Introduction
The renin-angiotensin system (RAS) is known for the
control of blood pressure and fluid homeostasis but also for its
engagement in the development of prostate cancer (1). The main peptide of RAS, angiotensin
II (AngII) acts via G-protein coupled AT1 and AT2 receptors (AT1R,
AT2R). AT1R is present in the healthy prostate and is upregulated
in prostate cancer cells (2,3).
Expression of AT2R is commonly lower than AT1R and its upregulation
is related to tissue damage, due to its role in protection and
regeneration of tissues (4).
AngII may transmit signals through protein tyrosine kinases (PTKs),
enzymes involved in cell proliferation as well as cell apoptosis
(5). Moreover, inhibitors of
tyrosine kinases have been recently developed as molecular-targeted
drugs for prostate cancer (6).
The physiological effects of AngII are dependent on the receptor
type. Vasodilatation, cell cycle arrest, antiproliferation and
apoptosis are mediated by AT2R, whereas action via AT1 causes
vascular contraction, proliferation and cell growth (7). AngII affects the proliferation of
prostate cancer cells (8) mainly
due to the local tissue RAS, also depending on receptor type.
Synthesis and function of AngII can be regulated by androgens in
the prostate gland (2).
Another RAS component, angiotensin 1–7 (Ang1-7) is a
product of AngII proteolysis (9).
Ang1-7 can bind to the AT1 and AT2 receptor or to its specific
G-protein coupled Mas receptor (10). The physiological effects of Ang1-7
are generally opposite to the effects of AngII (9), and it has been shown that Ang1-7
displays an antitumor effect in vivo and attenuates
cytopenia. Thus, it has potential in chemotherapy, and does not
cause hematologically toxic effects (11,12).
In Western countries prostate cancer is one of the
most common malignant diseases among men over 50 years of age
(13). In vitro tests of
potential treatments are frequently carried out using the
hormone-independent human prostate cancer DU145 human cell line
(14). Nevertheless, to date,
hormonal therapy for prostate cancer is possible only in the case
of androgen sensitivity of the tumor. Steroid hormones can evoke
changes in cells by binding to their own or other receptors both
intracellular and membrane. Thus, steroids can exhibit both genomic
and non-genomic action. The biological activity of testosterone
occurs mainly through binding with the intracellular specific
androgen receptor affecting gene expression but data indicate the
possibility of rapid, non-genomic action of testosterone, including
prostate cancer cells (15,16). These effects are considered to be
non-genomic, because they appear within minutes, too rapidly to
involve changes in gene transcription, and occur often in cells
that lack a functional AR, similar to DU145 (17). Additionally, testosterone can be
converted to its metabolite 17β-estradiol, which can be active in
prostate cells and can act both in genomic and non-genomic manners,
involving specific estradiol receptor or receptors of other factors
including angiotensin family peptides (18).
In the present study, we examined the effects of
testosterone and 17β-estradiol on angiotensin-induced changes in
human metastatic prostate cancer DU-145 cells. This line is
androgen receptor-negative, and does not respond to androgen
hormones in a classical way. We focused on the potential
non-genomic action of testosterone and 17β-estradiol, using short
periods of exposure. We verified changes in the activity of PTKs,
as they are important factors in cancer development and their
activity is upregulated in tumors, such as prostate cancer
(19). Furthermore, we elucidated
the participation of AT1 and AT2 receptors in 17β-estradiol-induced
or testosterone-induced changes in the angiotensin effect. We used
losartan and PD123319, specific antagonists of AT1 and AT2,
respectively.
Materials and methods
All chemicals used were of analytical grade. If not
stated, chemicals were purchased from Sigma Aldrich (St. Louis, MO,
USA).
Cell culture
The DU145 cell line was purchased from the American
Type Culture Collection (ATCC; Manassas, VA, USA). The cell line
was authenticated by short-tandem repeat (STR) DNA profiling. Cells
were cultured in Dulbecco's modified Eagle's medium (DMEM;
sigma-Aldrich; Merck Millipore, Darmstadt, Germany) supplemented
with 10% heat-inactivated fetal bovine serum (FBS; Biowest,
Nuaille, France), 2 mM L-glutamine, penicillin (50 U/ml),
streptomycin (50 μg/ml) and neomycin (100 μg/ml) in a
humidified atmosphere under 5% CO2 at 37°C.
Western blot analysis
Cells were incubated for 24 h with 5 nM AngII or
Ang1-7 with or without 1 h preincubation with 100 μM
17β-estradiol or testosterone. Cells were then washed once in PBS
and lysed in 60–100 μl of RIPA buffer supplemented with
protease and phosphatase inhibitors for 30 min at 4°C. Lysates were
centrifuged at 14,000 rpm for 20 min to remove insoluble material.
Protein concentration was measured using Bradford Protein Assay kit
(Bio-Rad, Berkeley, CA, USA). Lysates were incubated with Laemmli
buffer at 95°C for 5 min. Protein samples (30 μg) were
separated on a 10% polyacrylamide gel and transferred to
nitrocellulose membranes using a Mini-PROTEAN Tetra system
(Bio-Rad). Non-specific protein binding sites were blocked for 1 h
at room temperature, using 5% BSA in TBST buffer. Nitrocellulose
membranes were then incubated overnight at 4°C with anti-AT1,
anti-AT2 and GAPDH primary antibodies (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) and in alkaline phosphatase-labelled
secondary antibodies (Sigma) for 1 h at room temperature. The
positive bands were revealed using sigma-Fast BCIP/NBT reagent
(sigma-Aldrich). Blots were analyzed by densitometry using Quantity
One software (Bio-Rad). The relative expression of AT1R or AT2R was
determined by comparing their protein expression level to those of
GADPH.
PTK activity
Cells were suspended in medium containing 32 mM
sacharose, 50 μM EDTA, 10 mM Tris/HCl pH 7.4, 50 μM
PMSF and aprotinin 25 KIU/ml at 4°C. Samples were pretreated with
10 μM testosterone or 17β-estradiol for 15 min at 37°C.
After preincubation, AngII or Ang1-7 were added to both types of
samples. Concentrations of angiotensins were 50 pM, 500 pM and 5 nM
(5×10−11 to 5×10−9 M). Following 15 min, the
samples were lysed with 0.1% Triton X-100 for 15 min at 0°C. PTK
activity was assessed according to the method of Hirano et
al (20,21) with modifications.
γ-[32P]-ATP (200 mM; DuPont NEN, Boston, MA, USA) and
Poly(Glu, Tyr) (4:1) were used as substrates for phosphorylation by
PTKs or phosphorylation reaction. Poly(Glu, Tyr)-free samples were
taken as phosphorylation control. All samples were incubated for 7
min at 30°C. Radioactivity of 32P binding to the
substrate was measured as Cerencov radiation using a liquid
scintillation analyzer (Canberra-Packard, Bucharest, Romania). The
activity of PTKs was defined as the amount in pmol of
32P incorporated to Poly(Glu, Tyr) per 1 mg of protein
per 1 min (pmol/mg/min). Results were compared to the basal
activity of PTKs (control) obtained from the samples containing
only cyclodextrin which was a carrier of steroid hormones. The
degree of 32P incorporation to specific substrate by
PTKs in the control group (basal activity) was taken as 100%.
The inhibitors of angiotensin receptors, losartan
(Adamed Group, Czosnów, Poland) or PD319131 were added to samples
before tested hormones at the final concentrations during
preincubation 10−8 M (21).
Statistical analysis
Data are expressed as mean ± SEM of independent
experiments from at least 3–4 cultures; from each culture we
obtained 4–6 results. Statistical analysis was performed with the
t-test, using Statistica 12 software. P<0.05 was considered to
indicate a statistically significant difference.
Results
We used cyclodextrin-conjugated 17β-estradiol and
testosterone. Thus, in each experiment, the control culture medium
contained cyclodextrin. Cyclodextrin alone does not affect any
parameters which are subjects of the present study.
17β-estradiol and testosterone modify the
angiotensin-induced decrease in PTK activity
Cells were exposed to 50 pM, 500 pM and 5 nM AngII
or Ang1-7. These concentrations were chosen since they were
previously confirmed to be most efficient in altering the activity
of PTKs (21,22). Here, administration of AngII alone
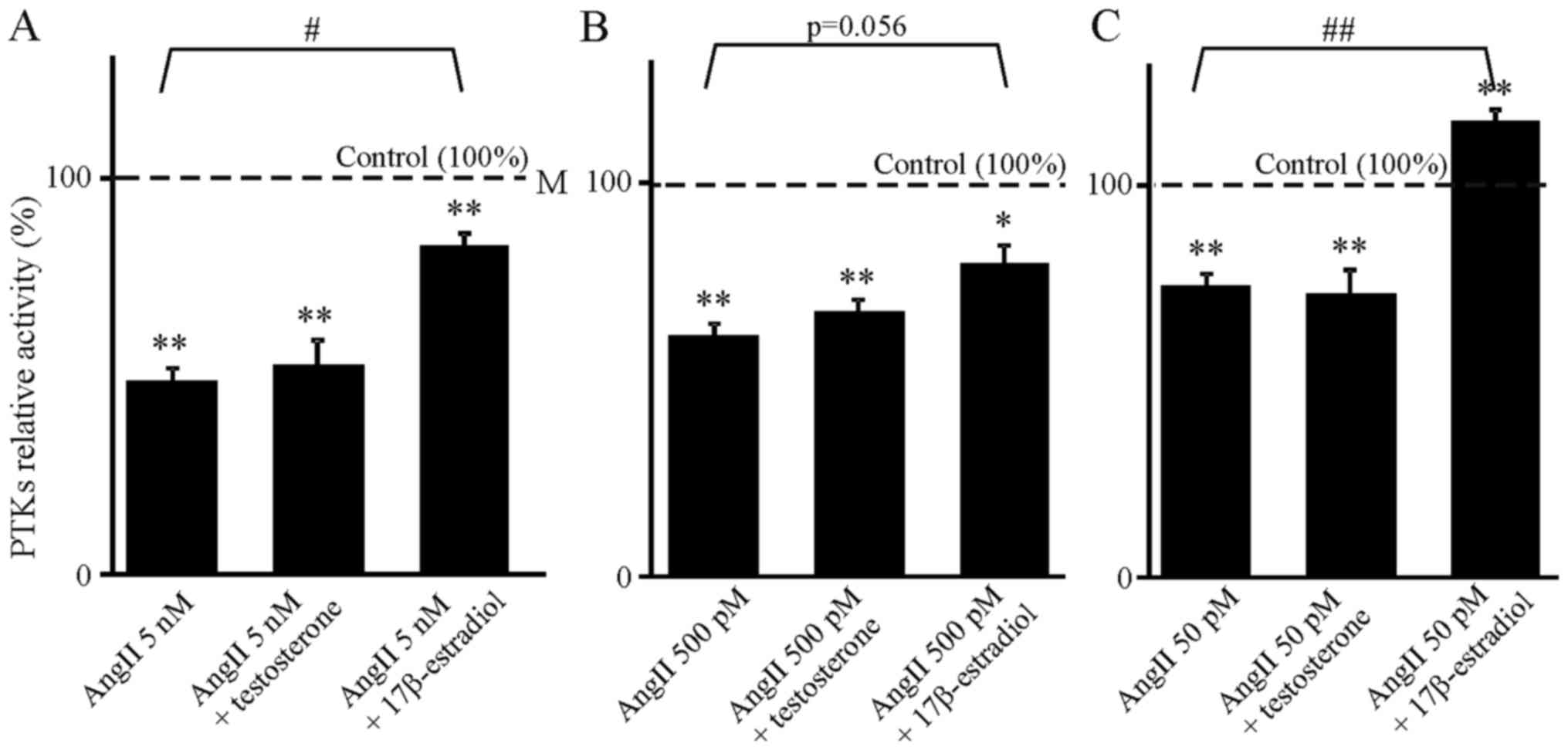
decreased the activity of the PTKs (Fig. 1). We observed a slight positive
correlation between the concentration of AngII and the strength of
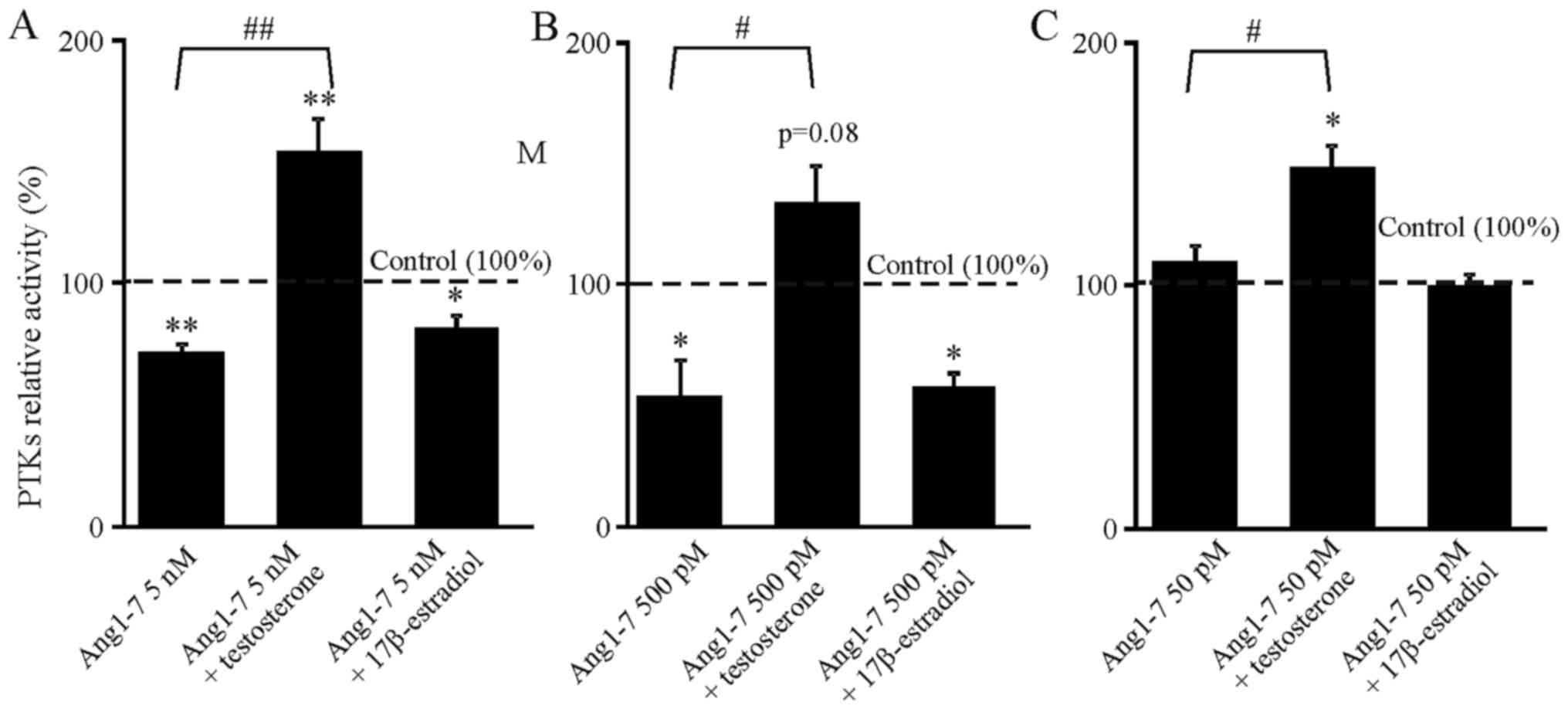
its inhibitory effect. Ang1-7 was effective only at higher
concentrations (Fig. 2).
Steroids alone did not induce significant change in
PTK activity (data not shown), but they acted to inverse the
inhibitory effect of angiotensin. Moreover, testosterone and
17β-estradiol (10 μM) showed clearly specific interaction;
estradiol reversed inhibition caused only by AngII (Fig. 1), and testosterone reversed
inhibition caused only by Ang1-7 (Fig. 2). In each case this effect was
statistically significant or close to achieving statistical
significance (Fig. 1B).
Furthermore, combination of 50 pM AngII with 17β-estradiol
increased PTK activity above the control value (Fig. 1C). The same applied to the
combination of Ang1-7 and testosterone (Fig. 2A–C).
Effect of steroid hormones on
AngII-induced enhancement of AT1R and AT2R expression
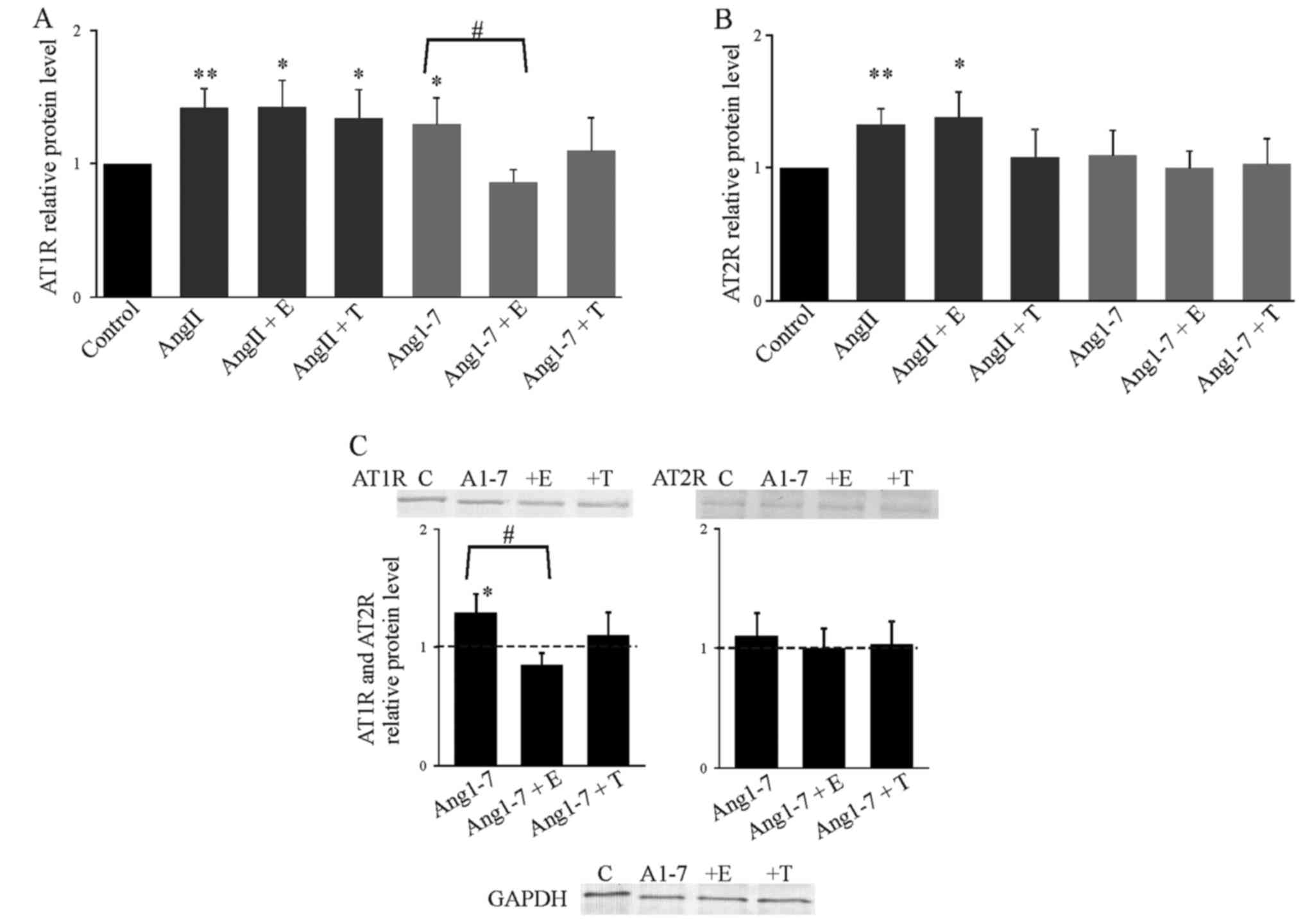
AngII induced expression of AT1R, but we observed an
additional significant increase in the AT2R level in the presence
of AngII (Fig. 3). This effect
was not affected by the hormones. Ang1-7 increased the expression
of AT1R and this effect was attenuated by 17β-estradiol, but not by
testosterone (Fig. 3C).
Expression of AT2R was affected neither by Ang1-7 alone, nor by
Ang1-7 together with 17β-estradiol or testosterone (Fig. 3).
Involvement of AT1R and AT2R in the
effect of steroids on angiotensin-induced PTK inhibition
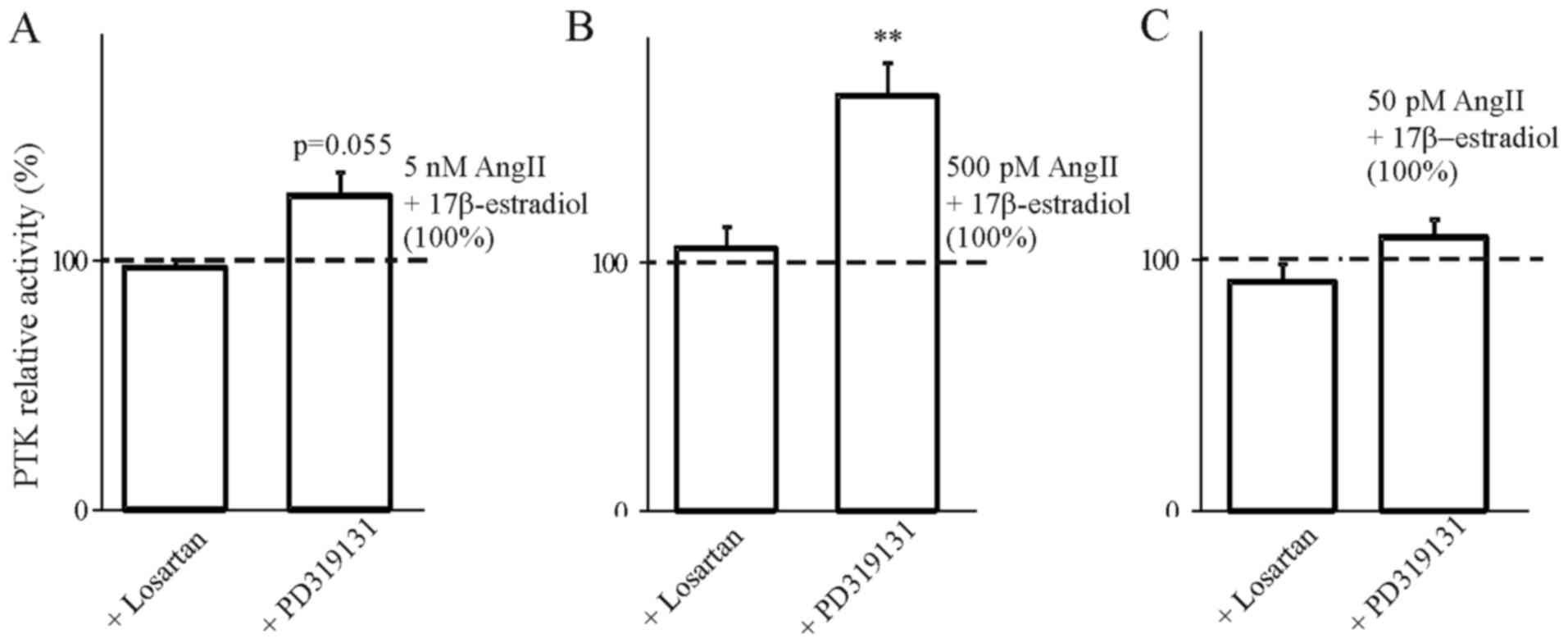
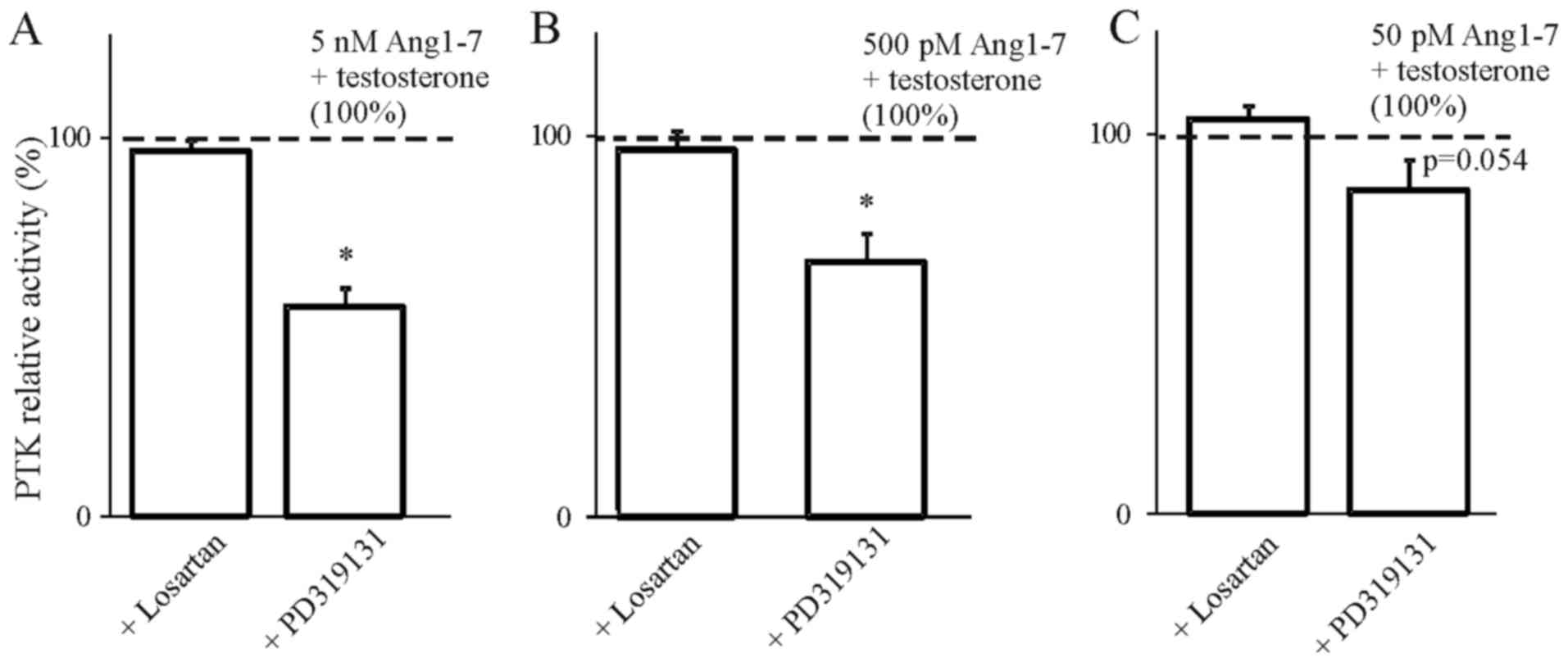
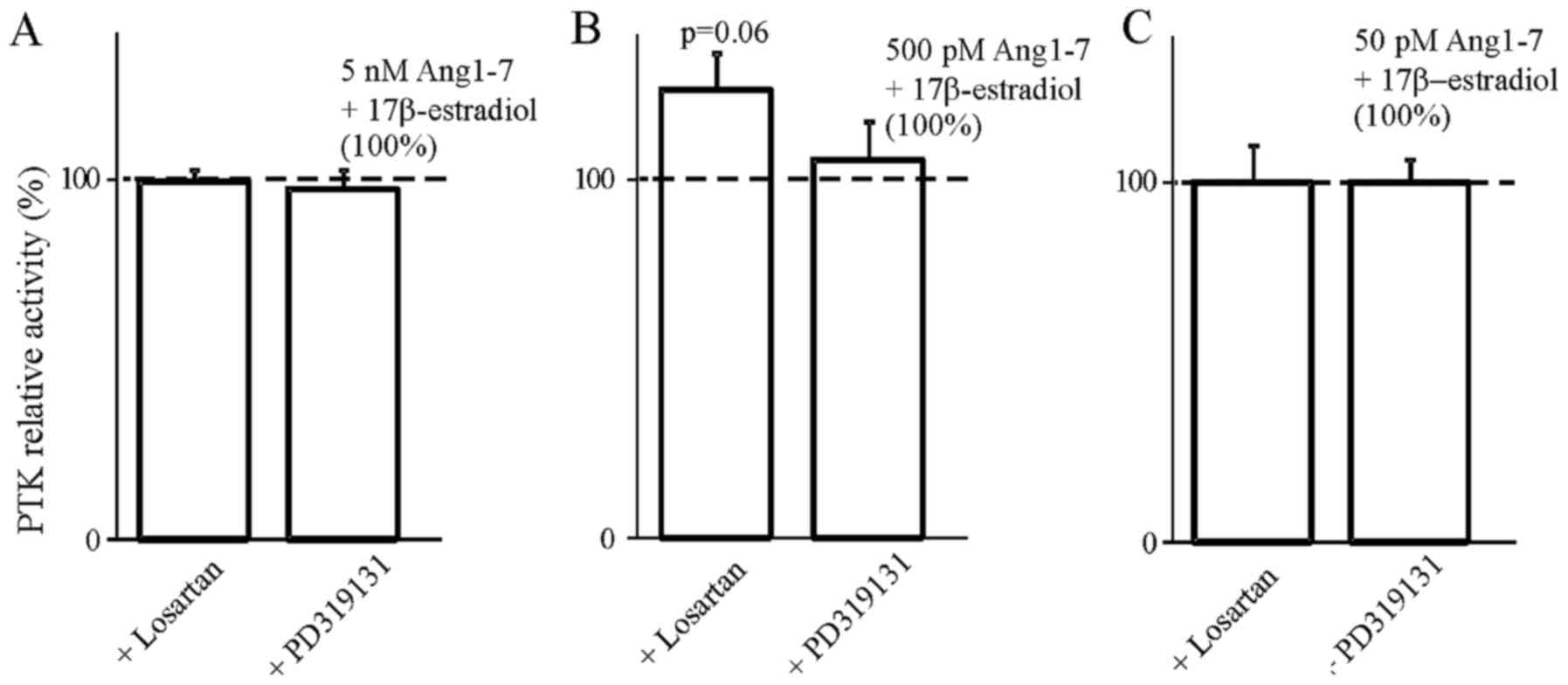
In order to determine the involvement of AT1 and AT2
receptors in the steroids we used selective blockers of both
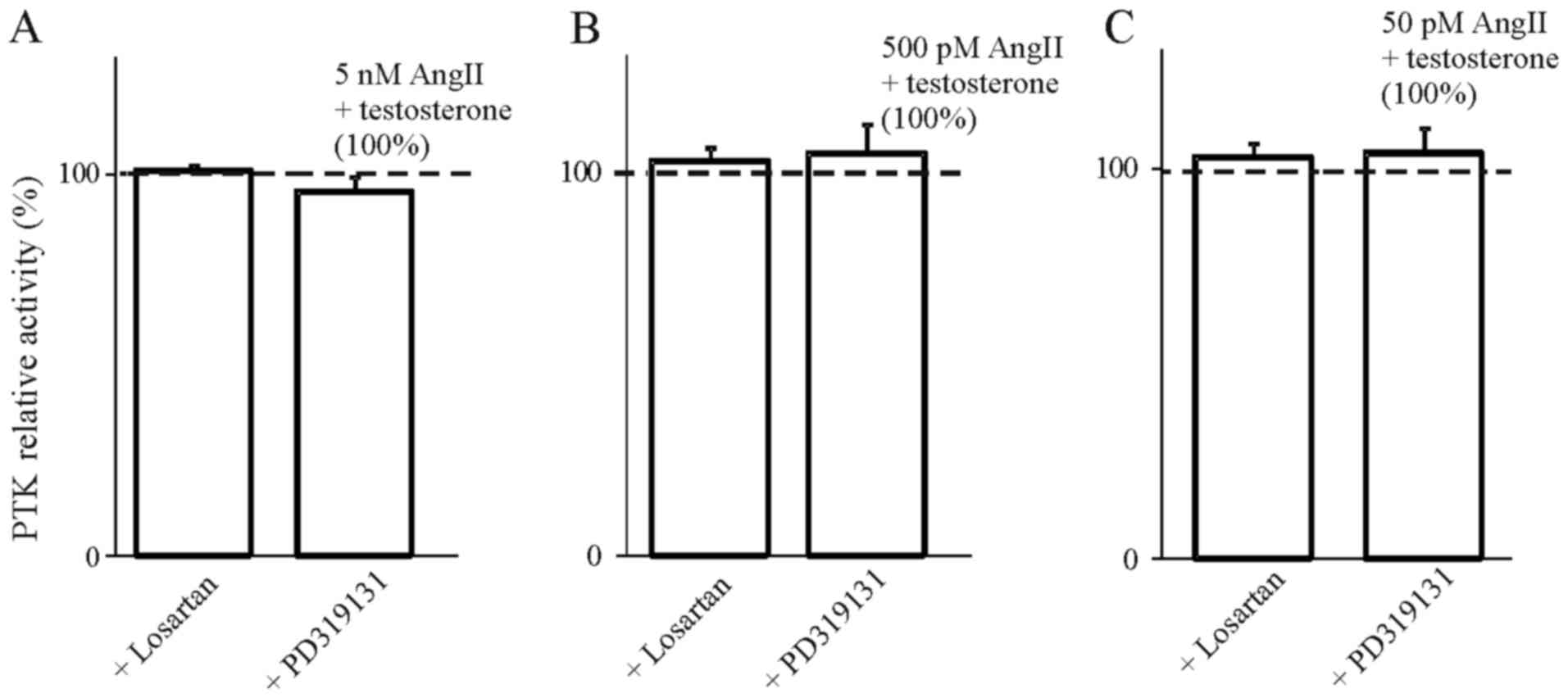
receptors. The results presented in Figs. 4Figure 5Figure 6–7 indicate that the use of the selective
inhibitor of AT2, PD319131 (10−8 M) caused the
inhibition of the simultaneous effect of Ang1-7 and testosterone at
all tested concentrations of angiotensin peptide. This suggests
that testosterone influences the interaction of Ang1-7 with the AT2
receptor. The lack of an inhibitory effect of receptor blockers on
the action of AngII in the presence of 17β-estradiol suggest that
the weak action of 17β-estradiol on AngII-induced inhibition of PTK
activity was caused by mechanisms independent of the AT1 and AT2
receptors.
Discussion
Prostate cancer is one of the most common diseases
and its mortality rate is increasing. The patient survival rate is
reduced in cases of late diagnosis when cells acquire metastatic
ability and become resistant to hormone treatment. One of the best
models in prostate cancer studies is the DU145 cell line,
characterized by lack or very low expression of the androgen
receptor (23). There are several
differences between androgen-insensitive cell lines and
androgen-responsive cells e.g., LNCaP, which are more susceptible
to hormonal treatment. However, the classical receptor-mediated
effect is not the only one possible of testosterone action. The
rapid membrane effect of testosterone mediated through
unconventional membrane androgen receptors was documented in LNCaP
and DU145 cells (24). Equally
important is the fact that this can be also mediated by
testosterone metabolite-17β-estradiol. Androgen-independent DU145
cells express both classical estradiol receptors, ERα and ERβ
(25), and show high activity of
5α-reductase and aromatase (26).
ERα is expressed in prostatic tissue mainly during fetal and early
neonatal life. Invalid activation of ERα causes late-life diseases,
including inflammation and pre-malignant pathologies. In contrast,
ERβ expression appears later, after ERα, primarily in the
epithelium. It seems to be an important factor of regulation of
cell proliferation and regulation of differentiation in adult
tissue and during puberty (25).
This isoform predominates in late, metastatic cells of prostate
cancer such as DU145 cells (27).
It is known that although the androgen level decreases with age,
the level of estrogens in males remains unchanged and even
increases. Androgens (including testosterone) are necessary for
normal prostate development and function, while in many cases
estradiol speeds up the development of prostate cancer (28). Yet, it appears that both synthetic
and natural (including exogenous) modulators of ERβ show protective
properties against cancer progression by inhibition of cell
proliferation and migration, and can be utilized in the therapy of
prostate cancer including androgen-independent cancer (27,29) Therefore, it is important to
determine the function of both steroids in DU145 cells.
There is evidence of a relationship between androgen
and RAS. In prostate cells this relationship is additively
complicated by the fact that angiotensins can interact with steroid
receptors and contrariwise steroids may bind to angiotensin
receptors. Other studies have shown that signaling transmission by
G-protein coupled receptors (including angiotensin receptors) can
induced androgen-independent androgen receptor activation causing
the conversion of androgen-dependent cells to androgen-independent
cells (30). On the other hand
some studies indicate that estrogens affect components of RAS,
e.g., decrease the expression of AT1R and increase AT2R expression
(31,32). Testosterone directly leads to
upregulation of RAS components including AT2 receptor expression
(32). The overlapping effects of
angiotensins and steroids can be detrimental to the cell and can be
the cause of cancer treatment failure.
Local RAS plays an important role in cancer
development (33,34). Elements of RAS exist in both
normal and cancer cells including cell lines (34,35). Local RAS includes
angiotensin-family peptides, enzymes involved in their synthesis
and metabolism, and angiotensin receptors. RAS is implicated in the
development of various cancers, and changes in the expression of
RAS components in most cases are correlated with cancer grade,
however they are not consistent and depend on tumor type and degree
of cancer possibly due to various cellular pathways initiated by
RAS (35). Elements of
intracellular signaling pathways, which are activated by
angiotensins, such as tyrosine kinases, can be also classified to
RAS (5). RAS and tyrosine kinases
are now targets in cancer therapies, including prostate cancer
(35–37).
Recent studies show that angiotensins may modulate
the proliferation and migration of cells, which promotes cancer
development and metastasis. Angiotensins can influence prostate
cancer cells (PC3, DU145 and LNCaP) in different ways, depending on
the type of cell (21,38,39). Data indicate the importance of the
angiotensin peptide type and time of exposure for progression of
prostate cancer. In addition, the action of angiotensin family
peptides in cancer cells is intricate and depends on cancer type
and development stage.
In most cases, in cancer cells the activity of
tyrosine kinases is much higher than that in healthy cells
(40). PTKs play important roles
in adverse, very fast proliferation of cells. But in some types of
cells, including cancer, tyrosine kinases can mediate apoptosis or
differentiation (41). Previous
studies indicate that angiotensin peptides modulate the tyrosine
kinase activity in pituitary (22,42), MCF and MDA cell lines (43) in a concentration-dependent manner.
Notable is the similarity of the inhibitory effect of AngII on PTK
activity in hormone-sensitive cell lines (LNCaP and MCF-7). In
hormone-independent lines (DU145 and MDA-MB-231) angiotensin had a
stronger inhibitory effect (43).
We found that both AngII and Ang1-7 decreased
tyrosine kinase activity. We rather expected that the action of
both peptides would be opposite and that AngII would stimulate PTKs
while Ang1-7 would decrease their activity. The DU145 cell line is
more advanced in terms of the development of cancer and may show
distinct properties and processes responsible for cancer
development in this line may be upset. Inhibition of tyrosine
kinases seems to be positive (protective) because of possible
simultaneous inhibition of proliferation. This unexpected
inhibition of PTK activity may be explained by the interaction of
AngII with the AT2 receptor as the mechanism of angiotensin action
depends on the type of receptor. Activation of AT1R causes the
pro-inflammatory and procancerogenic action of ligands, while
stimulation of AT2R leads to pro-apoptotic and anti-proliferative
effects of angiotensins.
Opposite physiological effects caused by both
peptides are considered to be associated with different affinity of
the two types of receptors to AngII and Ang1-7. T1R can bind both
AngII and Ang1-7, but has higher affinity to AngII in normal cells.
AT2R can also bind both peptides, but in normal cells, it
preferably binds Ang1-7. Aditionally, Ang1-7 can bind to a third
receptor, specific to Ang1-7 only (AT1-7/Mas receptor), which is
also found in testis (44).
Commonly, AngII preferably binds to AT1R causing unfavorable
effects, while antagonists of AT1R show strong antiproliferative
activity and inhibit angiogenesis in prostate cancer cells
(45). Ang1-7 has higher affinity
to AT2R, thus Ang1-7 can significantly decrease the size of tumors
as well as activity of kinases which participate in proliferation
(11).
As mentioned earlier, expression of AT1R and AT2R
was found in prostate cancer cells (46,47) while expression of both types of
receptors can differ in LNCap and DU145 cells (47). In androgen-positive LNCap cells
AT1R expression is higher than in late stage, androgen-independent
DU154 cells and in healthy cells (34). This may be due to increased
protein kinase activity, subsequent proliferation and finally the
rapid conversion to metastatic, aggressive form of prostate cancer.
Expression of angiotensin receptors at this late stage of prostate
depends on the state of severity as well. Research has shown that
in androgen-nonsensitive cells expression of AT2R is lower than
AT1R and lower than that in androgen-positive cells (46). Our results indicate that in DU145
cells expression of AT2R was increased in the presence of AngII.
Hence we suggest increased binding of AngII to AT2 receptor that
results in the subsequent decrease in PTK activity.
It is well established that G-protein coupled
receptors and tyrosine kinases can crosstalk and modulate each
other (48). We observed an
increase in PTK activity in the presence of Ang1-7 and testosterone
or AngII and 17β-estradiol. This may suggest that both steroids
have procancerogenic features and can contribute to cancer
progression. This is consistent with the fact that 17β-estradiol
facilitates prostate cancer growth.
It is widely known that the pharmacological effects
of steroids are linked with changes in gene transcription, but
there is also increasing data concerning their rapid non-genomic
actions. In the present study, we did not observe a significant
effect of testosterone or 17β-estradiol on AT1R and AT2R expression
in the presence of AngII or Ang1-7. Lack of a testosterone effect
is not surprising and can be explained by absence or low expression
of the androgen receptor, that eliminates a classical genomic mode
of action. Yet, the results indicate that testosterone does not
modify the stimulatory effect of angiotensins on receptor
expression in any other way. The inhibitory impact of 17β-estradiol
on AT1R expression in the presence of Ang1-7 was possible thanks to
the existence of estrogen receptors in the DU145 cells. But it
seems, that changes in angiotensin-induced inhibition of tyrosine
kinases are caused by direct interaction of hormones with existing
angiotensin receptors rather, not by changes in receptor number.
Additionally, the appearance of the effect after a quite short
time, precluding genomic action enforces this hypothesis.
Transcription alteration usually takes hours, but the effects of
those described as non-genomic direct pathways can be noticed in a
few minutes.
Withdrawal of changes triggered by testosterone
after using specific AT2R blockers (PD19234) indicates the wider
involvement of the AT2 receptor in this hormone action on
Ang1-7-induced changes. Probably, in the cell line lacking androgen
receptor, testosterone acts through AT2R characterized by a higher
affinity to Ang1-7. The observed rise in PTK activity in the
presence of AngII and 17β-estradiol after inhibition of AT2R by a
specific blocker probably was evoked by rapid interaction of both
17β-estradiol and AngII with other than angiotensin receptors,
e.g., membrane receptors of estradiol. All the more so, AT1R,
although found both in LNCaP and DU145, is only functional in
LNCaP. In PC3 cells which are similar to DU145, AT1R seems to be
non-functional (49).
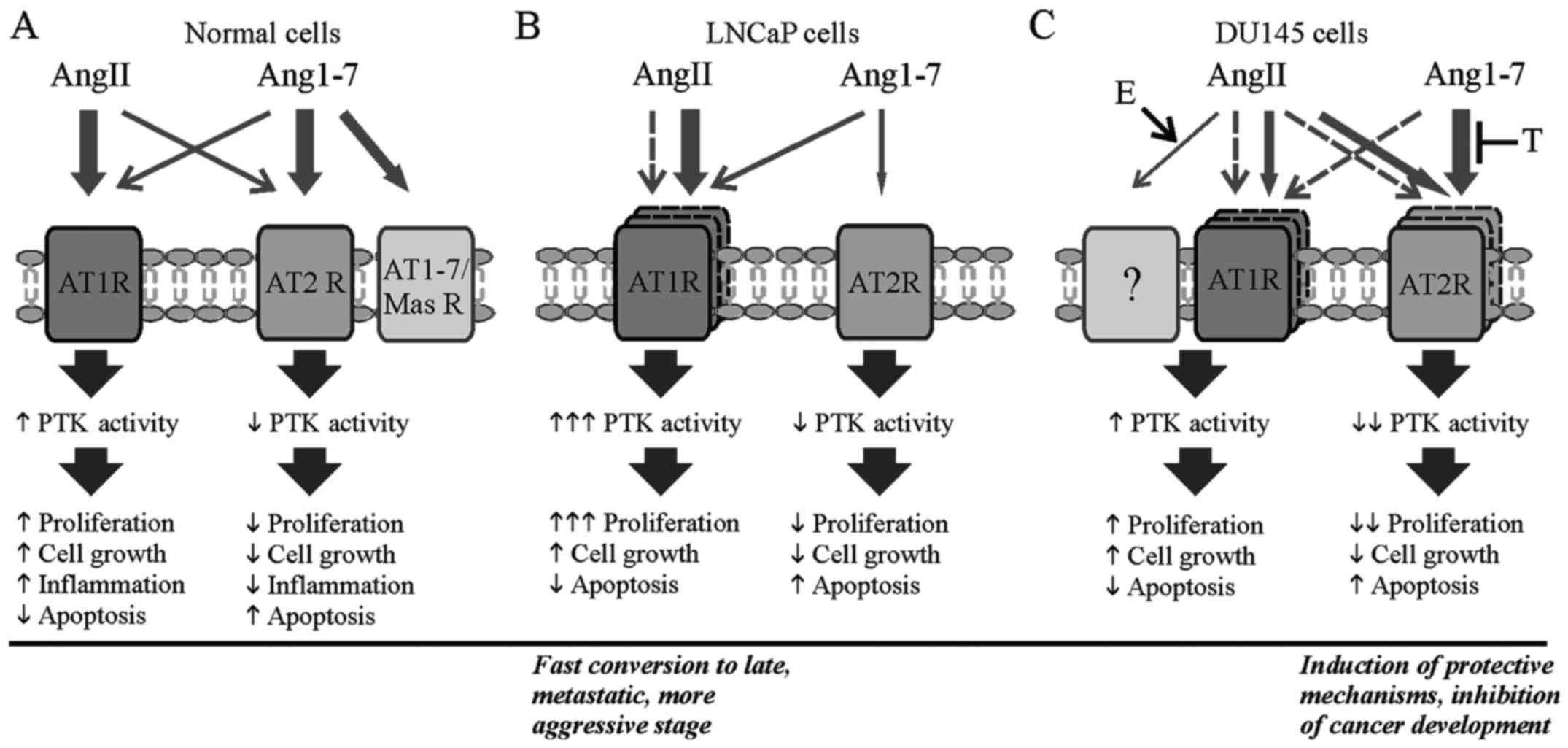
In conclusion, both our and cited results indicate
that local action of AngII and Ang1-7 on processes involved in
cancer progression depend on: the type of peptide, peptide
concentration, tissue, time of exposure and stage of cancer. In
late stage prostate cancer cell lines (DU145) an observed decrease
in protein kinase activity caused by both AngII and Ang1-7 may
protect against further development of cancer. This suggests that
treatment of RAS inhibitors for prostate cancer patients should be
prudent. Steroid hormones, testosterone and 17β-estradiol reverse
the action of angiotensins leading to adverse progression in
prostate cancer (Fig. 8).
Due to the high specificity of the described
interactions, further studies concerning the mechanism of action
are warranted and may be useful to explain advanced stage cancer
therapy failure. Further research is also warranted for the
development of new cancer therapies.
Acknowledgments
The present study was supported by the Medical
University of Lodz (Lodz, Poland; grant nos.
503/6-086-02/503-61-001; 502-03/0-078-04/502-04-030 and
502-03/6-086-02/502-64-109).
References
|
1
|
Uemura H, Hoshino K and Kubota Y:
Engagement of renin-angiotensin system in prostate cancer. Curr
Cancer Drug Targets. 11:442–450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
O'Mahony OA, Barker S, Puddefoot JR and
Vinson GP: synthesis and secretion of angiotensin II by the
prostate gland in vitro. Endocrinology. 146:392–398. 2005.
View Article : Google Scholar
|
|
3
|
Fujita M, Hayashi I, Yamashina S, Itoman M
and Majima M: Blockade of angiotensin AT1a receptor signaling
reduces tumor growth, angiogenesis, and metastasis. Biochem Biophys
Res Commun. 294:441–447. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Namsolleck P, Recarti C, Foulquier S,
Steckelings UM and Unger T: AT(2) receptor and tissue injury:
Therapeutic implications. Curr Hypertens Rep. 16:4162014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Haendeler J and Berk BC: Angiotensin II
mediated signal transduction. Important role of tyrosine kinases.
Regul Pept. 95:1–7. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McCarty MF: Targeting multiple signaling
pathways as a strategy for managing prostate cancer: Multifocal
signal modulation therapy. Integr Cancer Ther. 3:349–380. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rodrigues-Ferreira S and Nahmias C:
G-protein coupled receptors of the renin-angiotensin system: New
targets against breast cancer? Front Pharmacol. 6:242015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uemura H, Hasumi H, Ishiguro H, Teranishi
J, Miyoshi Y and Kubota Y: Renin-angiotensin system is an important
factor in hormone refractory prostate cancer. Prostate. 66:822–830.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moon JY: Recent update of
renin-angiotensin-aldosterone system in the pathogenesis of
hypertension. Electrolyte Blood Press. 11:41–45. 2013. View Article : Google Scholar
|
|
10
|
Santos RA, Ferreira AJ, Verano-Braga T and
Bader M: Angiotensin-converting enzyme 2, angiotensin-(1-7) and
Mas: New players of the renin-angiotensin system. J Endocrinol.
216:R1–R17. 2013. View Article : Google Scholar
|
|
11
|
Krishnan B, Torti FM, Gallagher PE and
Tallant EA: Angiotensin-(1-7) reduces proliferation and
angiogenesis of human prostate cancer xenografts with a decrease in
angiogenic factors and an increase in sFlt-1. Prostate. 73:60–70.
2013. View Article : Google Scholar
|
|
12
|
Krishnan B, Smith TL, Dubey P, Zapadka ME,
Torti FM, Willingham MC, Tallant EA and Gallagher PE:
Angiotensin-(1-7) attenuates metastatic prostate cancer and reduces
osteoclastogenesis. Prostate. 73:71–82. 2013. View Article : Google Scholar
|
|
13
|
Hsing AW, Tsao L and Devesa SS:
International trends and patterns of prostate cancer incidence and
mortality. Int J Cancer. 85:60–67. 2000. View Article : Google Scholar
|
|
14
|
Stone KR, Mickey DD, Wunderli H, Mickey GH
and Paulson DF: Isolation of a human prostate carcinoma cell line
(DU 145). Int J Cancer. 21:274–281. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun YH, Gao X, Tang YJ, Xu CL and Wang LH:
Androgens induce increases in intracellular calcium via a G
protein-coupled receptor in LNCaP prostate cancer cells. J Androl.
27:671–678. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Falkenstein E, Tillmann HC, Christ M,
Feuring M and Wehling M: Multiple actions of steroid hormones - a
focus on rapid, nongenomic effects. Pharmacol Rev. 52:513–556.
2000.PubMed/NCBI
|
|
17
|
Guo Z, Benten WP, Krücken J and Wunderlich
F: Nongenomic testosterone calcium signaling. Genotropic actions in
androgen receptor-free macrophages. J Biol Chem. 277:29600–29607.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yeh CR, Da J, Song W, Fazili A and Yeh S:
Estrogen receptors in prostate development and cancer. Am J Clin
Exp Urol. 2:161–168. 2014.PubMed/NCBI
|
|
19
|
Vlahovic G and Crawford J: Activation of
tyrosine kinases in cancer. Oncologist. 8:531–538. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hirano AA, Greengard P and Huganir RL:
Protein tyrosine kinase activity and its endogenous substrates in
rat brain: A subcellular and regional survey. J Neurochem.
50:1447–1455. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Domińska K, Piastowska-Ciesielska AW,
Lachowicz-Ochędalska A and Ochędalski T: similarities and
differences between effects of angiotensin III and angiotensin II
on human prostate cancer cell migration and proliferation.
Peptides. 37:200–206. 2012. View Article : Google Scholar
|
|
22
|
Rebas E, Zabczyńska J and Lachowicz A: The
effect of angiotensin 1–7 on tyrosine kinases activity in rat
anterior pituitary. Biochem Biophys Res Commun. 347:581–585. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alimirah F, Chen J, Basrawala Z, Xin H and
Choubey D: DU-145 and PC-3 human prostate cancer cell lines express
androgen receptor: Implications for the androgen receptor functions
and regulation. FEBS Lett. 580:2294–2300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Z, Liu L, Hou J, Wen D, Yan C, Pu J,
Ouyang J and Pan H: Rapid membrane effect of testosterone in LNCaP
cells. Urol Int. 81:353–359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McPherson SJ, Ellem SJ and Risbridger GP:
Estrogen-regulated development and differentiation of the prostate.
Differentiation. 76:660–670. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Negri-Cesi P, Colciago A, Poletti A and
Motta M: 5α-reductase isozymes and aromatase are differentially
expressed and active in the androgen-independent human prostate
cancer cell lines DU145 and PC3. Prostate. 41:224–232. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Piccolella M, Crippa V, Messi E, Tetel MJ
and Poletti A: Modulators of estrogen receptor inhibit
proliferation and migration of prostate cancer cells. Pharmacol
Res. 79:13–20. 2014. View Article : Google Scholar
|
|
28
|
Bonkhoff H and Berges R: The evolving role
of oestrogens and their receptors in the development and
progression of prostate cancer. Eur Urol. 55:533–542. 2009.
View Article : Google Scholar
|
|
29
|
Ho SM: Estrogens and anti-estrogens: Key
mediators of prostate carcinogenesis and new therapeutic
candidates. J Cell Biochem. 91:491–503. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hoshino K, Ishiguro H, Teranishi J,
Yoshida S, Umemura S, Kubota Y and Uemura H: Regulation of androgen
receptor expression through angiotensin II type 1 receptor in
prostate cancer cells. Prostate. 71:964–975. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baiardi G, Macova M, Armando I, Ando H,
Tyurmin D and Saavedra JM: Estrogen upregulates renal angiotensin
II AT1 and AT2 receptors in the rat. Regul Pept. 124:7–17. 2005.
View Article : Google Scholar
|
|
32
|
Hilliard LM, Sampson AK, Brown RD and
Denton KM: The 'his and hers' of the renin-angiotensin system. Curr
Hypertens Rep. 15:71–79. 2013. View Article : Google Scholar
|
|
33
|
Marchiani S, Tamburrino L, Nesi G,
Paglierani M, Gelmini S, Orlando C, Maggi M, Forti G and Baldi E:
Androgen-responsive and -unresponsive prostate cancer cell lines
respond differently to stimuli inducing neuroendocrine
differentiation. Int J Androl. 33:784–793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Uemura H, Ishiguro H, Nakaigawa N,
Nagashima Y, Miyoshi Y, Fujinami K, Sakaguchi A and Kubota Y:
Angiotensin II receptor blocker shows antiproliferative activity in
prostate cancer cells: A possibility of tyrosine kinase inhibitor
of growth factor. Mol Cancer Ther. 2:1139–1147. 2003.PubMed/NCBI
|
|
35
|
Ager EI, Neo J and Christophi C: The
renin-angiotensin system and malignancy. Carcinogenesis.
29:1675–1684. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ullén A, Farnebo M, Thyrell L, Mahmoudi S,
Kharaziha P, Lennartsson L, Grandér D, Panaretakis T and Nilsson S:
Sorafenib induces apoptosis and autophagy in prostate cancer cells
in vitro. Int J Oncol. 37:15–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brooks C, Sheu T, Bridges K, Mason K,
Kuban D, Mathew P and Meyn R: Preclinical evaluation of sunitinib,
a multi-tyrosine kinase inhibitor, as a radiosensitizer for human
prostate cancer. Radiat Oncol. 7:154–163. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ławnicka H, Potocka AM, Juzala A,
Fournie-Zaluski MC and Pawlikowski M: Angiotensin II and its
fragments (angiotensins III and IV) decrease the growth of DU-145
prostate cancer in vitro. Med Sci Monit. 10:BR410–bR413.
2004.PubMed/NCBI
|
|
39
|
Domińska K, Piastowska-Ciesielska AW,
Płuciennik E, Lachowicz-Ochędalska A and Ochedalski T: A comparison
of the effects of Angiotensin IV on androgen-dependent and
androgen-independent prostate cancer cell lines. J Renin
Angiotensin Aldosterone Syst. 14:74–81. 2013. View Article : Google Scholar
|
|
40
|
Brand TM, Iida M, Li C and Wheeler DL: The
nuclear epidermal growth factor receptor signaling network and its
role in cancer. Discov Med. 12:419–432. 2011.PubMed/NCBI
|
|
41
|
Montero JC, Seoane S, Ocaña A and
Pandiella A: Inhibition of SRC family kinases and receptor tyrosine
kinases by dasatinib: Possible combinations in solid tumors. Clin
Cancer Res. 17:5546–5552. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rebas E and Lachowicz-Ochedalska A: The
effect of angiotensin III on protein tyrosine kinase activity in
rat pituitary. Regul Pept. 130:14–18. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lewandowska U, Lachowicz-Ochędalska A,
Domińska K, Kaszewska D and Rębas E: Angiotensin II as a factor
modulating protein tyrosine kinase activity in two breast cancer
lines -MCF-7 and MDA-MB-231. Endokrynol Pol. 62:151–158. 2011.
|
|
44
|
Reis AB, Araújo FC, Pereira VM, Dos Reis
AM, Santos RA and Reis FM: Angiotensin (1–7) and its receptor Mas
are expressed in the human testis: Implications for male
infertility. J Mol Histol. 41:75–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Uemura H, Nakaigawa N, Ishiguro H and
Kubota Y: Antiproliferative efficacy of angiotensin II receptor
blockers in prostate cancer. Curr Cancer Drug Targets. 5:307–323.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chow L, Rezmann L, Catt KJ, Louis WJ,
Frauman AG, Nahmias C and Louis SNS: Role of the renin-angiotensin
system in prostate cancer. Mol Cell Endocrinol. 302:219–229. 2009.
View Article : Google Scholar
|
|
47
|
Pawlikowski M, Minias R, Sosnowski M and
Zieliński KW: Immu no histochemical detection of angiotensin AT 1
and AT 2 receptors in prostate cancer. Cent European J Urol.
64:252–255. 2011. View Article : Google Scholar
|
|
48
|
Waters C, Pyne S and Pyne NJ: The role of
G-protein coupled receptors and associated proteins in receptor
tyrosine kinase signal transduction. Semin Cell Dev Biol.
15:309–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chow L, Rezmann L, Imamura K, Wang L, Catt
K, Tikellis C, Louis WJ, Frauman AG and Louis SN: Functional
angiotensin II type 2 receptors inhibit growth factor signaling in
LNCaP and PC3 prostate cancer cell lines. Prostate. 68:651–660.
2008. View Article : Google Scholar : PubMed/NCBI
|