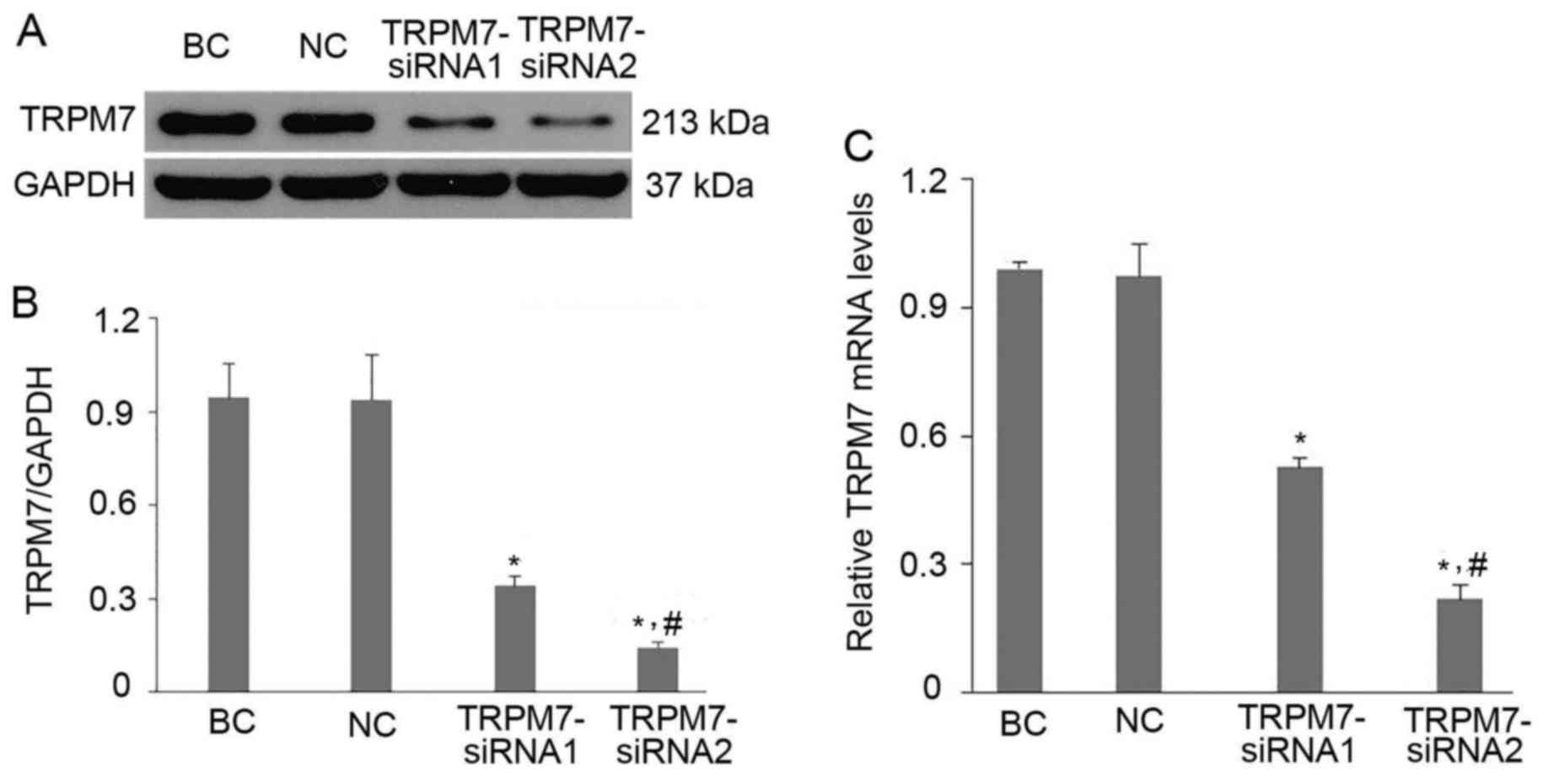
Gene silencing efficiency of TRPM7 in
CFs
In order to knockdown TRPM7 channel protein in CFs,
we used an adenovirus vector carrying an siRNA targeting the rat
TRPM7 gene (Ad-TRPM7-siRNA) as described in Materials and methods.
As revealed in Fig. 2B and C,
TRPM7 expression in the BC and NC groups had no statistical
difference (p>0.05, NC vs. BC) indicating that the adenovirus
vector carrying GFP only did not affect the expression of TRPM7
channels in the CFs. TRPM7-siRNA1 and TRPM7-siRNA2 obviously
reduced the protein expression level of the TRPM7 channel by
63.5±2.3 and 84.5±4.2%, respectively compared to the NC group
(p<0.01 vs. NC) (Fig. 2B). The
results of RT-qPCR were consistent with the western blot analysis
indicating that TRPM7-siRNA1 and TRPM7-siRNA2 significantly
decreased the mRNA level of TRPM7 in the CFs by 45.6±6.0 and
77.2±4.8% respectively (p<0.01 vs. NC) (Fig. 2C). Hence, TRPM7-siRNA2 was
selected for our subsequent gene silencing experiments due to its
higher efficiency (p<0.05 vs. TRPM7-siRNA1; Fig. 2B) (p<0.01 vs. TRPM7-siRNA1;
Fig. 2C).
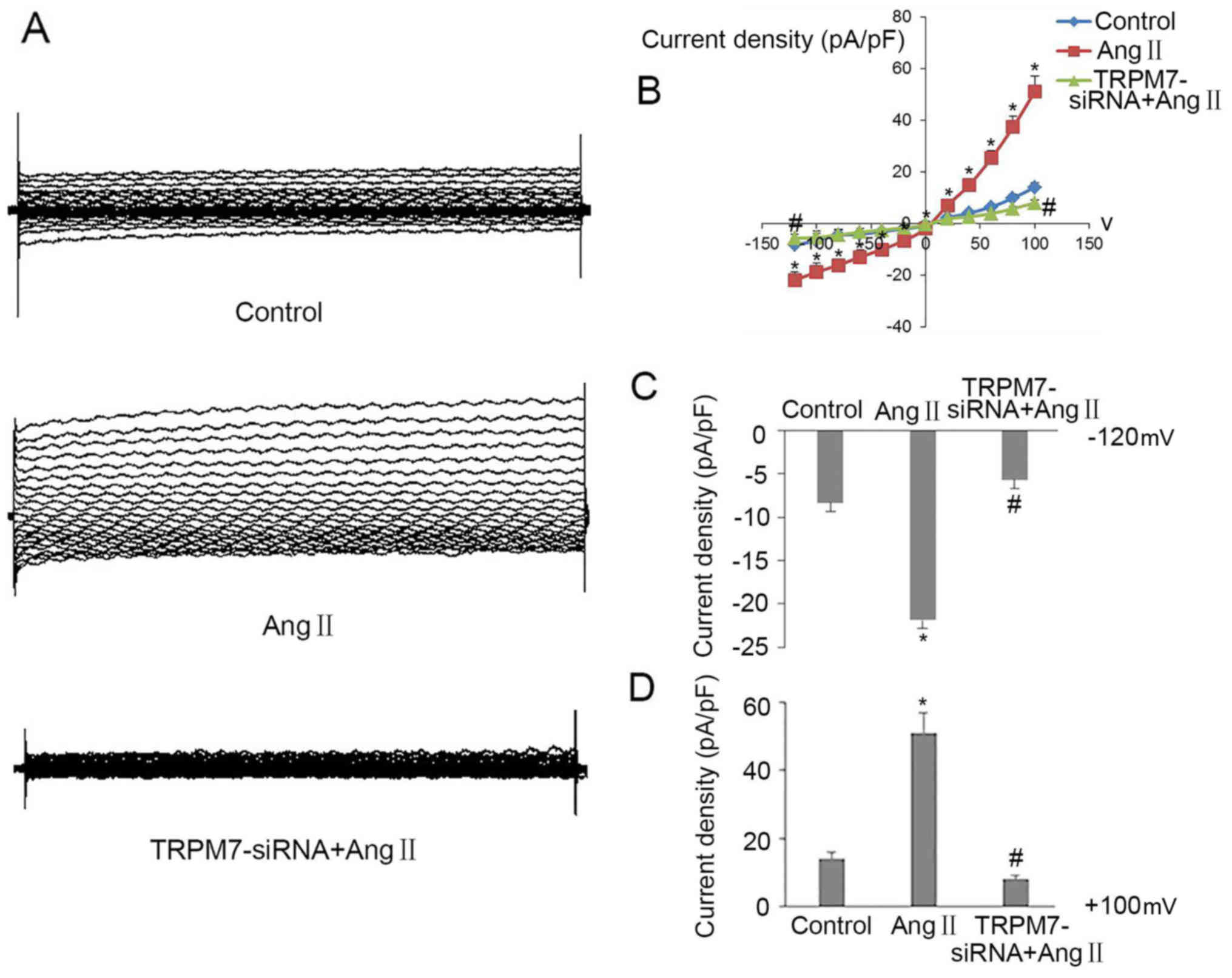
Silencing of TRPM7 attenuates Ang
II-induced elevation in TRPM7 currents in CFs
To further demonstrate the expression of TRPM7
channels increased by Ang II in CFs, we performed whole-cell
patch-clamp technique using a voltage step protocol (the stimulus
voltage ranging from −120 to +100 mV) to detect the TRPM7 current
amplitude on the CF membrane under basal condition, following
treatment with Ang II and Ad-TRPM7-siRNA. As shown in Fig. 3A, an inward and outward rectifying
current was recorded immediately in CFs after the whole-cell
configuration was established. At −120 mV, the current was
inward-rectifying, its amplitude gradually weakened and turned into
an outward-rectifying current at 0 mV around, then was gradually
elevated and reached the highest level at +100 mV, in ~500 msec. In
addition, the current-voltage relationships (I–V curves) (Fig. 3B) showed that the inward and
outward rectifying current density of TRPM7 in the CFs was small in
the basal condition, but dramatically increased after CFs were
treated with Ang II for 24 h (the inward current density was
~2-fold higher compared to the control, the outward current density
was ~3-fold higher compared to control, n=6, p<0.05 vs. control)
then impaired when Ang II-induced CFs were intervened with
Ad-TRPM7-siRNA (the inward current density was ~75% impaired
compared with that of Ang II, the outward current density was ~90%
impaired than that of Ang II, n=6, p<0.05 vs. Ang II). The
quantative current density values at −120 and +100 mV exhibited the
same change trends among the three groups (n=6, p<0.01 vs.
control; n=6, p<0.01 vs. Ang II) (Fig. 3C and D). These
electrophysiological results combined with the result of the above
experiments illustrated that Ang II increased and Ad-TRPM7-siRNA
decreased Ang II-induced expression of TRPM7 and TRPM7 currents in
the CFs.
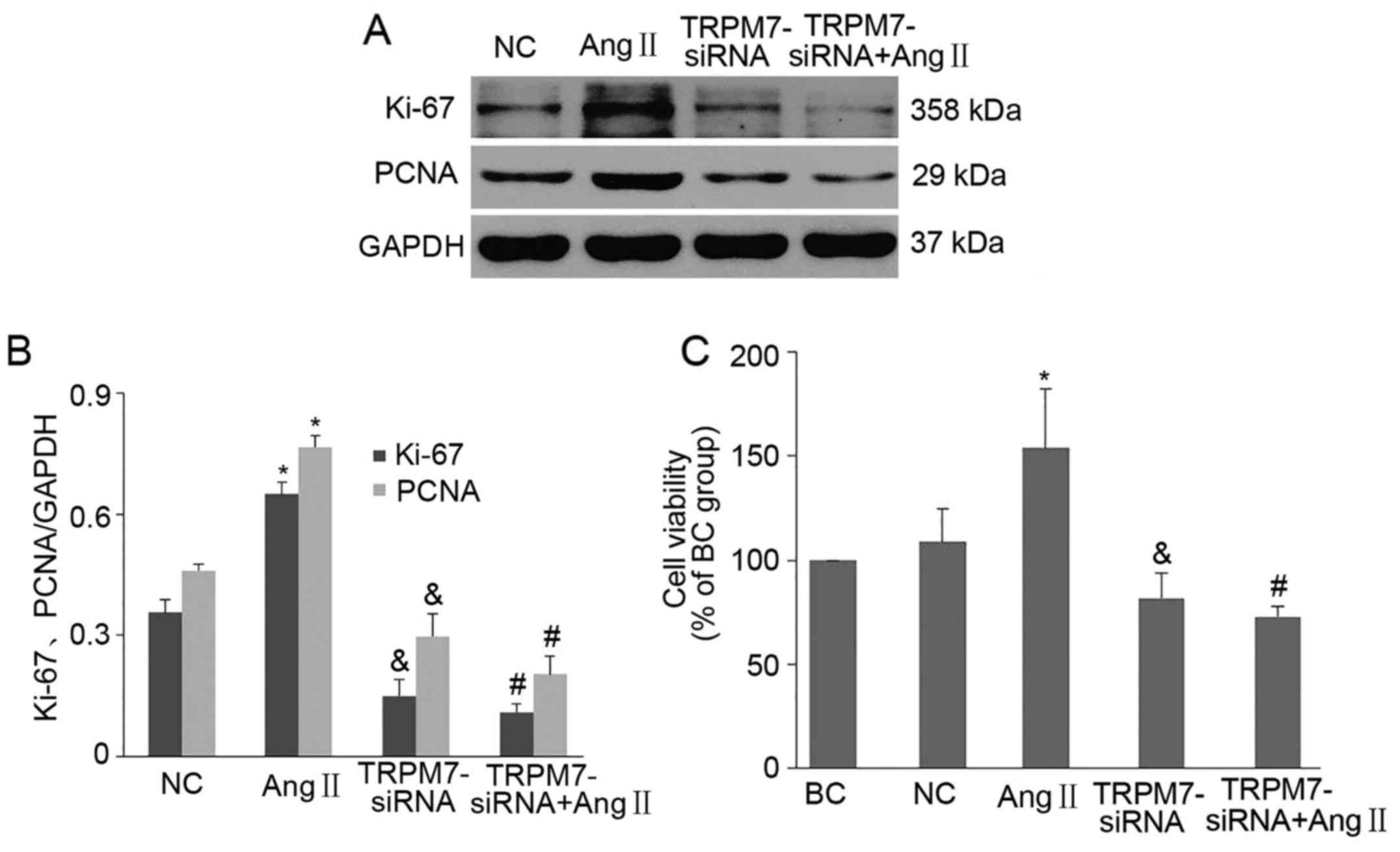
Knockdown of TRPM7 inhibits the
proliferation induced by Ang II in CFs
It was reported that TRPM7 channels influence the
growth and proliferation of many cell types (13). Here, we aimed to ascertain for the
first time whether TRPM7 channels play a critical role in the
proliferation of CFs. Firstly, we used CCK-8 assay to detect the
proliferation abilities of CFs under various conditions. We found
that pretreatment of CFs with 1 µM/l Ang II for 24 h
significantly increased the proliferative ability of CFs (p<0.05
vs. NC) (Fig. 4C). Downregulation
of TRPM7 in CFs with Ad-TRPM7-siRNA markedly reduced Ang II-induced
CF proliferation (p<0.05 vs. Ang II) (Fig. 4C). To further confirm the role of
TRPM7 channels in CF proliferation, cell cycle-related regulatory
protein Ki-67 and proliferating cell nuclear antigen (PCNA) were
employed to assess the CF proliferative rate. Fig. 4A and B shows that Ang II
intervention prominently promoted Ki-67 and PCNA protein expression
and treatment with Ad-TRPM7-siRNA notably inhibited Ang II-elicited
protein expression in the CFs (p<0.01 vs. NC; p<0.01 vs. Ang
II) (Fig. 4B). In addition, we
found that TRPM7-siRNA not only inhibited Ang II-elicited CF
proliferation (p<0.01 vs. Ang II) (Fig. 4B and C) but also suppressed the
proliferation of CFs in basal condition (p<0.05 vs. NC)
(Fig. 4B and C). Together, these
results indicated that TRPM7 channels are involved in the
proliferation of CFs, whereas knockdown of TRPM7 by siRNA inhibited
the proliferation induced by Ang II.
Downregulation of TRPM7 suppresses the
differentiation of CFs induced by Ang II
The pathophysiological process of CF differentiation
into hypersecretory, excessive ECM-formating myofibroblasts plays a
key role during cardiac fibrosis (3). Therefore, we decided to perform
immunofluorescence assay and western blot technique to detect the
expression of α-SMA indirectly reflecting the differentiation
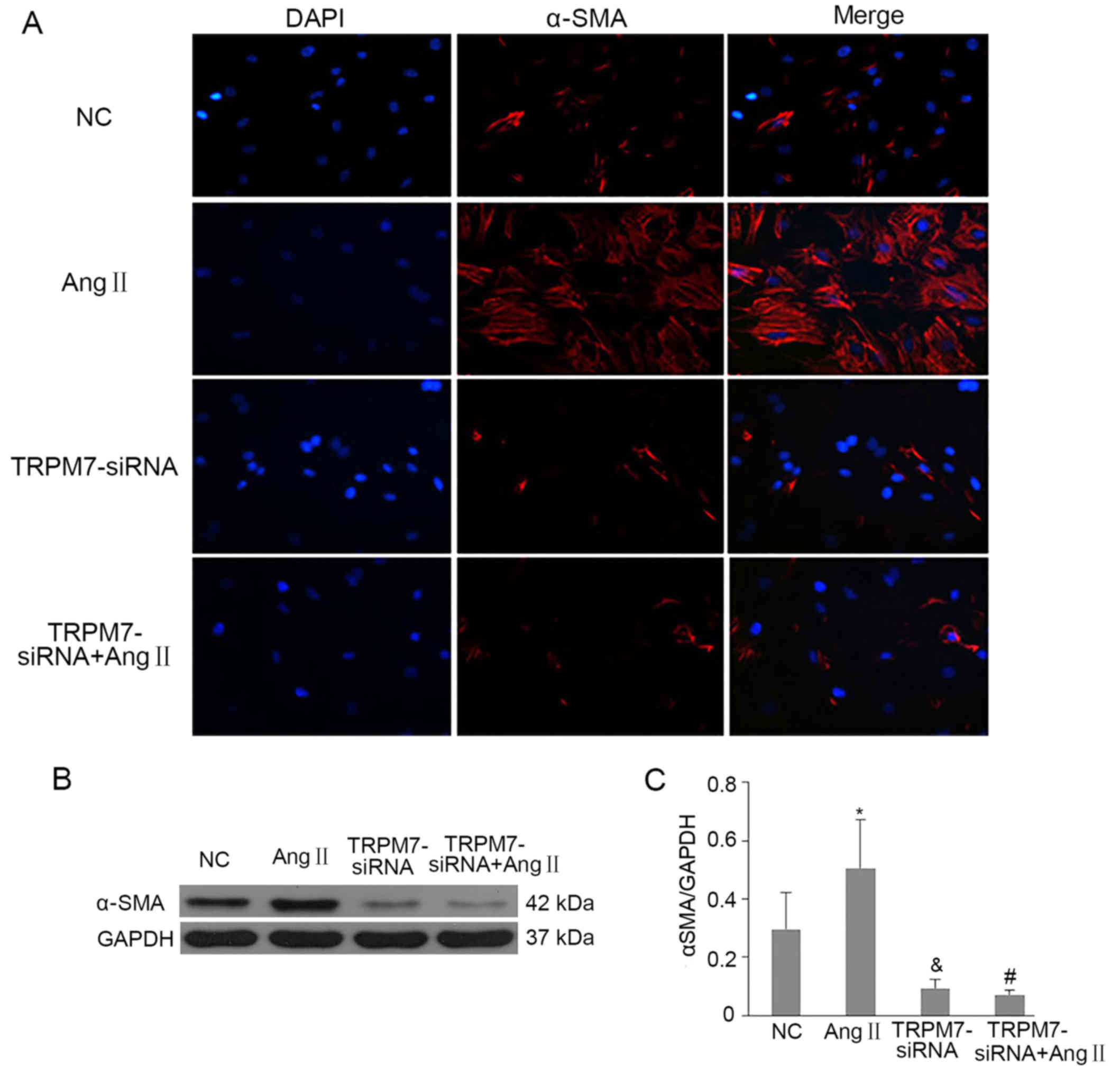
ability of CFs. As shown in Fig.
5A, CFs passaged for 3–5 generations expressed a low level of
α-SMA while Ang II treatment obviously increased the expression of
α-SMA which was reduced in CFs pretreated with Ad-TRPM7-siRNA. The
variation trends of α-SMA protein acquired from western blot
technology were consistent with the immunofluorescence assay
(p<0.05 vs. NC; p<0.05 vs. Ang II) (Fig. 5C). Furthermore, we found that
TRPM7-siRNA inhibited Ang II-elicited CF differentiation (p<0.05
vs. Ang II) (Fig. 5C) as well as
the differentiation of CFs in basal condition (p<0.05 vs. NC)
(Fig. 5C). These data clarified
that TRPM7 channels were also associated with basal and Ang
II-caused CF differentiation effect while silencing TRPM7
observably weakened the differentiation ability of the CFs.
Inhibitory effect of TRPM7-silencing on
collagen synthesis of CFs induced by Ang II
In the pathological process of cardiac fibrosis, ECM
proteins are considerably produced and excessively deposited in
normal heart tissues when the heart responds to various
pathological stimuli such as pressure overload or acute myocardium
infarction. Collagen I and III are two major components of ECM
proteins, thus they were chosen as fibrosis biomarkers in the
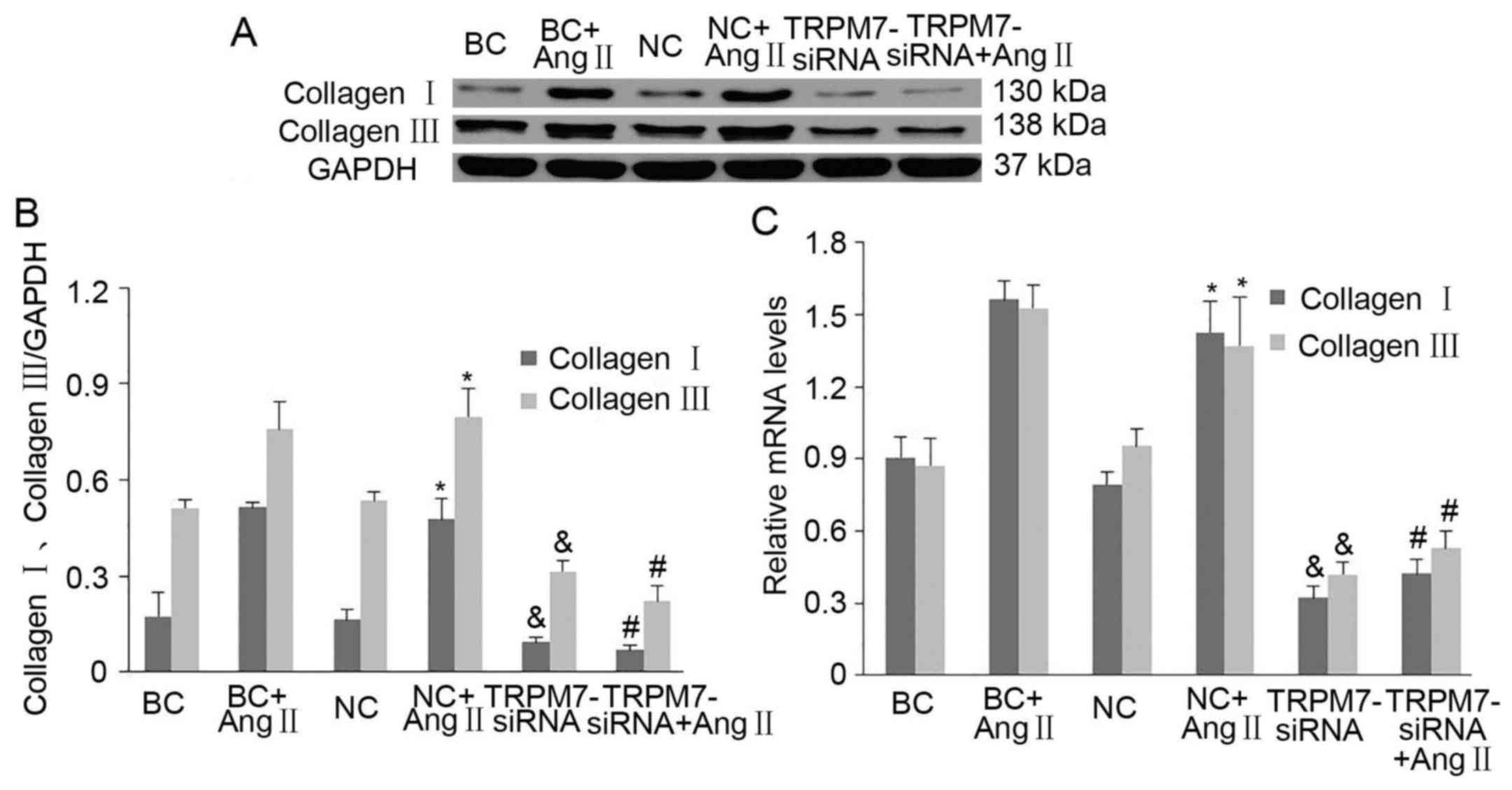
present study. Coincubation with Ang II for 24 h significantly
increased the expression levels of collagen I and III in the CFs
which were markedly lowered by Ad-TRPM7-siRNA (p<0.05 vs. NC;
p<0.01 vs. NC+Ang II) (Fig. 6A and
B). Quantification of the western blot data found that Ang II
increased collagen I and III levels by 207±108 and 48.9±9.8%,
respectively (p<0.05 vs. NC) (Fig.
6B) and TRPM7-siRNA reduced Ang II-induced collagen I and III
synthesis by 85.0±5.3 and 71.6±9.6%, respectively (p<0.05 vs.
NC+Ang II) (Fig. 6B).
Quantification of the RT-qPCR data revealed a similar tend: Ang II
increased collagen I and III mRNA levels by 80.9±26.3 and
44.6±20.9%, respectively (p<0.05 vs. NC) (Fig. 6C) while transfection of CFs with
Ad-TRPM7-siRNA reduced AngII-elicited collagen I and III mRNA
levels by 69.9±7.2 and 60.3±11.2%, respectively (p<0.05 vs.
NC+Ang II) (Fig. 6C). We also
found that TRPM7-siRNA inhibited Ang II-elicited CF collagen
synthesis (p<0.05 vs. NC+Ang II) (Fig. 6B and C) as well as the collagen
synthesis of CFs in basal condition (p<0.05 vs. NC) (Fig. 6B and C). Taken together, the
findings indicated that Ang II evoked an increase in collagen
synthesis by CFs resulting in cardiac fibrosis while inhibition of
TRPM7 expression prohibited the pro-fibrogenic effect of Ang
II.
Discussion
The transient receptor potential (TRP) channel
superfamily is a series of unique cation channels (14), firstly cloned in Drosophila
(15), containing 28 TRP channels
classified into six subfamilies: TRPC, TRPV, TRPM, TRPA, TRPML and
TRPP. They are expressed in various tissues and involved in
manifold basal cellular functions such as Ca2+
signaling, cell contraction, proliferation and death (16). Recently, the melastatin subfamily,
member seven (TRPM7) ion channel has attracted great attention due
to its distinctive structure that possesses a fusion which couples
a functional C-terminal α-type serine/threonine protein kinase
region to a cation channel (17).
The channel domain and the kinase region are responsible for
regulating transmembrane cation influx
(Ca2+/Mg2+) and downstream signaling pathways
respectively. Moreover, TRPM7 kinase activity can influence its
channel function (18). Hence,
TRPM7 is a remarkable cation channel with unique kinase activity.
TRPM7 channels are highly expressed in CFs and are the major
Ca2+-conducting channels in mouse and human CFs
(1). Ca2+ signaling is
closely implicated in the initiation and development of cardiac
fibrosis. Thus, it has been suggested that TRPM7 channels are
indispensable for the pro-fibrogenic effect of CFs. Although a
variety of studies have explored the relationship between the
cation channel TRPM7 and cardiac fibrosis, the present study
systematically described for the first time that TRPM7 channels
participate in the cardiac fibrosis process via mediating
functional changes in CFs. Our primary findings in the present
study are as follows. First, we confirmed that the Ca2+
permeable TRPM7 cation channels were expressed in dissociated
primary CFs from 1- to 3-day-old SD rat pup hearts. Ang II
time-dependently increased TRPM7 mRNA and protein levels and
amplified TRPM7 inward-outward rectifying currents in CFs.
Secondly, we demonstrated that downregulation of TRPM7 using siRNA
interference markedly reduced the Ang II-elicited increase in TRPM7
expression and TRPM7 current enhancement on the CF membrane.
Thirdly, we clarified that TRPM7 channels play a pivotal role in
Ang II-evoked CF proliferation, differentiation and ECM protein
synthesis and silencing of TRPM7 markedly inhibited Ang II-induced
CF proliferation, differentiation and ECM protein formation
capability. Taken together, our findings suggest that TRPM7
channels act as an essential factor in the Ang II-induced
functional change of CFs associated with cardiac fibrosis. The
pathological process can be summarized as follows. Under normal
physiological conditions, CFs remain quiescent, but are rapidly
activated and produce Ca2+ internal flow on the cell
membrane upon a variety of pathological damages such as myocardium
lesion, pressure overload and oxidative stress. Then they gain
contractibility, become proliferative, differentiate into
myofibroblasts and generate more pro-fibrogenic factors and ECM
proteins ultimately resulting in cardiac fibrosis.
TRPM7 modulates numerous cellular processes in
particular the mediation of cell proliferation. As reported
previously, TRPM7 was found to modulate the proliferation of
malignant human glioma cells (19), mouse cortical astrocytes (20), Ang II-induced rat aortic smooth
muscle cells (13) and
ox-LDL-induced vascular smooth muscle cells (21). A review by Thodeti et al
(3) reported that some TRP
channels are associated with CF differentiation. They stated that
TRPC3 and TRPM7 predominantly participate in atrial fibroblast
differentiation whereas TRPV4 and TRPC6 seemingly are mainly
involved in ventricular differentiation (3). However, there appears to be no
investigation concentrated on the role of TRPM7 channels in CF
proliferation and differentiation. In our in vitro study, we
observed that CFs passaged to 3–5 generations exhibited a certain
capacity for proliferation (assessed by monitoring CCK-8 and the
expression level of Ki-67 and PCNA) and differentiation (assessed
by monitoring the expression level of α-SMA using
immunofluorescence assay and western blot technique) which was
markedly increased after Ang II stimulation along with an increase
in TRPM7 channel mRNA and protein expression (Figs. 4 and 5). After intervention with
Ad-TRPM7-siRNA, the increase in Ang II-elicited proliferation and
differentiation in CFs was evidently diminished (Figs. 4 and 5). In addition, using
electrophysiological approaches, we found that the TRPM7 currents
recorded in the CF cell membrane also manifested a similar trend of
change (Fig. 3). These results
provided a direct relation between Ang II-induced CF proliferation,
differentiation and TRPM7 currents and TRPM7 channels and therefore
demonstrated that TRPM7 channels mediate Ang II-induced CF
proliferation and differentiation by regulating TRPM7 currents.
The ECM protein synthesis levels of CFs under
different conditions were previously investigated. Yu and Xu
(22) reported that organ
fibrosis including cardiac fibrosis can be deemed as erroneous ECM
'turnover' i.e. imbalance between ECM production (increased) and
ECM degradation (reduced). Collagens are the most abundant
constructional element of the ECM in the heart, consisting of five
types (types I, III, IV, V and VI) found in the myocardium
(22). Collagen I and III are the
main components of ECM proteins (22). In one of our previous studies, we
verified that addition of Ang II to cultured rat CFs notably
increased the expression of TRPM7 channels as well as TRPM7
currents and the expression of collagen I and III (4). To further observe the relationship
between the ECM protein formation of CFs and TRPM7 channels in the
cell membrane, we treated CFs with siRNA targeting TRPM7 in this
study. Our results demonstrated that the trend in variation among
TRPM7, collagen I and III protein expression levels following
treatment with Ang II were consistent with our previous study. The
increases in collagen I and III protein induced by Ang II were
completely abrogated by TRPM7-siRNA (Fig. 6). This finding together with the
above mentioned findings indicate that extracellular Ang II
activates TRPM7 channels, increases TRPM7 currents, causes CF
proliferation, differentiation, increased ECM protein synthesis and
eventually causes cardiac fibrosis.
In conclusion, the present study provides evidence
that TRPM7 channels are indispensable for Ang II-induced
proliferation, differentiation and collagen synthesis of CFs via
the mediation of TRPM7 currents in the CF cell membrane. More
importantly, our findings suggest that by modulating the functional
variations in CFs associated with cardiac fibrosis, TRPM7 may act
as a promising therapeutic strategy for the treatment of
fibrosis-related cardiac diseases. However, our data also
demonstrated that TRPM7-siRNA not only inhibited Ang II-elicited CF
proliferation, differentiation and collagen synthesis but also
suppressed the proliferation, differentiation and collagen
synthesis of CFs in a basal condition. Thus, we have to highlight
that in normal conditions, we cannot inhibit the expression of
TRPM7 channel and its function in CFs. We can consider inhibiting
the expression and function of TRPM7 channels in CFs in patients
with cardiac fibrosis-related cardiac diseases for therapeutic use
and patients with high risk factors for cardiac fibrosis for
prophylactic use. Our future research efforts are as follows: i) to
identify the downstream signaling pathways involved in the
regulation of the functional changes in CFs by TRPM7 channels; ii)
to identify various cardiac-specific TRPM7 inhibitors for future
therapeutic use; and iii) to carry out a series of in vivo
investigations to identify TRPM7 channels as promising therapeutic
targets for fibrosis-related cardiac diseases.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81170085), the
Natural Science Foundation of Hubei Province (no. 2016CFB162) and
the Fundamental Research Funds for the Central Universities (no.
2042016kf0074).
References
|
1
|
Du J, Xie J, Zhang Z, Tsujikawa H, Fusco
D, Silverman D, Liang B and Yue L: TRPM7-mediated Ca2+
signals confer fibrogenesis in human atrial fibrillation. Circ Res.
106:992–1003. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elnakish MT, Kuppusamy P and Khan M: Stem
cell transplantation as a therapy for cardiac fibrosis. J Pathol.
229:347–354. 2013. View Article : Google Scholar
|
|
3
|
Thodeti CK, Paruchuri S and Meszaros JG: A
TRP to cardiac fibroblast differentiation. Channels (Austin).
7:211–214. 2013. View Article : Google Scholar
|
|
4
|
Zhou Y, Yi X, Wang T and Li M: Effects of
angiotensin II on transient receptor potential melastatin 7 channel
function in cardiac fibroblasts. Exp Ther Med. 9:2008–2012.
2015.PubMed/NCBI
|
|
5
|
Yue L, Xie J and Nattel S: Molecular
determinants of cardiac fibroblast electrical function and
therapeutic implications for atrial fibrillation. Cardiovasc Res.
89:744–753. 2011. View Article : Google Scholar :
|
|
6
|
Berridge MJ, Bootman MD and Roderick HL:
Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev
Mol Cell Biol. 4:517–529. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
González A, López B and Díez J: Fibrosis
in hypertensive heart disease: Role of the
renin-angiotensin-aldosterone system. Med Clin North Am. 88:83–97.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Olson ER, Shamhart PE, Naugle JE and
Meszaros JG: Angiotensin II-induced extracellular signal-regulated
kinase 1/2 activation is mediated by protein kinase Cδ and
intracellular calcium in adult rat cardiac fibroblasts.
Hypertension. 51:704–711. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Manabe I, Shindo T and Nagai R: Gene
expression in fibroblasts and fibrosis: Involvement in cardiac
hypertrophy. Circ Res. 91:1103–1113. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Runnels LW, Yue L and Clapham DE:
TRP-PLIK, a bifunctional protein with kinase and ion channel
activities. Science. 291:1043–1047. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang YH, Sun HY, Chen KH, Du XL, Liu B,
Cheng LC, Li X, Jin MW and Li GR: Evidence for functional
expression of TRPM7 channels in human atrial myocytes. Basic Res
Cardiol. 107:2822012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li M, Jiang J and Yue L: Functional
characterization of homo-and heteromeric channel kinases TRPM6 and
TRPM7. J Gen Physiol. 127:525–537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang M, Zhao T, Lin J, Ju T and Zhang L:
Inhibition of TRPM7 attenuates rat aortic smooth muscle cell
proliferation induced by angiotensin II: Role of genistein. J
Cardiovasc Pharmacol. 66:16–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng J: Molecular mechanism of TRP
channels. Compr Physiol. 3:221–242. 2013.PubMed/NCBI
|
|
15
|
Montell C, Jones K, Hafen E and Rubin G:
Rescue of the Drosophila phototransduction mutation trp by germline
transformation. Science. 230:1040–1043. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nilius B: TRP channels in disease. Biochim
Biophys Acta. 1772:805–812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Visser D, Middelbeek J, van Leeuwen FN and
Jalink K: Function and regulation of the channel-kinase TRPM7 in
health and disease. Eur J Cell Biol. 93:455–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Desai BN, Krapivinsky G, Navarro B,
Krapivinsky L, Carter BC, Febvay S, Delling M, Penumaka A, Ramsey
IS, Manasian Y, et al: Cleavage of TRPM7 releases the kinase domain
from the ion channel and regulates its participation in Fas-induced
apoptosis. Dev Cell. 22:1149–1162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leng TD, Li MH, Shen JF, Liu ML, Li XB,
Sun HW, Branigan D, Zeng Z, Si HF, Li J, et al: Suppression of
TRPM7 inhibits proliferation, migration, and invasion of malignant
human glioma cells. CNS Neurosci Ther. 21:252–261. 2015. View Article : Google Scholar
|
|
20
|
Zeng Z, Leng T, Feng X, Sun H, Inoue K,
Zhu L and Xiong ZG: Silencing TRPM7 in mouse cortical astrocytes
impairs cell proliferation and migration via ERK and JNK signaling
pathways. PLoS One. 10:e01199122015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin J, Zhou S, Zhao T, Ju T and Zhang L:
TRPM7 channel regulates ox-LDL-induced proliferation and migration
of vascular smooth muscle cells via MEK-ERK pathways. FEBS Lett.
590:520–532. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu LM and Xu Y: Epigenetic regulation in
cardiac fibrosis. World J Cardiol. 7:784–791. 2015. View Article : Google Scholar : PubMed/NCBI
|