Introduction
Fibroblast growth factor 21 (FGF21) is a unique
member of the FGF superfamily, which is predominantly expressed in
the liver and exerts pleiotropic effects on metabolic processes
(1). FGF21 may activate FGF
receptors (FGFRs), including FGFR1-4, which all contain an
intracellular tyrosine kinase domain, and FGFR1 is the predominant
FGFR isotype in the heart and vessels (2). β-Klotho, as a co-receptor, is
necessary for FGF21 to bind with particular FGFRs and effectively
activate the FGF21 signaling pathway (3). Furthermore, among the various
isoforms of FGFRs, FGFR1 has been suggested as the primary form for
FGF21 in cardiomyocytes and the function of FGF21 was mediated
primarily by binding to FGFR1 in a β-Klotho-dependent manner
(2,4). A previous study identified that
FGF21 is upregulated and released into circulation by the liver
tissue in response to myocardial ischemia (5). Serum levels of FGF21 are also
elevated in patients with coronary artery disease, acute myocardial
infarction and heart failure (6–8).
Additionally, FGF21 may decrease ischemia-reperfusion injury of
cardiomyocytes and improve the antioxidant capacity of endothelial
cells (9,10), indicating that FGF21 is a critical
protective factor for the cardiovascular system.
Accumulation of unfolded or misfolded proteins at
the reticulum transmembrane causes endoplasmic reticulum (ER)
stress, activating an adaptive response termed the unfolded protein
response (UPR) (11). The UPR
involves the PKR-like ER kinase (PERK), inositol-requiring kinase
1α (IRE1α) and activating transcription factor 6 (ATF6) (12), which all regulate the
transcription of a variety of associated genes encoding ER
chaperone proteins to clear the unfolded protein aggregation at the
initial stage of ER stress. When ER stress is prolonged and/or
excessive, the UPR fails to control the level of unfolded or
misfolded proteins in the ER, subsequently leading to the
activation of apoptotic pathways (11–13). Proteins downstream of the PERK and
IRE1 signaling pathways have been identified as having
pro-apoptotic roles. PERK phosphorylates eIF2α to induce the
translation of ATF4. ATF4 modulates a wide range of genes involved
in the adaptation to ER stress and induces CCAAT/-enhancer-binding
protein homologous protein (CHOP) to initiate the apoptosis
signaling pathway (14,15). IRE1α mediates the accumulation of
misfolded proteins, and induces downstream molecular X-box binding
protein 1 (XBP1) and c-Jun N-terminal kinase (JNK). However,
IRE1α-dependent apoptotic signaling occurs via phosphorylating JNK
(16), which induces cell death
via the induction of various proteins from the B-cell lymphoma-2
(Bcl-2) family and the activation of cysteinyl aspartate specific
proteinase (caspases) (17,18).
Evidence indicates that the apoptosis induced by ER
stress is regarded as a precursor to the inflammatory reaction, and
is a central mechanism, which leads to the development and
progression of cardiovascular diseases, including atherosclerosis,
ischemic heart diseases and heart failure (19–21), which all exhibit markedly
increased FGF21 expression levels. However, whether the enhanced
expression level of FGF21 is associated with ER stress in
cardiomyocytes, and the functional role and molecular mechanism of
FGF21 in ER stress-induced myocardial apoptosis remain unknown.
Materials and methods
Reagents
Fetal bovine serum (FBS) and Dulbecco's modified
Eagle's medium (DMEM) were purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The rabbit anti β-Klotho (cat.
no. AV53325) and mouse anti-c-myc (cat. no. M5546) antibodies were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The
rabbit anti-FGF21 (cat. no. Ab171941), rabbit anti-phosphorylated
(p)-FGFR1 (cat. no. Ab59194) and rabbit anti-p-IRE1α (cat. no.
Ab124945) antibodies were obtained from Abcam (Cambridge, MA, USA).
The rabbit anti-FGFR1 (cat. no. 9740), rabbit anti-extracellular
signal-regulated kinases (ERK)1/2 (cat. no. 4695), rabbit
anti-p-ERK1/2 (cat. no. 4370), rabbit anti-PERK (cat. no. 3192),
rabbit anti-p-PERK (cat. no. 3179), rabbit anti-ATF4 (cat. no.
11815), rabbit anti-IRE1α (cat. no. 3294), rabbit anti-JNK (cat.
no. 9252), rabbit anti-p-JNK (cat. no. 9251), rabbit anti-eIF2α
(cat. no. 5324), rabbit anti-p-eIF2α (cat. no. 3398) and rabbit
anti-cleaved-caspase-3 (c-caspase-3; cat. no. 9661) antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
The rabbit anti-CHOP (cat. no. SC-7351) and mouse anti-β-actin
(cat. no. SC-47778) antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The rabbit anti-Bcl-2 (cat.
no. BA0315) and rabbit anti-Bcl-2 associated X protein (cat. no.
BA0412) antibodies were purchased from Wuhan Boster Biological
Technology, Ltd. (Wuhan, China). Anti-mouse IgG (cat. no. 7076) and
anti-rabbit IgG (cat. no. 7074), PD166866 and PD98059 were
purchased from Cell Signaling Technology. Tunicamycin (TM) was
purchased from Sangon Biotech Co., Ltd. (Shanghai, China).
Cell culture
Rat H9c2 cardiomyocytes (Chinese Academy of Sciences
Cell Bank, Shanghai, China) were cultured in high glucose DMEM
supplemented with 10% (v/v) FBS and 2 mM L-glutamine at 37°C under
a 5% CO2 atmosphere. For the present study H9c2 cells
were plated at ~60% density and incubated overnight to reach 70–80%
confluence at 37°C before experimentation.
Recombinant plasmid construction and
transfection
The cDNA encoding full-length rat FGF21 (627 bp;
accession no. NM_ 130752.1) was cloned and inserted into the
XhoI and EcoRI restriction sites of pcDNA4/myc-his
(5,151 bp) to generate the expression plasmid for rat FGF21
(pcDNA4-FGF21). The pcDNA4-FGF21 plasmid was verified by special
digestion analysis and DNA sequencing and transfected into cells
using X-tremeGENE HP DNA Transfection reagent (Roche Diagnostics,
Laval, QC, Canada) for 48 h.
Experimental protocols
The cultured H9c2 cardiomyocytes were randomly
divided into different groups. In the control group, the H9c2
cardiomyocytes were incubated under normal conditions. In the
TM-treated group, the H9c2 cardiomyocytes were treated with various
doses (1, 5, 10, 20 and 50 µM) of TM for 24 h. In the FGF21
overexpression-treated group (pcDNA4-FGF21 + TM), H9c2
cardiomyocytes were transfected with pcDNA4-FGF21 plasmid for 48 h
and subsequently exposed to TM (10 µM) treatment for a
further 24 h. Inhibitor-treated groups were processed the same as
the pcDNA4-FGF21 + TM group, but the cells were co-incubated with
the specific FGFR1 inhibitor, PD166866 (100 nM) or ERK1/2
inhibitor, PD98059 (20 µM) for 1 h before treatment with the
pcDNA4-FGF21 plasmid. Three experimental categories were included:
i) the control and TM (1, 5, 10, 20 and 50 µM) groups; ii)
the control, TM (10 µM), pcDNA4-FGF21 + TM (10 µM),
and pcDNA4 + TM (10 µM) groups; iii) the TM (10 µM),
pcDNA4-FGF21 + TM (10 µM), pcDNA4-FGF21 + TM (10 µM)
+ PD166866, and pcDNA4-FGF21 + TM (10 µM) + PD98059
groups.
Cell viability assay
Cell viability was measured using Cell Counting
Kit-8 (CCK-8, cat. no. CK04; Dojindo Laboratories, Kumamoto, Japan)
according to the manufacturer's instructions. Cells were dispensed
in 96-well plates at a density of 7×103 cells/well and
incubated overnight at 4°C. After the designated treatment, cells
were washed in DMEM and 10 µl CCK-8 solution was added to
each well for a 4-h incubation. Subsequently, the optical density
(OD) of each well was measured at a wavelength of 450 nm using a
spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA).
Cell viability (%) = (ODsample −
ODblank)/(ODcontrol − ODblank) ×
100%.
FGF21 secretion level measurement
H9c2 cells were maintained in medium containing 0.2%
BSA rather than 10% FBS and treated with various concentrations of
TM for 24 h. FGF21 released in the culture medium under ER stress
conditions was determined using an ELISA kit (cat. no. MF2100;
R&D Systems, Inc., Minneapolis, MN, USA) according to the
manufacturer's instructions. All reactions were performed in
triplicate using 96-well ELISA plates.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
TUNEL assay was performed using a One Step TUNEL
apoptosis kit (cat. no. KGA4052; Nanjing KeyGen Biotech Co., Ltd.,
Nanjing, China) according to the manufacturer's instruction. Cells
were plated on glass coverslips at a density of 1×105
cells/well. Following treatment, the cells were fixed in 4%
formaldehyde for 30 min and permeabilized in 0.5% Triton X-100 for
10 min. The cells were labeled with dUTP and TDT enzymes in a
humidified box at 37°C for 1 h, subsequently incubated in
streptavidin-fluorescein (SF) at room temperature for 30 min and
counterstained with 4′,6-diamidino-2-phenylindole for 10 min before
observation under a laser scanning confocal microscope (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). TUNEL-positive cells
exhibited a bright green fluorescence and >5 non-overlapping
fields in each group were quantified.
Protein extraction and western blot
analysis
Total protein was extracted according to the methods
used by Kurian et al (22). Protein concentration was
determined using a BCA protein assay kit (cat. no. P0012; Beyotime
Institute of Biotechnology, Haimen, China) with BSA as standard.
Total proteins (30 µg) were fractionated by 8–12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto a polyvinylidene difluoride (PVDF) membrane
(Immobilon P; Millipore, Bedford, MA, USA) at 90 V constant for 100
min. The blots were blocked with 5% BSA for 1 h and the membranes
were incubated with antibodies diluted in 5% BSA at 4°C overnight.
The primary antibodies used in the present study were as follows:
FGF21 (1:500 dilution), FGFR1 (1:250 dilution), p-FGFR1 (1:200
dilution), β-Klotho (1:500 dilution), c-myc (1:1,000 dilution),
ERK1/2 (1:3,000 dilution), p-ERK1/2 (1:3,000 dilution), PERK
(1:1,000 dilution), p-PERK (1:1,000 dilution), eIF2α (1:750
dilution), p-eIF2α (1:1,000 dilution), ATF4 (1:5,000 dilution),
CHOP (1:1,000 dilution), IRE1α (1:1,000 dilution), p-IRE1α (1:1,000
dilution), JNK(1:1,000 dilution), p-JNK (1:1,000 dilution), Bcl-2
(1:200 dilution), Bax (1:200 dilution), c-caspase-3 (1:200
dilution) and β-actin (1:2,000 dilution).
After washing three times with 0.1% Tris-buffered
saline with Tween-20 (TBST) and incubating with HRP-conjugated
secondary antibodies [anti-mouse IgG (1:5,000 dilution) and
anti-rabbit IgG (1:5,000 dilution)] for 1 h at room temperature,
specific bands were visualized by enhanced chemiluminescence
detection (cat. no. 34077; Thermo Fisher Scientific, Inc.). Bands
were analyzed by the ImageJ software (National Institutes of
Health, Bethesda, MD, USA). Each experiment was repeated at least
three times.
Statistical analysis
Statistical analysis was performed using the SPSS
19.0 software (SPSS, Inc., Chicago, IL, USA). Significant
differences between the groups were analyzed by the unpaired
Student's t-test or the one-way ANOVA and P<0.05 was considered
to indicate a statistically significant difference. Data were
presented as means ± standard deviation.
Results
TM inhibits the viability of H9c2 cells
and induces ER stress-associated signaling pathways
To establish whether the ER stress model had
successfully been established by TM, H9c2 cells were treated with
increased concentrations (1, 5, 10, 20 and 50 µM) of TM for
24 h. Initially, the effect of TM on the cell viability of H9c2
cells was examined. As shown in Fig.
1A, TM up to 5 µM did not significantly reduce the
viability of H9c2 cells, but TM became cytotoxic to H9c2 cells at a
dose of >10 µM and cell viability reduced with increased
concentrations of TM. Thus, 10 µM TM was used in subsequent
experiments for induction of ER stress. In addition, the
PERK-eIF2α-AFT4-CHOP signaling pathway and IRE1α-JNK mediated
signaling pathway are well known ER stress-associated signaling
pathways for inducing apoptosis. Following treatment with TM up to
5 µM, mild ER stress partly activated these signaling
pathways, which was evidenced by induction of p-PERK, p-eIF2α, ATF4
and CHOP in a dose-dependent manner (Fig. 1B). In addition, mild ER stress
caused upregulated expression of p-IRE1α, p-JNK, c-caspase-3 and
Bax while it reduced the level of anti-apoptosis protein, Bcl-2
(Fig. 1C). However, at a high
concentration (≥10 µM) of TM, this increasing level reached
a maximum at a concentration of 10 µM TM and plateaued with
the increasing concentration, this indicated that excessive ER
stress (≥10 µM TM) fully activated ER stress-associated
signaling pathways to induce further cell apoptosis.
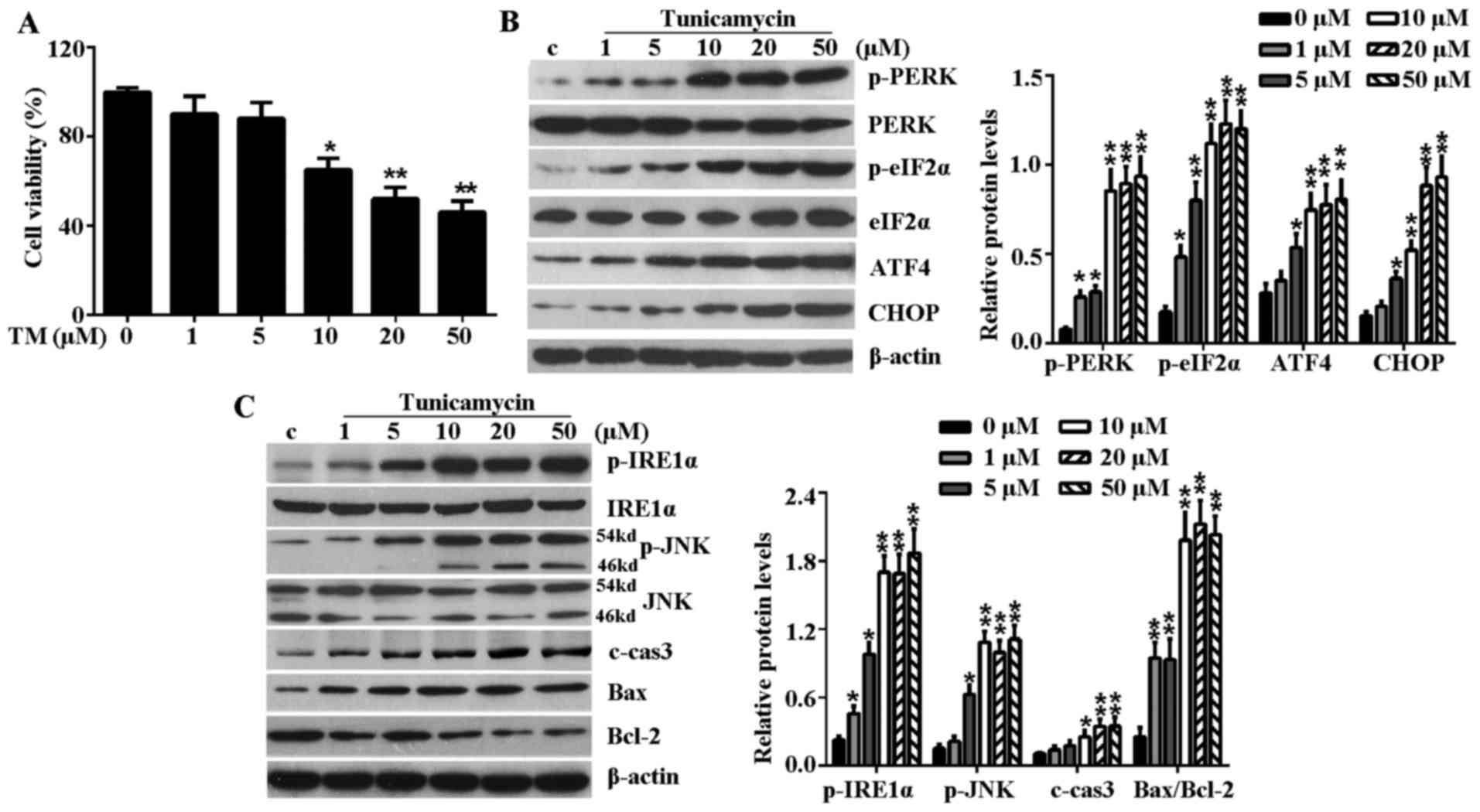 | Figure 1TM inhibited the cell viability of
H9c2 cells and induced ER stress-associated signaling pathways.
H9c2 cells were treated with various concentrations (1, 5, 10, 20
and 50 µM) of TM for 24 h. (A) Cell viability was detected
by Cell Counting Kit-8 assay. (B) Western blot analysis of the
expression levels of PERK-eIF2α-ATF4-CHOP signaling
pathway-associated key proteins, including p-PERK, PERK, p-eIF2α,
eIF2α, ATF4 and CHOP. (C) Western blot analysis of IRE1α-JNK
signaling pathway-associated proteins, including p-IRE1α, IRE1α,
p-JNK, JNK, c-caspase-3, Bax and Bcl-2. β-actin served as a loading
control. Error bars demonstrate the means. *P<0.05 or
**P<0.01 vs. control. TM, tunicamycin; ER,
endoplasmic reticulum; PERK, PKR-like ER kinase; eIF2α, eukaryotic
translational initiation factor 2α; ATF4, activating transcription
factor 4; CHOP, CCAAT/-enhancer-binding protein homologous protein;
p, phosphorylated; JNK, c-Jun N-terminal kinase; IRE1α,
inositol-requiring kinase 1α; c-cas3, cleaved caspase-3; Bax, Bcl-2
associated X protein; Bcl-2, B-cell lymphoma-2; c, control. |
Mild ER stress elevates the expression
levels of FGF21 and its main receptor in H9c2 cells
TM treatment significantly induced endogenous FGF21,
p-FGFR1 and β-Klotho expression levels in H9c2 cells (Fig. 2A). Furthermore, increased levels
of secreted FGF21 protein were detected from the culture medium of
TM-treated H9c2 cells (Fig. 2B).
However, the elevated expression level and secretion of these
proteins were not positively correlated with the degree of ER
stress-induced cell damage. Notably, in mild ER stress (≤5
µM TM), the protein expression level of FGF21, p-FGFR1 and
β-Klotho increased and peaked at a concentration of 5 µM TM.
However, in excessive ER stress (≥10 µM TM), the expression
level of FGF21 and its main receptors decreased gradually with
aggravation of the ER stress level. These results indicated that
the elevated expression of FGF21 and its main receptors may be a
compensatory response to injury in mild ER stress.
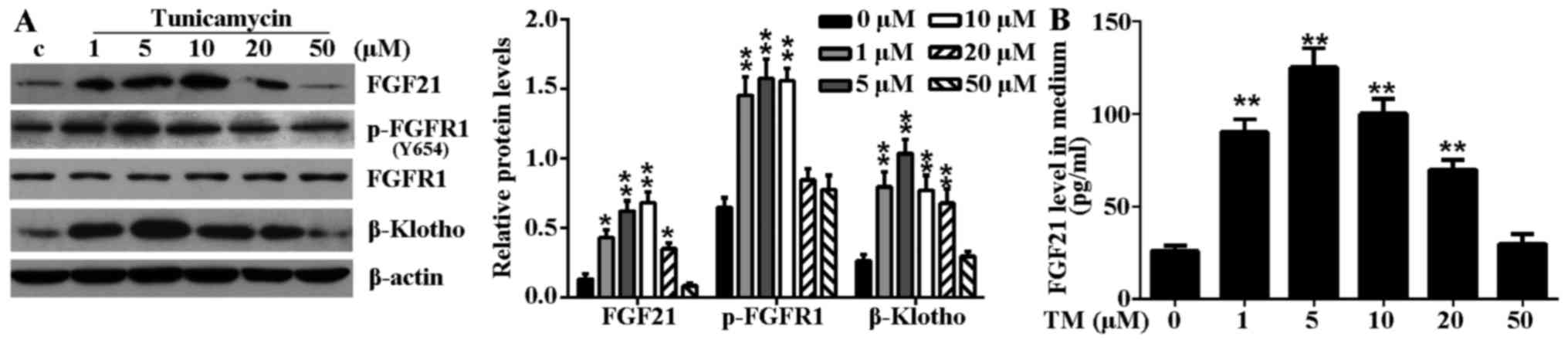 | Figure 2TM-induced ER stress induced FGF21
expression and increased circulating FGF21 expression levels in
H9c2 cells. (A) H9c2 cells were treated with various concentrations
(1, 5, 10, 20 and 50 µM) of TM for 24 h. The expression
levels of FGF21, FGFR1, p-FGFR1 and β-Klotho in H9c2 cells in
response to TM-induced ER stress were evaluated by western blot
analysis. β-actin served as a loading control. (B) Secreted FGF21
levels in culture medium following ER stress stimulation were
measured by ELISA assay. *P<0.05 or
**P<0.01 vs. control. TM, tunicamycin; ER,
endoplasmic reticulum; FGF21, fibroblast growth factor 21; FGFR1,
fibroblast growth factor receptor 1; p, phosphorylated; c,
control. |
FGF21 prevents the ER stress-induced
apoptosis of H9c2 cells
FGF21 was overexpressed from the pcDNA4-FGF21
eukaryotic expression vector in H9c2 cells (Fig. 3A and B). To demonstrate the
cardioprotective effect of FGF21 on ER stress-induced cardiomyocyte
injury, H9c2 cells overexpressing FGF21 were exposed to TM (10
µM) treatment for 24 h. The result demonstrated that FGF21
overexpression significantly reversed the decreased cell viability
caused by TM treatment, indicating that FGF21 protected H9c2 cells
from ER stress-mediated cell death (Fig. 3C). In addition, the TUNEL assay
indicated that ER stress-induced apoptosis was significantly
reduced by FGF21 overexpression (Fig.
3D).
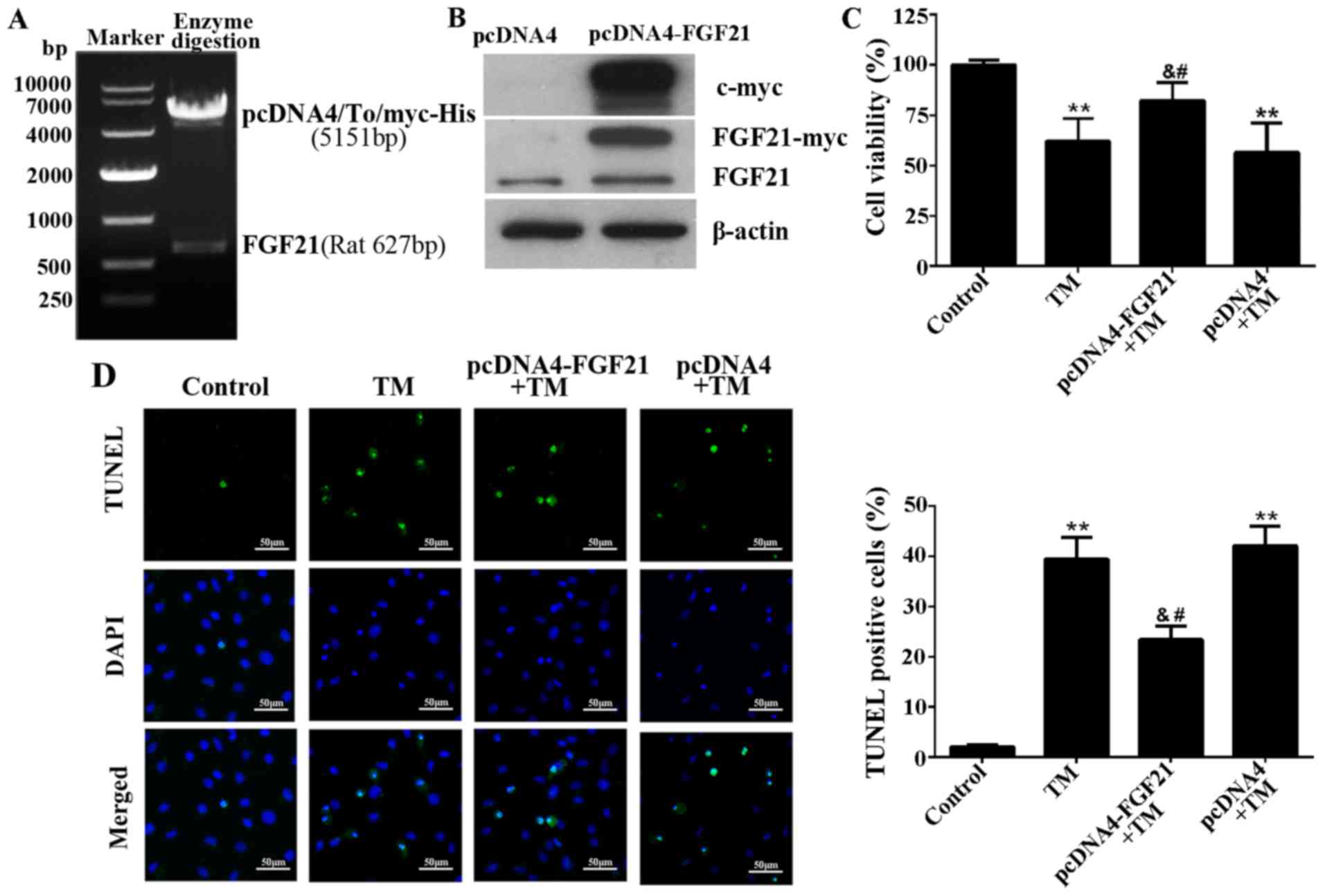 | Figure 3Overexpressed FGF21 protected H9c2
cells from ER stress-induced apoptosis. (A) The recombinant plasmid
pcDNA4-FGF21 was constructed by pcDNA4/To/myc-his (5,151 bp) and
full-length rat FGF21 (627 bp), confirmed by agarose gel
electrophoresis and gene sequencing. (B) Western blot analysis
demonstrating the overexpression of myc-labelled FGF21 protein from
the transfected pcDNA4-FGF21 plasmid in H9c2 cells using a c-myc
tag and FGF21 antibodies. (C) H9c2 cells were transfected with
pcDNA4-FGF21 plasmid for 48 h, and exposed to TM (10 µM)
treatment for a further 24 h, the cell viability was detected by
Cell Counting Kit-8 assay. (D) Fluorescence microscopy of TUNEL
staining of H9c2 cells. Cell nucleus was stained with DAPI (blue).
Scale bar, 50 µm. The percentage of apoptotic cells in each
group was calculated by counting the condensed nuclei.
**P<0.01 vs. control, &P<0.05 vs.
TM-treated cells, #P<0.05 vs. pcDNA4 + TM group.
FGF21, fibroblast growth factor 21; ER, endoplasmic reticulum; TM,
tunicamycin; TUNEL, terminal deoxynucleotidyl-transferase-mediated
dUTP nick end labeling; DAPI, 4′,6-diamidino-2-phenylindole. |
FGF21 inhibits ER stress-related
signaling pathways
The effects of FGF21 overexpression on the key
proteins of the PERK-eIF2α-AFT4-CHOP signaling pathway and
IRE1α-JNK signaling pathway were determined in H9c2 cells under ER
stress. Western blotting revealed that the expression level of
p-PERK, p-eIF2α, ATF4 and CHOP were markedly increased following 24
h of TM treatment compared with the untreated cells. However, FGF21
overexpression significantly reduced these inductions (Fig. 4A). In addition, as shown in
Fig. 4B, the upregulation of
p-IRE1α, p-JNK, c-caspase-3, Bax and Bcl-2 were markedly reversed
by FGF21 overexpression, indicating that FGF21 overexpression
inhibited these ER stress evoked pro-apoptotic signaling events in
H9c2 cells.
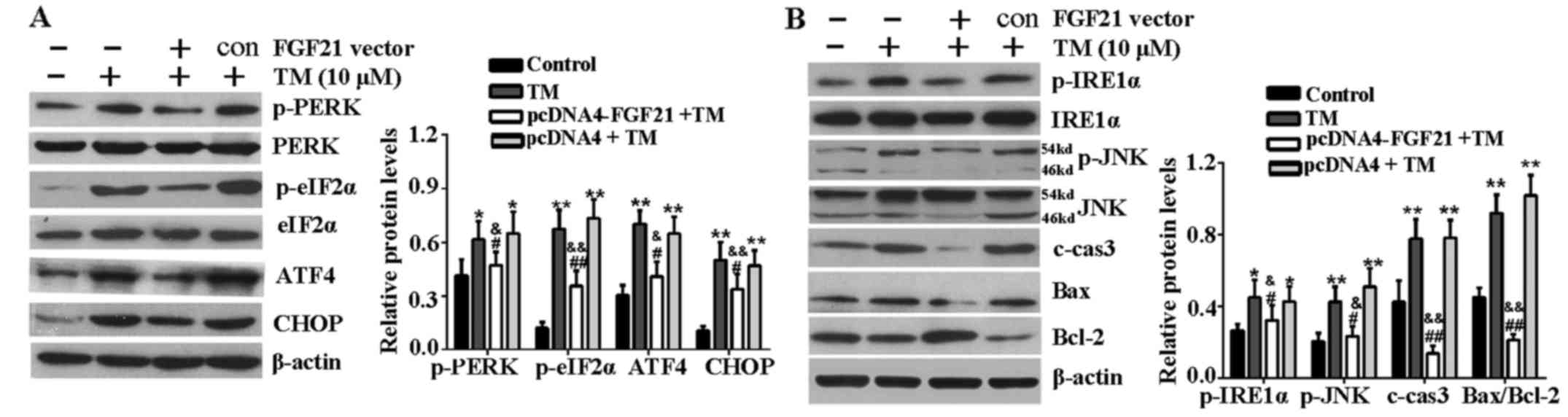 | Figure 4FGF21 inhibited ER stress-mediated
apoptosis signaling pathways. (A) H9c2 cells were treated with TM
(10 µM) following transfection with pcDNA4-FGF21 plasmid for
48 h, the expression of PERK-eIF2α-ATF4-CHOP signaling
pathway-associated key proteins, including p-PERK, PERK, p-eIF2α,
eIF2α, ATF4 and CHOP were analyzed by western blotting. (B) Western
blot analysis of IRE1α-JNK signaling pathway-associated proteins,
including p-IRE1α, IRE1α, p-JNK, JNK, c-caspase-3, Bax and Bcl-2.
β-actin served as a loading control. Error bars represent standard
deviation. *P<0.05 or **P<0.01 vs.
control, &P<0.05 or
&&P<0.01 vs. TM group, #P<0.05
or ##P<0.01 vs. pcDNA4 + TM group. FGF21, fibroblast
growth factor 21; ER, endoplasmic reticulum; TM, tunicamycin; PERK,
PKR-like ER kinase; eIF2α, eukaryotic translational initiation
factor 2α; ATF4, activating transcription factor 4; CHOP,
CCAAT/-enhancer-binding protein homologous protein; p,
phosphorylated; JNK, c-Jun N-terminal kinase; IRE1α,
inositol-requiring kinase 1α; c-cas3, cleaved caspase-3; Bax, Bcl-2
associated X protein; Bcl-2, B-cell lymphoma-2; c, control. |
FGF21 inhibits ER stress-induced
apoptosis via FGFR1 and ERK1/2 phosphorylation
The expression levels of FGF21, p-FGFR1 and p-ERK1/2
significantly increased in H9c2 cells transfected with pcDNA4-FGF21
compared with those transfected with an empty vector (Fig. 5A), indicating that FGF21 may
positively regulate the expression level of p-FGFR1 and p-ERK1/2 to
attenuate ER stress-induced injury. To determine whether
FGF21-induced FGFR1 and ERK1/2 activation is responsible for its
cardioprotective effect, H9c2 cells were treated with FGFR1
inhibitor, PD166866 or ERK1/2 inhibitor, PD98059 before being
transfected with pcDNA4-FGF21 and the addition of TM. The results
demonstrate that the improvement of cell viability mediated by
FGF21 overexpression was significantly reduced (Fig. 5B). In addition, the TUNEL assay
indicated that FGFR1 and ERK1/2 inhibitors attenuated the FGF21
overexpression-mediated anti-apoptotic effect on ER stress injury
(Fig. 5C).
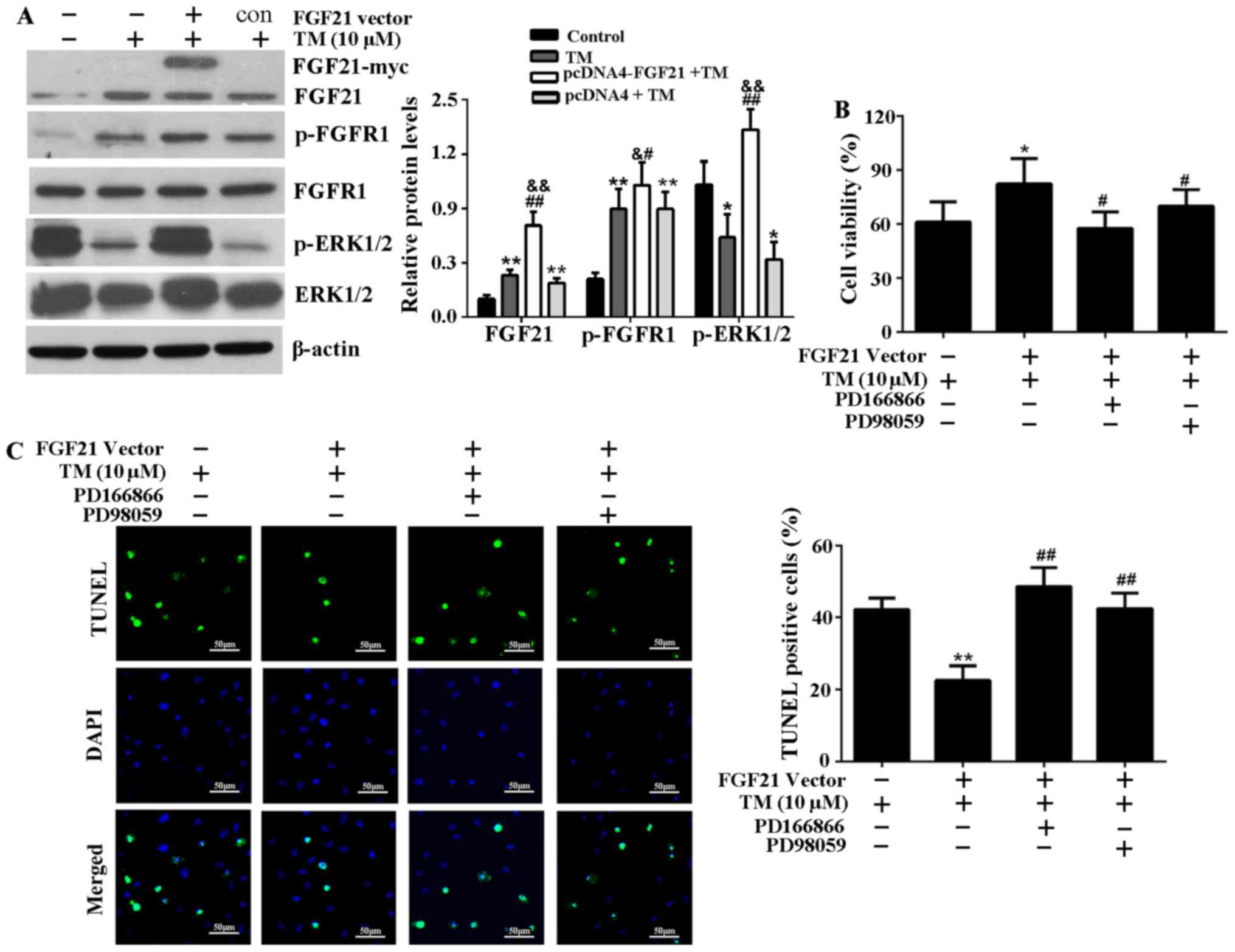 | Figure 5FGF21 overexpression activated FGFR1
and ERK1/2 in H9c2 cells, and the inhibitors of FGFR1 and ERK1/2
attenuate cardioprotective effect of FGF21 on ER stress injury. (A)
Subsequent to transfection with the pcDNA4-FGF21 plasmid, the
expression levels of FGF21, p-FGFR1, FGFR1, p-ERK1/2 and ERK1/2
were analyzed by western blotting and their relative protein
expression levels were compared. *P<0.05 or
**P<0.01 vs. control, &P<0.05 or
&&P<0.01 vs. TM group, #P<0.05
or ##P<0.01 vs. pcDNA4 + TM group. (B) H9c2 cells
were pretreated with the specific FGFR1 inhibitor, PD166866 (100
nM) or ERK1/2 inhibitor, PD98059 (20 µM) for 1 h, then
transfected with pcDNA4-FGF21 plasmids for 48 h. TM (10 µM)
was added to the media and incubated at 37°C for 24 h. The cell
viability was evaluated by Cell Counting Kit-8 assay.
*P<0.05 vs. TM group, #P<0.05 vs.
pcDNA4-FGF21 + TM group. (C) Fluorescence microscopy of TUNEL
staining of H9c2 cells. The cell nucleus was stained with DAPI
(blue). Scale bar, 50 µm. The percentage of apoptotic cells
in each group was calculated by counting the condensed nuclei.
**P<0.05 vs. TM group, ##P<0.05 vs.
pcDNA4-FGF21 + TM group. FGF21, fibroblast growth factor 21; FGFR1,
fibroblast growth factor receptor 1; ERK, extracellular
signal-regulated kinases; TM, tunicamycin; ER, endoplasmic
reticulum; p, phosphorylated; TUNEL, terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling; c,
control. |
FGFR1 and ERK1/2 inhibitors attenuate
FGF21-mediated inhibition of ER stress-associated signaling
pathways
Subsequently the effects of FGFR1 and ERK1/2
inhibitors on the activation of FGFR1 and ERK1/2, and the
inhibition of ER stress-associated signaling pathways induced by
FGF21 overexpression were investigated. The results indicated that
phosphorylation of FGFR1 and ERK1/2 were stimulated by FGF21
overexpression, and FGFR1 inhibitor, PD166866 attenuated the
stimulated effect of FGF21 on phosphorylated ERK1/2 (Fig. 6A), indicating that ERK1/2 should
be activated during the phosphorylation of FGFR1, as stimulated by
FGF21. Furthermore, the decrease of key proteins associated with
the PERK-eIF2α-ATF4-CHOP signaling pathway, including p-PERK,
p-eIF2α, ATF4 and CHOP mediated by FGF21 overexpression were
reversed following treatment with FGFR1 and ERK1/2 inhibitors
(Fig. 6B). In addition, FGFR1 and
ERK1/2 inhibitors reversed the reduced expression level of p-IRE1α,
p-JNK, c-caspase-3 and Bax, and the increased expression level of
Bcl-2 as a result of FGF21 overexpression (Fig. 6C). Therefore, the inhibitory
effect of FGF21 on the ER stress-associated signaling pathways was
at least in part via the FGFR1-ERK1/2 signaling pathway.
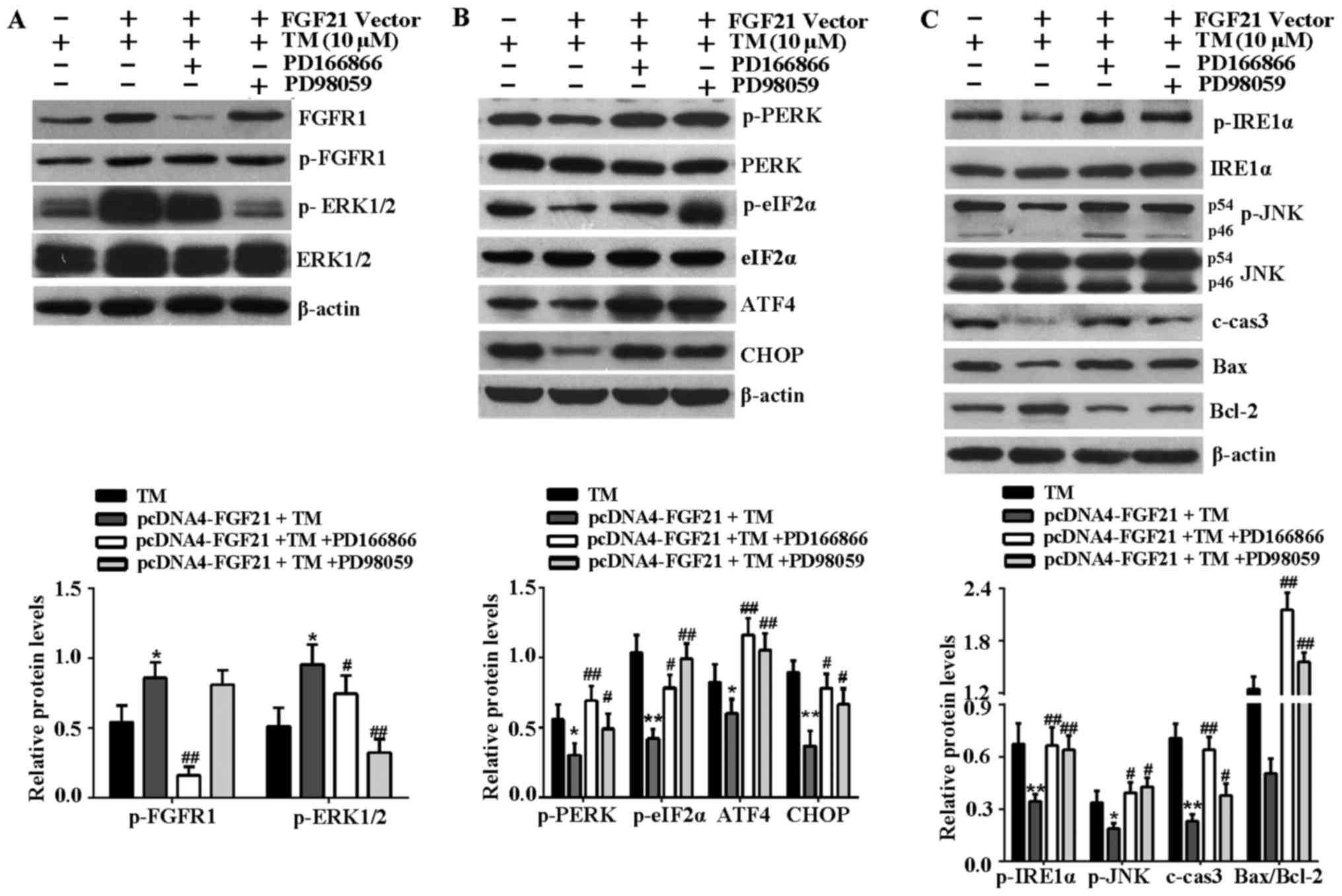 | Figure 6FGFR1 and ERK1/2 inhibitors attenuate
FGF21-mediated inhibition of ER stress-induced apoptosis signaling
pathways. (A) To confirm the effects of PD166866 and PD98059 on the
activation of FGFR1 and ERK1/2, the cell lysates were analyzed by
western blotting to detect the expression levels of p-ERK1/2,
ERK1/2, p-FGFR1 and FGFR1 in H9c2 cells. (B) Western blotting was
performed to detect the expression levels of the
PERK-eIF2α-ATF4-CHOP signaling pathway-associated key proteins,
including p-PERK, PERK, p-eIF2α, eIF2α, ATF4 and CHOP. (C) Western
blot analysis of IRE1α-JNK signaling pathway-associated proteins,
including p-IRE1α, IRE1α, p-JNK, JNK, c-caspase-3, Bax and Bcl-2.
β-actin served as a loading control. *P<0.05 or
**P<0.01 vs. TM group, #P<0.05 or
##P<0.01 vs. pcDNA4-FGF21 + TM group. FGFR1,
fibroblast growth factor receptor 1; ERK, extracellular
signal-regulated kinases; FGF21, fibroblast growth factor 21; TM,
tunicamycin; ER, endoplasmic reticulum; p, phosphorylated; PERK,
PKR-like ER kinase; eIF2α, eukaryotic translational initiation
factor 2α; ATF4, activating transcription factor 4; CHOP,
CCAAT/-enhancer-binding protein homologous protein; p,
phosphorylated; JNK, c-Jun N-terminal kinase; IRE1α,
inositol-requiring kinase 1α; c-cas3, cleaved caspase-3; Bax, Bcl-2
associated X protein; Bcl-2, B-cell lymphoma-2; c, control. |
Discussion
In the present study, ER stress was demonstrated to
induce the expression of FGF21 and its main receptors in rat H9c2
cardiomyocytes, although the enhanced expression levels were not
positively correlated with the degree of ER stress. A recent study
found that FGF21, as a target gene of ATF4, was induced by ER
stress via the PERK-eIF2α-ATF4 signaling pathway in mouse
hepatocytes (23). Furthermore,
ER stress caused an upregulation of hepatic β-Klotho via an
ATF4-dependent signaling pathway (24). Therefore, the present study
concluded that in mild ER stress (≤5 µM TM), the elevated
expression level of FGF21 and its main receptors may be due to the
partly activated PERK-eIF2α-ATF4 signaling pathway. In the early
stage of ER stress, UPR is an important feature to protect cells
against environmental interference, through a compensatory
mechanism, to reduce cell damage and maintain ER homeostasis
(11,12), so the induced FGF21 expression and
secretion may be the case of a native compensatory mechanism in
response to ER stress.
However, in the case of excessive ER stress or an
inadequate adaptation to stress, apoptotic signaling pathways are
activated. In excessive ER stress (≥10 µM TM), the
PERK-eIF2α-ATF4 and IRE1a-JNK signaling pathways were fully
activated to induce a large number of proapoptotic proteins,
including CHOP, caspase-3 and Bax and decrease the levels of
anti-apoptosis protein, Bcl-2. Previous reports have shown that the
activation of JNK induces apoptosis by activation of caspase-3 and
Bax, and inhibition of Bcl-2 in ER stress (18,25,26). CHOP, as the key protein of ER
stress to induced cell apoptosis, could predominantly be induced by
the PERK-eIF2α-ATF4 signaling pathway (27). The expression levels of FGF21 and
CHOP are dependent on the ATF4 signaling pathway, although more
CHOP was induced by ATF4 in excessive ER stress, which may lead to
competitive inhibition of FGF21, which may explain the reduced
FGF21 at TM concentrations >10 µM. Thus, the present
study concluded that FGF21, as a cardioprotective cytokine, is
expressed and secreted by cardiomyocytes in response to ER stress
injury.
To further investigate the effect of FGF21 on ER
stress injury, endogenous FGF21 expression was increased in H9c2
cells. The results demonstrated that FGF21 overexpression
alleviated TM-induced ER stress and apoptosis in cardiomyocytes by
interfering with the PERK-eIF2α-ATF4-CHOP and IRE1a-JNK apoptotic
pathways, which in turn suppressed the activity of pro-apoptotic
cytokines, thereby reducing cardiomyocyte apoptosis and supporting
survival. Therefore, in the context of ER stress, the
anti-apoptotic action of FGF21 may prolong the phase in which ER
stress is resolved before apoptotic pathways are activated.
The present result demonstrated that the FGF21
overexpression stimulated tyrosine phosphorylation of FGFR1 in H9c2
cells, this result was consistent with a previous study reporting
that the mRNA expression levels of FGFR1 and β-Klotho were induced
by FGF21 overexpression (28).
Recent studies have demonstrated that FGF21 exerts its biological
actions via transcriptional activation of FGFRs (1,2,4).
FGF21 binds with high affinity and activates only FGFR1-β-Klotho,
which, as a receptor complex, may regulate the function of FGF21.
As a result of FGF21 overexpression, the increased expression level
of p-FGFR1 is induced to meet the need of FGF21 in alleviating
cardiomyocyte injury and restoring cardiac function.
In the present study, the phosphorylation of ERK1/2
was significantly induced by FGF21 overexpression, indicating that
FGF21 may activate the ERK1/2 signaling pathway. ERK1/2, a member
of the mitogen-activated protein kinases family, mediates cell
proliferation and survival. Previous studies demonstrated that
ERK1/2 protects the myocardium from ischemic injury (29,30). Yang et al (31) identified that FGF19 and FGF21 bind
to the integrative FGFR1-β-Klotho complex and activate downstream
ERK1/2 signaling in 3T3L1 adipocytes. Certain studies have
confirmed that FGF21 reduces cell apoptosis via activation of the
ERK1/2 signaling pathway (15,32–34). Combining previous studies and the
current results, the present study speculated that the protective
effect of FGF21 on ER stress-induced myocardial injury was
potentially via the FGFR1/β-Klotho-ERK1/2 signaling pathways. In
order to investigate this hypothesis, H9c2 cells were treated with
FGFR1 and ERK1/2 inhibitors, and it was found that the
anti-apoptotic effect mediated by FGF21 overexpression was
attenuated, and the inhibition of FGF21 overexpression-mediated ER
stress-associated signaling pathways was reversed. Furthermore, the
FGFR1 inhibitor attenuated the stimulated effect of FGF21 on
p-ERK1/2, indicating that the ERK1/2 signaling pathway may be
controlled by the phosphorylation of FGFR1. Collectively, these
results indicate that the protective effect of FGF21 on ER
stress-induced myocardial injury, at least in part, is mediated by
FGFR1-ERK1/2 signaling pathways.
In conclusion, the present study confirmed the
elevated expression level of FGF21/FGFR1/β-Klotho in a mild ER
stress model of rat cardiomyocytes, and that FGF21 exerted a
cardio-protective role against ER stress-induced cell apoptosis at
least in part via activation of the FGFR1-ERK1/2 signaling pathway.
These findings indicate that FGF21 therapeutic interventions may
present a novel approach to preventing ER stress injury and
treating cardiovascular diseases. Further studies investigating the
pro-survival role of FGF21 in the context of cellular stress using
FGF21 knock out mice may provide a deeper understanding of this
protein.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81571636), the
Natural Science Foundation of Shangdong, China (grant no.
ZR2015HM058), the Yantai Project of Science and Technology
Development Plan (grant nos. 2013WS224 and 2015WS021), the Yantai
Yuhuangding Hospital Youth Scientist Research Foundation (grant no.
201526), and the Research Award Fund for Outstanding Young and
Middle-aged Scientists of Shandong Province (grant no.
BS2013SW043). All experiments were performed at the Shandong
Research Center of Stem Cell Engineering, Yantai Yuhuangding
Hospital (Yantai, China).
Abbreviations:
|
FGF21
|
fibroblast growth factor 21
|
|
ER
|
endoplasmic reticulum
|
|
UPR
|
unfolded protein response
|
|
PERK
|
PKR-like ER kinase
|
|
IRE1
|
inositol-requiring kinase 1α
|
|
ATF6
|
activating transcription factor 6
|
|
eIF2α
|
eukaryotic translational initiation
factor 2α
|
|
ATF4
|
activating transcription factor 4
|
|
XBP1
|
X-box binding protein 1
|
|
JNK
|
c-Jun N-terminal kinase
|
|
CHOP
|
CCAAT/-enhancer-binding protein
homologous protein
|
|
c-caspase-3
|
cleaved caspase-3
|
|
Bcl-2
|
B-cell lymphoma-2
|
|
Bax
|
Bcl-2 associated X, apoptosis
regulator
|
|
ERK
|
extracellular signal-regulated
kinases
|
|
TM
|
tunicamycin
|
References
|
1
|
Kharitonenkov A, Shiyanova TL, Koester A,
Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers
JS, Owens RA, et al: FGF-21 as a novel metabolic regulator. J Clin
Invest. 115:1627–1635. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fon Tacer K, Bookout AL, Ding X, Kurosu H,
John GB, Wang L, Goetz R, Mohammadi M, Kuro-o M, Mangelsdorf DJ, et
al: Research resource: Comprehensive expression atlas of the
fibroblast growth factor system in adult mouse. Mol Endocrinol.
24:2050–2064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ogawa Y, Kurosu H, Yamamoto M, Nandi A,
Rosenblatt KP, Goetz R, Eliseenkova AV, Mohammadi M and Kuro-o M:
BetaKlotho is required for metabolic activity of fibroblast growth
factor 21. Proc Natl Acad Sci USA. 104:7432–7437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Foltz IN, Hu S, King C, Wu X, Yang C, Wang
W, Weiszmann J, Stevens J, Chen JS, Nuanmanee N, et al: Treating
diabetes and obesity with an FGF21-mimetic antibody activating the
βKlotho/FGFR1c receptor complex. Sci Transl Med. 4:162ra1532012.
View Article : Google Scholar
|
|
5
|
Liu SQ, Roberts D, Kharitonenkov A, Zhang
B, Hanson SM, Li YC, Zhang LQ and Wu YH: Endocrine protection of
ischemic myocardium by FGF21 from the liver and adipose tissue. Sci
Rep. 3:27672013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shen Y, Ma X, Zhou J, Pan X, Hao Y, Zhou
M, Lu Z, Gao M, Bao Y and Jia W: Additive relationship between
serum fibroblast growth factor 21 level and coronary artery
disease. Cardiovasc Diabetol. 12:1242013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang W, Chu S, Ding W and Wang F: Serum
level of fibroblast growth factor 21 is independently associated
with acute myocardial infarction. PLoS One. 10:e01297912015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Di Lisa F and Itoh N: Cardiac Fgf21
synthesis and release: An autocrine loop for boosting up
antioxidant defenses in failing hearts. Cardiovasc Res. 106:1–3.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lü Y, Liu JH, Zhang LK, Du J, Zeng XJ, Hao
G, Huang J, Zhao DH, Wang GZ and Zhang YC: Fibroblast growth factor
21 as a possible endogenous factor inhibits apoptosis in cardiac
endothelial cells. Chin Med J (Engl). 123:3417–3421. 2010.
|
|
10
|
Cong WT, Ling J, Tian HS, Ling R, Wang Y,
Huang BB, Zhao T, Duan YM, Jin LT and Li XK: Proteomic study on the
protective mechanism of fibroblast growth factor 21 to
ischemia-reperfusion injury. Can J Physiol Pharmacol. 91:973–984.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Faitova J, Krekac D, Hrstka R and Vojtesek
B: Endoplasmic reticulum stress and apoptosis. Cell Mol Biol Lett.
11:488–505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chaudhari N, Talwar P, Parimisetty A,
Lefebvre d'Hellencourt C and Ravanan P: A molecular web:
Endoplasmic reticulum stress, inflammation, and oxidative stress.
Front Cell Neurosci. 8:2132014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee WK, Chakraborty PK, Roussa E, Wolff NA
and Thévenod F: ERK1/2-dependent bestrophin-3 expression prevents
ER-stress-induced cell death in renal epithelial cells by reducing
CHOP. Biochim Biophys Acta. 1823:1864–1876. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
16
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brozzi F, Gerlo S, Grieco FA, Juusola M,
Balhuizen A, Lievens S, Gysemans C, Bugliani M, Mathieu C,
Marchetti P, et al: Ubiquitin D regulates IRE1α/c-Jun N-terminal
kinase (JNK) protein-dependent apoptosis in pancreatic beta cells.
J Biol Chem. 291:12040–12056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Joo H, Lee HJ, Shin EA, Kim H, Seo KH,
Baek NI, Kim B and Kim SH: c-Jun N-terminal kinase-dependent
endoplasmic reticulum stress pathway is critically involved in
arjunic acid Induced apoptosis in non-small cell lung cancer cells.
Phytother Res. 30:596–603. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Minamino T and Kitakaze M: ER stress in
cardiovascular disease. J Mol Cell Cardiol. 48:1105–1110. 2010.
View Article : Google Scholar
|
|
20
|
Brahma MK, Adam RC, Pollak NM, Jaeger D,
Zierler KA, Pöcher N, Schreiber R, Romauch M, Moustafa T, Eder S,
et al: Fibroblast growth factor 21 is induced upon cardiac stress
and alters cardiac lipid homeostasis. J Lipid Res. 55:2229–2241.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hotamisligil GS: Endoplasmic reticulum
stress and atherosclerosis. Nat Med. 16:396–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kurian MV, Hamilton L, Keeven J, Mehl P
and Mullins JM: Enhanced cell survival and diminished apoptotic
response to simulated ischemia-reperfusion in H9c2 cells by
magnetic field preconditioning. Apoptosis. 17:1182–1196. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schaap FG, Kremer AE, Lamers WH, Jansen PL
and Gaemers IC: Fibroblast growth factor 21 is induced by
endoplasmic reticulum stress. Biochimie. 95:692–699. 2013.
View Article : Google Scholar
|
|
24
|
Dong K, Li H, Zhang M, Jiang S, Chen S,
Zhou J, Dai Z, Fang Q and Jia W: Endoplasmic reticulum stress
induces up-regulation of hepatic β-Klotho expression through ATF4
signaling pathway. Biochem Biophys Res Commun. 459:300–305. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang C, Lu T, Wang GD, Ma C and Zhou YF:
Costunolide, an active sesquiterpene lactone, induced apoptosis via
ROS-mediated ER stress and JNK pathway in human U2OS cells. Biomed
Pharmacother. 80:253–259. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mhaidat NM, Thorne R, Zhang XD and Hersey
P: Involvement of endoplasmic reticulum stress in Docetaxel-induced
JNK-dependent apoptosis of human melanoma. Apoptosis. 13:1505–1512.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen YJ, Su JH, Tsao CY, Hung CT, Chao HH,
Lin JJ, Liao MH, Yang ZY, Huang HH, Tsai FJ, et al: Sinulariolide
induced hepatocellular carcinoma apoptosis through activation of
mitochondrial-related apoptotic and PERK/eIF2α/ATF4/CHOP pathway.
Molecules. 18:10146–10161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li K, Li L, Yang M, Liu H, Boden G and
Yang G: The effects of fibroblast growth factor-21 knockdown and
over-expression on its signaling pathway and glucose-lipid
metabolism in vitro. Mol Cell Endocrinol. 348:21–26. 2012.
View Article : Google Scholar
|
|
29
|
Lips DJ, Bueno OF, Wilkins BJ, Purcell NH,
Kaiser RA, Lorenz JN, Voisin L, Saba-El-Leil MK, Meloche S,
Pouysségur J, et al: MEK1-ERK2 signaling pathway protects
myocardium from ischemic injury in vivo. Circulation.
109:1938–1941. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bourke L, McCormick J, Taylor V,
Pericleous C, Blanchet B, Costedoat-Chalumeau N, Stuckey D, Lythgoe
MF, Stephanou A and Ioannou Y: Hydroxychloroquine protects against
cardiac ischaemia/reperfusion injury in vivo via enhancement of
ERK1/2 phosphorylation. PLoS One. 10:e01437712015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang C, Jin C, Li X, Wang F, McKeehan WL
and Luo Y: Differential specificity of endocrine FGF19 and FGF21 to
FGFR1 and FGFR4 in complex with KLB. PLoS One. 7:e338702012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Z, Wang Y, Ye J, Lu X, Cheng Y, Xiang
L, Chen L, Feng W, Shi H, Yu X, et al: bFGF attenuates endoplasmic
reticulum stress and mitochondrial injury on myocardial
ischaemia/reperfusion via activation of PI3K/Akt/ERK1/2 pathway. J
Cell Mol Med. 19:595–607. 2015. View Article : Google Scholar
|
|
33
|
Hu P, Han Z, Couvillon AD and Exton JH:
Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in
counteracting endoplasmic reticulum stress-induced cell death. J
Biol Chem. 279:49420–49429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang X, Zhang C, Xin Y, Huang Z, Tan Y,
Huang Y, Wang Y, Feng W, Li X, Li W, et al: Protective effect of
FGF21 on type 1 diabetes-induced testicular apoptotic cell death
probably via both mitochondrial- and endoplasmic reticulum
stress-dependent pathways in the mouse model. Toxicol Lett.
219:65–76. 2013. View Article : Google Scholar : PubMed/NCBI
|














