Introduction
Ewing sarcoma (ES) is a highly malignant bone and
soft tissue tumor that most often occur in children, teenagers and
young adults with a peak incidence between the ages of 14 and 20
years (1,2). While the histogenesis of ES remains
enigmatic, 85% of cases are defined by the t(11;22)(q24;q12) chromosomal translocation,
resulting in a pathognomonic chimeric fusion gene which encodes the
EWS-FLI1 protein (3). EWS-FLI1
induces massive deregulation of protein expression by either
transcriptionally inducing or repressing specific target genes,
many of which are involved in the oncogenic process, including cell
proliferation (4), transformation
(5) and in vivo tumor
growth (6), but the function of
EWS-FLI1 in autophagy still remains unknown.
Macroautophagy (hereafter referred to as autophagy)
has proven to be an evolutionarily conserved catabolic pathway
within all eukaryotic cells that plays a critical function for
cellular homeostasis to remove senescent or damaged proteins and
organelles through lysosome-mediated degradation (7,8).
In addition to removing dysfunctional proteins and organelles,
autophagy could also provide amino acids, monosaccharides, nucleic
acids and lipids during times of nutrient deprivation (9,10).
Autophagy can be stimulated by various stressors, such as
starvation, oxidative stress and pharmaceutical compound treatment
(11). Besides maintaining normal
cellular homeostasis, autophagy was also illustrated to play
crucial roles in infectious, inflammatory, neurodegenerative and
metabolic diseases (12–15). Moreover, autophagy was identified
to have dual roles in tumors (16). In some cases, autophagy was found
to serve a tumor suppressor function (17,18). Paradoxically, however,
accumulating evidence strongly suggests that autophagy promotes
tumor growth and resistance to chemotherapy in established tumors
(19,20). These two distinct functions may be
mainly due to different genes induced by autophagy in different
tumors and conditions (21).
At least 36 autophagy-related (ATG) genes are
primarily involved in the progression of autophagy from phagophore
initiation to autophagosome formation in mammalian cells (22). Among these, ATG7 and ATG3
conjugate mammalian light chain 3 (LC3) homologues to phosphatidyl
ethanolamine (PE), and ATG7 and ATG10 conjugate ATG12 to ATG5
(23,24). The cysteine protease ATG4
contributes to this chain of events by cleaving the LC3 C-terminal
domain to generate LC3-I (25).
Consequently, LC3-I is converted by ATG7 and ATG3 to LC3-II, which
is essential for phagophore and autophagosome formation (22,26). And the LC3-II on the cytoplasmic
location of autolysosome is recycled following delipidation of PE,
which is also performed by ATG4 (27). There are four different ATG4
paralogs expressed in mammals (ATG4A, ATG4B, ATG4C and ATG4D) with
ATG4B being crucial for the autophagic process and having the
broadest substrate spectrum for different LC3 forms and homologs
(28,29).
In the present study, the authors investigated the
function of EWS-FLI1 in autophagy in ES cells. It was identified
that autophagy was promoted by overexpressing EWS-FLI1 in ES cells,
as evidenced by the decreases in the amount of p62 (SQSTM1) and
increases in the amount of LC3B-II, two important markers of
autophagy, as well as the increased LC3 puncta in cells.
Furthermore, ATG4B was significantly upregulated by EWS-FLI1
overexpression, and ATG4B gene may be a transcript target of
EWS-FLI1. To the best of the authors' knowledge, the present study
is the first to have examined the function of ATG4B in ES cells.
ATG4B was shown to greatly modulate the autophagy process and
ATG4B-potentiated autophagy is required for ES cells survival. In
conclusion, the authors revealed the effect of EWS-FLI1 and ATG4B
on autophagy in ES cells in the present study, and suggested
EWS-FLI1 and ATG4B as potential therapeutic targets for ES.
Materials and methods
Cell culture
NIH3T3, A673 and TC71 cells were obtained from
American Type Culture Collection (Manassas, VA, USA). All the cells
were cultured in RPMI-1640 (Gibco, Life Technologies, Shanghai,
China) containing 10% fetal calf serum (Gibco, Life Technologies,
Mulgrave, VIC, Australia) for normal condition. Cells were cultured
in serum-free medium for starvation for indicated time to induce
autophagy.
Lentivirus and plasmid
Lentiviruses containing the following were
constructed and obtained from MDL Biotechnology (Beijing, China):
An empty plasmid or expression plasmid of EWS-FLI1, control small
interfering (si)RNA and EWS-FLI1 siRNAs, control plasmid and ATG4B
expression plasmid, short hairpin (sh)RNA-control plasmid and
shRNA-ATG4B plasmid. The lentivirus particle was used to infect
NIH3T3 for 3 days at 50 MOI with the presence of polybrene,
puromycin selection was performed to establish the overexpression
cell lines. Lipofectamine 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to
transfect plasmids and siRNAs into ES cells.
Western blot analysis
The cells were harvested by using
radioimmunoprecipitation assay buffer (MDL Biotechnology), and the
protein concentration was measured using Bio-Rad quantification
assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Proteins
were separated using 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 3 g non-fat dry milk in TBST (50 mM
Tris-HCl, 150 mM NaCl, 0.1% Tween-20, pH 7.4) for 2 h. Primary
antibodies including anti-p62 (1:500, #sc-101542), anti-LC3 (1:500,
#sc-398822), anti-FLI1 (1:500, #sc-22808), anti-cyt c
(1:500, #sc-13561), anti-glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) (1:1,000, #sc-32233) were bought from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA) and anti-ATG4B (1:500,
#13507), anti-PUMA (1:500, #4976) were obtained from Cell Signaling
Technology (Beverly, MA, USA). The antibodies were added and
incubated overnight at 4°C. After washing, the membranes was
incubated with anti-mouse secondary antibody (1:2,000, #sc2055) or
anti-rabbit secondary antibody (1:2,000, #sc2054) (both from Santa
Cruz Biotechnology, Inc.) at room temperature for 1 h. The target
protein was visualized by enhanced chemiluminescence (Thermo Fisher
Scientific, Inc.). The results from western blot analysis were
quantitatively analyzed by optical density using ImageJ
software.
RNA Isolation, reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis and CHIP assay
Total RNA was extracted with TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's
instructions. A LightCycler (ABI PRISM 7000; Applied Biosciences;
Thermo Fisher Scientific, Inc.) and a SYBR RT-PCR kit (Takara
Biotechnology Co., Ltd., Dalian, China) were used for RT-qPCR
analysis. GAPDH was used as the internal control, thermocycling
conditions were 1 cycle (95°C, 5 min) and 40 cycles (95°C, 15 sec;
57°C, 30 sec; 72°C, 30 sec) and the 2−ΔΔCt method
(30) was used to evaluate the
relative quantities of each amplified product in the samples. CHIP
assay was performed as previously reported and the primer sequences
for qPCR and CHIP-qPCR were used as described (31).
Dual-luciferase reporter gene assay
ATG4B promoter region was cloned into pGL3-based
vectors to construct the luciferase reporter plasmid. The phRL-TK
plasmid was used as an internal control. The luciferase activities
were measured on a SpectraMax M5 reader (Molecular Devices LLC,
Sunnyvale, CA, USA) using the Dual Luciferase Reporter assay system
(Promega Corp., Madison, WI, USA).
Confocal microscopic analysis
The experiment was performed as described (31). All slides were observed under a
confocal laser microscopy (LSM780; Carl Zeiss AG, Oberkochen,
Germany).
Cell viability assay and detection of
cell apoptosis
Cell viability was examined by CCK8 kit and cell
apoptosis percent was detected by flow cytometry as described
(32). The caspase-3 activity of
cell extracts (~106 cells) was determined using the
synthetic substrate, acetyl-Asp-Glu-Val-Asp-7-amino-4-methyl
coumarin (DEVD-AMC; Peptron, Inc., Daejeon, Korea) according to the
manufacturer's instructions. The AMC fluorescence released by
active caspase-3 was measured at an excitation wavelength of 360 nm
and an emission wavelength of 460 nm.
Statistical analysis
All data are presented as mean ± standard deviation
of three independent experiments. Statistical significance was
determined with the two-tailed Student's t-test to compare two
groups. One-way analysis of variance (ANOVA) was performed to
compare three or more groups. If the ANOVA analysis was
significant, the Newman-Keuls test was applied for comparison
between each two groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
EWS-FLI1 promotes autophagy in ES
cells
In order to investigate the function of EWS-FLI1 in
autophagy in ES, the authors used in vitro cultured NIH3T3
cells and ES tumor-derived cell lines to perform the study. First,
the lentivirus containing EWS-FLI1 expressing plasmid was used to
construct NIH3T3-EWS-FLI1 stable overexpression cell lines. As
presented in Fig. 1A, EWS-FLI1
expression was significantly increased after EWS-FLI1-lentivirus
infection compared to control-lentivirus infection. Most important,
the expression of p62 was decreased and the level of LC3-II was
increased in EWS-FLI1 overexpressed NIH3T3 cells (Fig. 1A and B). Furthermore, the authors
used EWS-FLI1 specific siRNA to knockdown the expression of
EWS-FLI1 in A673 cells. As presented in Fig. 1D, the effect of the two siRNAs was
examined and EWS-FLI1 expression was much weaker in siRNA-2 treated
A673 cells compared to cells transfected with siRNA-1 (Fig. 1D and E). Interestingly, the
expression of p62 was significantly enhanced and LC3-II expression
was markedly decreased in both siRNAs treated cell, consistent with
the knockdown efficiency (Fig. 1D and
E). The effect of EWS-FLI1 on autophagy was also verified by
immunofluorescence to show the formation of autophagosomes labeled
the by anti-LC3 antibody, and the observed stable overexpression of
EWS-FLI1 in NIH3T3 greatly increased the formation of
autophagosomes, accordant results were obtained in EWS-FLI1
silencing A673 cells (Fig. 1C and
F).
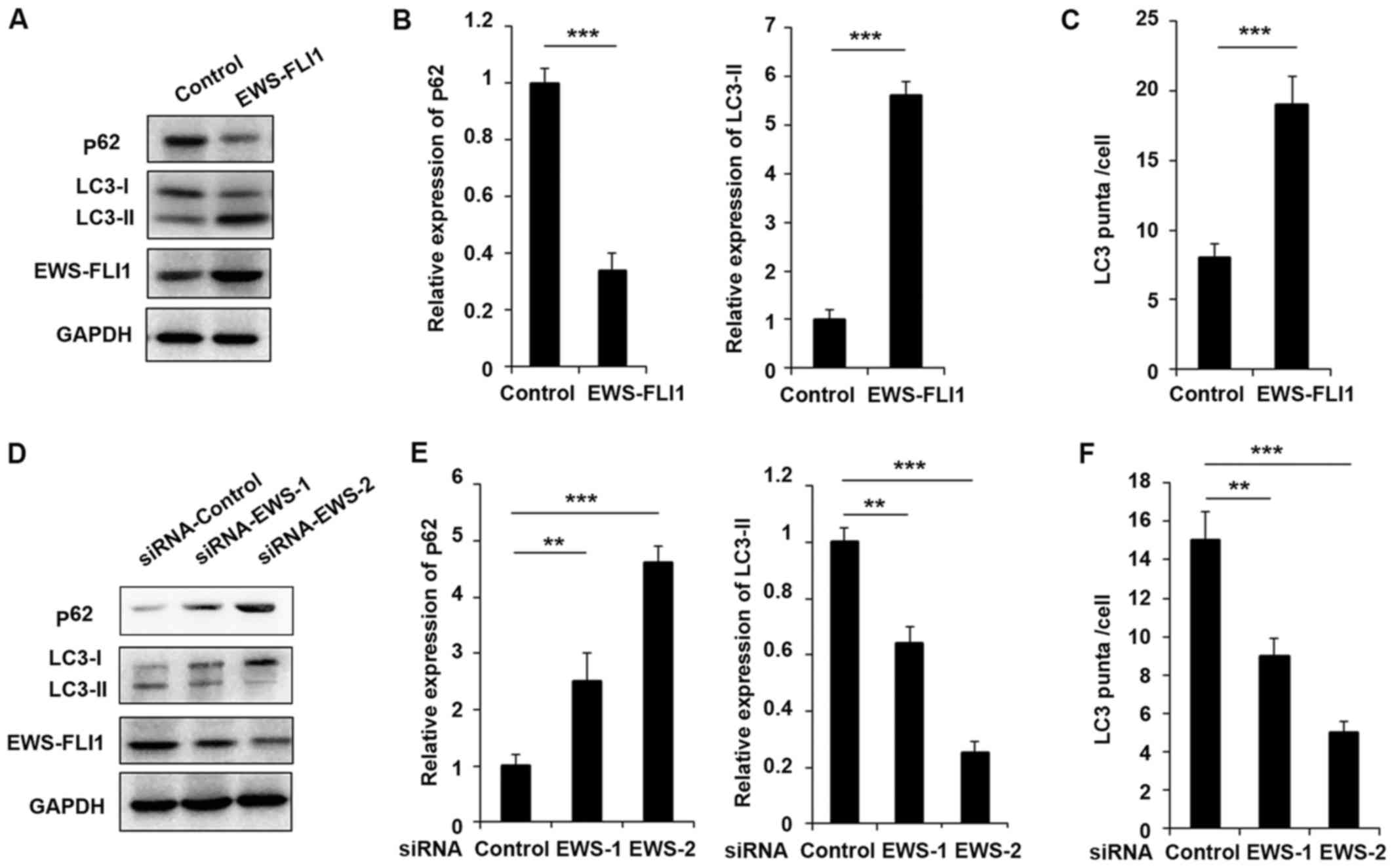 | Figure 1EWS-FLI1 promotes autophagy in ES
cells. (A) Western blot analysis of p62, LC3-I, LC3-II, EWS-FLI1
and GAPDH in NIH3T3 cells stable over-expressing control plasmid or
EWS-FLI1 expression plasmid following starvation for 2 h. (B) Band
intensity of p62 and LC3-II was quantified and normalized to the
GAPDH in (A). (C) After starvation for 2 h, LC3 puncta in NIH3T3
cells stable overexpressing control plasmid or EWS-FLI1 expression
plasmid was detected by confocal laser microscopy and quantified
using the ImageJ program, 30 to 60 cells chosen in random were
counted. (D) Western blot analysis of p62, LC3-I, LC3-II, EWS-FLI1
and GAPDH in control-siRNA or EWS-FLI1-siRNAs treated A673 cells
after starvation for 2 h. (E) Band intensity of p62 and LC3-II was
quantified and normalized to the GAPDH in (D). (F) After starvation
for 2 h, LC3 puncta in control-siRNA or EWS-FLI1-siRNAs treated
A673 cells were detected by confocal laser microscopy and
quantified using the ImageJ program, 30 to 60 cells chosen in
random were counted. Data are representative of three independent
experiments (mean ± standard deviation). **P<0.01 and
***P<0.001 as indicated. ES cells, Ewing sarcoma
cells; siRNA, small interfering RNA; GAPDH, glyceraldehyde
3-phosphate dehydrogenase. |
ATG4B is a transcript target of
EWS-FLI1
EWS-FLI1 induces massive deregulation of protein
expression, therefore the authors aimed to examine whether EWS-FLI1
could regulate the expression of ATG proteins. The expression of
various ATG proteins was investigated including ATG2A, ATG4B,
ATG4C, ATG5, ATG7 and ATG9A, and only ATG4B expression was
significantly upregulated by EWS-FLI1 overexpression (Fig. 2A). Consistently, the CHIP-qPCR
result demonstrated that EWS-FLI1 has the ability to interact with
ATG4B promoter but not others (Fig.
2B), which meant that ATG4B gene may be a transcript
target of EWS-FLI1. In addition, ATG4B activation was significantly
increased in EWS-FLI1 stable overexpressed NIH3T3 cells, especially
in starvation conditions (Fig.
2C). Moreover, the authors infected NIH3T3 cells with
increasing titer of EWS-FLI1-lentivirus, and the expression of
ATG4B was upregulated in a dose-dependent manner in both protein
and mRNA levels (Fig. 2D and F).
Consistent with the overexpression data, knockdown the expression
of EWS-FLI1 also decreased the mRNA and protein levels of ATG4B
(Fig. 2E and G).
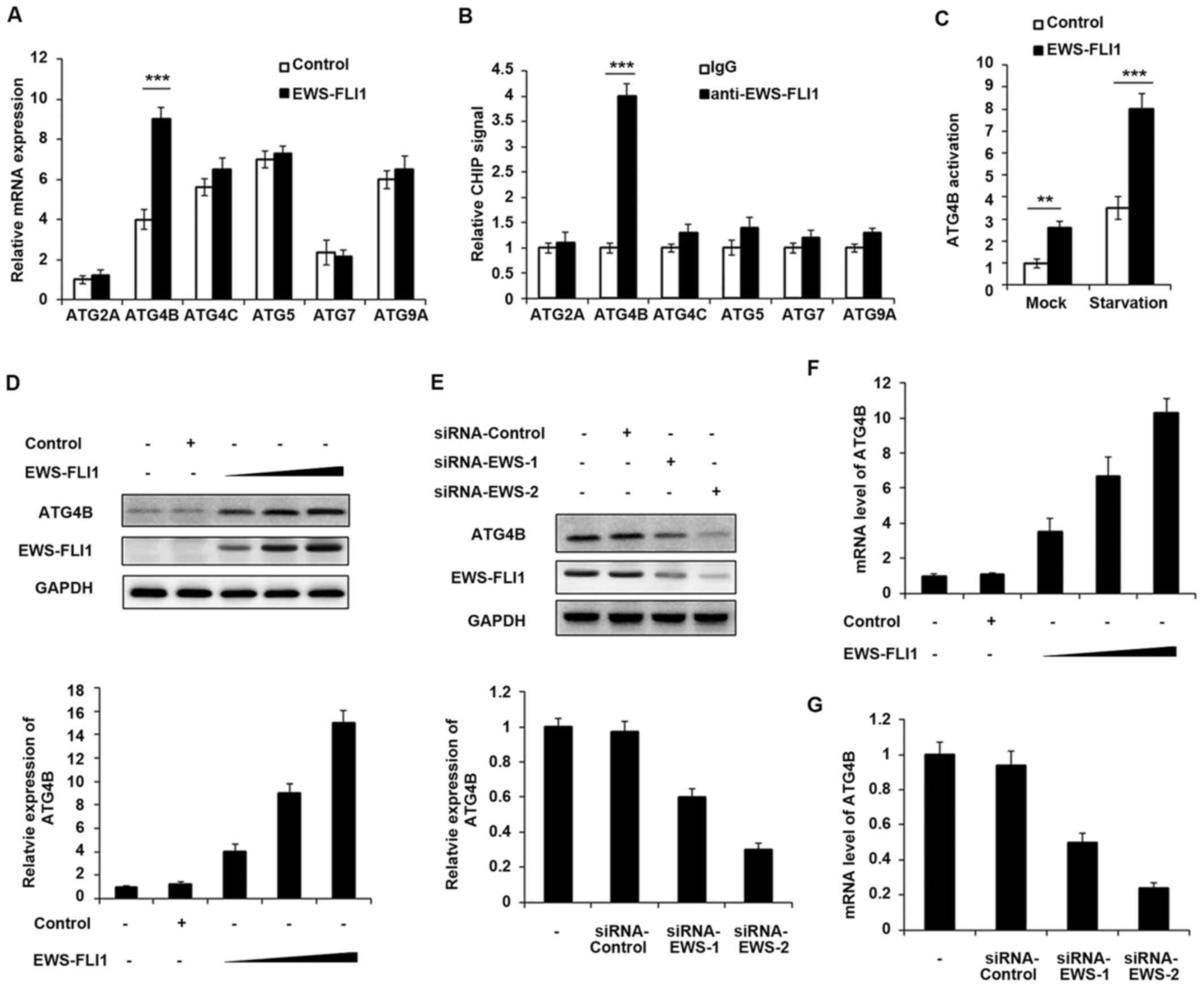 | Figure 2ATG4B is a transcript target of
EWS-FLI1. (A) RT-qPCR analysis of mRNA expression of ATG2A, ATG4B,
ATG4C, ATG5, ATG7 and ATG9A in NIH3T3 cells stable overexpressing
control plasmid or EWS-FLI1 expression plasmid after starvation for
2 h. (B) CHIP assay and RT-qPCR analysis were performed with
fragmented chromatin from NIH3T3 cells stable overexpressing
control plasmid or EWS-FLI1 expression plasmid after starvation for
2 h. (C) Dual-luciferase reporter gene assay was performed to
investigate the activation of ATG4B promoter in NIH3T3 cells stable
overexpressing control plasmid or EWS-FLI1 expression plasmid in
normal or starvation condition for 2 h. (D) NIH3T3 cells were
infected with increasing titer of EWS-FLI1-lentivirus, the protein
levels of ATG4B and EWS-FLI1 were examined by western blotting, and
band intensity of ATG4B was quantified and normalized to the GAPDH.
(E) A673 cells were treated with control-siRNA or EWS-FLI1 siRNAs,
the protein levels of ATG4B and EWS-FLI1 were examined by western
blot, and band intensity of ATG4B was quantified and normalized to
the GAPDH. (F and G) RT-qPCR analysis of mRNA level of ATG4B in (D
and E). Data are representative of three independent experiments
(mean ± standard deviation). **P<0.01 and
***P<0.001 vs. control. RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; siRNA, small
interfering RNA; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
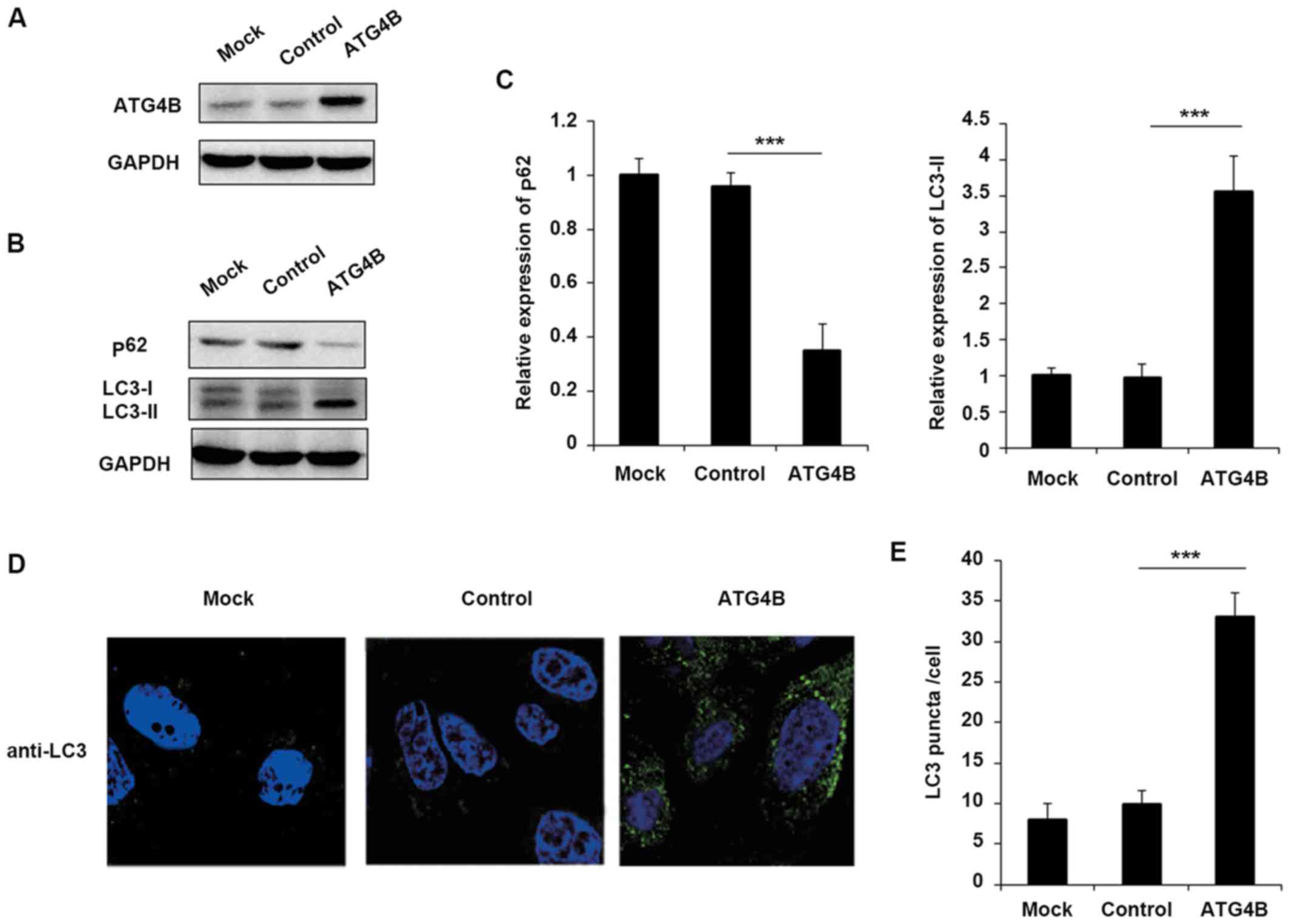
Overexpression of ATG4B promotes
autophagy in ES cells
Although ATG4B has been reported that serve
essential roles in autophagy in numerous cells, the function of
ATG4B in ES cells still remains unknown. To confirm that EWS-FLI1
affected autophagy through ATG4B, the authors further examined the
effect of ATG4B on autophagy in ES cells. Overexpression of ATG4B
in TC71 cells was confirmed as presented in Fig. 3A. Consistent with the results of
EWS-FLI1 overexpression, ATG4B overexpression also decreased the
level of p62 and enhanced the expression of LC3-II (Fig. 3B and C), as well as the increased
formation of autophagosomes (Fig. 3D
and E).
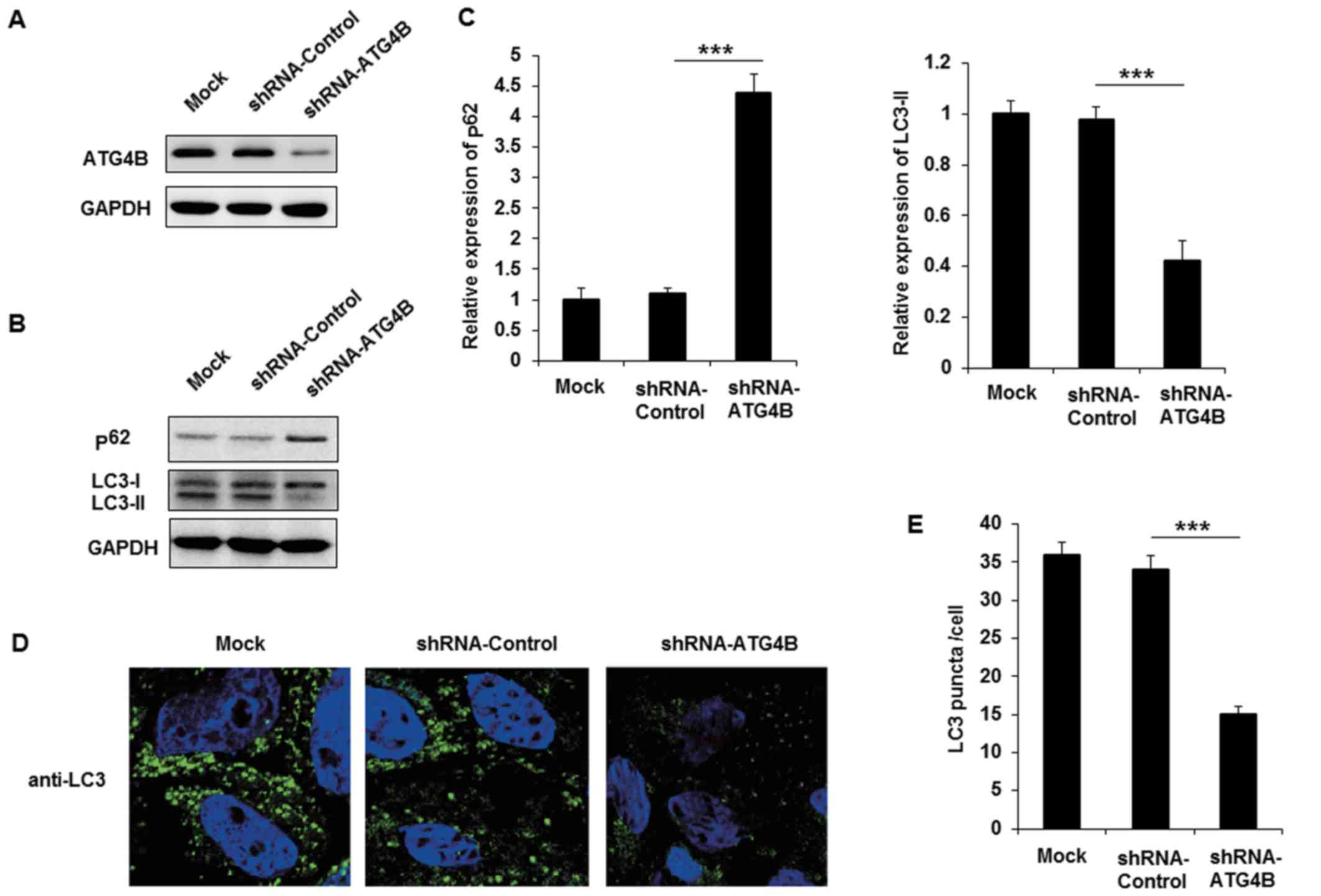
Silencing of ATG4B inhibits autophagy in
ES cells
The authors purchased shRNA plasmid of ATG4B to
knockdown the expression of ATG4B in A673 cells, and the effect of
ATG4B specific shRNA was examined (Fig. 4A). Following transfection with
shRNA-ATG4B, the expression of p62 was markedly increased and
LC3-II level was decreased (Fig. 4B
and C). Similar to the EWS-FLI1 silencing results, knockdown
the expression of ATG4B also decreased autophagosomes formation in
A673 cells. Together with the data in ATG4B overexpressed TC71
cells, these results indicated that ATG4B indeed promoted the
autophagy process in ES cells.
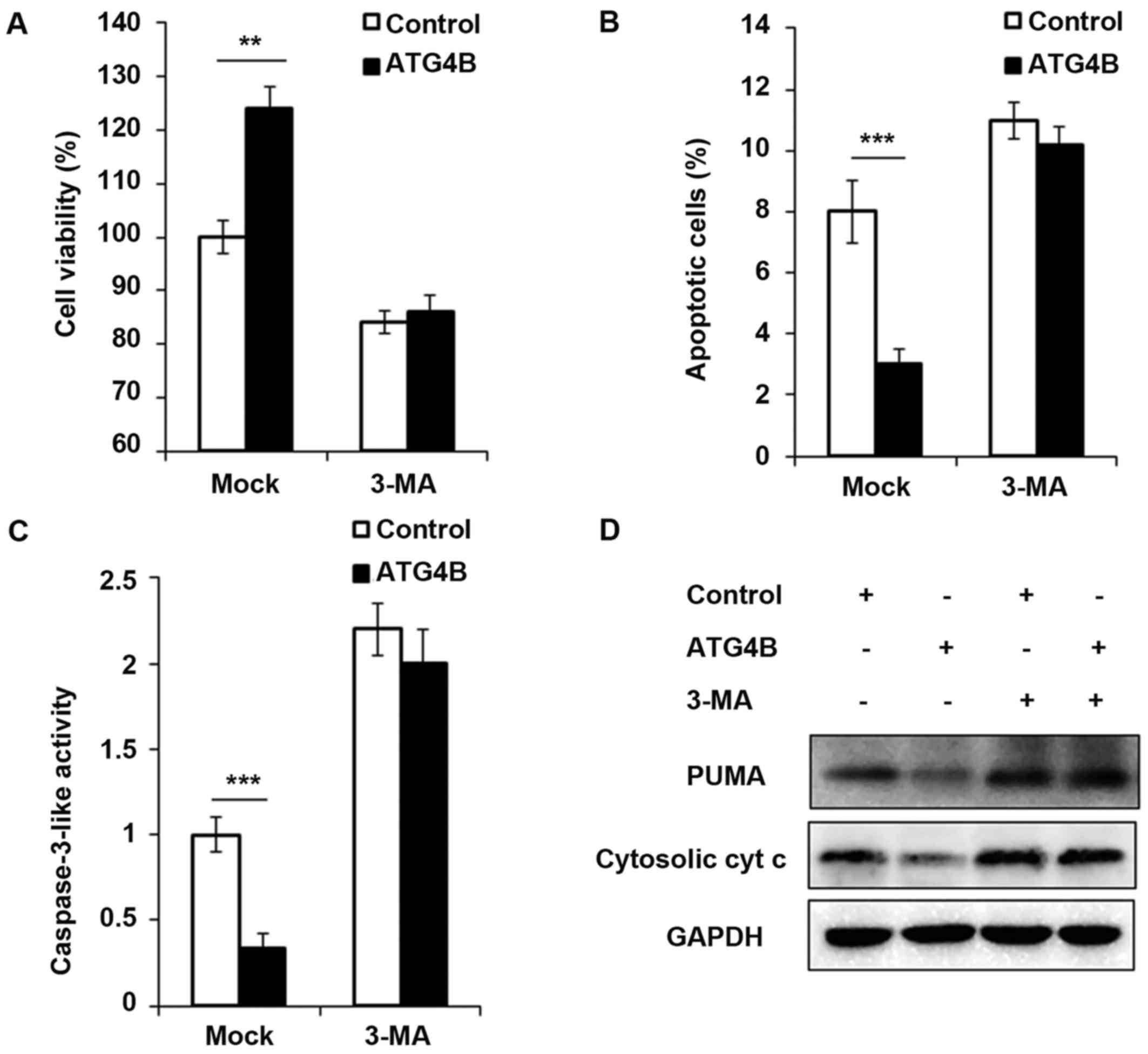
Cell viability is increased and apoptosis
is attenuated in ATG4B overexpressed ES cells
In addition, the authors investigated the
contribution of ATG4B-mediated autophagy to tumor cell viability
and apoptosis. As presented in Fig.
5A, ATG4B overexpressed TC71 cells with high level of autophagy
grew significantly faster than TC71 cells transfected with control
plasmid. However, the blockage of autophagy by 3-methyladenine
(3-MA), an important agent to block autophagy, retarded the cell
viability of ATG4B overexpressed cells. Apoptosis is suppressed by
autophagy in various cancer cells (33,34). Recently, it has been reported that
ATG4B protease and autophagy serve crucial roles in protecting
epithelial cells against bleomycin-induced pulmonary fibrosis and
apoptosis (35). Therefore, the
authors investigated the effect of ATG4B-induced autophagy on
apoptosis in ES cells. As presented in Fig. 5B and C, the number of apoptotic
cells and caspase-3 activity was greatly decreased in
ATG4B-overexpressed cells, treatment of 3-MA increased the
apoptosis process as previous study reported (36), however after 3-MA treatment, there
was no significant difference of apoptosis between ATG4B
overexpressed cells and control group (Fig. 5B and C). Furthermore, the authors
examined the expression of PUMA and cytosolic cytochrome c,
and both the protein levels of PUMA and cytosolic cytochrome
c was found decreased in ATG4B overexpressed cells without
3-MA challenge, nevertheless, the downregulated expression of PUMA
and cytosolic cytochrome c modulated by ATG4B overexpression
was all observed to be reversed following 3-MA treatment.
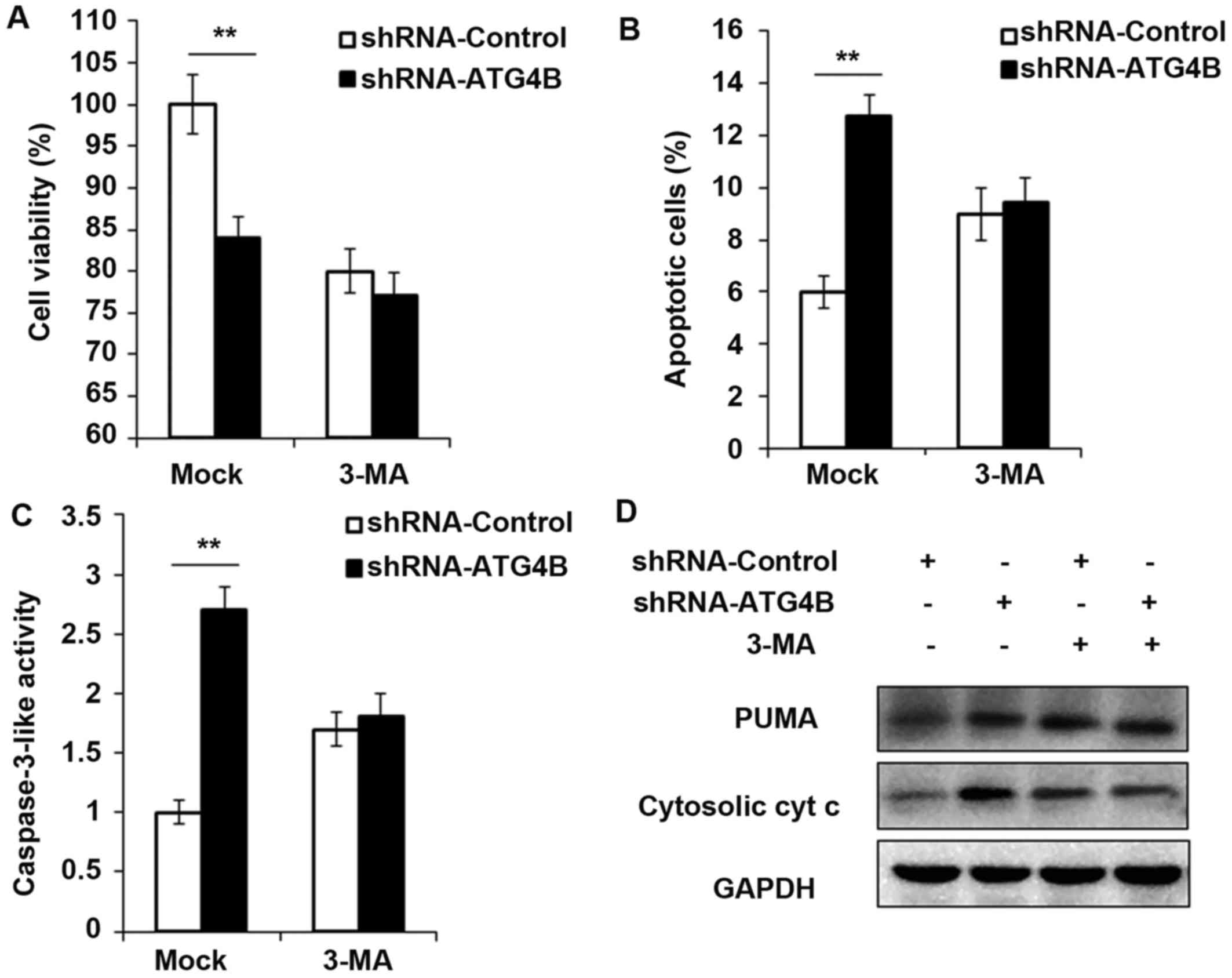
Cell viability is decreased and apoptosis
is potentiated in ATG4B silenced ES cells
To ensure the results in Fig. 5, shRNA-ATG4B was used to repeat
the experiments, and consistent with the ATG4B overexpression
results, knockdown the expression of ATG4B indeed decreased the
cell viability and increased the apoptotic cell number (Fig. 6A and B), as well as the activity
of caspase-3 and expression of PUMA and cytosolic cytochrome
c (Fig. 6C and D). But all
these processes were found reversed after autophagy was blocked by
3-MA.
Discussion
In the present study, the authors investigated the
function of EWS-FLI1 in autophagy in ES. The present study may
extend our understanding of the pathogenesis of ES, and provided a
new approach for the related drug designs.
Described for the first time in 1921 by Ewing
(37), ES primarily arises at the
pelvic bones, the diaphysis of the lower extremities' long bones
and the chest wall's bones. The overall survival of ES patients
remains poor: 25% of patients with localized tumor and 75% of
patients with metastasis do not have durable therapeutic responses
(38). ES is characterized by
unique chromosomal translocations and EWS-FLI1 is the most commonly
fusion protein observed in ES (39). Transfection of EWS-FLI1 into
NIH3T3 cells was reported to lead to the phenotypic characteristics
of ES (40). In the present
study, to extend our understanding of EWS-FLI1 in autophagy, a
lentivirus containing a EWS-FLI1 expression plasmid was used to
build a EWS-FLI1 stable expression NIH3T3 cell line. Overexpression
of EWS-FLI1 was identified in NIH3T3 cells leading to the
attenuated p62 expression and enhanced LC3-II expression, as well
as the autophagosomes number is significantly increased. Silencing
EWS-FLI1 resulted in the consistent data. Based on these results,
the authors supposed that EWS-FLI1 could regulate the autophagy
process in ES.
Autophagy facilitates the clearance of long-lived
proteins, aggregates and damaged organelles, and dysregulation of
autophagy is highly associated with cancer, particularly in
tumorigenesis and chemotherapy resistance (39,41). The cysteine protease ATG4B serves
a role in key steps of the autophagy process as cleaving proLC3
isoforms to form LC3-I for subsequent lipidation to form LC3-II and
autophagosome membrane insertion. A previous study demonstrated
that ATG4B was essential for starvation-induced autophagy, and
inhibition of ATG4B activity by anti-autophagy compound NSC185058
greatly attenuated osteosarcoma tumorigenesis (42). In addition, ATG4B was reported to
promote colorectal cancer growth and suggested as a potential
biomarker and therapeutic target in CML stem/progenitor cells
(43,44). Nevertheless, the study of ATG4B in
ES remains largely unknown. In the present study, the authors found
that EWS-FLI1 induced the expression of ATG4B and EWS-FLI1 was
observed to interact with the ATG4B promoter, which suggested that
ATG4B as a transcriptional target of EWS-FLI1. In addition,
overexpression of ATG4B significantly promoted the autophagy in ES
cells, consistently, silencing of ATG4B markedly suppressed the
progression of autophagy. These results supposed that the
overexpression of EWS-FLI1 may also lead to the increased
expression of ATG4B in ES tissues, however, we still need to make
further studies to confirm the exact conclusion.
The profound relationship between autophagy and
apoptosis has been widely reported. Autophagy was found to promote
apoptosis in response to specific stimuli (45), but accumulated evidences showed
that autophagy could protect against apoptosis (46,47). In the present study, the authors
also examined the function of ATG4B in cell viability. Consistent
with the previous results in other cell types (43,44), overexpression of ATG4B was found
to promote cell growth in ES cells, but blockade of ATG4B-dependent
autophagic flux was insufficient to accelerate cell growth in ES
cells. Furthermore, the role of ATG4B on apoptosis was investigated
and ATG4B dramatically suppressed the apoptosis process in ES
cells. In addition, it was found that ATG4B-dependent autophagic
flux is also necessary for the inhibition of apoptosis. Moreover,
the authors examined the level of PUMA and cytosolic cytochrome
c, two important pro-apoptotic proteins, and both the
expression of PUMA and cytosolic cytochrome c was
significantly decreased in ATG4B overexpressed ES cells, but the
processes were found reversed after autophagy was blocked by 3-MA.
Taken together, these findings suggested that ATG4B-dependent
autophagy plays great roles in regulating cell viability and cell
apoptosis in ES cells.
In conclusion, in the present study, the authors
revealed the promoting effect of EWS-FLI1 and ATG4B on autophagy in
ES cells, and these findings may provide new venues for
anti-autophagy drugs and new targets for the treatment of ES.
Acknowledgments
This study was supported by grants from the Natural
Science Foundation of Shandong Province (No. ZR2015HQ018).
References
|
1
|
Mackintosh C, Madoz-Gúrpide J, Ordóñez JL,
Osuna D and Herrero-Martín D: The molecular pathogenesis of Ewing's
sarcoma. Cancer Biol Ther. 9:655–667. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burningham Z, Hashibe M, Spector L and
Schiffman JD: The epidemiology of sarcoma. Clin Sarcoma Res.
2:142012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Delattre O, Zucman J, Plougastel B,
Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau
G, et al: Gene fusion with an ETS DNA-binding domain caused by
chromosome translocation in human tumours. Nature. 359:162–165.
1992. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanaka K, Iwakuma T, Harimaya K, Sato H
and Iwamoto Y: EWS-Fli1 antisense oligodeoxynucleotide inhibits
proliferation of human Ewing's sarcoma and primitive
neuroectodermal tumor cells. J Clin Invest. 99:239–247. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Joo J, Christensen L, Warner K, States L,
Kang HG, Vo K, Lawlor ER and May WA: GLI1 is a central mediator of
EWS/FLI1 signaling in Ewing tumors. PLoS One. 4:e76082009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mateo-Lozano S, Gokhale PC, Soldatenkov
VA, Dritschilo A, Tirado OM and Notario V: Combined transcriptional
and translational targeting of EWS/FLI-1 in Ewing's sarcoma. Clin
Cancer Res. 12:6781–6790. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mortimore GE, Pösö AR and Lardeux BR:
Mechanism and regulation of protein degradation in liver. Diabetes
Metab Rev. 5:49–70. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mortimore GE, Lardeux BR and Heydrick SJ:
Mechanism and control of protein and RNA degradation in the rat
hepatocyte: Two modes of autophagic sequestration. Revis Biol
Celular. 20:79–96. 1989.PubMed/NCBI
|
|
10
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y and Qin ZH: Coordination of
autophagy with other cellular activities. Acta Pharmacol Sin.
34:585–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deretic V: Autophagy in infection. Curr
Opin Cell Biol. 22:252–262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang S, Dupont N, Castillo EF and Deretic
V: Secretory versus degradative autophagy: Unconventional secretion
of inflammatory mediators. J Innate Immun. 5:471–479. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Janda E, Isidoro C, Carresi C and Mollace
V: Defective autophagy in Parkinson's disease: Role of oxidative
stress. Mol Neurobiol. 46:639–661. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ren SY and Xu X: Role of autophagy in
metabolic syndrome-associated heart disease. Biochim Biophys Acta.
1852:225–231. 2015. View Article : Google Scholar
|
|
16
|
Choi KS: Autophagy and cancer. Exp Mol
Med. 44:109–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Czyzyk-Krzeska MF, Meller J and Plas DR:
Not all autophagy is equal. Autophagy. 8:1155–1156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ogier-Denis E and Codogno P: Autophagy: A
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
20
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ávalos Y, Canales J, Bravo-Sagua R,
Criollo A, Lavandero S and Quest AF: Tumor suppression and
promotion by autophagy. BioMed Res Int. 2014:6039802014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ichimura Y, Kirisako T, Takao T, Satomi Y,
Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi
M, et al: A ubiquitin-like system mediates protein lipidation.
Nature. 408:488–492. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Geng J and Klionsky DJ: The Atg8 and Atg12
ubiquitin-like conjugation systems in macroautophagy. 'Protein
modifications: Beyond the usual suspects' review series. EMBO Rep.
9:859–864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohsumi Y: Molecular dissection of
autophagy: Two ubiquitin-like systems. Nat Rev Mol Cell Biol.
2:211–216. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tanida I, Ueno T and Kominami E: Human
light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121
to expose Gly120 for lipidation and targeting to autophagosomal
membranes. J Biol Chem. 279:47704–47710. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kabeya Y, Mizushima N, Yamamoto A,
Oshitani-Okamoto S, Ohsumi Y and Yoshimori T: LC3, GABARAP and
GATE16 localize to autophagosomal membrane depending on form-II
formation. J Cell Sci. 117:2805–2812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanida I, Sou YS, Ezaki J,
Minematsu-Ikeguchi N, Ueno T and Kominami E:
HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three
human Atg8 homologues and delipidates microtubule-associated
protein light chain 3- and GABAA receptor-associated
protein-phospholipid conjugates. J Biol Chem. 279:36268–36276.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mariño G, Uría JA, Puente XS, Quesada V,
Bordallo J and López-Otín C: Human autophagins, a family of
cysteine proteinases potentially implicated in cell degradation by
autophagy. J Biol Chem. 278:3671–3678. 2003. View Article : Google Scholar
|
|
29
|
Hemelaar J, Lelyveld VS, Kessler BM and
Ploegh HL: A single protease, Apg4B, is specific for the
autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3,
GABARAP, and Apg8L. J Biol Chem. 278:51841–51850. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Guo L, Huang JX, Liu Y, Li X, Zhou SR,
Qian SW, Liu Y, Zhu H, Huang HY, Dang YJ, et al: Transactivation of
Atg4b by C/EBPβ promotes autophagy to facilitate adipogenesis. Mol
Cell Biol. 33:3180–3190. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He C, Zhu H, Zhang W, Okon I, Wang Q, Li
H, Le YZ and Xie Z: 7-Ketocholesterol induces autophagy in vascular
smooth muscle cells through Nox4 and Atg4B. Am J Pathol.
183:626–637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
c and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harhaji-Trajkovic L, Vilimanovich U,
Kravic-Stevovic T, Bumbasirevic V and Trajkovic V: AMPK-mediated
autophagy inhibits apoptosis in cisplatin-treated tumour cells. J
Cell Mol Med. 13:3644–3654. 2009. View Article : Google Scholar
|
|
35
|
Cabrera S, Maciel M, Herrera I, Nava T,
Vergara F, Gaxiola M, López-Otín C, Selman M and Pardo A: Essential
role for the ATG4B protease and autophagy in bleomycin-induced
pulmonary fibrosis. Autophagy. 11:670–684. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Song L, Liu H, Ma L, Zhang X, Jiang Z and
Jiang C: Inhibition of autophagy by 3-MA enhances endoplasmic
reticulum stress-induced apoptosis in human nasopharyngeal
carcinoma cells. Oncol Lett. 6:1031–1038. 2013.PubMed/NCBI
|
|
37
|
Ewing J: Classics in oncology. Diffuse
endothelioma of bone. James Ewing. Proceedings of the New York
Pathological Society, 1921. CA Cancer J Clin. 22:95–98. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stahl M, Ranft A, Paulussen M, Bölling T,
Vieth V, Bielack S, Görtitz I, Braun-Munzinger G, Hardes J, Jürgens
H, et al: Risk of recurrence and survival after relapse in patients
with Ewing sarcoma. Pediatr Blood Cancer. 57:549–553. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar
|
|
40
|
Thompson AD, Teitell MA, Arvand A and
Denny CT: Divergent Ewing's sarcoma EWS/ETS fusions confer a common
tumorigenic phenotype on NIH3T3 cells. Oncogene. 18:5506–5513.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma XH, Piao S, Wang D, McAfee QW,
Nathanson KL, Lum JJ, Li LZ and Amaravadi RK: Measurements of tumor
cell autophagy predict invasiveness, resistance to chemotherapy,
and survival in melanoma. Clin Cancer Res. 17:3478–3489. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Akin D, Wang SK, Habibzadegah-Tari P, Law
B, Ostrov D, Li M, Yin XM, Kim JS, Horenstein N and Dunn WA Jr: A
novel ATG4B antagonist inhibits autophagy and has a negative impact
on osteosarcoma tumors. Autophagy. 10:2021–2035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu PF, Leung CM, Chang YH, Cheng JS, Chen
JJ, Weng CJ, Tsai KW, Hsu CJ, Liu YC, Hsu PC, et al: ATG4B promotes
colorectal cancer growth independent of autophagic flux. Autophagy.
10:1454–1465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rothe K, Lin H, Lin KB, Leung A, Wang HM,
Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM and Jiang X:
The core autophagy protein ATG4B is a potential biomarker and
therapeutic target in CML stem/progenitor cells. Blood.
123:3622–3634. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gump JM and Thorburn A: Autophagy and
apoptosis: What is the connection? Trends Cell Biol. 21:387–392.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Boya P, González-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Thorburn J, Andrysik Z, Staskiewicz L,
Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM and Thorburn A:
Autophagy controls the kinetics and extent of mitochondrial
apoptosis by regulating PUMA levels. Cell Rep. 7:45–52. 2014.
View Article : Google Scholar : PubMed/NCBI
|