1. Introduction
It is known that alcohol-related liver disease is a
major cause of morbidity and mortality worldwide. Chronic alcohol
abuse leads to liver damage, simple steatosis, alcoholic liver
disease (ALD) and alcoholic steatohepatitis (ASH) and sometimes
leads to liver cirrhosis or hepatocellular carcinoma (HCC).
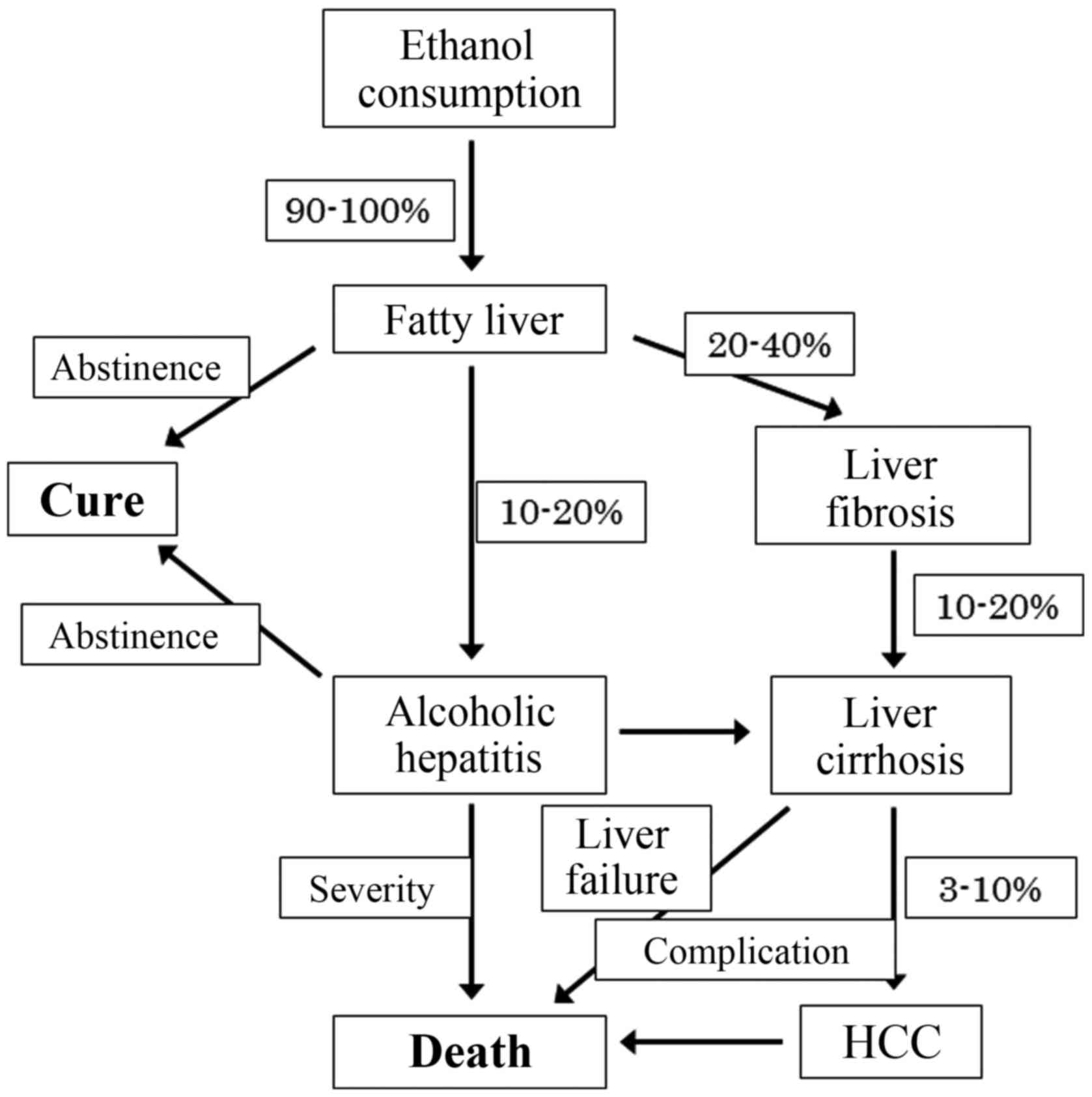
Although most heavy drinkers develop steatosis, 10 to 20% of those
with steatosis progress to ASH and develop cirrhosis (1,2).
Steatosis is usually asymptomic and is rapidly reversible with
abstinence (2). However,
continuous alcohol abuse in some patients leads to liver
inflammation characterized by the infiltration of polymorphonuclear
leukocytes. Hepatocyte damage often described as ballooning or
Mallory bodies is characteristic of ASH. Twenty to forty percent of
the patients develop liver fibrosis, and 10 to 20% of patients
develop cirrhosis with various complications, such as ascites,
variceal bleeding and hepatic encephalopathy (Fig. 1) (3,4).
The prognosis of ALD is poor, especially in liver cirrhosis
(5) or HCC (6). The pathogenesis of ALD is not fully
understood, but some factors, such as the metabolism of alcohol
into toxic products, oxidative stress, acetaldehyde adducts,
abnormal methionine metabolism, malnutrition, endotoxin activation,
and impaired hepatic regeneration, are involved (7). Kupffer cells, macrophages residing
in the liver, play a role in the innate immune system and produce
various cytokines, which may lead to liver disease (8). Tumor necrosis factor-α (TNF-α) is
mainly involved in acute alcoholic liver injury (9). By drinking alcohol, the permeability
of the intestinal membrane is augmented and the portal blood
endotoxin [lipopolysaccharide (LPS)] concentration continuously
increases (10). The Kupffer
cells are activated by phagocytozing apoptotic cells, which
increases their inflammatory cytokine production (11). Chronic alcohol abuse leads to
hepatocyte injury by TNF-α after phagocytosis by Kupffer cells.
Alcohol consumption promotes hepatic inflammation by increasing the
translocation of gut-derived endotoxins and activating Kupffer
cells through Toll-like receptor 4 (TLR4) signaling. ALD is
associated with imbalanced immune responses and increased
production of proinflammatory cytokines or chemokines (12). Hepatoprotective cytokines, such as
interleukin-6 (IL-6), and anti-inflammatory cytokines, such as
IL-10, protect against the development of ALD (13). In contrast, hepatic stellate cells
(HSCs) are the main source of transforming growth factor-β (TGF-β)
which is associated with liver fibrosis. Liver fibrosis can be
defined as a result of progressive accumulation and decreased
remodeling of the extracellular matrix (ECM). ECM remodeling is the
main source of homeostasis during liver fibrosis. This review
mainly discusses the molecular mechanisms associated with liver
inflammation and liver fibrosis that may represent future
therapeutic targets.
2. Metabolism of alcohol
When alcohol is consumed, it passes from the stomach
and intestine into the blood by the process of absorption. Next,
alcohol enters into the liver through the portal vein. The
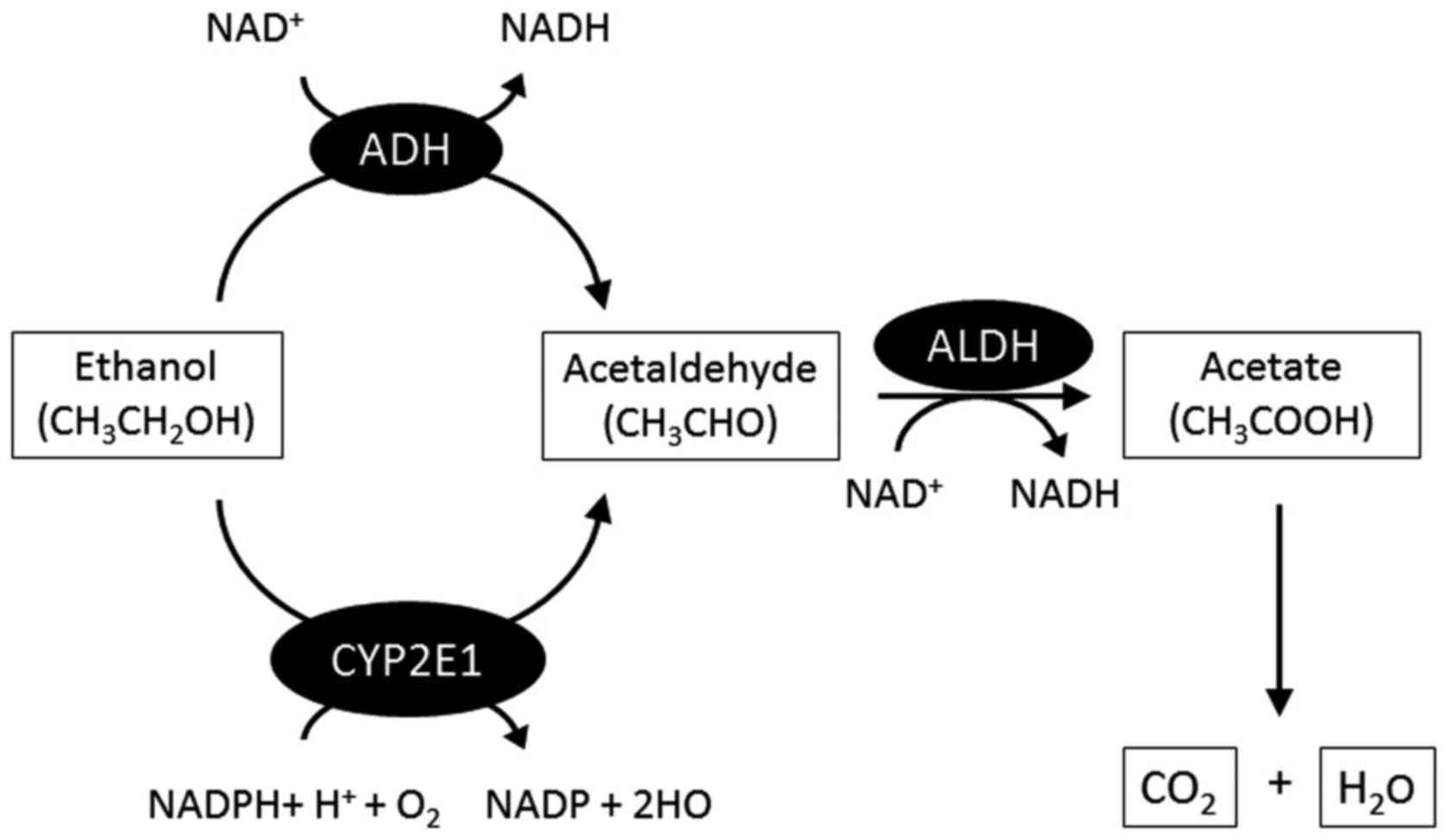
mechanism of alcohol metabolism is shown in Fig. 1. In brief, in the liver, alcohol
dehydrogenase (ADH), the main enzyme in alcohol metabolism
(14), mediates the development
of alcohol to acetaldehyde. Acetaldehyde is rapidly converted to
acetate by acetaldehyde dehydrogenase and is eventually metabolized
to carbon dioxide and water in muscle tissues. The microsomal
ethanol oxidizing system is another pathway of alcohol metabolism,
which is independent of ADH (15). Alcohol is metabolized in the liver
by the enzyme cytochrome P450 2E1 (CYP2E1), which is mainly
expressed in the liver. CYP2E1 is mainly located within the
endoplasmic reticulum, but it is also expressed in mitochondria
(16), and its expression is
increased after chronic alcohol consumption (17), with an increase in acetaldehyde.
Acetaldehyde has a stronger toxicity than ethanol and can lead to
liver injury. Most of the alcohol consumed is metabolized in the
liver, but the small quantity that remains unmetabolized is
excreted in breath and urine (Fig.
2).
3. Cytokines
TNF-α
TNF-α is a cytokine involved in systemic
inflammation and is a member of a cytokine family that stimulates
acute inflammation. TNF-α is mainly produced by Kupffer cells in
the liver and is also an important mediator of inflammation, cell
proliferation and apoptosis (9).
TNF-α functions as a critical inflammatory cytokine in the
progression of ALD (4). However,
the mechanism of the enhancing effect of alcohol on TNF-α has not
yet been clarified. Kupffer cells secrete inflammatory cytokines
(18) and reactive oxygen species
(19), which activate
hepatocytes, HSCs and endothelial cells (20). In alcoholic hepatitis (AH),
inflammatory cytokines, such as TNF-α, IL-6, IL-8 and IL-18 induce
liver injury (21). Serum TNF-α
is increased in patients with ALD and correlates with mortality.
After chronic alcohol consumption, Kupffer cells exhibit enhanced
sensitivity to LPS-stimulated TNF-α production (22). Administration of excessive ethanol
to TNF-α-knockout mice does not result in liver injury, and in both
ALD and non-alcoholic steatohepatitis (NASH) (23), TNF-α is responsible for the
development of liver injury. Treatment with pentoxifylline, an
inhibitor of TNF-α, was found to improve the survival of patients
with severe AH (24). Anti-TNF-α
antibodies were found to prevent inflammation and necrosis in an
alcohol-fed rat model (25), and
infliximab, an anti-TNF-α antibody, was also effective in patients
with severe AH (26). The
multiple cytokine modulator, Y-40138, inhibits the production of
inflammatory cytokines, such as TNF-α or IL-6, and enhances
anti-inflammatory cytokine production, such as IL-10. Our
experimental research showed that Y-40138 reduced the production of
inflammatory cytokines in ALD (27). These facts suggest that TNF-α
plays an important role in the progression of ALD.
IL-6 and IL-10
The role of IL-6 in ALD is complex and is not well
understood. IL-6 appears to have some beneficial effects on the
liver by possibly protecting against hepatocyte apoptosis and
participating in mitochondrial DNA repair following alcoholic liver
injury (28). IL-10 is an
anti-inflammatory cytokine that controls the endogenous production
of TNF-α during endotoxemia and reduces LPS stimulation when added
exogenously (29). The liver is
the main organ of IL-10 production (30), and Kupffer cells and lymphocytes
are the main producers of IL-10. IL-10 is stimulated by LPS and
downregulates the release of TNF-α and IL-6. IL-10 also exerts a
hepatoprotective effect on liver proliferation and fibrosis
(31). IL-6 may promote human
Th17 cell differentiation and IL-17 production. Therefore, IL-6
contributes to ethanol-induced liver inflammation. After alcohol
consumption, IL-6 is released along with IL-10, TNF-α and other
cytokines by Kupffer cells. Both IL-6 and IL-10 play roles in
reducing alcoholic liver injury and inflammation through the
activation of signal transducer and activator of transcription 3
(STAT3) (13). IL-6 is elevated
in chronically alcohol-fed animals and in alcoholics (32). In contrast, IL-6-knockout
chronically alcohol-fed mice were found to have increased liver fat
accumulation, lipid peroxidation, mitochondrial DNA damage, and
sensitization of hepatocytes to TNF-α-induced apoptosis (28,33). Blocking of IL-6 and IL-10
signaling in the mice reduced neutrophil and mononuclear cell
infiltration and inflammation (34). IL-10 decreases the production of
TNF-α, IL-1β and IL-6 from activated Kupffer cells and monocytes.
IL-10-deficient mice exhibited increased liver inflammation after
being fed ethanol (35).
IL-10-knockout mice have elevated IL-6 and STAT3 activation in the
liver, leading to steatosis and hepatocellular damage. These
findings suggest that both IL-6 and IL-10 have a protective effect
in the early phase of ALD. On the other hand, IL-10-knockout mice
exhibit reduced fatty liver change and lower serum aspartate
aminotransferase and alanine transaminase levels after ethanol
feeding than wild-type mice (35). Recently, it has been suggested
that IL-10 may play a biphasic role. First, IL-10 inhibits
inflammatory cytokines (LPS, TNF-α, IL-6), reducing steatosis and
liver damage; second, IL-10 blocks IL-6 production, enhancing liver
damage. The overall effect of IL-10 on hepatic steatosis or liver
injury may be determined by the balance between proinflammatory
cytokines that promote liver injury and hepatoprotective cytokines
that prevent liver injury.
Other cytokines
Nuclear regulatory factor-κB (NF-κB) is a protein
complex that controls the transcription of DNA and is a central
regulator of cellular stress in all liver cell types. NF-κB plays a
key role in regulating the immune response to infection and acute
and chronic inflammation. Activation of NF-κB in rats can induce
IL-1β expression, which increases the expression of proinflammatory
molecules (36). IL-1β and IL-6
appear essential for the induction of Th17 lymphocyte
differentiation from human naive CD4+ T cells (37). Furthermore, LPS-stimulated human
monocytes induce Th17 polarization of naive CD4+ T cells
in an IL-1β signaling-dependent manner. IL-8 is produced by
macrophages and is a critical proinflammatory cytokine involved in
the mobilization of neutrophils. IL-8 is induced by TNF-β and TLRs
via the activation of NF-κB. Serum IL-8 is highly elevated in
patients with AH and is linked to infiltration with neutrophils.
However, IL-8 is only moderately elevated in alcoholic cirrhosis
patients and alcoholics. IL-17 is a cytokine that acts as a potent
mediator in delayed-type reactions by increasing chemokine
production in various tissues to recruit monocytes and neutrophils
to the inflammation site and activate NF-κB or induce IL-8. IL-17
plays a key role in autoimmune diseases (38). IL-17 stimulates multiple types of
non-parenchymal hepatic cells to produce proinflammatory cytokines
and chemokines such as TNF-β (39). Plasma IL-17 levels are higher in
patients with ALD than those in controls (40). The functions of Th17 cells are
also mediated via IL-22, a member of the IL-10 family, playing an
important role in promoting hepatocyte survival and proliferation
(41). IL-22 administration to
alcohol-fed mice also prevented liver steatosis and liver injury
through the activation of hepatic STAT3 (42).
4. Chemokines and inflammasomes
Members of the CXC family of chemokines include IL-8
and growth-regulated α-protein (Gro-α). These mediators attract
polymorphonuclear leukocytes, which are predominant inflammatory
cells infiltrating the livers of patients with ALD. In patients
with AH, expression of these chemokines in the liver correlates
with the severity of portal hypertension and patient survival
(43). CCL2 [monocyte
chemoattractant peptide-1 (MCP-1)] is a member of the CC chemokine
family. Its expression can be induced by inflammatory cells,
hepatocytes and HSCs. CCR2 is the only known receptor for CCL2 and
is expressed on monocytes, T lymphocytes and basophils (44). MCP-1 regulates adhesion molecules
and proinflammatory cytokines, such as TNF-α, IL-1β and IL-6
(45). The pivotal role of MCP-1
in ALD was recognized by observing higher amounts of MCP-1 than
those of other CC chemokines and macrophage inflammatory protein-1α
in the liver and mononuclear cells in patients with AH (46). MCP-1 is important in the
modulation of proinflammatory cytokines (47). Blockage of MCP-1 protects mice
against ALD, independently of CCR2, by inhibiting proinflammatory
cytokines and induction of fatty acid oxidation, linking chemokines
to hepatic lipid metabolism (48). In the liver, HSCs express a large
number of chemokines (49),
including CXC chemokines (CXCL8, CXCL9, CXCL10 and CXCL12) and CC
chemokines (CCL2, CCL3 and CCL5) (49). These chemokines have been related
to liver fibrosis in chronic liver diseases (49,50). CXC chemokines drive angiogenesis
during fibrosis initiation and progression (49,50).
The inflammasome is a multiprotein oligomer
consisting of caspase-1, an apoptosis-associated speck-like protein
containing caspase recruitment domain, and NOD-like receptor family
pyrin domain-containing 3 (NLRP3) that mediate the response to
cellular danger signals activating and recruiting inflammatory
cells (51). IL-1β is produced
following inflammasome activation. NLRP3 activates inflammatory
caspase, caspase-1, which accelerates the aging process through the
impairment of autophagy, leading to cell death.
Inflammasomes are activated by two steps. The first
step is upregulation of pro-IL-1β expression and inflammasome
components. The second step is triggered by ligands of the NLR
sensor in the inflammasome, resulting in cleavage of procaspase-1
into active caspase-1 that cleaves pro-IL-1β into the mature,
secreted IL-1β (52). Increased
IL-1β upregulates caspase-1 activity and inflammasome activation.
Alcohol results in release of sterile danger signals, uric acid,
and extracellular adenosine triphosphate, which are activators of
the NLRP3 inflammasome (53).
Inflammasomes are activated in bone marrow (BM)-derived Kupffer
cells in alcohol-fed mice through IL-1 signaling (54). IL-1β increases the activity of
MCP-1 in hepatocytes and contributes to increased TLR4-dependent
proinflammatory signaling in macrophages.
5. Toll-like receptors
TLRs comprise a family of pattern-recognition
receptors, which contribute to the production of antimicrobial
peptides against microorganism invasion. Endogenous components
derived from dying host cells, termed damage-associated molecular
patterns (DAMPs), can also activate TLRs (55). To date, 11 and 13 TLRs have been
identified in humans and mice, respectively (56). TLRs recognize pathogen-derived
molecules, such as structural components unique to bacteria, virus,
parasites, and fungi and activate inflammatory cytokines and type I
interferon (IFN) production. TLRs are expressed on the surface of
immune cells, such as macrophages, dendritic cells and epithelial
cells. After binding to its ligands, TLRs transduce signals via
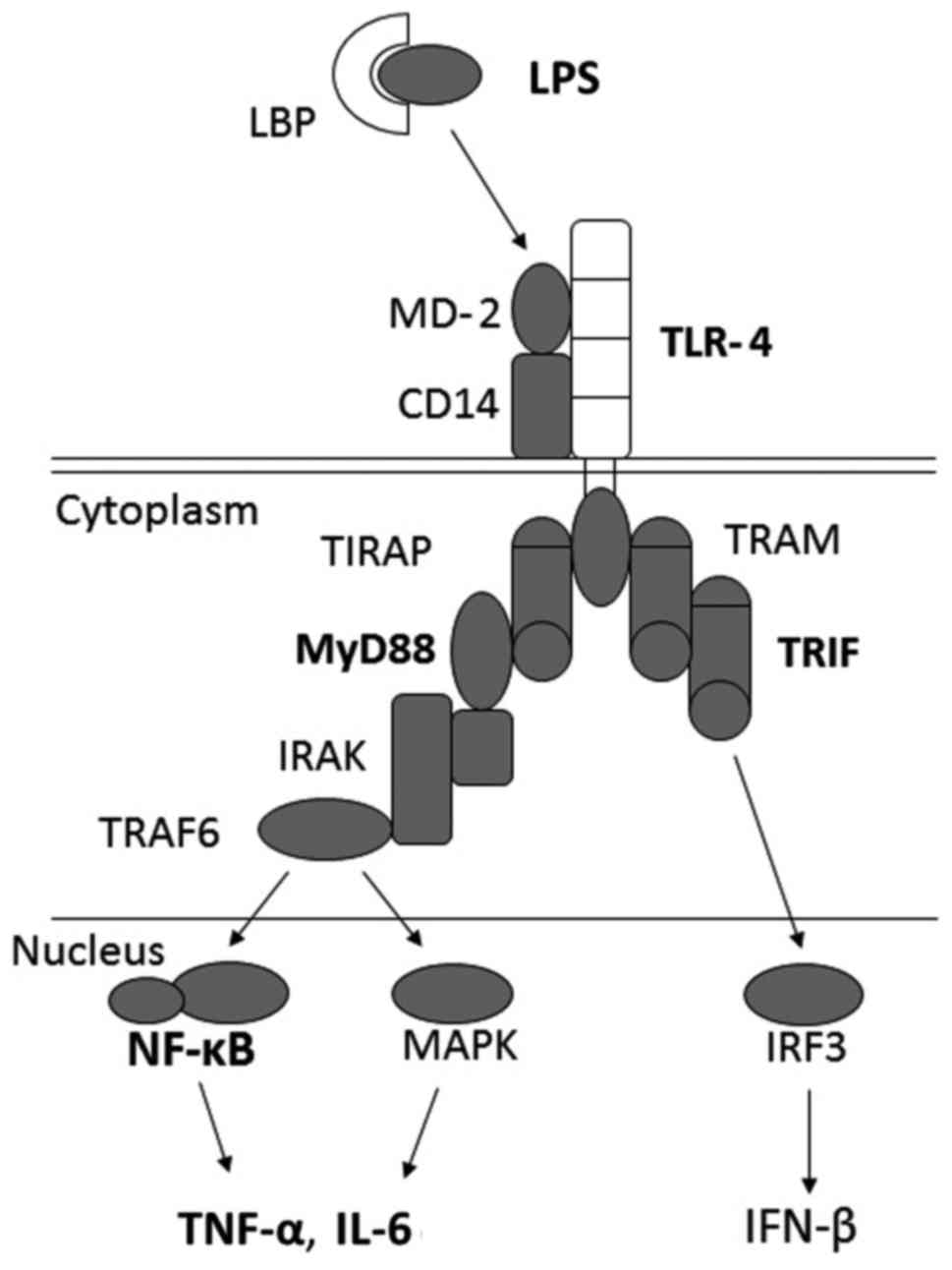
myeloid differentiation factor 88 (MyD88). TLR4 is expressed on the
surface of Kupffer cells and various other types of cells that
transmit endotoxin signals. LPS is the ligand of TLR4, and cluster
of differentiation 14 (CD14), a component of the innate immune
system, binds LPS and subsequently presents it to TLR4 and MD-2,
which activate MyD88. This leads to the activation of NF-κB
(57) and results in the
production of various proinflammatory cytokines, such as TNF-α and
IL-6. Both MyD88 and TRIF (MyD88-independent) signaling modulate
TLR4 (Fig. 3). Mice deficient in
TLR4 expression are protected from alcohol-induced liver
inflammation and hepatocyte injury (58). In the liver, TLR4 is expressed not
only on innate immune cells, such as Kupffer cells, but also on
hepatocytes, HSCs, sinusoidal endothelial cells, and biliary
epithelial cells. The TLR4-TRIF-dependent pathway is important in
the development of ASH (59).
Blocking of TLR4 or CD14 reduced liver pathology and inflammation
in a mouse model of alcoholic liver injury (60,61). LPS recognition by TLR4 expressed
on HSCs and sinusoidal epithelial cells also contribute to the
progression of ALD (61). Alcohol
stimulates Kupffer cells and monocytes to produce increased TNF-α
in response to endotoxins (62).
Our group showed that endotoxemia plays an important role in the
initiation and aggravation of ALD through the enhancement of
proinflammatory cytokines, including IL-6, IL-8 and TNF-α (63,64). Hepatic expression of TLR1, 2, 4,
6, 7, 8 and 9 mRNA was increased in chronically alcohol-fed mice
(65). Alcohol feeding also
resulted in sensitization to liver damage and inflammation because
administration of TLR1, 2, 4, 6, 7, 8 and 9 ligands resulted in
increased expression of TNF-α mRNA (66). Acute alcohol exposure inhibited
TLR4 signaling in macrophages following alcohol treatment in mice,
leading to decreased LPS-induced TNF-α production. It is clear that
alcohol abuse leads to the activation of innate immunity via TLR
signaling. This suggests that TLRs are important in ALD.
6. Gut-derived bacteria
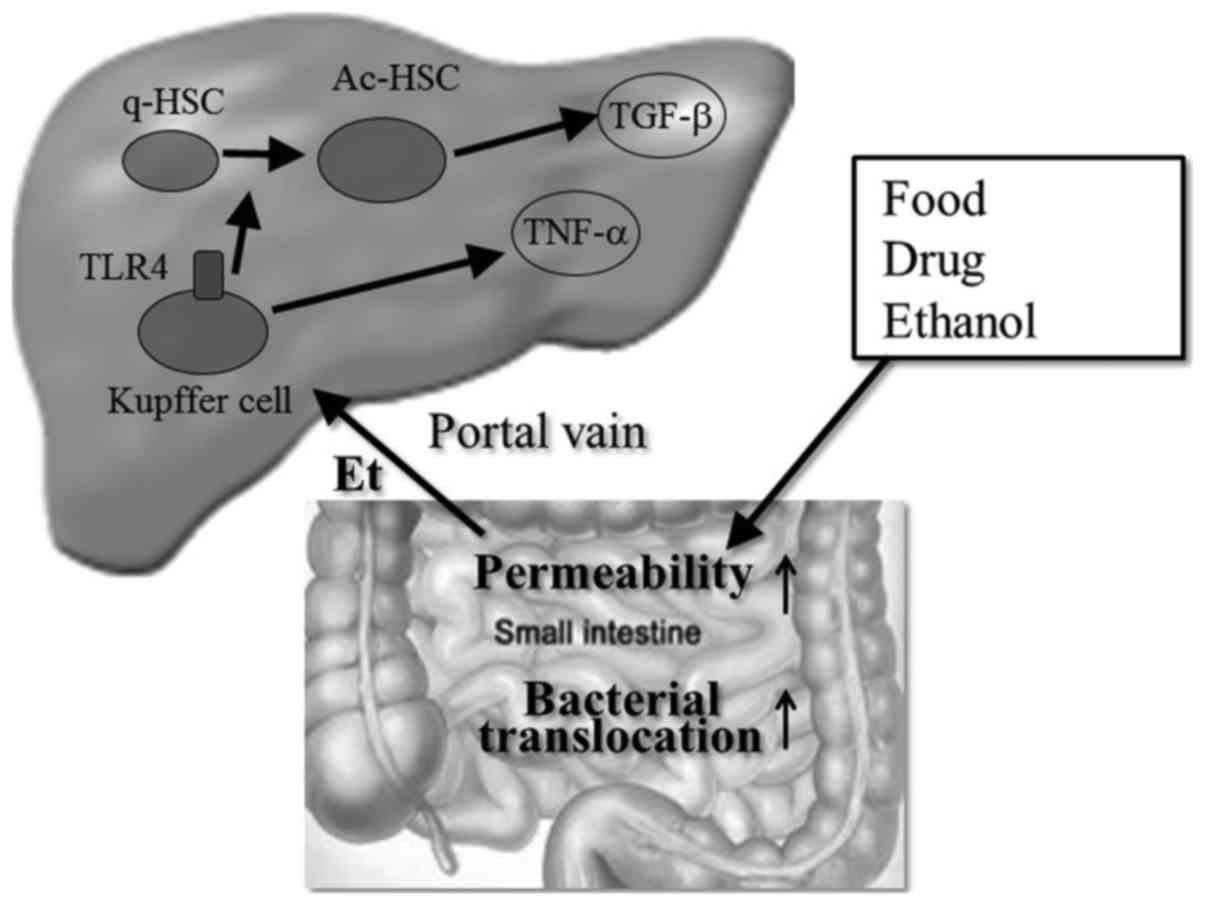
Recently, the importance of bacterial translocation
in the pathogenesis of ALD has been shown in many studies (67). Dysfunction of intestinal tight
junctions (68,69) or bacterial proliferation (70) by alcohol or its metabolites
enhance bacterial translocation into the liver, which induces the
activation of Kupffer cells to release various proinflammatory
cytokines and chemokines (71)
(Fig. 4). Chronic alcohol feeding
causes structural changes in the gastrointestinal tract that may
contribute to LPS translocation (72) and results in intestinal bacterial
overgrowth and enteric dysbiosis, which is due to alcohol-induced
downregulation of several intestinal antimicrobial molecules
(73). ALD also results in
quantitative alterations in intestinal microbiota. Small intestinal
bacterial overgrowth is another cause of bacterial translocation.
Chronic alcohol consumption elevates the growth of Gram-negative
bacteria causing a decrease in both Bacteroidetes and
Firmicutes and an increase in Actinobacteria and
Proteobacteria (74). In
chronic alcohol consumption, the intestinal flora releases a larger
amount of endotoxins due to hyperpermeability, which is responsible
for the altered intestinal barrier and activation of inflammatory
processes, leading to the progression of ALD (75), cirrhosis and HCC (76). In the intestine, the disruption of
tight junctions may lead to increased permeability to pathogens,
which is a common mechanism involved in the pathogenesis of ALD
(77). Zonula occludens-1 (ZO-1),
occludin and claudin-4 are transmembrane proteins expressed at
tight junctions. Alcohol-treated mice showed loss of ZO-1,
occludin, and claudin-4 expression in the colon. Alcohol-induced
'leaky gut' results in the translocation of Gram-negative bacteria
from the intestinal lumen into the portal blood, elevating LPS
levels and triggering significant inflammation and liver injury
(78), and activation of Kupffer
cells is involved in the pathogenesis of ALD (79). Treatment with antibiotics also
prevents alcohol-induced liver injury (80). Probiotics modulate intestinal
microbiota, which leads to decreased bacterial translocation and
endotoxin production. Moreover, probiotics improve the intestinal
barrier integrity, thereby improving ALD. These factors imply that
there are interactions between the liver and intestinal bacteria in
ALD.
7. Angiogenesis
Angiogenesis is an active, growth factor-dependent,
and hypoxia-induced event that takes place in several organs during
growth and repair of injured tissues (81,82). Chronic ethanol consumption induces
inflammation in the liver parenchyma, triggering both fibrogenesis
and angiogenesis (83).
Angiogenesis mainly contributes to the development of liver
complications such as portal hypertension and formation of
portal-systemic collaterals. Moreover, angiogenesis contributes to
the progression of fibrosis in chronic liver disease. In contrast,
intrahepatic vascular remodeling with capillarization of sinusoids
and alcohol-related central zone steatosis of lobuli alter hepatic
oxygen supply leading to hypoxia with formation of new vessels;
thus, creating a viscous circle (84,85). It has been suggested that
angiogenesis may contribute to the progression of fibrosis during
the wound healing process in chronic liver damage (84,86). Moreover, wound healing is also
defined by an increase in the expression of several cytokines and
growth factors with pro-angiogenic action (84,87,88). HSCs may constitute a crossroad at
the interaction between inflammation, angiogenesis and fibrosis.
Hepatic angiogenesis takes place in chronic liver diseases that are
characterized by inflammation and progressive fibrosis.
Anti-angiogenic therapy is efficient in the prevention of fibrosis
in chronic liver diseases. Angiogenesis is activated by hypoxia and
mediated by hypoxia-inducible factors (HIFs) (87,88). HIFs are heterodimers formed by an
oxygen-sensitive and inducible α subunit and an oxygen-independent
β subunit (87). HIF-1α, HIF-2α,
and HIF-3α have been described, and all bind to a common β subunit
named the aryl hydrocarbon nuclear receptor translocator. Vascular
endothelial growth factor (VEGF) is the most potent stimulator of
pathological angiogenesis and wound healing. It also induces
vascular permeability inside injured tissues. VEGF is released
under hypoxic conditions and regulated by HIF-1α. Chronic ethanol
consumption is related to angiogenesis by means of finely
coordinated action of various mediators in the liver (89). Ethanol upregulates VEGF and VEGF
receptor-2 and stimulates angiogenesis in the rat liver (89). Tyrosine kinase receptors (Tie1 and
Tie2) and their ligands, the angiopoietins (Ang-1, Ang-2, Ang-3 and
Ang-4), play a key role during the late phase of angiogenesis and
are responsible for the maturation of newly formed vascular
structures. In patients with ALD, concentrations of Ang-2 and
VEGF-A were found to be increased relative to controls. It has also
been observed that alcohol induces angiogenesis via platelet
endothelial cell adhesion molecule-1 (89). This insight may contribute to the
development of new anti-angiogenic treatment strategies.
8. Fibrosis
The course from chronic liver disease to cirrhosis
is complex. It is modulated by the influence of various
disease-specific, host-specific, and environmental factors. The
development of liver fibrosis in alcoholics has been linked to
oxidation of ethanol to the highly reactive compound acetaldehyde.
After alcohol consumption, acetaldehyde stimulates not only type IV
but also type I collagen synthesis and gene transcription in
cultured rat and human HSCs through the activation of protein
kinase C (90). Liver fibrosis is
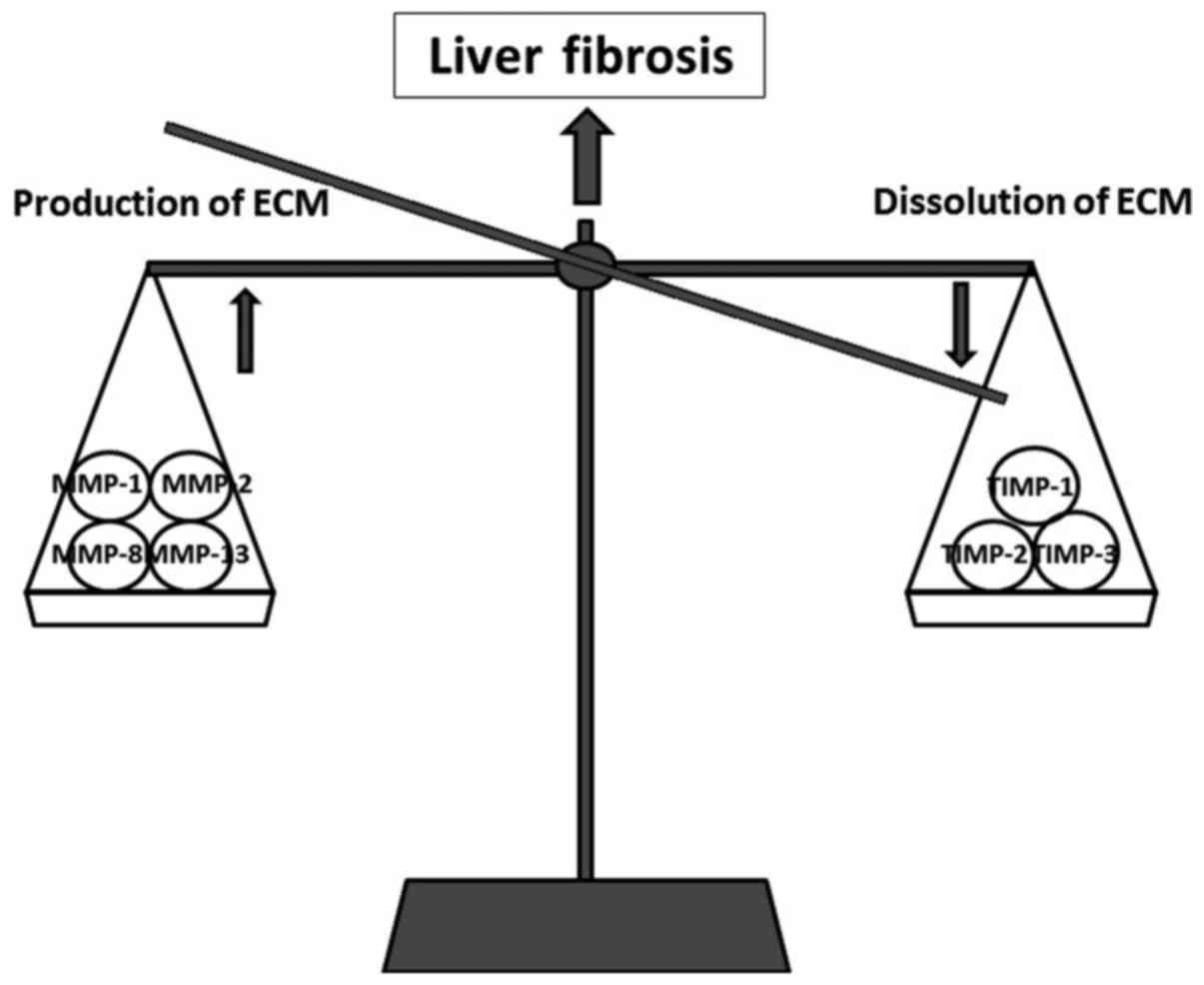
a dynamic process that results from an imbalance between the
production and dissolution of ECM. This again depends on the
balance between matrix metalloproteinases (MMPs) and tissue
inhibitors of matrix metalloproteinases (TIMPs). The increase in
ECM is controlled by MMPs (MMP-1, MMP-2, MMP-8 and MMP-13), while
the progression of fibrosis is correlated with an increase in TIMPs
(TIMP-1 and TIMP-2) (91)
(Fig. 5). A critical event in
liver fibrogenesis is that ECM is a dynamic structure and even
advanced fibrosis may be reversible. Multiple interactions between
ECM, HSCs and immune cells have been identified. These findings
have advanced the understanding of the pathogenesis of liver
inflammation and liver fibrosis and present opportunities for novel
therapeutic approaches for the management of ALD and liver
fibrosis.
9. Treatment for ALD
Abstinence from alcohol is the essential treatment
for ALD (92). However, in most
patients with ALD, abstinence is difficult to maintain. On the
other hand, there is little pharmacological treatment for ALD.
Thus, there is an urgent need to develop novel therapeutic
interventions. There are many animal models of ALD, all having
limits when it comes to mimicking human ALD. In the clinical
setting, AH and alcoholic liver cirrhosis have identifiable
symptoms and receive treatment. Patients with severe AH have a high
mortality rate, approximately 50%, and those who survive have a 70%
probability of developing liver cirrhosis. Nutrition
supplementation is necessary for patients with AH because of the
prevalence of malnutrition. Corticosteroids and pentoxifylline are
used in the treatment of AH, both aiming to reduce inflammatory
conditions. Corticosteroids reduce cytokine production through
transcriptional regulation, and pentoxifylline has a similar effect
through the inhibition of phosphodiesterases. These two agents have
relatively strong side effects; thus, it is mainly used for severe
AH. On the other hand, probiotics are effective in ALD.
Lactobacillus reduced endotoxemia and improved liver damage
in a rat ALD model (93).
Moreover, some clinical studies indicate the effectiveness of
probiotics combined with antibiotic treatment. Rifaximin, a
non-absorbable antibiotic that alters the gut microbiota, is
efficacious in the treatment of hepatic encephalopathy and could
have a role in modulating ALD (94). Inhibition of LPS-induced TLR4 or
TNF-α signaling has been suggested as a target for novel therapies
(95). It is suggested that
blockade of angiogenesis could be a promising therapeutic option in
patients with advanced fibrosis. Determination of proangiogenic
factors may be a useful non-invasive approach in the follow-up of
liver fibrosis progression and response to therapy. The main event
in fibrogenesis appears to be the activation of HSCs, which
indicates multiple potential sites for therapeutic interventions.
Anti-angiogenic therapy may be promising in the prevention of
fibrosis in chronic liver disease, however, it should be well
balanced since an excessive blockage may prevent wound healing
response. Specific, effective and safe antifibrotic therapies have
not yet been developed that can mediate the progression of hepatic
fibrogenesis.
10. Future therapies for ALD
As discussed in this review, future treatment
strategies for ALD are summarized in Table I. Currently, the medical
management of ALD is limited to steroids. Use of steroids can be
associated with significant adverse reactions, such as infection
and bone fractures. In addition, the long-term benefit of steroids
remains to be established. Targeting gut microbes and their
products, targeting hepatic inflammation and fibrosis, and immune
modulation, help improve liver regeneration. These are the most
promising areas for research and future clinical trials should
focus on these areas in developing new therapies in the treatment
for ALD.
 | Table IPotential future treatments for
alcoholic liver disease. |
Table I
Potential future treatments for
alcoholic liver disease.
| Target | Medication |
|---|
| Microbiota | Antibiotics |
| Prebiotics and
probiotics |
| LPS | Blockade of
LPS |
| Immune
modulation |
| Inflammation | Chemokines (CCL20
inhibition) |
| Inhibition of IL-8,
IL-17 |
| Recombinant IL-6,
IL-10 |
| TNF-α superfamily
receptor modulation (Y-40138) |
| Blockade of
inflammasome activation |
| Fibrosis | Blockade of
angiogenesis |
11. Conclusion
Alcohol is one of the most common causes of chronic
liver diseases worldwide (93).
In the development of ALD or alcoholic liver cirrhosis, various
factors, such as inflammation, oxidative stress, innate immunity,
angiogenesis, or fibrosis, are important. Inflammatory cytokines,
such as TNF-α, play an important role in ALD. The progression of
alcohol-induced liver injury involves immune cells and hepatocytes
through the release of cytokines or chemokines. Kupffer cells play
an important role in early-stage ALD, producing TNF-α through TLR4.
In the current understanding of the pathogenesis of ALD, TNF-α
appears to be a key to developing new approaches for its treatment.
In contrast, ECM plays a key role in liver fibrogenesis, and
multiple interactions between ECM, HSCs, and endothelial cells have
been identified. Activation of HSCs is the central event in
fibrogenesis, and TGF-β is a key to developing new approaches for
its treatment. The treatment of ALD has advanced only little after
the introduction of corticosteroid therapy. The development of
targeted therapies for ALD and alcoholic liver cirrhosis are
hampered by poor knowledge of the molecular mechanisms involved in
its development, particularly in humans and by the perception that
it is an addictive and a self-inflicting disease. We propose that
further reseach is warranted to increase the understanding of the
pathogenesis of liver inflammation and fibrosis to discover novel
therapeutic agents for patients with ALD.
References
|
1
|
Teli MR, Day CP, Burt AD, Bennett MK and
James OF: Determinants of progression to cirrhosis or fibrosis in
pure alcoholic fatty liver. Lancet. 346:987–990. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lefkowitch JH: Morphology of alcoholic
liver disease. Clin Liver Dis. 9:37–53. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cojocariu CE, Trifan AV, Girleanu I and
Stanciu C: Alcoholic liver disease - epidemiology and risk factors.
Rev Med Chir Soc Med Nat Iasi. 118:910–917. 2014.
|
|
4
|
Stickel F: Alcoholic cirrhosis and
hepatocellular carcinoma. Adv Exp Med Biol. 815:113–130. 2015.
View Article : Google Scholar
|
|
5
|
Browning JD and Horton JD: Molecular
mediators of hepatic steatosis and liver injury. J Clin Invest.
114:147–152. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
MacSween RN and Burt AD: Histologic
spectrum of alcoholic liver disease. Semin Liver Dis. 6:221–232.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haber PS, Warner R, Seth D, Gorrell MD and
McCaughan GW: Pathogenesis and management of alcoholic hepatitis. J
Gastroenterol Hepatol. 18:1332–1344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsujimoto T, Kuriyama S, Yamazaki M,
Nakatani Y, Okuda H, Yoshiji H and Fukui H: Augmented
hepatocellular carcinoma progression and depressed Kupffer cell
activity in rat cirrhotic livers. Int J Oncol. 18:41–47. 2001.
|
|
9
|
Kitazawa T, Nakatani Y, Fujimoto M, Tamura
N, Uemura M and Fukui H: The production of tumor necrosis
factor-alpha by macrophages in rats with acute alcohol loading.
Alcohol Clin Exp Res. 27(Suppl 8): 72S–75S. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rao RK, Seth A and Sheth P: Recent
advances in alcoholic liver disease I. Role of intestinal
permeability and endotoxemia in alcoholic liver disease. Am J
Physiol Gastrointest Liver Physiol. 286:G881–G884. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Canbay A, Feldstein AE, Higuchi H,
Werneburg N, Grambihler A, Bronk SF and Gores GJ: Kupffer cell
engulfment of apoptotic bodies stimulates death ligand and cytokine
expression. Hepatology. 38:1188–1198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McClain CJ, Song Z, Barve SS, Hill DB and
Deaciuc I: Recent advances in alcoholic liver disease. IV.
Dysregulated cytokine metabolism in alcoholic liver disease. Am J
Physiol Gastrointest Liver Physiol. 287:G497–G502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao B: Cytokines, STATs and liver disease.
Cell Mol Immunol. 2:92–100. 2005.PubMed/NCBI
|
|
14
|
Garcin F, Lau You Hin G, Cote J,
Radouco-Thomas S, Chawla S and Radouco-Thomas C: Aldehyde
dehydrogenase in Drosophila: developmental and functional aspects.
Alcohol. 2:85–89. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lieber CS and DeCarli LM: The role of the
hepatic microsomal ethanol oxidizing system (MEOS) for ethanol
metabolism in vivo. J Pharmacol Exp Ther. 181:279–287.
1972.PubMed/NCBI
|
|
16
|
Neve EP and Ingelman-Sundberg M: Molecular
basis for the transport of cytochrome P450 2E1 to the plasma
membrane. J Biol Chem. 275:17130–17135. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lieber CS: Metabolic consequences of
ethanol. Endocrinologist. 4:127–139. 1994. View Article : Google Scholar
|
|
18
|
Bilzer M, Roggel F and Gerbes AL: Role of
Kupffer cells in host defense and liver disease. Liver Int.
26:1175–1186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Diehl AM: Recent events in alcoholic liver
disease V. Effects of ethanol on liver regeneration. Am J Physiol
Gastrointest Liver Physiol. 288:G1–G6. 2005. View Article : Google Scholar
|
|
20
|
Hansen J, Cherwitz DL and Allen JI: The
role of tumor necrosis factor-alpha in acute endotoxin-induced
hepatotoxicity in ethanol-fed rats. Hepatology. 20:461–474. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aldred A and Nagy LE: Ethanol dissociates
hormone-stimulated cAMP production from inhibition of TNF-alpha
production in rat Kupffer cells. Am J Physiol. 276:G98–G106.
1999.PubMed/NCBI
|
|
22
|
Kawaratani H, Tsujimoto T, Kitazawa T,
Kitade M, Yoshiji H, Uemura M and Fukui H: Innate immune reactivity
of the liver in rats fed a choline-deficient L-amino-acid-defined
diet. World J Gastroenterol. 14:6655–6661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Crespo J, Cayon A, Fernandez-Gil P,
Hernandez-Guerra M, Mayorga M, Dominguez-Diez A,
Fernandez-Escalante JC and Pons-Romero F: Gene expression of tumor
necrosis factor alpha and TNF-receptors, p55 and p75, in
nonalcoholic steatohepatitis patients. Hepatology. 34:1158–1163.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tilg H, Jalan R, Kaser A, Davies NA,
Offner FA, Hodges SJ, Ludwiczek O, Shawcross D, Zoller H, Alisa A,
et al: Anti-tumor necrosis factor-alpha monoclonal antibody therapy
in severe alcoholic hepatitis. J Hepatol. 38:419–425. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Akriviadis E, Botla R, Briggs W, Han S,
Reynolds T and Shakil O: Pentoxifylline improves short-term
survival in severe acute alcoholic hepatitis: a double-blind,
placebo-controlled trial. Gastroenterology. 119:1637–1648. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Iimuro Y, Gallucci RM, Luster MI, Kono H
and Thurman RG: Antibodies to tumor necrosis factor alfa attenuate
hepatic necrosis and inflammation caused by chronic exposure to
ethanol in the rat. Hepatology. 26:1530–1537. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawaratani H, Tsujimoto T, Kitazawa T,
Yoshiji H, Uemura M and Fukui H: Therapeutic effects of cytokine
modulator Y-40138 in the rat alcoholic liver disease model. J
Gastroenterol Hepatol. 26:775–783. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hong F, Kim WH, Tian Z, Jaruga B, Ishac E,
Shen X and Gao B: Elevated interleukin-6 during ethanol consumption
acts as a potential endogenous protective cytokine against
ethanol-induced apoptosis in the liver: involvement of induction of
Bcl-2 and Bcl-x(L) proteins. Oncogene. 21:32–43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mathurin P, Deng QG, Keshavarzian A,
Choudhary S, Holmes EW and Tsukamoto H: Exacerbation of alcoholic
liver injury by enteral endotoxin in rats. Hepatology.
32:1008–1017. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hawrylowicz CM and O'Garra A: Potential
role of interleukin-10-secreting regulatory T cells in allergy and
asthma. Nat Rev Immunol. 5:271–283. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Louis H, Le Moine O, Goldman M and Deviere
J: Modulation of liver injury by interleukin-10. Acta Gastroenterol
Belg. 66:7–14. 2003.PubMed/NCBI
|
|
32
|
Latvala J, Hietala J, Koivisto H, Jarvi K,
Anttila P and Niemela O: Immune responses to ethanol metabolites
and cytokine profiles differentiate alcoholics with or without
liver disease. Am J Gastroenterol. 100:1303–1310. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang X, Tachibana S, Wang H, Hisada M,
Williams GM, Gao B and Sun Z: Interleukin-6 is an important
mediator for mitochondrial DNA repair after alcoholic liver injury
in mice. Hepatology. 52:2137–2147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rabe B, Chalaris A, May U, Waetzig GH,
Seegert D, Williams AS, Jones SA, Rose-John S and Scheller J:
Transgenic blockade of interleukin 6 transsignaling abrogates
inflammation. Blood. 111:1021–1028. 2008. View Article : Google Scholar
|
|
35
|
Miller AM, Wang H, Bertola A, Park O,
Horiguchi N, Ki SH, Yin S, Lafdil F and Gao B:
Inflammation-associated interleukin-6/signal transducer and
activator of transcription 3 activation ameliorates alcoholic and
nonalcoholic fatty liver diseases in interleukin-10-deficient mice.
Hepatology. 54:846–856. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jura J, Wegrzyn P, Korostyński M, Guzik K,
Oczko- Wojciechowska M, Jarzab M, Kowalska M, Piechota M,
Przewłocki R and Koj A: Identification of interleukin-1 and
interleukin-6-responsive genes in human monocyte-derived
macrophages using microarrays. Biochim Biophys Acta. 1779:383–389.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Acosta-Rodriguez EV, Napolitani G,
Lanzavecchia A and Sallusto F: Interleukins 1beta and 6 but not
transforming growth factor-beta are essential for the
differentiation of interleukin 17-producing human T helper cells.
Nat Immunol. 8:942–949. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weaver CT, Hatton RD, Mangan PR and
Harrington LE: IL-17 family cytokines and the expanding diversity
of effector T cell lineages. Annu Rev Immunol. 25:821–852. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lafdil F, Miller AM, Ki SH and Gao B: Th17
cells and their associated cytokines in liver diseases. Cell Mol
Immunol. 7:250–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lemmers A, Moreno C, Gustot T, Marechal R,
Degre D, Demetter P, de Nadai P, Geerts A, Quertinmont E,
Vercruysse V, et al: The interleukin-17 pathway is involved in
human alcoholic liver disease. Hepatology. 49:646–657. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nanji AA, Zhao S, Sadrzadeh SM, Dannenberg
AJ, Tahan SR and Waxman DJ: Markedly enhanced cytochrome P450 2E1
induction and lipid peroxidation is associated with severe liver
injury in fish oil-ethanol-fed rats. Alcohol Clin Exp Res.
18:1280–1285. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ki SH, Park O, Zheng M, Morales-Ibanez O,
Kolls JK, Bataller R and Gao B: Interleukin-22 treatment
ameliorates alcoholic liver injury in a murine model of
chronic-binge ethanol feeding: role of signal transducer and
activator of transcription 3. Hepatology. 52:1291–1300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dominguez M, Miquel R, Colmenero J, Moreno
M, Garcia-Pagan JC, Bosch J, Arroyo V, Gines P, Caballeria J and
Bataller R: Hepatic expression of CXC chemokines predicts portal
hypertension and survival in patients with alcoholic hepatitis.
Gastroenterology. 136:1639–1650. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Simpson KJ, Henderson NC, Bone-Larson CL,
Lukacs NW, Hogaboam CM and Kunkel SL: Chemokines in the
pathogenesis of liver disease: so many players with poorly defined
roles. Clin Sci (Lond). 104:47–63. 2003. View Article : Google Scholar
|
|
45
|
Jiang Y, Beller DI, Frendl G and Graves
DT: Monocyte chemoattractant protein-1 regulates adhesion molecule
expression and cytokine production in human monocytes. J Immunol.
148:2423–2428. 1992.PubMed/NCBI
|
|
46
|
Fisher NC, Neil DA, Williams A and Adams
DH: Serum concentrations and peripheral secretion of the beta
chemokines monocyte chemoattractant protein 1 and macrophage
inflammatory protein 1alpha in alcoholic liver disease. Gut.
45:416–420. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lu B, Rutledge BJ, Gu L, Fiorillo J,
Lukacs NW, Kunkel SL, North R, Gerard C and Rollins BJ:
Abnormalities in monocyte recruitment and cytokine expression in
monocyte chemoattractant protein 1-deficient mice. J Exp Med.
187:601–608. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mandrekar P, Ambade A, Lim A, Szabo G and
Catalano D: An essential role for monocyte chemoattractant
protein-1 in alcoholic liver injury: regulation of proinflammatory
cytokines and hepatic steatosis in mice. Hepatology. 54:2185–2197.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sahin H and Wasmuth HE: Chemokines in
tissue fibrosis. Biochim Biophys Acta. 1832:1041–1048. 2013.
View Article : Google Scholar
|
|
50
|
Strieter RM, Burdick MD, Gomperts BN,
Belperio JA and Keane MP: CXC chemokines in angiogenesis. Cytokine
Growth Factor Rev. 16:593–609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Alfonso-Loeches S, Ureña-Peralta JR,
Morillo-Bargues MJ, Oliver-De La Cruz J and Guerri C: Role of
mitochondria ROS generation in ethanol-induced NLRP3 inflammasome
activation and cell death in astroglial cells. Front Cell Neurosci.
8:2162014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Szabo G and Csak T: Inflammasomes in liver
diseases. J Hepatol. 57:642–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Petrasek J, Iracheta-Vellve A, Saha B,
Satishchandran A, Kodys K, Fitzgerald KA, Kurt-Jones EA and Szabo
G: Metabolic danger signals, uric acid and ATP, mediate
inflammatory cross-talk between hepatocytes and immune cells in
alcoholic liver disease. J Leukoc Biol. 98:249–256. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Petrasek J, Bala S, Csak T, Lippai D,
Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA and Szabo G:
IL-1 receptor antagonist ameliorates inflammasome-dependent
alcoholic steatohepatitis in mice. J Clin Invest. 122:3476–3489.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal
T, Junger W, Brohi K, Itagaki K and Hauser CJ: Circulating
mitochondrial DAMPs cause inflammatory responses to injury. Nature.
464:104–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Medzhitov R, Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila Toll protein
signals activation of adaptive immunity. Nature. 388:394–397. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yamamoto M, Sato S, Hemmi H, Hoshino K,
Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et
al: Role of adaptor TRIF in the MyD88-independent toll-like
receptor signaling pathway. Science. 301:640–643. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Petrasek J, Mandrekar P and Szabo G:
Toll-like receptors in the pathogenesis of alcoholic liver disease.
Gastroenterol Res Pract. 2010:7103812010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Szabo G, Mandrekar P, Petrasek J and
Catalano D: The unfolding web of innate immune dysregulation in
alcoholic liver injury. Alcohol Clin Exp Res. 35:782–786. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yin M, Bradford BU, Wheeler MD, Uesugi T,
Froh M, Goyert SM and Thurman RG: Reduced early alcohol-induced
liver injury in CD14-deficient mice. J Immunol. 166:4737–4742.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Paik YH, Schwabe RF, Bataller R, Russo MP,
Jobin C and Brenner DA: Toll-like receptor 4 mediates inflammatory
signaling by bacterial lipopolysaccharide in human hepatic stellate
cells. Hepatology. 37:1043–1055. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fujimoto M, Uemura M, Nakatani Y, Tsujita
S, Hoppo K, Tamagawa T, Kitano H, Kikukawa M, Ann T, Ishii Y, et
al: Plasma endotoxin and serum cytokine levels in patients with
alcoholic hepatitis: relation to severity of liver disturbance.
Alcohol Clin Exp Res. 24(Suppl 4): 48S–54S. 2000.PubMed/NCBI
|
|
64
|
Fukui H, Brauner B, Bode JC and Bode C:
Plasma endotoxin concentrations in patients with alcoholic and
non-alcoholic liver disease: reevaluation with an improved
chromogenic assay. J Hepatol. 12:162–169. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gustot T, Lemmers A, Moreno C, Nagy N,
Quertinmont E, Nicaise C, Franchimont D, Louis H, Devière J and Le
Moine O: Differential liver sensitization to toll-like receptor
pathways in mice with alcoholic fatty liver. Hepatology.
43:989–1000. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mandrekar P, Catalano D, White B and Szabo
G: Moderate alcohol intake in humans attenuates monocyte
inflammatory responses: inhibition of nuclear regulatory factor
kappa B and induction of interleukin 10. Alcohol Clin Exp Res.
30:135–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hartmann P, Seebauer CT and Schnabl B:
Alcoholic liver disease: the gut microbiome and liver cross talk.
Alcohol Clin Exp Res. 39:763–775. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Keshavarzian A, Holmes EW, Patel M, Iber
F, Fields JZ and Pethkar S: Leaky gut in alcoholic cirrhosis: a
possible mechanism for alcohol-induced liver damage. Am J
Gastroenterol. 94:200–207. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bala S, Marcos M, Kodys K, Csak T,
Catalano D, Mandrekar P and Szabo G: Up-regulation of microRNA-155
in macrophages contributes to increased tumor necrosis factor α
(TNFα) production via increased mRNA half-life in alcoholic liver
disease. J Biol Chem. 286:1436–1444. 2011. View Article : Google Scholar
|
|
70
|
Kavanaugh MJ, Clark C, Goto M, Kovacs EJ,
Gamelli RL, Sayeed MM and Choudhry MA: Effect of acute alcohol
ingestion prior to burn injury on intestinal bacterial growth and
barrier function. Burns. 31:290–296. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Thakur V, McMullen MR, Pritchard MT and
Nagy LE: Regulation of macrophage activation in alcoholic liver
disease. J Gastroenterol Hepatol. 22(Suppl 1): S53–S56. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Rao R: Endotoxemia and gut barrier
dysfunction in alcoholic liver disease. Hepatology. 50:638–644.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yan AW, Fouts DE, Brandl J, Stärkel P,
Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA and
Schnabl B: Enteric dysbiosis associated with a mouse model of
alcoholic liver disease. Hepatology. 53:96–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bull-Otterson L, Feng W, Kirpich I, Wang
Y, Qin X, Liu Y, Gobejishvili L, Joshi-Barve S, Ayvaz T, Petrosino
J, et al: Meta-genomic analyses of alcohol induced pathogenic
alterations in the intestinal microbiome and the effect of
Lactobacillus rhamnosus GG treatment. PLoS One. 8:e530282013.
View Article : Google Scholar
|
|
75
|
Forsyth CB, Farhadi A, Jakate SM, Tang Y,
Shaikh M and Keshavarzian A: Lactobacillus GG treatment ameliorates
alcohol-induced intestinal oxidative stress, gut leakiness, and
liver injury in a rat model of alcoholic steatohepatitis. Alcohol.
43:163–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gratz SW, Mykkanen H and El-Nezami HS:
Probiotics and gut health: a special focus on liver diseases. World
J Gastroenterol. 16:403–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Choudhry MA, Fazal N, Goto M, Gamelli RL
and Sayeed MM: Gut-associated lymphoid T cell suppression enhances
bacterial translocation in alcohol and burn injury. Am J Physiol
Gastrointest Liver Physiol. 282:G937–G947. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Szabo G and Bala S: Alcoholic liver
disease and the gut-liver axis. World J Gastroenterol.
16:1321–1329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hoek JB and Pastorino JG: Ethanol,
oxidative stress, and cytokine-induced liver cell injury. Alcohol.
27:63–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Adachi Y, Moore LE, Bradford BU, Gao W and
Thurman RG: Antibiotics prevent liver injury in rats following
long-term exposure to ethanol. Gastroenterology. 108:218–224. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Semenza GL: Hypoxia-inducible factors in
physiology and medicine. Cell. 148:399–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Nath B and Szabo G: Alcohol-induced
modulation of signaling pathways in liver parenchymal and
nonparenchymal cells: implications for immunity. Semin Liver Dis.
29:166–177. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Valfrè di Bonzo L, Novo E, Cannito S,
Busletta C, Paternostro C, Povero D and Parola M: Angiogenesis and
liver fibrogenesis. Histol Histopathol. 24:1323–1341.
2009.PubMed/NCBI
|
|
85
|
Novo E, Povero D, Busletta C, Paternostro
C, di Bonzo LV, Cannito S, Compagnone A, Bandino A, Marra F,
Colombatto S, et al: The biphasic nature of hypoxia-induced
directional migration of activated human hepatic stellate cells. J
Pathol. 226:588–597. 2012. View Article : Google Scholar
|
|
86
|
Ciupińska-Kajor M, Hartleb M, Kajor M,
Kukla M, Wyleżoł M, Lange D and Liszka L: Hepatic angiogenesis and
fibrosis are common features in morbidly obese patients. Hepatol
Int. 7:233–240. 2013. View Article : Google Scholar
|
|
87
|
Sanz-Cameno P, Trapero-Marugán M, Chaparro
M, Jones EA and Moreno-Otero R: Angiogenesis: from chronic liver
inflammation to hepatocellular carcinoma. J Oncol. 2010:2721702010.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Marra F and Tacke F: Roles for chemokines
in liver disease. Gastroenterology. 147:577–594.e1. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Raskopf E, Gonzalez Carmona MA, Van
Cayzeele CJ, Strassburg C, Sauerbruch T and Schmitz V: Toxic damage
increases angiogenesis and metastasis in fibrotic livers via
PECAM-1. Biomed Res Int. 2014:7128932014. View Article : Google Scholar :
|
|
90
|
Svegliati-Baroni G, Ridolfi F, Di Sario A,
Saccomanno S, Bendia E, Benedetti A and Greenwel P: Intracellular
signaling pathways involved in acetaldehyde-induced collagen and
fibronectin gene expression in human hepatic stellate cells.
Hepatology. 33:1130–1140. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Das SK and Vasudevan DM: Genesis of
hepatic fibrosis and its biochemical markers. Scand J Clin Lab
Invest. 68:260–269. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gao B and Bataller R: Alcoholic liver
disease: pathogenesis and new therapeutic targets.
Gastroenterology. 141:1572–1585. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Nanji AA, Khettry U and Sadrzadeh SM:
Lactobacillus feeding reduces endotoxemia and severity of
experimental alcoholic liver (disease). Proc Soc Exp Biol Med.
205:243–247. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bass NM, Mullen KD, Sanyal A, Poordad F,
Neff G, Leevy CB, Sigal S, Sheikh MY, Beavers K, Frederick T, et
al: Rifaximin treatment in hepatic encephalopathy. N Engl J Med.
362:1071–1081. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mencin A, Kluwe J and Schwabe RF:
Toll-like receptors as targets in chronic liver diseases. Gut.
58:704–720. 2009. View Article : Google Scholar : PubMed/NCBI
|