Introduction
Liver cirrhosis is a consequence of diverse
mechanisms of liver injury that lead to a wound-healing reponse
characterized by necroinflammation and fibrogenesis (1,2).
Hepatic stellate cells (HSCs) play a critical role in the
transition from chronic liver injury to cirrhosis (3). Upon liver injury, HSCs
transdifferentiate from quiescent cells to activated,
myofibroblast-like cells, resulting in proliferation, excessive
production and secretion of extracelluar matrix (4,5).
Thus, the suppression of activated HSCs has been recognized as an
effective pathway through which to attenuate liver fibrosis.
Autophagy is a degradation pathway by which the cell
self-digests its own components by forming autophagosomes and
autolysosomes (4). This
self-digestion process occurs in all types of cells at a low basal
level, serving a homeostatic function to recycle proteins and
organelles. When the cell is going to rid itself of starvation,
damaged organelles and superfluous or misfolded proteins, autophagy
is rapidly upregulated (6).
Moreover, recent studies have revealed that autophagy could be
instigated by the upregulation of endoplasmic reticulum (ER) stress
(7,8).
ER is a type of flattened sac or tube-like organelle
in eukaryotic cells, serving many essential functions, such as
correctly folding newly made protein molecules and a
Ca2+ reservoir (9,10).
Increased protein synthesis load and accumulation of unfolded or
misfolded proteins in ER may disturb ER homeostasis, thereby
imposing ER stress and the unfolded protein response (UPR)
(11,12). The UPR, initially, compensates for
the damage caused by ER stress and maintains celluar homeostasis
via enhancing the protein-folding ability of the ER and
facilitating proteasomal degradation of unfolded or misfolded
proteins. However, if ER stress is excessive or prolonged that
transcends the protective ability of UPR, the high-intensitive UPR
eventually results in cell apoptosis (13–15).
Caffeine, the major compound in coffee, has been
identified to have a potential beneficial effect against liver
fibrosis (16,17). Recent findings have revealed that
caffeine-mediated attenuation of liver fibrosis is probably
associated with HSC apoptosis (18). Another study found that caffeine
induced apoptosis of SH-SY5Y, PC12D and HeLa cells by stimulating
autophagic flux by inhibiting the PI3K/AKT/mTOR pathway (19). However, little is known concerning
the mechanism by which caffeine regulates the apoptosis of
HSCs.
Here, we report that caffeine-enhanced autophagic
flux in HSCs was stimulated by ER stress through the IRE1-α
pathway. Furthermore, enhanced autophagy attenuated the expression
of α-SMA by instigating HSC apoptosis. Therefore, our results
provide new insight into the mechanism of caffeine's anti-fibrotic
effects.
Materials and methods
Cell culture and compounds
LX-2 cells, a type of immortalized human HSC line,
was purchased from the Cell Center of Xiangya School of Medicine,
Central South University (Hunan, China), cultured in Dulbecco's
modified Eagle's medium (DMEM) and supplemented with 10% fetal
bovine serum (FBS) (both from Gibco, Grand Island, NY, USA), 100
IU/ml penicillin and 100 µg/ml streptomycin at 37° C in 5%
CO2. To measure the autophagic flux of LX-2 cells after
being treated with caffeine, LX-2 cells were transfected with
RFR-GFR-hLC3 lentivirus as previously described elsewhere (20). Caffeine (Sigma-Aldrich, St. Louis,
MO, USA) was dissolved and diluted with DMEM to the desired
concentrations which we selected according to other articles
(16,18,19). 3-Methyladenine (MA)
(Sigma-Aldrich) was dissolved in phosphate-buffered saline (PBS)
and diluted with DMEM to the concentration of 5 mM.
Western blotting
LX-2 cells were lysed in RIPA (Solarbio, Beijing,
China) supplemented with protease inhibitor cocktail
(Sigma-Aldrich) and the protein samples were centrifuged for 30 min
at 10,000 × g (4°C). The supernatants were collected and the BCA
protein assay kit (Pierce, Rockford, IL, USA) was applied to detect
the protein concentration. The protein samples were separated by
12.5% SDS-PAGE and subsequently electrotransferred onto PVDF
membranes. The membranes were blocked with 5% defatted milk for 1.5
to 2 h and incubated with the primary antibody overnight at 4°C.
The primary antibodies were LC3 (4108; Cell Signaling Technology,
Inc., Danvers, MA, USA), SQSTM1 (ab109012; Abcam, Cambridge, UK),
α-SMA (A7249; ABclonal, Cambridge, MA, USA), Gpr78 (ab21685) and
IRE1-α (ab124945) (both from Abcam) and CHOP (2895; Cell Signaling
Technology, Inc., Danvers, MA, USA). Afterwards, the membranes were
incubated with the HRP-conjugated secondary antibodies (SA00001-1
and SA00001-2; Proteintech Group, Inc., Rosemont, IL, USA) at room
temperature for 1.5 h. Finally the results were visualized by
enhanced chemiluminescence.
Flow cytometry assay
Annexin V-FITC Apoptosis Detection kit (BD
Pharmingen, Franklin Lakes, NJ, USA) was used to detect the
apoptosis of cells. In brief, both floating and adherent cells were
collected. After being washed and resuspended with PBS twice, the
cells were stained with 3 µl Annexin V-FITC and 5 µl
PI for 20 min in the dark at room temperature. Then 150 µl
binding buffer was added to each sample of cells, after which the
cells were analyzed by flow cytometry.
RNA interference
In order to reduce the expression of Atg7 and IRE1-α
in LX-2 cells, small interfering RNAs (siRNAs) targeted against
human Atg7 and IRE1-α were synthesized by GenePharma (Shanghai,
China).
The sequence of Atg7-siRNA was
5′-GCCGUGGAAUUGAUGGUAUTT-3′. The sequence of IRE1-α was
5′-CTACTGGATGATAAATTTGCTTCA-3′. The negative control siRNA sequence
was 5′-UUCUUCGAACGUGUCACGUTT-3′. Then LX-2 cells were transfected
with Atg7-siRNA, IRE1-α-siRNA or nC-siRNA, respectively, using
Lipofectamine 2000 (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the production's instruction manual.
Cell viability assay
The effects of caffeine on the viability of LX-2
cells were evaluated by Cell Counting Kit-8 (CCK-8) assay (Dojindo
Laboratories, Kumamoto, Japan), as previously described in another
article (21).
Intracellular Ca2+
detection
The levels of intracellular Ca2+ were
detected using Fluo-3 AM (Beyotime Institute of Biotechnology,
Shanghai, China). In brief, LX-2 cells were treated with different
concentrations of caffeine (0, 10 and 30 mM) for 24 h, and the
cells were harvested and washed twice using Ca2+-free
PBS. Subsequently, the cells were resuspended in 5 µm Fluo-3
AM for 30 min in the dark at 37°C, after which the cells were
washed twice using Ca2+-free PBS. To ensure that the
Fluo-3 AM was completely transformed into Fluo-3 within the cells
and to enable the Fluo-3 to more closely combine with
Ca2+, the cells were placed quietly for 30 min in the
dark at room temperature. Finally, the fluorescence intensities of
Fluo-3 combined with Ca2+ were detected by flow
cytometery.
Electron microscopy
Transmission electron microscopy was used to
identify the ultrastructure of autophagosomes/autolysosomes as
previously described elsewhere (22).
Statistical analysis
All data are presented as means ± SD from at least
triplicate parallel experiments. The Student's t-test was used to
analyze the differences between two groups by SPSS version 23.0
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered as
statistically significant.
Results
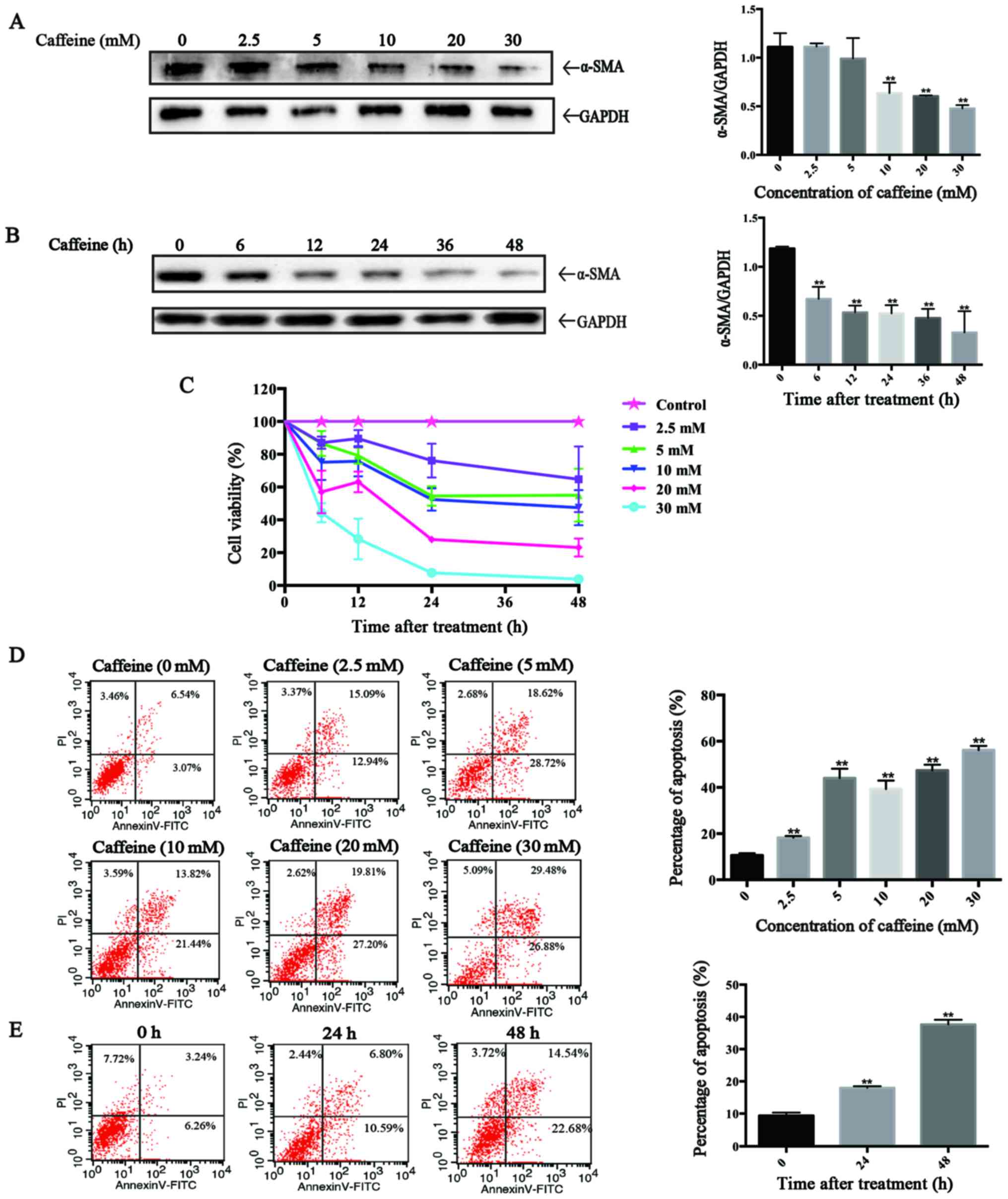
Caffeine inhibits the viability of LX-2
cells and induces cell apoptosis
To evaluate the direct effects of caffeine on HSCs
in vitro, the LX-2 cell line, a type of immortal human HSCs,
was applied in our study. After incubation with various
concentrations of caffeine (0, 2.5, 5, 10, 20 and 30 mM) for 48 h
or 20 mM caffeine for the indicated times (0, 6, 12, 24, 36 and 48
h), western blotting was used to detect the expression of α-smooth
muscle actin (α-SMA), a marker of activated HSCs. We found that the
level of α-SMA was inversely proportional to the caffeine
concentration and to the duration of exposure. Particularly,
caffeine decreased the α-SMA/GAPDH ratio at concentrations as low
as 5 mM (Fig. 1A) and at times as
early as 6 h (Fig. 1B) in LX-2
cells. Furthermore, the results of the CCK-8 assay showed that the
cell viability of the LX-2 cells was markedly reduced in a
dose-dependent and time-dependent manner after treatment with
caffeine (Fig. 1C). To
investigate whether the caffeine-inhibited viability of LX-2 cells
is associated with apoptosis, we examined the effects of caffeine
on the apoptotic activity of cells by flow cytometry. The
percentage of apoptotic cells which is the sum of early apoptotic
cells (the low right region) and late apoptotic cells (the upper
right region) was significantly increased in both a dose-dependent
(Fig. 1D) and time-dependent
manner (Fig. 1E) by incubation
with caffeine.
Caffeine induces ER stress in the LX-2
cell line
Here we further investigated whether
caffeine-induced HSC apoptosis is mediated by increasing ER stress.
From the images taken by transmission electron microscopy, we
identified many dilated cytoplasmic vacuoles in the LX-2 cells
after caffeine (20 mM) treatment for 48 h. However, there were few
dilated cytoplasmic vacuoles in the LX-2 cells cultured with DMEM
not containing caffeine (Fig.
2A). These dilated cytoplasmic vacuoles can be recognized as
dilated ER lumens as previously described (23), which indicates an increase in ER
stress. Furthermore, we detected the expression of GRP78/BiP,
IRE1-α and CHOP which all are markers of UPR. Notably, the ratio of
GRP78/GAPDH, IRE1-α/GAPDH and CHOP/GAPDH were parallel to the
increase in caffeine concentration (Fig. 2B and C) and the duration of
exposure (Fig. 2D and E). In
addition, Fluo-3 AM was used to label cytoplasmic calcium in the
LX-2 cells since calcium disturbance is another agent that induces
ER stress. The results showed that caffeine significantly elevated
the levels of cytoplasmic calcium in the LX-2 cells (Fig. 2F). Collectively, all these data
demonstrated that caffeine induced ER stress in LX-2 cells.
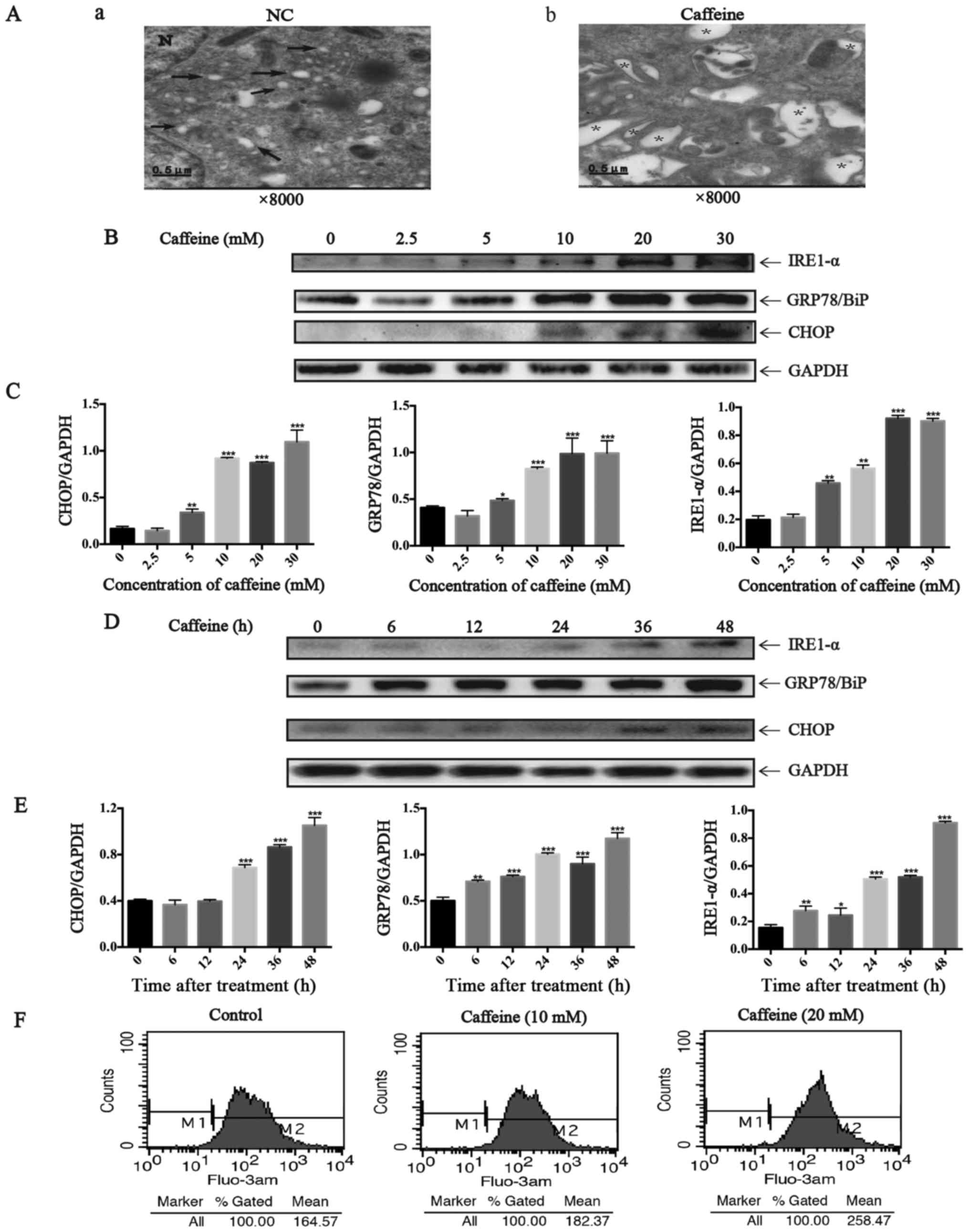 | Figure 2Endoplasmic reticulum (ER) stress
induced by caffeine in the LX-2 cell line. (A) Electron microscopy
(×8,000) shows dilated ER cavities in LX-2 cells in the presence of
caffeine (20 mM/48 h). (a) Control cells and (b) caffeine-incubated
cells are shown. →, indicates normal ER; *, represents dilated ER
cavity; N, indicates the nucleus. (B) Western blotting and (C)
densitometric analysis show a dose response of inositol requiring
enzyme 1 (IRE1)-α, GRP78/BiP, and CHOP accumulation in LX-2 cells
incubated with various concentrations of caffeine (0, 2.5, 5, 10,
20 and 30 mM) for 48 h. Bars represent the means ± SD of three
independent experiments. **P<0.01,
***P<0.001. (D) Western blotting and (E)
densitometric analysis show time-dependent changes in the
expression of IRE1-α, GRP78/BiP and CHOP in LX-2 cells incubated
with 20 mM caffeine for the indicated times. Bars represent the
means ± SD of three independent experiments.
**P<0.01, ***P<0.001. (F) LX-2 cells
were treated with various concentrations of caffeine (0, 10, 30 mM)
for 48 h, after which cells were loaded with Fluo-3 AM for 30 min.
Flow cytometry was used to detect the changes in cytosolic calcium
in LX-2 cells. |
Autophagic flux in the LX-2 cell line is
increased after caffeine treatment
Four different methods were applied to explore
autophagic flux. Firstly, we detected the expression of LC3II, a
good indicator for autophagosome formation, in LX-2 cells after
caffeine treatment. Our results showed that the expression of LC3II
was parallel to the increase in caffeine concentration (Fig. 3A and B) or the duration of
exposure to caffeine (Fig. 3C and
D). Secondly, we measured the accumulation of p62. p62 is a
marker of autophagic flux whose level increases when autophagy is
inhibited and decreases when autophagic flux is triggered.
Interestingly, we found that caffeine-incubated LX-2 cells
exhibited significant reduction in the level of p62 in a dose- and
time-dependent manner. The results indicated that autophagy in LX-2
cells was induced by caffeine. Thirdly, autophagy is a dynamic
process. The increase in LC3II level can be due to an increased
autophagy presence or a result of the accumulation of
autophagosomes caused by late autophagy inhibition. For the purpose
of distinguishing autophagy induction from autophagy inhibition, a
type of lentivirus encoding the RFP-GFP-LC3 fusion gene was used to
our study. In this study, the autophagosomes are labeled with
yellow dots (the overlay of green and red fluorescence), while
autolysosomes are labeled with red dots only. If autophagic flux is
upregulated, yellow and red dots are both increased, while only
yellow dots are increased if the process of autophagosomes fusing
with lysosomes is inhibited. It is the different numbers of red
dots that represent changes in autophagic flux. To evaluate whether
autophagic flux is upregulated by caffeine in LX-2 cells, we
incubated GFP-RFP-hLC3 lentivirus-transfected LX-2 cells with
various concentrations of caffeine (0, 10 and 30 mM). At 48 h
points, fluorescence images were captured and quantification of the
number of yellow and red dots was carried out. As shown in the
fluorescence images, the number of yellow dots and red dots alone
were both increased (Fig. 3E and
F). Finally, electron microscopic evaluation showed an
increased amount of autophagosomes/autolysosomes in the cells
treated with caffeine (Fig. 3G).
Taken together, these findings strongly demonstrated that
autophagic flux in LX-2 cells was induced by caffeine.
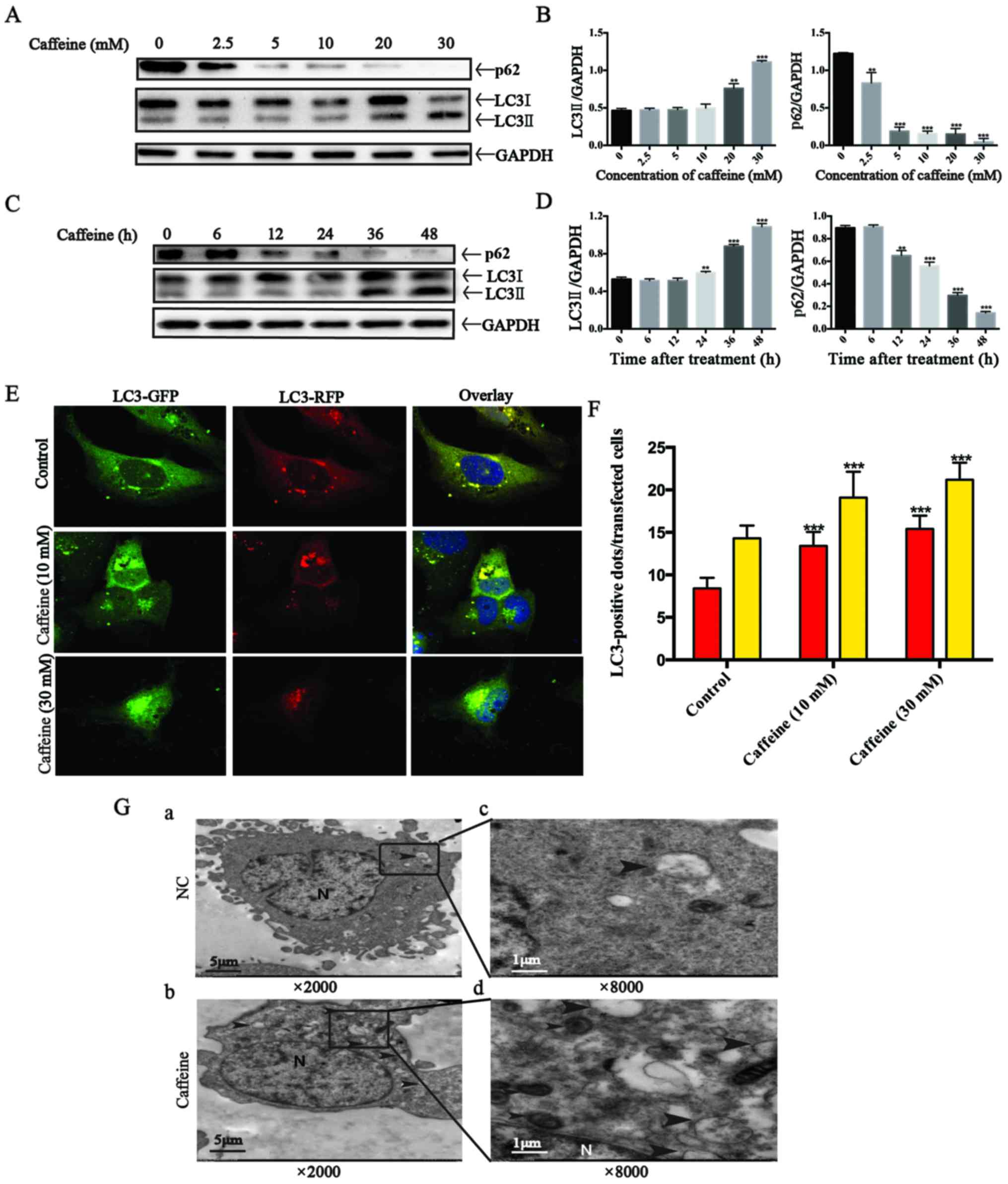 | Figure 3Autophagy flux is stimulated by
caffeine in the LX-2 cell line. (A) Western blotting and (B)
densitometric analysis show the dose response of p62 and LC3II
accumulation in LX-2 cells incubated with various concentrations of
caffeine (0, 2.5, 5, 10, 20 and 30 mM) for 48 h. Bars represent the
means ± SD of three independent experiments.
**P<0.01, ***P<0.001. (C) Western
blotting and (D) densitometric analysis show time-dependent changes
in the expression of p62 and LC3II in LX-2 cells incubated with 20
mM caffeine for the indicated times. Bars represent the means ± SD
of three independent experiments. **P<0.01,
***P<0.001. (E and F) Representative images and
quantification of early-autophagosomes (yellow dots generated from
overlapping GFP and RFP puncta) shown as yellow bars and late
autolysosomes (red dots generated from RFP puncta) shown as red
bars after 48 h of 10 and 30 mM caffeine treatment vs. the controls
(treated with 0 mM caffeine) in LX-2 cells transfected with the
RFP-GFP-hLC3 lentivirus. Error bar, standard deviation (n=30, from
a total of independent three experiments),
***P<0.001. (G) Electron microscopy shows increased
autophagosomes/autolysosomes in LX-2 cells incubated with 20 mM
caffeine for 48 h (b and d) vs. the control cells (a and c). Arrows
indicate autophagosome or autolysosome structures; n, indicates the
nucleus. Magnification (×2,000 and ×8,000). |
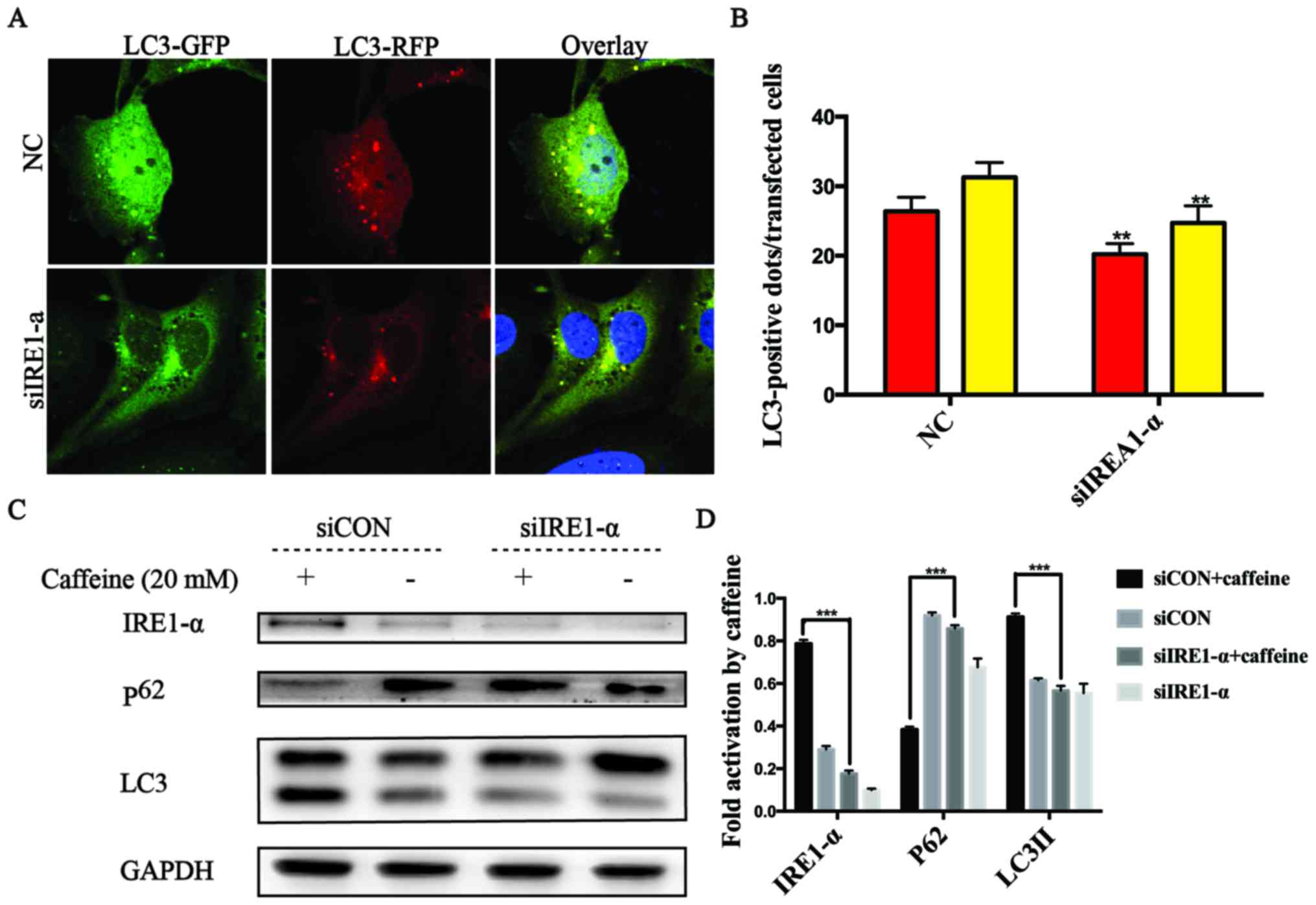
The IRE1-α signaling pathway is required
for activation of caffeine-induced autophagy in the LX-2 cell
line
It has been reported that autophagy can be triggered
by ER stress via the IRE1-α signaling pathway (19,24). Herein, we found that caffeine
contributed to autophagy and ER stress, as well as increased the
expression levels of IRE1-α and CHOP in LX-2 cells. These findings
prompted us to explore whether UPR acts as an upstream event of
autophagy and the crucial role of IRE1-α in caffeine-treated LX-2
cells. To confirm the hypothesis, specific siRNAs directly against
IRE1-α were used in the RFP-GFP-hLC3-transfected LX-2 cells. In
these co-transfected cells, we observed that the numbers of yellow
or red dots alone in the negative siRNA-transfected cells were both
higher than those in the IRE1-α siRNA-transfected cells (Fig. 4A and B). We also found that the
level of p62 accumulation in the IRE1-α siRNA-transfected cells was
higher than that in the negative siRNA-transfected cells.
Similarly, the accumulation of LC3II was decreased in the IRE1-α
siRNA-transfected cells compared with the negative
siRNA-transfected cells after caffeine treatment (Fig. 4C and D). All these data indicated
that the IRE1-α pathway was an upstream event of caffeine-induced
autophagy in the LX-2 cell line.
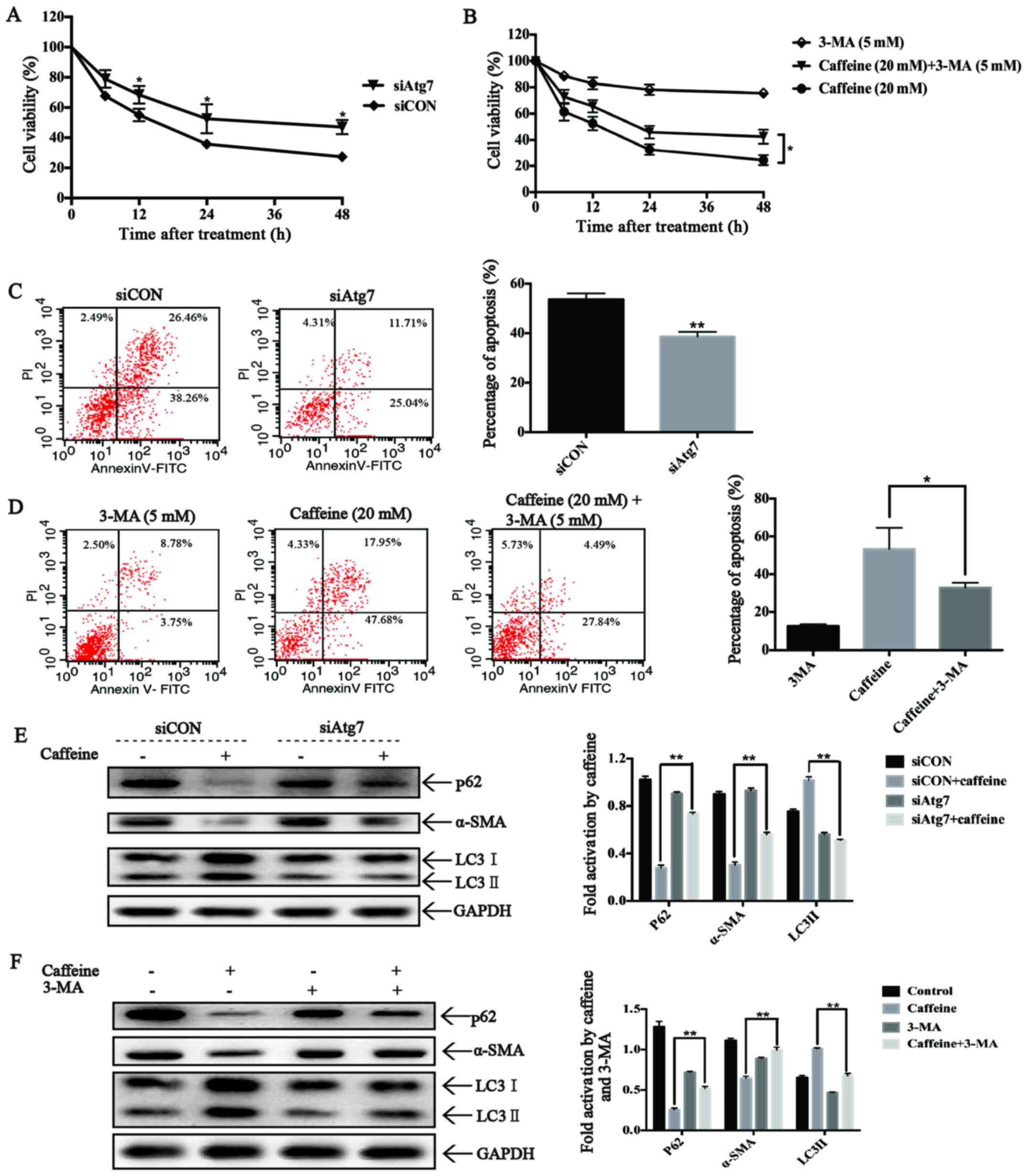
Autophagy triggered by caffeine inhibits
cell viability, induces apoptosis and suppresses activation of LX-2
cells
To investigate the roles of autophagy on cell
viability in caffeine-treated LX-2 cells, we used Atg7-siRNAs to
block autophagosome formation and 3-MA to inhibit the activation of
autophagic flux in LX-2 cells. As expected, Atg7-knockdown reduced
LC3II accumulation and increased the expression level of p62, which
significantly increased the cell viability of the LX-2 cells after
caffeine treatment (Fig. 5A).
These findings strongly suggest the involvement of autophagy in
caffeine-inhibited viability of LX-2 cells. Furthermore,
Atg7-knockdown also strongly decreased the extent of apoptotic
death of the LX-2 cells after caffeine incubation (Fig. 5C), suggesting that autophagy is
essential for caffeine-induced apoptosis of LX-2 cells.
Additionally, Atg7-knockdown rescued the expression of α-SMA,
indicating a direct link between autophagy and LX-2 activation.
Similar results were also found in 3-MA and caffeine co-treated
LX-2 cells (Fig. 5B, D and F).
All these results indicated that autophagy induced apoptosis,
inhibited cell viability and suppressed cell activation in
caffeine-treated LX-2 cells.
Discussion
Recently, many studies have demonstrated that the
long-term consumption of caffeine is associated with lower risks
for chronic liver diseases, such as nonalcoholic fatty disease and
liver fibrosis (25–27). Moreover, other studies have
reported that caffeine attenuates liver fibrosis by directly
inducing apoptosis of HSCs (18).
However, a better understanding of the pathways implicated in this
process remains to be characterized. In the present study, our
findings established autophagy mediated by ER stress as a novel
pathway implicated in caffeine-induced HSC apoptosis.
To the best of our knowledge, liver fibrosis is
triggered by hepatocyte apoptosis, while induction of activated HSC
apoptosis can reverse the progression of liver fibrosis (28–30). Importantly, our study showed that
caffeine markedly reduced the cell viability of activated HSCs and
downregulated the expression of α-SMA in vitro, suggesting
caffeine as a suppressor of HSC activation. Furthermore, in
agreement with other studies, we established the role of caffeine
as an important driver of stellate cell apoptosis. The results
above correspond to the principle that augmentation of HSC
apoptosis is a potential approach through which to promote the
resolution of liver fibrosis (31), therefore, epitomizing an
anti-fibrotic function of caffeine.
Morphological analysis found that caffeine induced
an increase in vacuoles which were recognized as enlarged ERs in
HSCs. One reasonable explanation for these enlarged vacuoles is
induction of the UPR, a self-protective process initially occurring
after ER stress (22,24). Moreover, treatment with caffeine
caused upregulated expression of GRP78/BiP. GRP78/BiP is an ER
chaperone that normally binds with three transmembrane proteins,
IRE1-α, activating transcription factor (ATF)-6α and RNA-activated
protein kinase-like ER kinase (PERK) (32,33). Upon ER stress, GRP78/BiP
dissociates with the three transmembrane proteins described above,
causing the activation of PERK and IRE1-α by
trans-autophosphorylation as well as ATF6α via proteolytic
processing (34,35). The IRE1-α/CHOP pathway is the most
conserved branch of the UPR among the three (7). Notably, our study showed that the
accumulation of IRE1-α and CHOP in HSCs was rapidly increased after
caffeine treatment. Collectively, these events strongly
demonstrated that the ER stress-dependent UPR signaling pathway is
triggered in caffeine-treated HSCs.
Although the UPR is an adaptive process that
compensates for the damage caused by ER stress, prolonged and
massive ER stress that cannot be alleviated by UPR will induce cell
apoptosis (12,36). In the present study, we revealed
that caffeine inhibited the cell viability of HSCs by inducing
apoptosis; meanwhile, ER stress was stimulated after caffeine
treatment in HSCs. These results can be explained by the principle
that treatment with caffeine induces prolonged and sustained ER
stress in HSCs and the ER stress-related damage cannot be
eliminated by UPR, eventually, resulting in apoptotic death
(22,37). Additionally, we also found that
the level of cytosolic calcium was increased after caffeine
incubation in HSCs. To date, perturbations in ER calcium have been
demonstrated to have a close associate with apoptosis effectors
(38). In certain cell lines, ER
stress inducers promote calcium to be released from the ER
accompanied by some related apoptosis effectors released from
mitochondria (33,39). Although the exact functional role
of calcium mobilization in caffeine-mediated ER stress remains
unclear, the possibility that the calcium released from the ER
triggers ER stress and apoptosis seems reasonable.
Recent research of the mechanisms by which caffeine
ameliorates the severity of NAFLD has revealed that caffeine
markedly decreased intracellular lipids via upregulating autophagic
flux in hepatocytes (27).
Analogously, in the present research, we demonstrated that the
biochemical hallmarks of autophagic flux in HSCs were induced, for
example, LC3II accumulation and GFP-RFP-LC3 redistribution in early
and late autophagosomes. Furthermore, many double-membrane vacuolar
structures, the morphological pattern of autophagosomes, were found
in caffeine-treated HSCs, while few were observed in the controls.
Notably, we also showed that GFP-RFP-LC3 redistribution in early
and late autophagosomes and LC3II accumulation were rapidly
decreased after knockdown of IRE1-α. In addition, accumulation of
p62 was significantly compromised in the IRE1-α siRNA-transfected
HSCs. These data not only demonstrated that caffeine induced the
activation of autophagy in HSCs, but also established the role of
ER stress as an important driver of autophagic flux induced by
caffeine in HSCs.
The primary role of autophagy in cells is
complicated, sometimes paradoxical. Whether autophagy protects
cells from death or promotes cell death depends on the cellular
context, the duration and strength of stimuli (40,41). Previously, one study revealed that
caffeine induced apoptosis in SH-SY5Y, PC12D and HeLa cells by
enhancement of autophagy (19).
Similarly, the data presented here showed that both of the genetic
and chemical autophagy inhibitors, Atg7-siRNAs and 3-MA, alleviated
caffeine-induced apoptosis. Coincident with the alleviation of cell
death by autophagy, the cell viability was increased in the
Atg7-knockdown and 3-MA-pretreated HSCs after caffeine treatment.
These data strongly demonstrate that autophagy induced by caffeine
promoted apoptotic cell death rather than cell survival in the
HSCs. To date, many studies have reported that autophagy removes
superabundant proteins when the accumulation of unfolded or
misfolded proteins surpasses the ability of the degradation system
mediated by the proteasome (42,43). The protective function of
autophagy was contradictory with our results. One plausible
explanation is that the outcome of ER stress-mediated autophagy
depends on the type of stimulants and the cellular context.
Notably, our findings rightly identify with the anti-fibrosis
mechanism of caffeine. In brief, caffeine suppresses activation of
HSCs by induction of autophagy via the IRE1-α pathway. And once the
activated HSCs are eliminated, the progression of liver fibrosis
can be attenuated and reversed. Viewed this way, our finding is
consistent with previous studies on the induction of apoptotic cell
death by caffeine and provides novel insight into the mechanism
involved in the anti-fibrotic function of caffeine.
Additionally, it is worth noting that in our study
the concentrations of caffeine that induced apoptosis of HSCs were
in a high range. The results disclosed that only a high
concentration of caffeine attenuated liver fibrosis, which is in
accordance with previous research that individuals who consume a
large amount of caffeine every day have the lowest risk of liver
disease (44).
In conclusion, our data highlight the importance of
autophagy mediated by ER stress in caffeine-treated HSCs as a main
driver of apoptosis, extending the mechanisms of the attenuation of
liver fibrosis by caffeine. We speculate that moderate to high
caffeine intake can be useful in the treatment of liver
fibrosis.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81070358), (http://www.nsfc.gov.cn) to JQY.
References
|
1
|
Tsochatzis EA, Bosch J and Burroughs AK:
Liver cirrhosis. Lancet. 383:1749–1761. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman SL: Hepatic stellate cells:
protean, multifunctional, and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mederacke I, Hsu CC, Troeger JS, Huebener
P, Mu X, Dapito DH, Pradere JP and Schwabe RF: Fate tracing reveals
hepatic stellate cells as dominant contributors to liver fibrosis
independent of its aetiology. Nat Commun. 4:28232013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thoen LF, Guimarães EL, Dollé L, Mannaerts
I, Najimi M, Sokal E and van Grunsven LA: A role for autophagy
during hepatic stellate cell activation. J Hepatol. 55:1353–1360.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parola M, Marra F and Pinzani M:
Myofibroblast-like cells and liver fibrogenesis: emerging concepts
in a rapidly moving scenario. Mol Aspects Med. 29:58–66. 2008.
View Article : Google Scholar
|
|
6
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hernández-Gea V, Hilscher M, Rozenfeld R,
Lim MP, Nieto N, Werner S, Devi LA and Friedman SL: Endoplasmic
reticulum stress induces fibrogenic activity in hepatic stellate
cells through autophagy. J Hepatol. 59:98–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ding WX and Yin XM: Sorting, recognition
and activation of the misfolded protein degradation pathways
through macroautophagy and the proteasome. Autophagy. 4:141–150.
2008. View Article : Google Scholar
|
|
9
|
Bernales S, Papa FR and Walter P:
Intracellular signaling by the unfolded protein response. Annu Rev
Cell Dev Biol. 22:487–508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Momoi T: Conformational diseases and ER
stress-mediated cell death: apoptotic cell death and autophagic
cell death. Curr Mol Med. 6:111–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koo JH, Lee HJ, Kim W and Kim SG:
Endoplasmic reticulum stress in hepatic stellate cells promotes
liver fibrosis via PERK-mediated degradation of HNRNPA1 and
up-regulation of SMAD2. Gastroenterology. 150:181–193.e8. 2016.
View Article : Google Scholar
|
|
12
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meares GP, Mines MA, Beurel E, Eom TY,
Song L, Zmijewska AA and Jope RS: Glycogen synthase kinase-3
regulates endoplasmic reticulum (ER) stress-induced CHOP expression
in neuronal cells. Exp Cell Res. 317:1621–1628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi AY, Choi JH, Yoon H, Hwang KY, Noh
MH, Choe W, Yoon KS, Ha J, Yeo EJ and Kang I: Luteolin induces
apoptosis through endoplasmic reticulum stress and mitochondrial
dysfunction in Neuro-2a mouse neuroblastoma cells. Eur J Pharmacol.
668:115–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hsu SJ, Lee FY, Wang SS, Hsin IF, Lin TY,
Huang HC, Chang CC, Chuang CL, Ho HL, Lin HC, et al: Caffeine
ameliorates hemodynamic derangements and portosystemic collaterals
in cirrhotic rats. Hepatology. 61:1672–1684. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gressner OA, Lahme B, Rehbein K, Siluschek
M, Weiskirchen R and Gressner AM: Pharmacological application of
caffeine inhibits TGF-β-stimulated connective tissue growth factor
expression in hepatocytes via PPARgamma and SMAD2/3-dependent
pathways. J Hepatol. 49:758–767. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shim SG, Jun DW, Kim EK, Saeed WK, Lee KN,
Lee HL, Lee OY, Choi HS and Yoon BC: Caffeine attenuates liver
fibrosis via defective adhesion of hepatic stellate cells in
cirrhotic model. J Gastroenterol Hepatol. 28:1877–1884. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri
S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K,
et al: Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar :
|
|
20
|
Yan X, Shan Z, Yan L, Zhu Q, Liu L, Xu B,
Liu S, Jin Z and Gao Y: High expression of zinc-finger protein
X-linked promotes tumor growth and predicts a poor outcome for
stage II/III colorectal cancer patients. Oncotarget. 7:19680–19692.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ou B, Zhao J, Guan S, Wangpu X, Zhu C,
Zong Y, Ma J, Sun J, Zheng M, Feng H, et al: Plk2 promotes tumor
growth and inhibits apoptosis by targeting Fbxw7/Cyclin E in
colorectal cancer. Cancer Lett. 380:457–466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke
AW, Wang XY, Dai Z, Peng YF, Gu CY, et al: Targeting autophagy
enhances sorafenib lethality for hepatocellular carcinoma via ER
stress-related apoptosis. Autophagy. 7:1159–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar
|
|
24
|
Xu WH, Liu ZB, Hou YF, Hong Q, Hu DL and
Shao ZM: Inhibition of autophagy enhances the cytotoxic effect of
PA-MSHA in breast cancer. BMC Cancer. 14:2732014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Friedrich K, Smit M, Wannhoff A, Rupp C,
Scholl SG, Antoni C, Dollinger M, Neumann-Haefelin C, Stremmel W,
Weiss KH, et al: Coffee consumption protects against progression in
liver cirrhosis and increases long-term survival after liver
transplantation. J Gastroenterol Hepatol. 31:1470–1475. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wadhawan M and Anand AC: Coffee and liver
disease. J Clin Exp Hepatol. 6:40–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sinha RA, Farah BL, Singh BK, Siddique MM,
Li Y, Wu Y, Ilkayeva OR, Gooding J, Ching J, Zhou J, et al:
Caffeine stimulates hepatic lipid metabolism by the
autophagy-lysosomal pathway in mice. Hepatology. 59:1366–1380.
2014. View Article : Google Scholar
|
|
28
|
Elsharkawy AM, Oakley F and Mann DA: The
role and regulation of hepatic stellate cell apoptosis in reversal
of liver fibrosis. Apoptosis. 10:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jeschke MG, Gauglitz GG, Song J, Kulp GA,
Finnerty CC, Cox RA, Barral JM, Herndon DN and Boehning D: Calcium
and ER stress mediate hepatic apoptosis after burn injury. J Cell
Mol Med. 13(8B): 1857–1865. 2009. View Article : Google Scholar
|
|
30
|
Ji C: Dissection of endoplasmic reticulum
stress signaling in alcoholic and non-alcoholic liver injury. J
Gastroenterol Hepatol. 23(Suppl 1): S16–S24. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Gao J, Zhang D, Zhang J, Ma J and
Jiang H: New insights into the antifibrotic effects of sorafenib on
hepatic stellate cells and liver fibrosis. J Hepatol. 53:132–144.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shimada Y, Kobayashi H, Kawagoe S, Aoki K,
Kaneshiro E, Shimizu H, Eto Y, Ida H and Ohashi T: Endoplasmic
reticulum stress induces autophagy through activation of p38 MAPK
in fibroblasts from Pompe disease patients carrying c.546G>T
mutation. Mol Genet Metab. 104:566–573. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Malhi H and Kaufman RJ: Endoplasmic
reticulum stress in liver disease. J Hepatol. 54:795–809. 2011.
View Article : Google Scholar
|
|
34
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oikawa D, Kimata Y, Kohno K and Iwawaki T:
Activation of mammalian IRE1alpha upon ER stress depends on
dissociation of BiP rather than on direct interaction with unfolded
proteins. Exp Cell Res. 315:2496–2504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hwang MS and Baek WK: Glucosamine induces
autophagic cell death through the stimulation of ER stress in human
glioma cancer cells. Biochem Biophys Res Commun. 399:111–116. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Deniaud A, Sharaf el dein O, Maillier E,
Poncet D, Kroemer G, Lemaire C and Brenner C: Endoplasmic reticulum
stress induces calcium-dependent permeability transition,
mitochondrial outer membrane permeabilization and apoptosis.
Oncogene. 27:285–299. 2008. View Article : Google Scholar
|
|
39
|
Lawless MW, Mankan AK, White M, O'Dwyer MJ
and Norris S: Expression of hereditary hemochromatosis C282Y HFE
protein in HEK293 cells activates specific endoplasmic reticulum
stress responses. BMC Cell Biol. 8:302007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Criollo A, Maiuri MC, Tasdemir E, Vitale
I, Fiebig AA, Andrews D, Molgó J, Díaz J, Lavandero S, Harper F, et
al: Regulation of autophagy by the inositol trisphosphate receptor.
Cell Death Differ. 14:1029–1039. 2007.PubMed/NCBI
|
|
43
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hu G, Tuomilehto J, Pukkala E, Hakulinen
T, Antikainen R, Vartiainen E and Jousilahti P: Joint effects of
coffee consumption and serum gamma-glutamyltransferase on the risk
of liver cancer. Hepatology. 48:129–136. 2008. View Article : Google Scholar : PubMed/NCBI
|