Introduction
Atherosclerosis (AS) is a complex process involving
numerous cell types and important cell-to-cell interactions that
ultimately lead to progression from the 'fatty streak' to formation
of more complex atherosclerotic plaques (1–4).
The initiation of AS is multifactorial and caused by a collection
of risk factors (3–5). The precise initiating event is
unknown; however, dysfunction within the endothelium is thought to
be an important early contributor and results in the earliest
detectable changes in the life history of an atherosclerotic lesion
(1,5–9).
The endothelium is crucial for maintenance of vascular homeostasis,
ensuring that a balance remains between vasoactive factors
controlling its permeability, adhesiveness, and integrity (1,5,7).
Endothelial cell (EC) activation or injury by a variety of stimuli
including pro-inflammatory cytokines, certain bacterial endotoxins
and hemodynamic factors, leads to local thrombosis, loss of vessel
barrier function, and rapid and robust leukocyte recruitment
(5,10,11). If unchecked, these alterations can
contribute to cardiovascular diseases including AS,
ischemia/reperfusion injury, rheumatoid arthritis and allograft
rejection (5,6). One of the important links between AS
and pro-inflammatory endothelial activation is the intrinsic
capacity of activated vascular endothelium to synthesize and
secrete chemokines, such as tumor necrosis factor-α (TNF-α),
interleukin-1β (IL-1β), thus generating localized, intercellular
autocrine and paracrine signaling loops within the vessel wall
(1,12,13). Indeed, cytokines are produced by
and act on almost all cells involved in the pathogenesis of AS,
participating in all steps of the process, from the early
endothelial dysfunction to the late formation and disruption of a
vulnerable plaque (14,15). Thus detecting early injury or
pro-inflammatory endothelial activation will shed light on
mechanisms that are responsible for inflammation/injury-initiated
AS.
In recent years, it has been shown that many
cellular regulatory processes depend on the post-translational
functions of ubiquitin and ubiquitin-like proteins (Ubls),
including transcription, DNA repair, signal transduction,
autophagy, cell proliferation, differentiation, apoptosis,
endoplasmic reticulum (ER) regulation, inflammation, antigen
processing and stress responses (16–18). Ubiquitin-fold modifier 1 (Ufm1)
has recently been identified as a novel Ubl with a molecular mass
of 9.1 kDa, and it appears to have a similar tertiary structure to
ubiquitin, despite having little (16%) amino acid sequence identity
(19). Similar to the process of
protein ubiquitination, Ufm1 is first synthesized in a pro-form and
is cleaved at the C-terminus by the specific cysteine proteases,
UfSP1 and UfSP2, to expose the conserved glycine residue that is
essential for its subsequent conjugating reactions (20,21). The mature form of Ufm1 is
activated via an E1-activating enzyme, Uba5, and is then conjugated
by an E2 enzyme, Ufc1. With the assistance of E3 ligase, Ufm1 is
presumed to modify its protein targets (19,21,22). These findings suggest that protein
ufmylation is orchestrated through a sophisticated enzymatic
pathway, and it represents a novel potential mechanism to regulate
protein interaction, localization and function (21).
Ufm1 and its system have been demonstrated to play a
significant role in erythroid differentiation (22), cellular growth and development and
ER functions (21). It is also
suggested that Ufm1 is involved in pathological conditions or
diseases, such as tumorigenesis (23–25), ischemic heart diseases (26) and diabetes (27). Despite, to date, the function of
Ufm1 remains poorly understood. Previously, both our group
(28) and other studies (27,29) indicate that Ufm1 is involved in ER
stress, which is a key process of macrophage differentiation and
cholesterol deposition in the development of AS (30–32). Our follow-up study also showed
that Ufm1 is markedly upregulated under AS conditions and Ufm1
suppresses foam cell formation via the LXRα-dependent pathway
(33).
Although the relationship of Ufm1 in ER stress
induced-macrophage foam cells, the late stage of AS, has been
uncovered, its potential role in initial EC dysfunction or
inflammatory responses, the early event in AS, remains unclear. The
expression pattern is also unknown. Here, in this study, we
evaluated Ufm1 expression in human umbilical vein endothelial cells
(HUVECs), as well as the effects of Ufm1 on lipopolysaccharide
(LPS)-induced HUVEC inflammation. We found that Ufm1 was expressed
in HUVECs and localized in the nucleus and cytoplasm. In
LPS-induced HUVEC inflammation, Ufm1 mRNA and protein levels were
upregulated. Moreover, Ufm1 overexpression significantly inhibited
the expression of LPS-induced inflammatory cytokines by targeting
nuclear factor-κB (NF-κB) nuclear translocation. Thus, our results
demonstrated that Ufm1 plays a significant role in suppressing
inflammatory responses via the NF-ĸB signaling pathway.
Materials and methods
Cell culture and treatment
HUVECs were obtained from the American Type Culture
Collection (ATCC, Rockville, MD, USA) and cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin.
The cells were incubated in a humidified incubator at 37°C with 5%
CO2. When the cells reached ~70–80% confluency, they
were dissociated with trypsinization and subcultured. All of the
transient transfections were performed with Lipofectamine 2000
reagent (Invitrogen). For the LPS stimulation, the HUVECs were
treated with 100 ng/ml LPS at a final concentration at the
indicated times.
Construction of the Ufm1 plasmid
The Ufm1 cDNA sequence was obtained from 293 cells
(obtained from ATCC) by reverse transcription of 293 cell mRNA and
PCR amplification as previously described (33). Then the Ufm1 sequence was
amplified and cloned into the pcDNA3.1 vector, along with a Flag
tag in its N-terminus.
Immunofluorescence
For the immunofluorescence assay, the HUVECs were
dissociated into single cells and the dissociated single HUVEC cell
suspension was plated onto chamber slides in a 24-well plate. After
being cultured for 2 days in complete medium, the HUVECs were then
fixed for 30 min at 4°C in 4% paraformaldehyde (PFA). Then the
cells were blocked in 1X phosphate-buffered saline (PBS) plus 0.3%
Triton X-100 and 10% normal donkey serum (122346; Jackson
ImmunoResearch, West Grove, PA, USA) for 60 min at room temperature
(RT), followed by incubation at 4°C overnight in primary antibodies
(diluted in blocking buffer). After 3 washes (10 min for each wash)
in 1X PBS plus 0.1% Triton X-100, the cells were incubated in the
fluorescence-conjugated secondary antibodies (1:500 dilution in 1X
PBS) plus DAPI for 1 h at RT. After 3 times washes, the chamber
slides were then mounted with mounting medium and imaged. Rabbit
anti-Ufm1 antibody (1:1,000; ab109305; Abcam, Cambridge, MA, USA)
was used as the primary antibody. Donkey anti-rabbit IgG conjugated
with AlexaFluor 594 (1:500; Life Technologies, Grand Island, NY,
USA) was used as the secondary antibody. For the negative control,
PBS was used to replace the primary antibody and other procedures
were identical. Images were captured using a fluorescence
microscope.
Real-time quantitative PCR (qRT-PCR)
Total RNA was extracted from cultured cells using
TRIzol reagent (Invitrogen), according to the manufacturer's
instructions. qRT-PCR was performed using SYBR Premix ExTaq™
(Takara, Tokyo, Japan) and an Applied Biosystems 7500 Fast
real-time PCR system. PCR primer sequences were as follows: Ufm1
forward, 5′-CCTGAAAGTACACCTTTCACAGC-3′ and reverse,
5′-CCAGCAGTCTGTGCAGGATT-3′; TNF-α forward,
5′-ATTGCCCTGTGAGGAGGAC-3′ and reverse, 5′-TGAGCCAGAAGAGGTTGAGG-3′;
IL-6 forward, 5′-CTTCGGTCCAGTTGCCTTCT-3′ and reverse,
5′-GTGAGTGGCTGTCTGTGTGG-3′; IL-12 forward,
5′-CTTGTGGCTACCCTGGTCCT-3′ and reverse, 5′-GAGTTTGTCTGGCCTTCTGG-3′;
IL-1β forward, 5′-GGATATGGAGCAACAAGTGG-3′ and reverse,
5′-ATGTACCAGTTGGGGAACTG-3′; monocyte chemoattractant protein-1
(MCP-1) forward, 5′-CCAATTCTCAAACTGAAGCTCGC-3′ and reverse
5′-CTTAGCTGCAGATTCTTGGGTTGTG-3′; and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) forward, 5′-TGCACCACCAACTGCTTAGC-3′ and
reverse, 5′-GGCAT GGACTGTGGTCATGAG-3′. Individual samples were run
in triplicate, and each experiment was repeated at least three
times. The 2−ΔΔCq method was used to analyze the
relative changes in gene expression. GAPDH was used as an
endogenous normalization control.
Western blot analysis
Western blot analysis was performed as previously
described (28,33). Briefly, the HUVECs were dissected,
homogenized, and solubilized at 4°C in Cell lysis buffer (P0013;
Beyotime, Shanghai, China) supplemented with 1 mM PMSF, 50 mM NaF,
1 mM Na3VO4 and protease inhibitor. The total
protein lysates were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed
by western blotting. The following antibodies were used: anti-Ufm1
antibody (1:1,000; ab109305; Abcam), anti-nuclear factor-κB (NF-κB)
p65 (1:1,000; 8242; Cell Signaling Technology, Danvers, MA, USA),
anti-GAPDH (1:5,000; G8795; Sigma-Aldrich, St. Louis, MO, USA),
anti-β-actin (1:5,000; MA5-15739; Thermo Fisher Scientific,
Waltham, MA, USA) and anti-histone H3 (1:3,000; 4499; Cell
Signaling Technology). HRP-conjugated anti-rabbit, anti-mouse and
anti-goat secondary antibodies (A0208, A0216 and A0181) were from
Beyotime. Analysis of the data was performed using NIH ImageJ
software. The mean density of each band was normalized to the actin
or GAPDH signal in the same sample.
For the nuclear and cytoplasmic protein separation
experiment (Fig. 5), the protein
in the nucleus and cytoplasm were extracted using a nuclear and
cytoplasmic protein extraction kit (P0028; Beyotime); then as
described above, the nuclear and cytoplasmic protein were analyzed
by western blotting with the indicated antibody. β-actin and
histone H3 were used as loading controls for protein in the
cytoplasm and nucleus respectively.
Statistical analysis
All experiments were performed at least three times
in triplicate. The results are presented as mean ± SEM. Statistical
differences were determined by the Student's t test for two-group
comparisons or ANOVA followed by Tukey's test for multiple
comparisons among more than two groups. P<0.05 was considered
statistically significant.
Results
Expression of Ufm1 in HUVECs
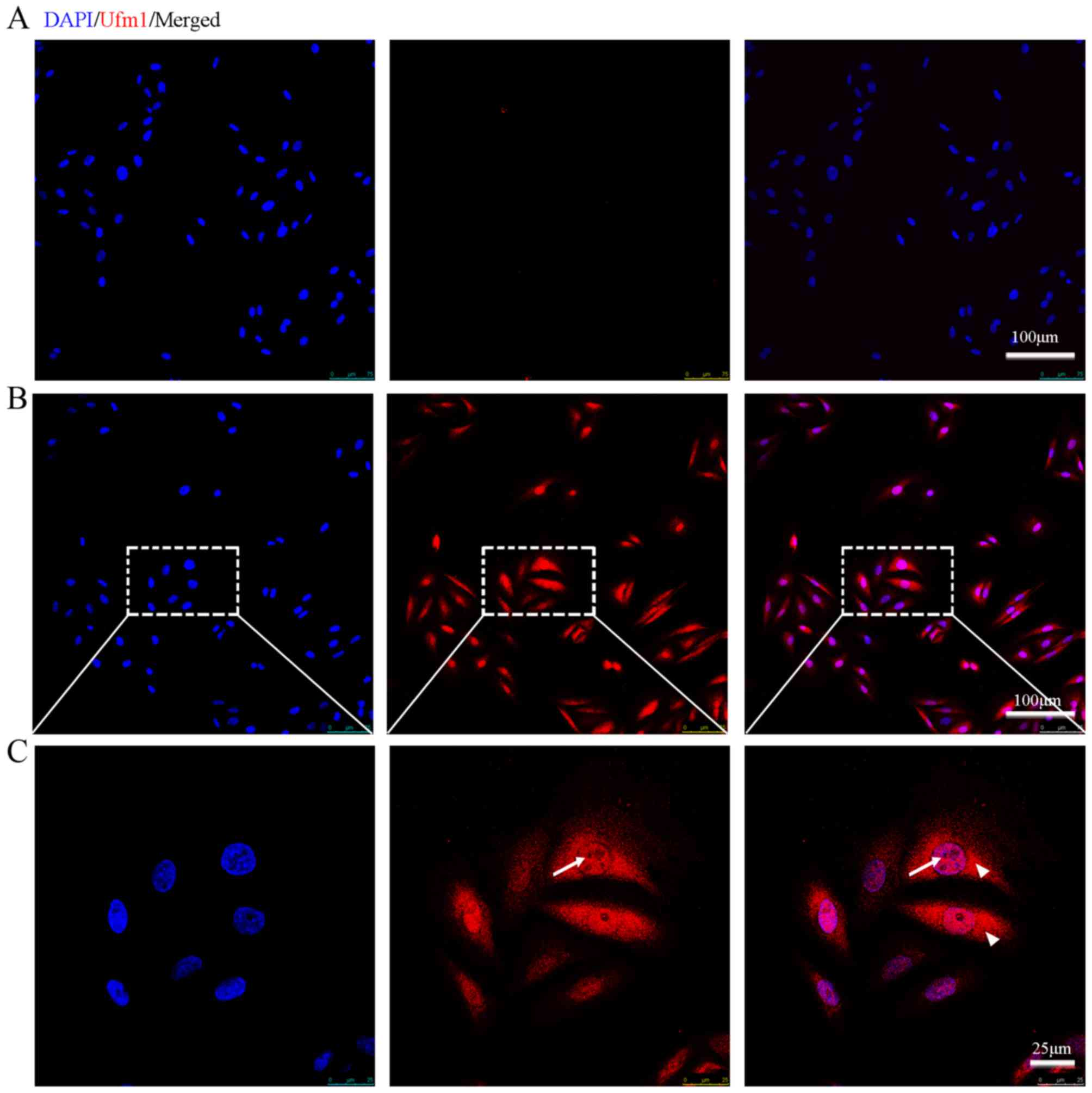
To identify whether Ufm1 is expressed in endothelial
cells, we performed immunofluorescence on the cultured HUVECs with
the Ufm1 antibody. Immunofluorescence assay revealed that
endogenous Ufm1 was highly expressed in the HUVECs and was
localized both in the cytoplasm and the nucleus (as indicated by
its co-localization with DAPI) (Fig.
1).
Ufm1 is involved in LPS-induced
inflammatory responses
Both our previous and other studies have
demonstrated that Ufm1 is involved in ER stress (21,26–29); however, whether Ufm1 participates
in inflammatory responses remains unknown. In this study, to
investigate the association between Ufm1 expression and endothelial
cell injury and inflammatory responses, we examined whether Ufm1
expression is affected in LPS-induced inflammation in HUVECs.
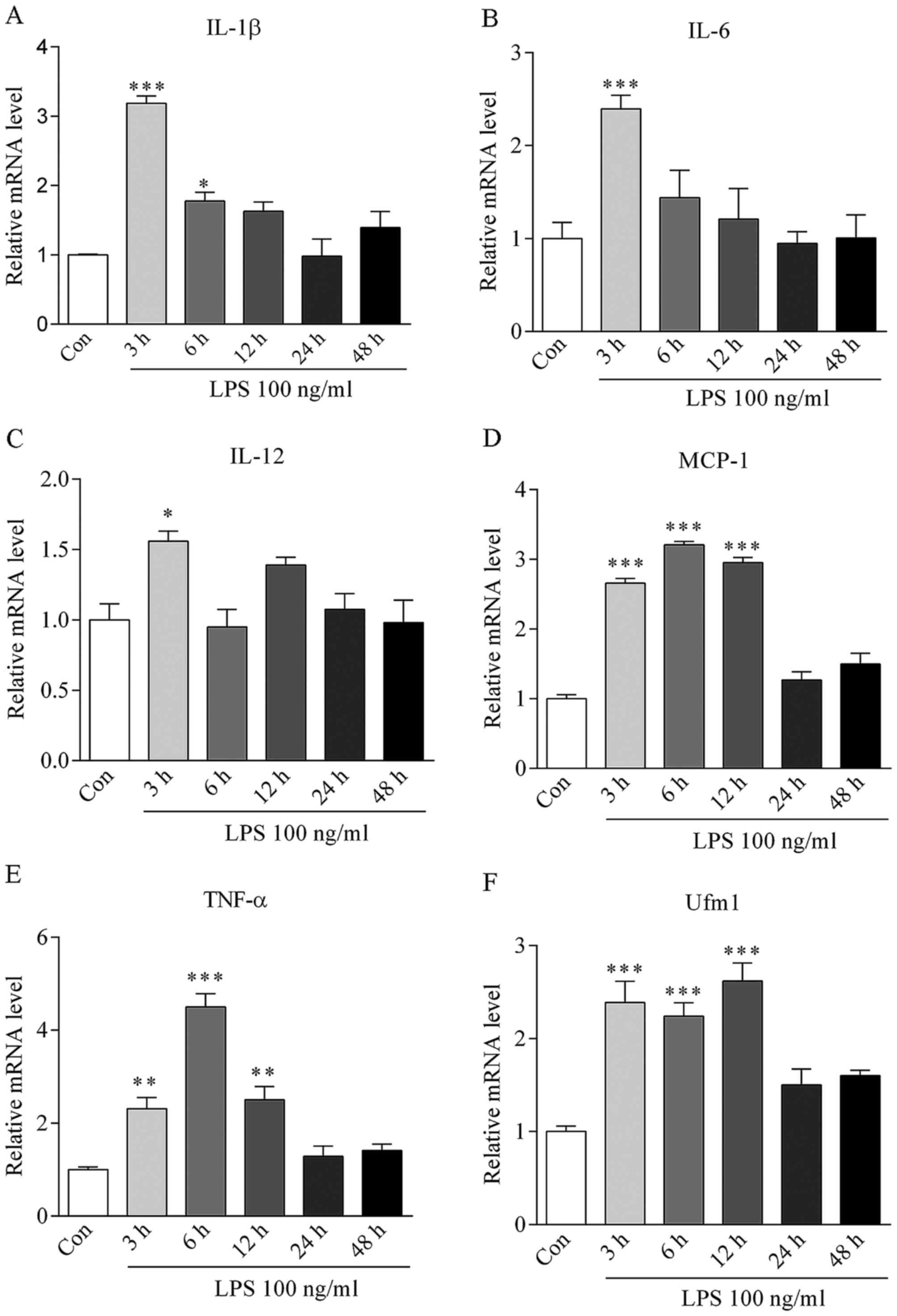
The concentration of 100 ng/ml LPS was used to
induce HUVEC inflammation in vitro. After stimulation for 0,
3, 6, 12, 24 and 48 h, the mRNA levels of inflammatory cytokines
TNF-α, IL-6, IL-1β, IL-12, MCP-1 were markedly increased and
reached a peak at 3–6 h (Fig.
2A–E), suggesting the successful induction of inflammation.
After LPS treatment for 3 h, Ufm1 mRNA expression
was increased by >2.5-fold in the HUVECs (P<0.001 vs.
control) (Fig. 2F). In addition,
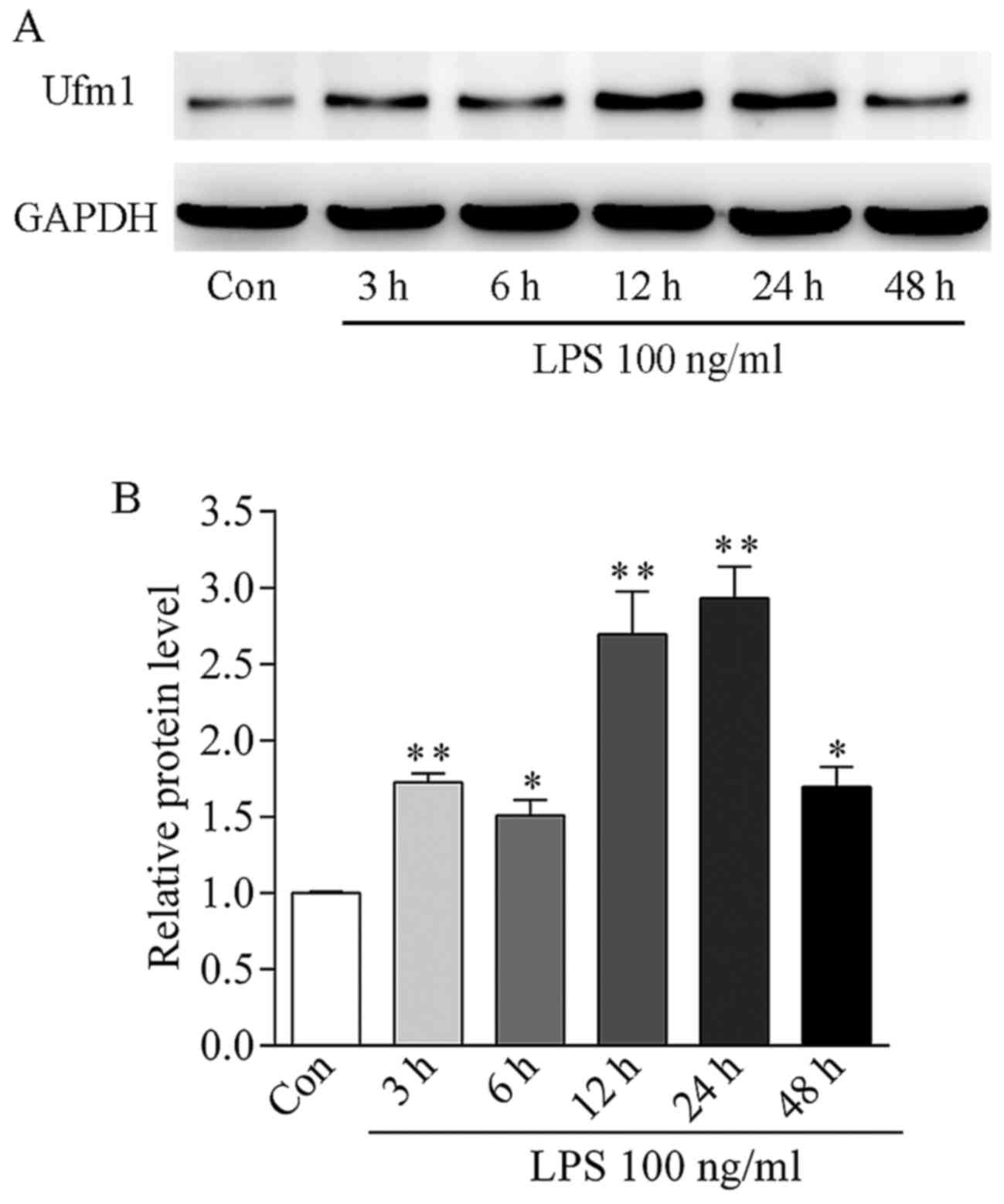
the Ufm1 protein level was gradually increased from 3 to 24 h and
reached a peak level at 24 h, which was ~3-fold high compared to
the control (Fig. 3).
Together, our data suggest that an upregulation in
Ufm1 expression may be associated with LPS-induced HUVEC
inflammatory responses.
Overexpression of Ufm1 inhibits
LPS-induced inflammation in HUVECs
To detect the potential role of Ufm1 in inflammatory
responses, we first constructed a Ufm1 overexpression plasmid.
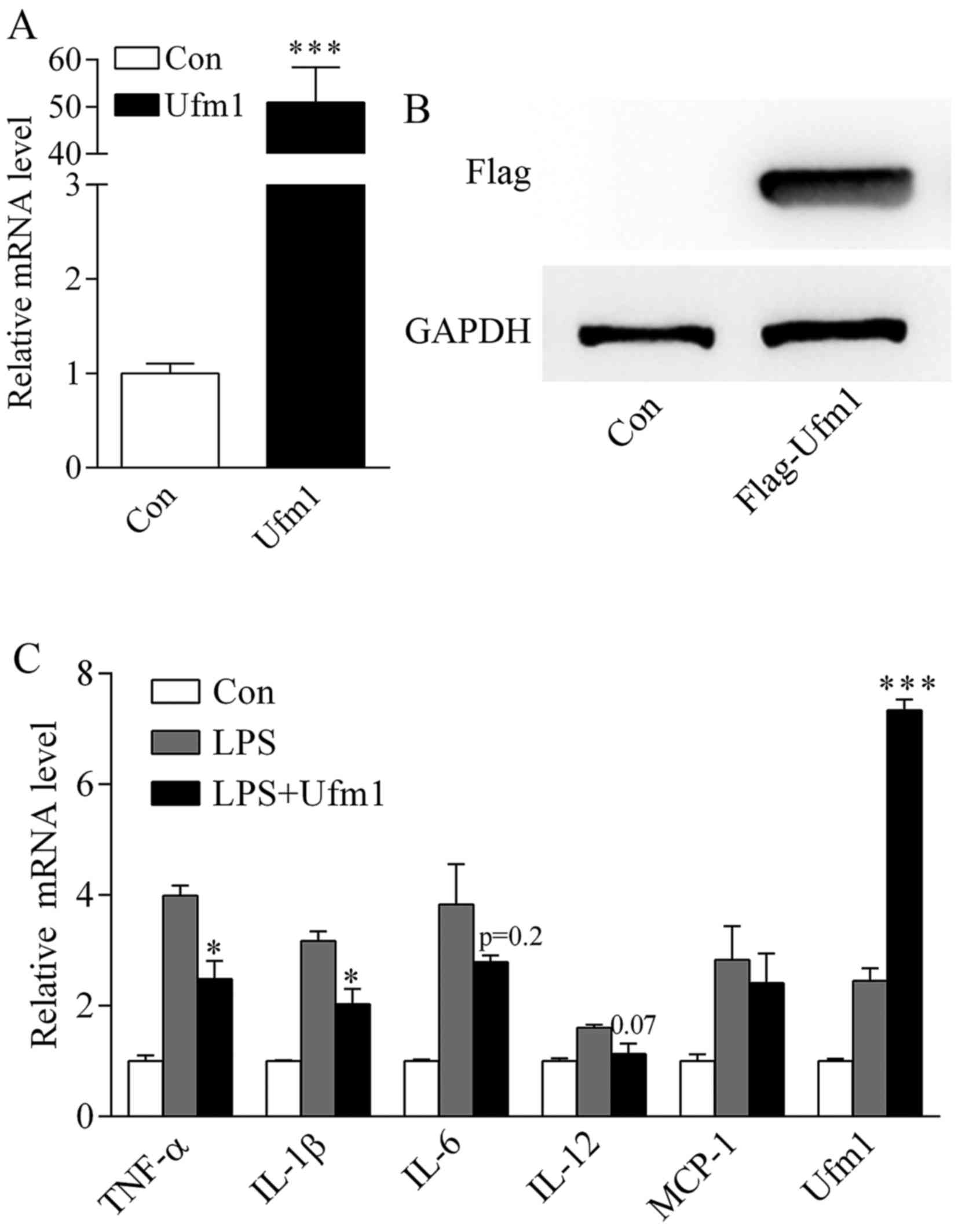
After transfection into HUVECs, the mRNA level of Ufm1 was
significantly increased compared to the control (transfected with
the plasmid vector) (Fig. 4A). We
also confirmed its protein expression by western blotting (WB)
using an anti-flag antibody (Fig.
4B).
Since Ufm1 is involved in LPS-induced inflammation
(Figs. 2 and 3), it prompted us to further investigate
its role in inflammatory responses. After transfection with the
Ufm1 overexpression plasmid or vector, the HUVECs were treated with
100 ng/ml LPS as mentioned above. The mRNA level of cytokines were
determined by real-time PCR. In comparison to the control, LPS
significantly increased the inflammatory cytokine expression
(Fig. 4C). After Ufm1
overexpression, the expression of the inflammatory cytokines was
markedly reduced, especially TNF-α, IL-1β and IL-12 (Fig. 4C).
Our data indicated that overexpression of Ufm1 can
inhibit LPS-induced inflammatory responses in HUVECs.
Ufm1 suppresses inflammatory responses
via the LPS-induced NF-κB pathway in HUVECs
As Ufm1 is involved in inflammation and Ufm1
overexpression can inhibit LPS-induced inflammatory responses in
HUVECs (Figs. 2 and 4), it raised the question of how Ufm1
exerts its function. Given the fact that LPS-induced inflammation
is through Toll-like receptor (TLR) signaling, and that NF-κB is
activated and enters into the nucleus to regulate the induced
transcription of pro-inflammatory genes (34), a process known as NF-κB nuclear
translocation, we analyzed the effect of Ufm1 on the cellular
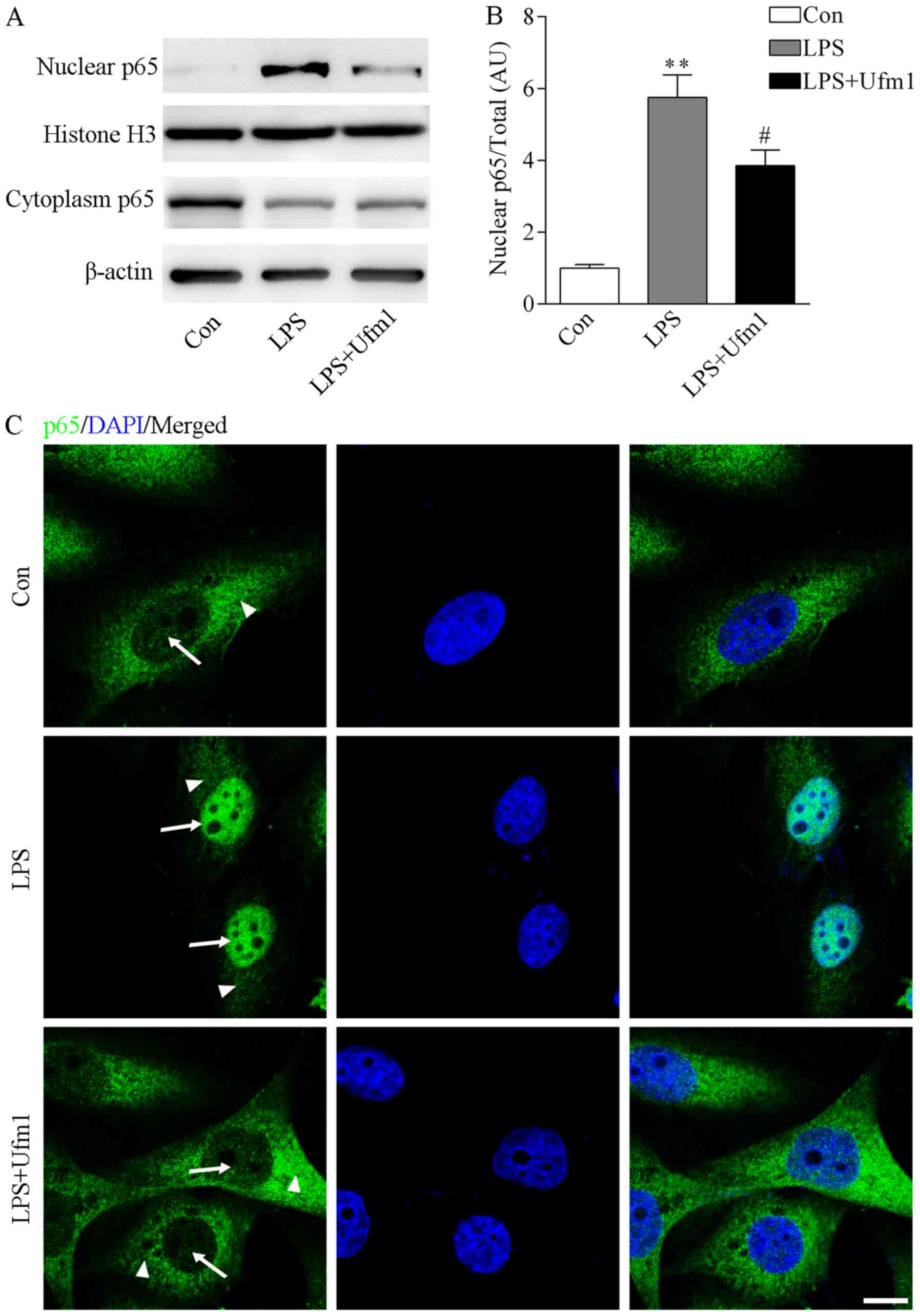
distribution of NF-κB. The cytoplasmic and nuclear proteins were
separated as indicated by β-actin and histone H3. Upon LPS
treatment, the protein level of NF-κB in the nucleus was
significantly increased while it was decreased in the cytoplasm
(Fig. 5A and B). Interestingly,
we found that Ufm1 overexpression reversed the role of LPS,
resulting in decreased expression of NF-κB in the nucleus (Fig. 5A and B). Consistently,
immunofluorescence staining of p65 also indicated that upon LPS
treatment, the NF-κB protein translocated to the nucleus (Fig. 5C). However, Ufm1 overexpression
abolished the phenotype induced by LPS, resulting in decreased
expression of NF-κB in the nucleus (Fig. 5C). Based on these findings, we
propose that Ufm1 can decrease the nuclear trans-location of NF-κB,
thus further inhibiting the expression of target inflammatory
cytokines.
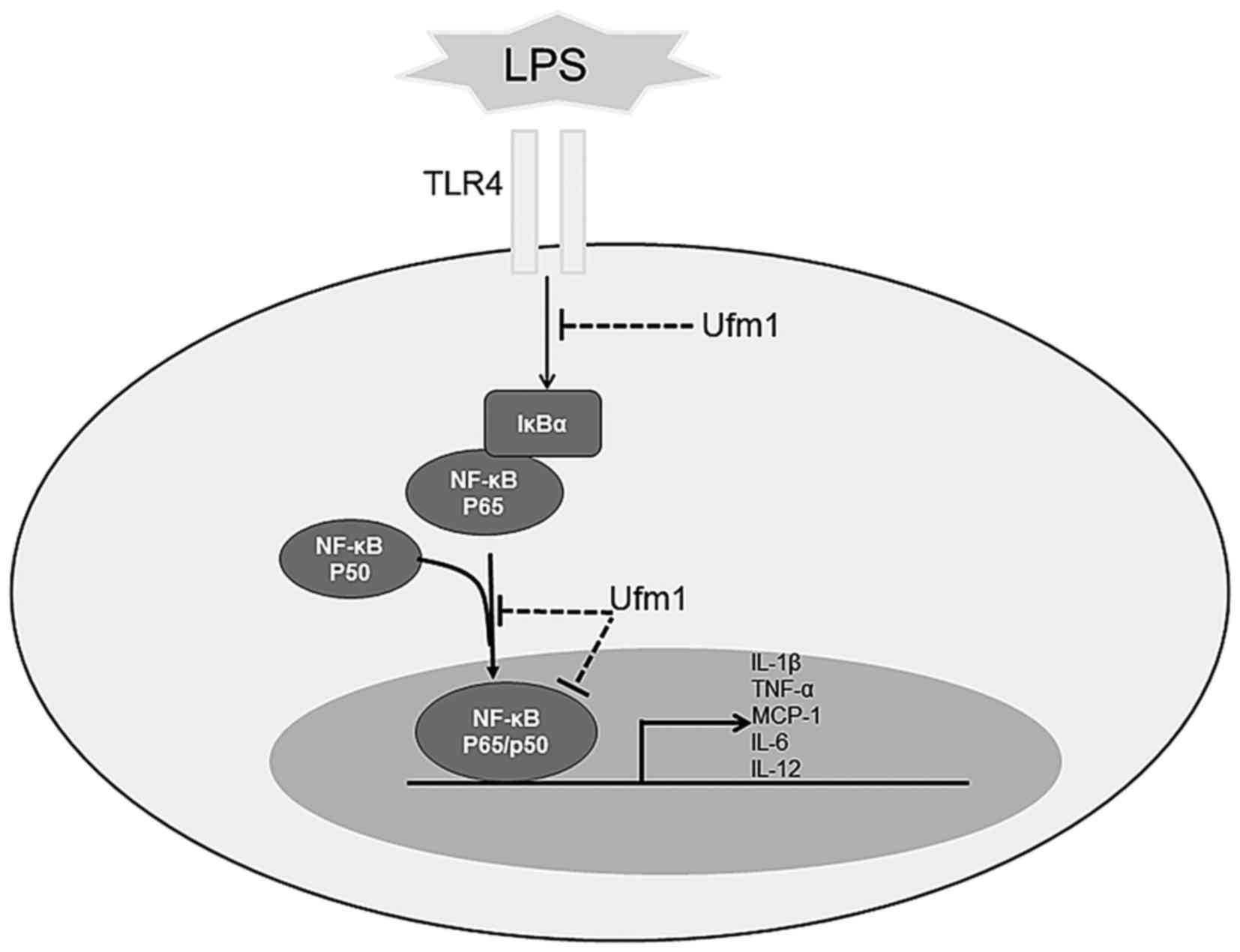
Discussion
In the present study, we investigated the expression
pattern and potential biological function of Ufm1 in ECs. We found
that Ufm1 was expressed in both the nucleus and the cytoplasm of
HUVECs. Along with the increase in inflammatory cytokines, Ufm1
expression was markedly upregulated in response to the inflammatory
response induced by LPS in the HUVECs. Further investigation
revealed that overexpression of Ufm1 attenuated the LPS-induced
inflammatory response. More importantly, we found that Ufm1
inhibited the expression of inflammatory cytokines through the
LPS-induced NF-κB pathway (Fig.
6).
Ufm1 is a new member of the Ubl family, whose
biological functions are poorly understood, particularly in ECs. It
has been reported that Ufm1 is expressed in many tissues and cell
lines (19,27) and that upregulation of Ufm1
expression is linked with the activation of the ER stress response
in AS and diabetes (27,28,33,35). Our previous studies also
demonstrated that Ufm1 is involved in AS conditions (28,33). In addition, we found that Ufm1
protects against ER stress-induced apoptosis in macrophages as well
as oxidized low-density lipoprotein (oxLDL)-induced foam cells
(28,33), both of which are key regulators in
macrophage differentiation and cholesterol deposition in the
development of AS (30–32). Here, we investigate Ufm1
expression and further uncovered a novel role of Ufm1 particularly
in EC inflammation, which provides new insight into the early stage
of AS.
LPS, a unique glycolipid contained in the outer wall
of Gram-negative bacteria, can cause endothelial dysfunction and
inflammation, which have been demonstrated to be associated with AS
(36–38). In this study, we used 100 ng/ml
LPS to induce inflammation and endothelial dysfunction. After LPS
treatment, Ufm1 expression was upregulated, together with an
upregulation of the expression of inflammatory genes including
TNF-α, IL-6, IL-1β, IL-12 and MCP-1 (Fig. 2), suggesting that Ufm1 is involved
in EC inflammation. The LPS-TLR4/NF-κB signaling pathway is a major
player in the regulation of diverse biological processes, including
immune responses, cell proliferation and inflammation (34,39–41). NF-κB is a complex of dimeric
subunits that belong to the NF-κB/Rel family: NF-κB1 (p50/p105),
NF-κB2 (p52/p100), p65 (RelA), RelB and c-Rel (34,39). Normally, NF-κB is inactive and
resides in the cytoplasm, where it is sequestered by inhibitors of
κB (IκB), of which the most important are IκBα, IκBβ and IκBε
(34,39,40). When cells are stimulated by
cytokines or LPS, they bind to TLR4 assisted by other proteins.
Then the TLR4 protein complex initiates recruitment of other
proteins (42,43), leading to the phosphorylation of
IκBs at specific serine residues by IκB kinase (39,44). Upon phosphorylation and
ubiquitin-dependent degradation of IκBα, NF-κB translocates to the
nucleus and functions as a transcription factor (39). These events lead to the activation
of the NF-κB cascade for the inflammatory response.
In this study, we found that after LPS treatment,
NF-κB was activated and translocated to the nucleus (Fig. 5), together with upregulation of
expression of its target inflammatory genes including TNF-α, IL-6,
IL-1β, IL-12 and MCP-1 (Fig. 2).
Moreover, overexpression of Ufm1 prevented the NF-κB nuclear
translocation indicated by the decreased ratio of p65 in the
nucleus to total (Fig. 5),
resulting in decreased pro-inflammatory cytokine expression
(Fig. 4). Together, our data
indicated that Ufm1 can inhibit pro-inflammatory responses through
the LPS-TLR4/NF-κB pathway in HUVECs. In addition, we also found
that in response to LPS treatment, Ufm1 expression was upregulated
(Figs. 2F and 3), suggesting a compensatory response in
EC inflammation. This is consistent with our previous study that
Ufm1 is also increased in ER stress and its overexpression plays a
protective role in suppressing foam cell formation and ER stress
(28,33).
Recently, Ufm1 modification (ufmylation) has emerged
as a novel post-translational modification that plays essential
roles in several physiological and pathological processes (21). However, there are still
limitations to our study. Because of the difficulty in identifying
substrate proteins, the study of the Ufm1 cascade is still in its
infancy and its functions are yet to be completely understood.
Regarding the fact that upon phosphorylation and
ubiquitin-dependent degradation of IκBα, NF-κB translocates to the
nucleus and functions as a transcription factor (34,39), it is possible that IκBα may be a
potential substrate of Ufm1 and Ufm1 inhibits the NF-κB nuclear
translocation through IκBα ufmylation. Or possibly NF-κB is the
target of Ufm1 and ufmylated NF-κB can inhibit its role in
promoting the transcription of target gene expression. Still we
cannot exclude other possibilities, since there are other
substrates of Ufm1 which may serve as a bridge in mediating Ufm1 to
NF-κB translocation. For example, in a recent study, Yoo et
al found that Ufm1 may also ufmylate LZAP (24), which is a putative tumor
suppressor and inhibits the NF-κB pathway (21,45,46). Thus, identification of the precise
ufmylated substrates involved in LPS-induced EC inflammation will
help us to better understand the role of Ufm1 in the initiation and
progression of AS. How Ufm1 exerts its role in the NF-κB pathway,
identification of ufmylated substrates and whether subsequent
ufmylation participates in the protective action of Ufm1 against
pro-inflammatory response in ECs require further investigation.
In conclusion, this study found that Ufm1 is
expressed in HUVECs and is localized in the nucleus and cytoplasm.
Furthermore, our study provides new insight into the
anti-inflammatory properties of Ufm1, which reduces
pro-inflammatory cytokine expression by regulating the NF-ĸB
signaling pathway. On the basis of our in vitro findings, we
anticipate that Ufm1 may serve as a potential molecular target for
developing novel therapeutic strategies that protect ECs from
dysfunction, suppress inflammation and subsequently suppress the
initial occurrence and development of AS. Moreover, in vivo
studies exploring the functions of Ufm1 in AS are still needed to
provide further evidence for the importance of Ufm1 in the
pathogenesis of AS.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81270908).
References
|
1
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 118:620–636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Libby P, Ridker PM and Hansson GK; Leducq
Transatlantic Network on Atherothrombosis: Inflammation in
atherosclerosis: From pathophysiology to practice. J Am Coll
Cardiol. 54:2129–2138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ross R: Atherosclerosis is an inflammatory
disease. Am Heart J. 138:S419–S420. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Otsuka F, Finn AV, Yazdani SK, Nakano M,
Kolodgie FD and Virmani R: The importance of the endothelium in
atherothrombosis and coronary stenting. Nat Rev Cardiol. 9:439–453.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pober JS and Sessa WC: Evolving functions
of endothelial cells in inflammation. Nat Rev Immunol. 7:803–815.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rao RM, Yang L, Garcia-Cardena G and
Luscinskas FW: Endothelial-dependent mechanisms of leukocyte
recruitment to the vascular wall. Circ Res. 101:234–247. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stary HC: Natural history and histological
classification of atherosclerotic lesions: An update. Arterioscler
Thromb Vasc Biol. 20:1177–1178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Virmani R, Kolodgie FD, Burke AP, Farb A
and Schwartz SM: Lessons from sudden coronary death: A
comprehensive morphological classification scheme for
atherosclerotic lesions. Arterioscler Thromb Vasc Biol.
20:1262–1275. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gimbrone MA Jr: Endothelial dysfunction,
hemodynamic forces, and atherosclerosis. Thromb Haemost.
82:722–726. 1999.PubMed/NCBI
|
|
11
|
Zhang C: The role of inflammatory
cytokines in endothelial dysfunction. Basic Res Cardiol.
103:398–406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pober JS, Bevilacqua MP, Mendrick DL,
Lapierre LA, Fiers W and Gimbrone MA Jr: Two distinct monokines,
interleukin 1 and tumor necrosis factor, each independently induce
biosynthesis and transient expression of the same antigen on the
surface of cultured human vascular endothelial cells. J Immunol.
136:1680–1687. 1986.PubMed/NCBI
|
|
13
|
Libby P, Ordovas JM, Auger KR, Robbins AH,
Birinyi LK and Dinarello CA: Endotoxin and tumor necrosis factor
induce interleukin-1 gene expression in adult human vascular
endothelial cells. Am J Pathol. 124:179–185. 1986.PubMed/NCBI
|
|
14
|
Tedgui A and Mallat Z: Cytokines in
atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev.
86:515–581. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tousoulis D, Oikonomou E, Economou EK,
Crea F and Kaski JC: Inflammatory cytokines in atherosclerosis:
Current therapeutic approaches. Eur Heart J. 37:1723–1732. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hochstrasser M: Origin and function of
ubiquitin-like proteins. Nature. 458:422–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kerscher O, Felberbaum R and Hochstrasser
M: Modification of proteins by ubiquitin and ubiquitin-like
proteins. Annu Rev Cell Dev Biol. 22:159–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weissman AM: Themes and variations on
ubiquitylation. Nat Rev Mol Cell Biol. 2:169–178. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Komatsu M, Chiba T, Tatsumi K, Iemura S,
Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T and Tanaka K: A
novel protein-conjugating system for Ufm1, a ubiquitin-fold
modifier. EMBO J. 23:1977–1986. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kang SH, Kim GR, Seong M, Baek SH, Seol
JH, Bang OS, Ovaa H, Tatsumi K, Komatsu M, Tanaka K, et al: Two
novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1
and UfSP2. J Biol Chem. 282:5256–5262. 2007. View Article : Google Scholar
|
|
21
|
Daniel J and Liebau E: The ufm1 cascade.
Cells. 3:627–638. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tatsumi K, Sou YS, Tada N, Nakamura E,
Iemura S, Natsume T, Kang SH, Chung CH, Kasahara M, Kominami E, et
al: A novel type of E3 ligase for the Ufm1 conjugation system. J
Biol Chem. 285:5417–5427. 2010. View Article : Google Scholar :
|
|
23
|
Shiwaku H, Yoshimura N, Tamura T, Sone M,
Ogishima S, Watase K, Tagawa K and Okazawa H: Suppression of the
novel ER protein Maxer by mutant ataxin-1 in Bergman glia
contributes to non-cell-autonomous toxicity. EMBO J. 29:2446–2460.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW,
Lee SW, Ka SH, Sou YS, Komatsu M, Tanaka K, et al: Modification of
ASC1 by UFM1 is crucial for ERα transactivation and breast cancer
development. Mol Cell. 56:261–274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim CH, Nam HS, Lee EH, Han SH, Cho HJ,
Chung HJ, Lee NS, Choi SJ, Kim H, Ryu JS, et al: Overexpression of
a novel regulator of p120 catenin, NLBP, promotes lung
adenocarcinoma proliferation. Cell Cycle. 12:2443–2453. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Azfer A, Niu J, Rogers LM, Adamski FM and
Kolattukudy PE: Activation of endoplasmic reticulum stress response
during the development of ischemic heart disease. Am J Physiol
Heart Circ Physiol. 291:H1411–H1420. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lemaire K, Moura RF, Granvik M,
Igoillo-Esteve M, Hohmeier HE, Hendrickx N, Newgard CB, Waelkens E,
Cnop M and Schuit F: Ubiquitin fold modifier 1 (UFM1) and its
target UFBP1 protect pancreatic beta cells from ER stress-induced
apoptosis. PLoS One. 6:e185172011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu X, Pang Q, Shen Q, Liu H, He J, Wang J,
Xiong J, Zhang H and Chen F: Ubiquitin-fold modifier 1 inhibits
apoptosis by suppressing the endoplasmic reticulum stress response
in Raw264.7 cells. Int J Mol Med. 33:1539–1546. 2014.PubMed/NCBI
|
|
29
|
Zhang Y, Zhang M, Wu J, Lei G and Li H:
Transcriptional regulation of the Ufm1 conjugation system in
response to disturbance of the endoplasmic reticulum homeostasis
and inhibition of vesicle trafficking. PLoS One. 7:e485872012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh J, Riek AE, Weng S, Petty M, Kim D,
Colonna M, Cella M and Bernal-Mizrachi C: Endoplasmic reticulum
stress controls M2 macrophage differentiation and foam cell
formation. J Biol Chem. 287:11629–11641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tabas I: The role of endoplasmic reticulum
stress in the progression of atherosclerosis. Circ Res.
107:839–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tabas I: Macrophage apoptosis in
atherosclerosis: Consequences on plaque progression and the role of
endoplasmic reticulum stress. Antioxid Redox Signal. 11:2333–2339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pang Q, Xiong J, Hu XL, He JP, Liu HF,
Zhang GY, Li YY and Chen FL: UFM1 Protects macrophages from
oxLDL-induced foam cell formation through a liver X receptor α
dependent pathway. J Atheroscler Thromb. 22:1124–1140. 2015.
View Article : Google Scholar
|
|
34
|
Tak PP and Firestein GS: NF-kappaB: A key
role in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu H, Yang Y, Allister EM, Wijesekara N
and Wheeler MB: The identification of potential factors associated
with the development of type 2 diabetes: A quantitative proteomics
approach. Mol Cell Proteomics. 7:1434–1451. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang B, Chen H and Fan M: Inhibition of
TLR4 signaling pathway: Molecular treatment strategy of
periodontitis-associated atherosclerosis. Med Hypotheses.
70:614–617. 2008. View Article : Google Scholar
|
|
37
|
Lin MI and Sessa WC: Vascular endothelial
growth factor signaling to endothelial nitric oxide synthase: More
than a FLeeTing moment. Circ Res. 99:666–668. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stoll LL, Denning GM and Weintraub L:
Potential role of endotoxin as a proinflammatory mediator of
atherosclerosis. Arterioscler Thromb Vasc Biol. 24:2227–2236. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baldwin AS Jr: The NF-kappa B and I kappa
B proteins: New discoveries and insights. Annu Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sen R and Baltimore D: Inducibility of
kappa immunoglobulin enhancer-binding protein Nf-kappa B by a
posttranslational mechanism. Cell. 47:921–928. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: A pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fitzgerald KA and Chen ZJ: Sorting out
Toll signals. Cell. 125:834–836. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li H and Sun B: Toll-like receptor 4 in
atherosclerosis. J Cell Mol Med. 11:88–95. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rakonczay Z Jr, Hegyi P, Takács T,
McCarroll J and Saluja AK: The role of NF-kappaB activation in the
pathogenesis of acute pancreatitis. Gut. 57:259–267. 2008.
View Article : Google Scholar
|
|
45
|
Gusarova GA, Wang IC, Major ML,
Kalinichenko VV, Ackerson T, Petrovic V and Costa RH: A
cell-penetrating ARF peptide inhibitor of FoxM1 in mouse
hepatocellular carcinoma treatment. J Clin Invest. 117:99–111.
2007. View Article : Google Scholar
|
|
46
|
Xi P, Ding D, Zhou J, Wang M and Cong YS:
DDRGK1 regulates NF-κB activity by modulating IκBα stability. PLoS
One. 8:e642312013. View Article : Google Scholar
|