Introduction
Amyloid-β (Aβ) are peptides of 36–43 amino acids in
length, known to elicit neurodegenerative Alzheimer's disease (AD)
(1). The peptides are generated
by the proteolytic cleavage of the amyloid precursor protein by α-,
β- and γ-secretases (2,3). Monomeric Aβ is prone to the change
in its conformation to β-sheet-rich intermediate structures. The
intermediates interact with each other to form multimeric
aggregates, such as oligomers, protofibrils and fibrils (4–6).
The aggregated Aβ peptides are progressively deposited in the brain
parenchyma and cerebral blood vessels (7). However, the soluble Aβ oligomers and
protofibrils have been found to be more toxic than the deposited
fibrils, suggesting that the oligomeric aggregates would be the
main factor for AD (4,8).
An essential role of apoptosis in eliciting Aβ
cytotoxicity has been proposed, as caspases are activated in cells
treated with the peptide (9–12).
Caspase, a hallmark enzyme of apoptosis, is synthesized as a
zymogen that is activated by the apoptotic signal. The signal of
receptor-mediated apoptosis or the extrinsic apoptotic pathway
(EAPW) leads to processing and catalytic activation of caspase-8
through the formation of the death-inducing signaling complex
(DISC) with other proteins, such as Fas-associated protein with
death domain (FADD) (13). On the
other hand, chemically-induced apoptosis or the caspase-dependent
intrinsic apoptoticpathway (IAPW) activates caspase-9, which is
associated with the adaptor protein, apoptotic protease activating
factor-1 (Apaf-1), dATP and cytochrome c released from the
mitochondria to form mutiprotein complexes known as apoptosomes
(12–14). Activated caspases-8 and -9 then
process effector caspases, including caspases-3, -6 and -7, which
subsequently transduce the death signal by cleaving other proteins
(15,16). The EAPW and IAPW are the two major
apoptotic processes.
The binding of Aβ to receptors, such as DR4, DR5 or
p75 neurotrophin receptor can trigger the activation of the EAPW
(13,17,18), while impaired autophagic
degradation of the damaged mitochondria during aging (19) may lead to the the accumulation of
Aβ in the mitochondrial membrane in neurons followed by the release
of cytochrome c (19,20), which can trigger the activation of
the caspase cascade of IAPW (19). On the other hand,
caspase-4-mediated apoptosis is also induced by the unfolded
protein response signaling pathway when endoplasmic reticulum
stress is prolonged by the peptide (21). The differential activation of each
pathway depends on the proteins or factors that interact with Aβ
(22,23) and the conformational states of Aβ
(oligomer vs. fibril) (12,24,25).
Although a number of studies have established a
major role for apoptosis in Aβ-induced cell death, it is frequently
observed that caspase activation is not potent in Aβ-treated cells
(see Results section) (26).
Furthermore, our recent findings (27) indicated that caspase activation
and cell death induced by staurosporine (STS), commonly employed to
induce the activation of the IAPW, are significantly reduced by
Aβ42. The inhibitory effect of Aβ42 on the apoptotic pathway is
associated with its interaction with procaspase-9 and the
consequent inhibition of Apaf-1 apoptosome assembly. Similarly, it
is also possible that Aβ42 interacts with other proteins involved
in apoptosis and disrupts their functions, resulting in low levels
of caspase activation. However, based on the studies regarding
Aβ-induced apoptosis, we hypothesized that these inhibitory effect
can be overcome in such a way that caspases can be activated. In
the present study, we aimed to detect a condition under which the
inhibitory effect of Aβ on apoptosis is overcome and caspases are
activated, as well as to identify the apoptotic pathways involved
in this condition.
Materials and methods
Materials
Fetal bovine serum (FBS) was purchased from Life
Technology Inc. (Grand Island, NY, USA). Dulbecco's modified
Eagle's medium, high glucose (DMEM/HG) was obtained from Welgene
(Daegu, Korea). N-Acetyl-LEHD-amino methyl coumarin (Ac-LEHD-AMC),
Ac-IETD-AMC, Ac-VEID-AMC and Ac-DEVD-AMC were from A.G. Scientific
Inc. (San Diego, CA, USA). Anti-caspase-9 (cat. no. sc-7885),
anti-caspase-3 (cat. no. sc-56053), anti-β-actin (cat. no.
sc-47778) and goat anti-rabbit IgG-HRP (cat. no. sc-2030)
antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz,
CA, USA). Anti-caspase-8 antibody (cat. no. 9746) was from Cell
Signaling Technology (Dedham, MA, USA). Anti-DFF45 antibody (cat.
no. 611036) was from BD Transduction Laboratory (San Diego, CA,
USA). Anti-caspase-6 (cat. no. YF-PA10680), anti-FADD (cat. no.
YF-MA16512) and anti-cytochrome c (cat. no. LF-MA0182)
antibodies were from AbFrontier (Seoul, Korea). Anti-Bid antibody
was developed in laboratory. Recombinant mouse TNF-α (cat. no.
410-MT) was obtained from R&D Systems (Minneapolis, MN, USA)
and actinomycin D (cat. no. A1410) from Sigma (St. Louis, MO, USA).
All other chemicals were obtained from Sigma, unless otherwise
stated.
Preparation of Aβ peptide
Aβ42 was purified from a fusion protein with GroES
as previously described (28).
After drying, the purified peptide powder was dissolved in 100%
1,1,1,3,3,3,-hexafluoro-2-propanol which was removed by evaporation
under a fume hood and then under a vacuum. Nitrogen gas was added
to the resulting Aβ42 powder and it was stored at −20°C.
Immediately before use, the peptide was dissolved in 0.1% NH4OH at
a concentration of 2 mg/ml and then diluted at the desired
concentration with the cell culture media without FBS. Aβ42
oligomers were prepared by dissolution of the peptide powder in
cell culture media at 100 µM and incubation at 4°C for 12 h
as previously described (29). To
prepare the fibrils, the Aβ42 powder at 100 µM was dissolved
in phosphate-buffered saline (PBS) supplemented with 0.02% sodium
azide and incubated at 37°C for 4 days.
Cell culture and cell death assay
The human epithelial HeLa cells (30) were cultured in DMEM (HG) medium
supplemented with 10% (v/v) FBS and 1% antibiotics
(penicillin/streptomycin) at 37°C under 5% CO2. For cell
death assay, the cells were seeded at a density of 15,000
cells/well in 96-well plates (Nunc, Roskilde, Denmark), cultured
for 24 h, serum-deprived for a further 12 h and treated with Aβ42
for the indicated periods of time or STS (0.5 µM) for 6 h.
Cell viability was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide
formazan (MTT) reduction test (27), in which each well was supplemented
with 20 µl of 5 mg/ml MTT solution in PBS, incubated for 2 h
and then with 100 µl of a solubilization buffer [20% sodium
dodecylsulfate (SDS) solution in 50% (v/v) DMF (pH 4.7)] for 12-16
h. For the assessment of cell viability, alamarBlue assay (31,32) was also employed, in which 10
µl of alamarBlue (Life Technologies, Carlsbad, CA, USA) were
added directly to the cells and the mixture was incubated for 4–16
h. In the both assays, the absorbance was recorded at 570 nm using
a microplate reader Spectra Max 190 (Molecular Devices, Sunnyvale,
CA, USA).
Measurement of caspase activity
Caspase activity was measured as previously
described (27). In brief, 20,000
cells/well were seeded in a 96-well plate, cultured for 24 h, and
serum-starved for an additional 12 h. Following treatment with Aβ42
preparations or other damaging agents, the cells were washed twice
with ice-cold PBS. Subsequently, 40 µl of lysis buffer (20
mM HEPES-NaOH, pH 7.0, 1 mM ethylenediaminetetraacetic acid (EDTA),
1 mM EGTA, 20 mM NaCl, 0.25% Triton X-100, 1 mM dithiothreitol, 1
mM PMSF, 10 µg/ml leupeptin, 5 µg/ml pepstatin A, 2
µg/ml aprotinin, 25 µg/ml N-acetyl-Leu-Leu-Norleu-al)
were added to each well. The mixture was incubated on ice for 20
min. Caspase assay buffer (20 mM HEPES-NaOH, pH 7.0, 20 mM NaCl,
1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA and 10 mM DTT) and
substrates were then added, and the release of AMC was monitored
for 2 h at 2-min intervals at excitation and emission wavelengths
of 360 and 480 nm, respectively, using a microplate
spectrofluorometer Gemini-XS (Molecular Devices). The results were
expressed as the slope of total readings vs. time.
Western blot analysis
The cells were washed twice with ice-cold PBS and
resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl,
1% Triton X-100, 5 mM EDTA, 5 mM EGTA, 1 mM PMSF, 10 µg/ml
leupeptin, 2 µg/ml pepstatin A and 2 µg/ml aprotinin)
for 20 min on ice, as preivously desribed (27). The extracts were obtained by a
microfuge centrifugation of the lysed cells at 13,000 rpm at 4°C
for 30 min. They were subjected to SDS-polyacrylamide gel
electrophoresis (SDS-PAGE), and the proteins on the gel were
transferred onto polyvinylidene difluoride membranes. The membranes
were immunoprobed with primary antibodies and horseradish
peroxidase-conjugated secondary antibodies. The blots were
visualized using West-Zol plus reagent (Intron Biotechnology Inc.,
Seoul, Korea). Optical densities of immunoreactive bands were
quantified using NIH ImageJ software (obtained in http://rsb.info.nih.gov/ij/), if necessary.
Analysis of cytochrome c release
The cells were washed twice with ice-cold PBS and
resuspended in digitonin buffer (75 mM NaCl, 1 mM
NaH2PO4, 8 mM Na2HPO4,
250 mM sucrose, and 190 µg/ml digitonin). After 5 min on
ice, the supernatants and pellets were obtained by microfuge
centrifugation at 14,000 rpm at 4°C for 5 min. The supernatants
were transferred to fresh tubes containing protease inhibitors (0.1
mM PMSF, 1 mM EDTA, 10 µg/ml leupeptin, 5 µg/ml
pepstatin A, 2 µg/ml aprotinin) and the pellets were
resuspended in a buffer containing 25 mM Tris-HCl (pH 8.0) and 1%
Triton X-100. 30 µg of protein from each sample was
subjected to western blot analysis for the examination of the
levels of cytochrome c as previously described (27).
Size exclusion column chromatography
(SEC)
The formation of the apoptosome and DISC were
determined by SEC analysis of the cell extracts as previously
described (33,34). For DISC formation study, cells
were treated with 100 µg/ml of actinomycin D for 2 h
followed by 50 nM of TNF-α for a further 24 h. The cell extracts
were prepared as follows: The harvested cells were washed with
ice-cold PBS twice and lysed in a lysis buffer containing 20 mM
HEPES-KOH, pH 7.5, 0.5 mM EDTA, 0.5 mM EGTA, 10 mM KCl, 1 mM
β-mercaptoethanol, 0.1 mM PMSF, 10 µg/ml leupeptin, 5
µg/ml pepstatin A, 2 µg/ml aprotinin and 25
µg/ml ALLN. The cell extracts were obtained by centrifuging
the lysed cells at 13,000 rpm for 1 h at 4°C. For SEC, cell
extracts were loaded onto a Superose 6 HR (10/30) column (Amersham
Pharmacia Biotech, Uppsala, Sweden) and 0.5 ml fractions were
collected. Proteins in the fractions were concentrated using a
10-kDa cut-off membrane filter (Merck Millipore, Billerica, MA,
USA) and analyzed by western blot analysis. The column was
calibrated using calibration kits (Amersham Pharmacia Biotech)
containing thyroglobulin (669 kDa), ferritin (440 kDa), catalase
(232 kDa) and aldolase (158 kDa).
Reconstitution of the cell-free apoptotic
process
Cell-free apoptotic processes were induced by
incubating cell extracts (70–1,000 µg) with 1 mM dATP, and
1–10 µM cytochrome c or purified caspases in a buffer
containing 20 mM HEPES-NaOH (pH 7.0), 20 mM NaCl, 1 mM EDTA, 1 mM
EGTA, 1.5 mM MgCl2 and 10 mM DTT at 30°C as previously
described (27). Cell extracts
were obtained as follows: The harvested cells were washed twice
with ice cold PBS, resuspended in the lysis buffer shown in the SEC
section on ice and douncehomogenized with 40–50 strokes. Cell
extracts were obtained from supernatants following microfuge
centrifugation of the disrupted cells at 13,000 rpm for 1 h at
4°C.
Construction and purification of caspases
and Bid
Caspases-3 and -6, prodomainless equivalent of the
p41/43 fragment of caspase-8 (p18-10) (see Results), and Bid were
prepared as previously described (27). Prodomainless caspase-8 double
mutant (D374A/D384A) mimicking the p30 fragment of caspase-8 (DM)
(see Results) was constructed by point mutations using
site-directed mutagenesis by polymerase chain reaction and
DpnI-mediated cleavage methods (27,30). In the polymerase chain reaction,
the template was caspase-8 cloned in the pET15b bacterial
expression vector (Novagen, Darmstadt, Germany) and the primers
were 5′-ATACC TGTTG AGACT GCTTC AGAGGA GCAA-3′ (sense)
and 5′-TTGCT CCTCT GAAGC AGTCT CAACA GGTAT-3′
(antisense) for D374A; 5′-TATTT AGAAA TGGCT TTATC ATCAC CTCAA-3′
(sense) and 5′-TTGAG GTGAT GATAA AGCCA TTTCT AAATA-3′
(antisense) for D384A. The underlined sequences refer to the
incorporated alanine sequence in each case. The mutated plasmid was
confirmed by sequencing both strands. The recombinant caspase was
expressed in a BL21 pLys Escherichia coli strain and
purified as previously described (27). Aliquots of the purified proteins
were stored at −80°C until use. The amount of protein was measured
through Bradford assay according to the instructions provided by
the manufacturer (Sigma).
Results
Single treatment with Aβ42 induces the
minimal activation of caspases-3, -8 and -9
We initially sought an experimental condition under
which caspase would be robustly activated in Aβ-treated cells to
probe the Aβ-induced apoptotic pathway. To achieve this, we sought
to use Aβ-treatment methods that result in the activation. In this
study, we examined the activities of caspases-3, -8 and -9, which
participate in the IAPW and EAPW. Their activities were measured
using three synthetic substrates: Ac-DEVD-AMC, Ac-IETD-AMC and
Ac-LEHD-AMC for caspase-3, -8 and -9, respectively. The cells were
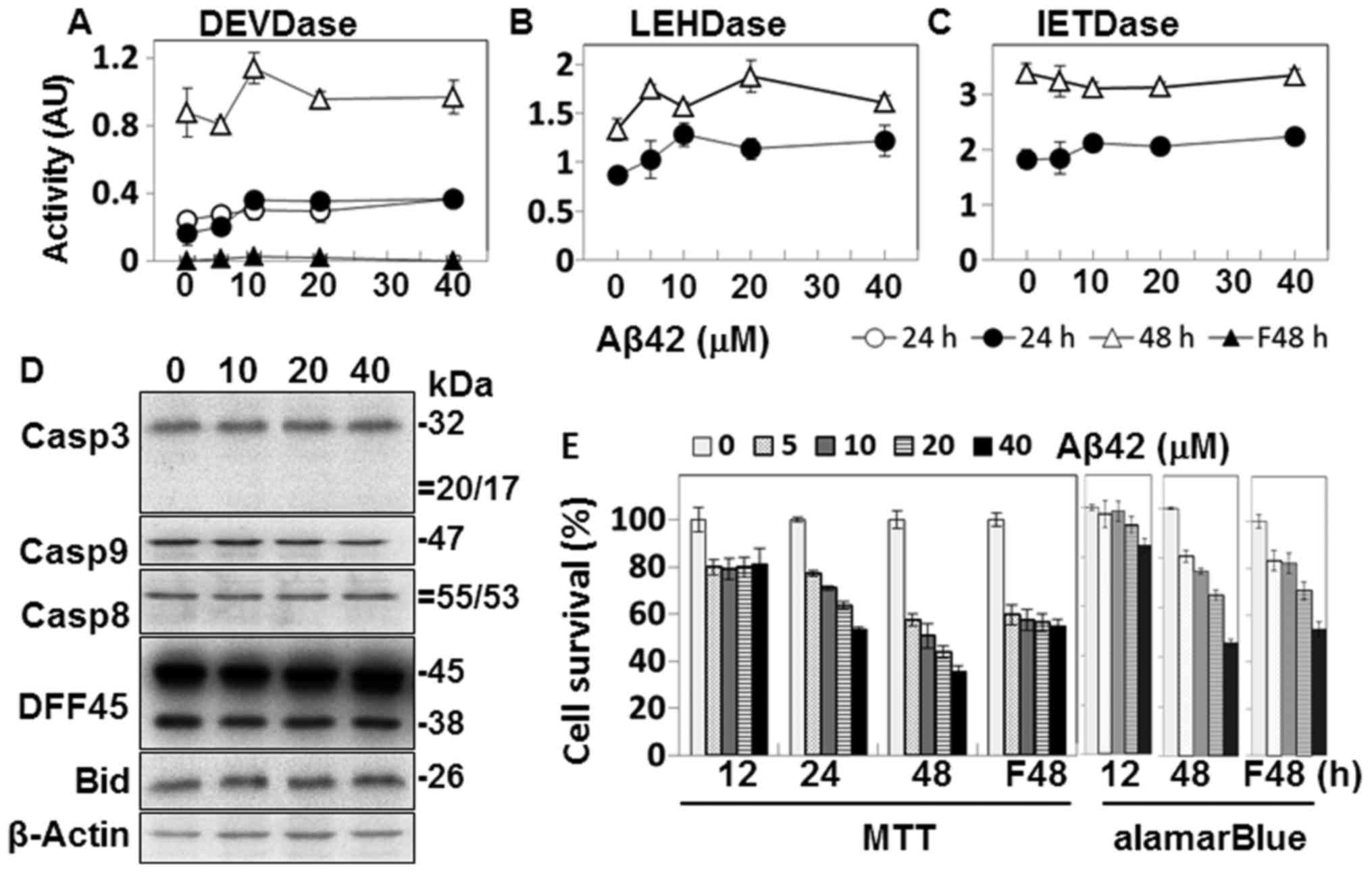
incubated with up to 40 µM Aβ42 for up to 48 h (Fig. 1). The oligomeric preparation of
Aβ42 was mainly used in the present study, as it is superior to the
monomer or fibrillar form in inducing caspase activity and cell
death (4,8). Unless otherwise indicated, Aβ42
refers to the oligomeric preparation of the peptide in the
following experiments. In this experiment, we employed human
epithelial HeLa cells that exhibited a relatively higher caspase
activity than other cell lines, such as human neuroblastoma SH-SY5Y
cells (26) in which death
occurred too readily upon Aβ treatment and the levels of activated
caspase were often varied (data not shown).
Some levels of caspase activity on the synthetic
substrates were detected in the Aβ42-treated cells, particularly
after 48 h of incubation (Fig.
1A–C). The activity levels were lower by several folds than
those induced by other damaging agents, such as STS (Fig. 7A and B) (26). Although a number of studies,
including ours have indicated the involvement of apoptosis and
accompanying caspase activation in Aβ-induced cell death (9–12,26), low activities of caspases are
observed, as shown in Fig. 1.
Moreover, we were not convinced that even the low activity was
caused by Aβ42-induced damage, as no Aβ42-concentration-dependent
activity was observed.
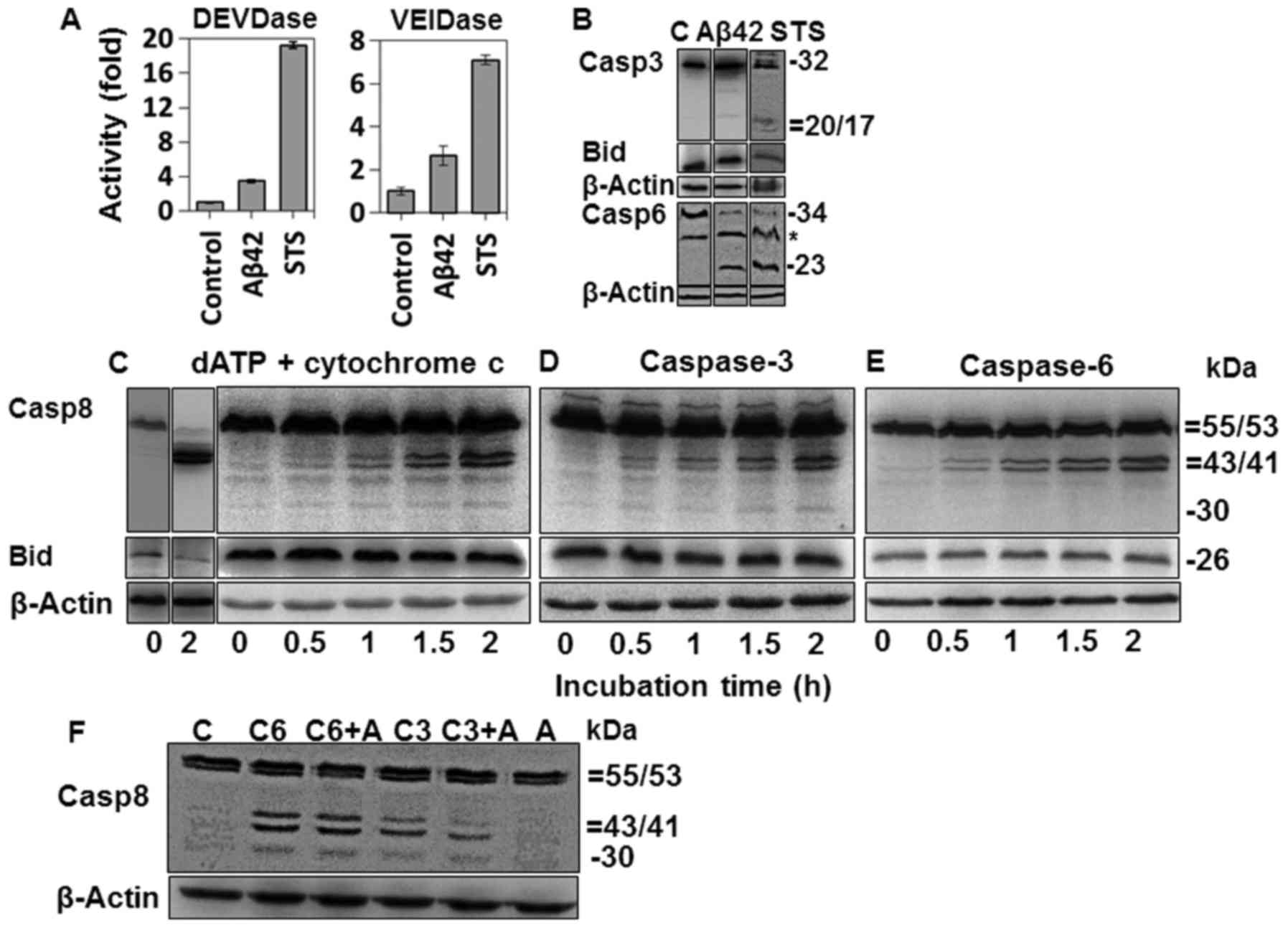 | Figure 7Activation of caspases-3 and -6 in
amyloid-β (Aβ)42-doubly-treated or staurosporine (STS)-treated
cells and fragmentation of procaspase-8 in cell-free apoptotic
process. (A) Activities of caspases-3 and -6 were measured for
cells treated with 20 µM Aβ42 for 2+22 h or with 0.1 mM STS
for 6 h, using 10 µM DEVD-AMC and 50 µM VEID-AMC as
substrates. Activity is indicated by fold increase over that of the
control and expressed as mean ± standard deviation of 3 independent
experiments. DEVDase data of the Aβ42 group was extracted from
Fig. 2A. (B) Cleavages of
procaspase-3, bid, and procaspase-6 were probed by western blot
analysis for the cells treated as described in (A). *, Denotes a
nonspecific band. C, indicates untreated control. (C–E) Cell
extracts were incubated with cytochrome c (10 µM for
lane 2 and 1 µM for 3–7 lanes) and 1 mM dATP (C), purified
caspase-3 (30 ng) (D) or purified caspase-6 (50 ng) (E) for the
indicated durations and analyzed by western blot analysis for
caspase-8 and bid. (F) Cell extracts (70 µg) were incubated
with purified caspase-6 (50 ng) or caspase-3 (30 ng) in the
presence or absence of 20 µM Aβ42, and analyzed for
procaspase-8 cleavage as in Fig.
6C–E. (B-F) β-actin was used as a loading control. The relative
sizes are indicated at the right side of the figures. The results
are a representative of at least 3 independent experiments. Lane 1
in (B–E) and in lane C of (F) are a control sample prepared from
cell extracts incubated without cytochrome c/dATP or
caspases. C3, C6 and A, indicate caspase-3, caspase-6 and Aβ42,
respectively. Casp, caspase. |
The activation of caspases and the processing of
DFF45 and Bid, substrates of caspases-3 and -8, respectively, were
further explored in the Aβ42-treated cells by western blot
analysis. Cleaved fragments or reduction in levels of the
pro-proteins was expected if they are activated (Fig. 2). No such fragment or reduction
was detected by western blot analysis (Fig. 1D). Taken together, our data show
no evidence of caspase activation in the cells treated with Aβ42
under the experimental conditions described above.
Aβ42 cytotoxicity was examined to confirm that the
low level of caspase activation was not due to faulty peptide
preparation. Cell death was mainly assessed by MTT assay (27). However, soluble Aβ can lead to a
decrease in MTT formazan production in the absence of overt cell
death when cells are incubated for longer durations (35). Thus, the alamarBlue assay was also
performed to complement the cell death experiments. Slight
reductions in MTT formazan production were observed in samples
after 12 h incubation (Fig. 1E).
However, it is questionable whether cell death occurred in the
samples, because they showed no Aβ42-dose dependency and the levels
of cell death in these samples were barely decreased in the
alamarBlue assay. On the contrary, in samples of 24- and 48-h
incubation, MTT formazan production was reduced in an
Aβ42-dose-dependent manner (Fig.
1E). Consistent with this, reduction in alamarBlue was observed
in the 48-h sample, although the levels were less than those of the
MTT assay (Fig. 1E). Based on the
results of the 24- and 48-h samples, we concluded that the Aβ42
peptide used herein was cytotoxic.
It has been reported that Aβ fibrils can induce the
activation of the EAPW, which leads to activation of caspase-8 and
subsequently, caspase-3 (12). In
the present study, caspase-3-like DEVDase activity was not detected
in the fibrillar Aβ42-treated cells (Fig. 1A). MTT formazan production was
reduced in the cells treated with the fibrillary form of Aβ42, but
it was not Aβ42-dose-dependent (Fig.
1E). It seems that the ambiguous results were caused by the
non-specific reduction of MTT formazan production. On the contrary,
the Aβ-dose-dependent reduction was clearly observed in the
alamarBlue assay on the same samples, suggesting that
Aβ42-dose-dependent cell death occurred (Fig. 1E). Taken together, these data
suggest that the fibrillar form of Aβ42 led to cell death in which
caspase-dependent apoptosis played a minimal role in the process.
In the following experiments, fibrillar Aβ42 was not used, due to
the lack of caspase activation in the peptide-treated cells
(Fig. 1A).
Double treatment with Aβ induces potent
caspase activation
As Aβ42-dependent caspase activation was not
prominent under the above-mentioned treatment conditions, we sought
other conditions for the robust activation of the enzyme. We
previously observed that the 'double' treatment of the peptide
could lead to higher levels of caspase activation (27). Others have reported that
nucleation-dependent polymerization in the doubly treated cells was
essential for Aβ-mediated cell death (31,36). Thus, we hypothesized that the
polymerization process in the doubly treated samples may provide a
different or stronger signal to activate caspases. In the present
study, we systematically tested the double treatment method using
different combinations of conditions to find an optimal condition
under which caspases could be potently activated. After testing
different time points, a 2-h initial treatment of Aβ42 at the
indicated concentrations was selected, as it resulted in the
highest caspase activity (other time point data not shown). At the
end of the first incubation, culture medium containing the peptide
was removed, and subsequently, the cells were further incubated
with a new preparation of Aβ42 peptide at the same concentration
for 10, 22, 34 or 46 h (2+10 h, 2+22 h, 2+34 h, or 2+46 h sample,
respectively). It is noted that in a previous study (36), cells were treated with the
fibrillar form of Aβ42 for 1 h followed by treatment with soluble
Aβ42; however, in this study, we used only soluble oligomeric Aβ42,
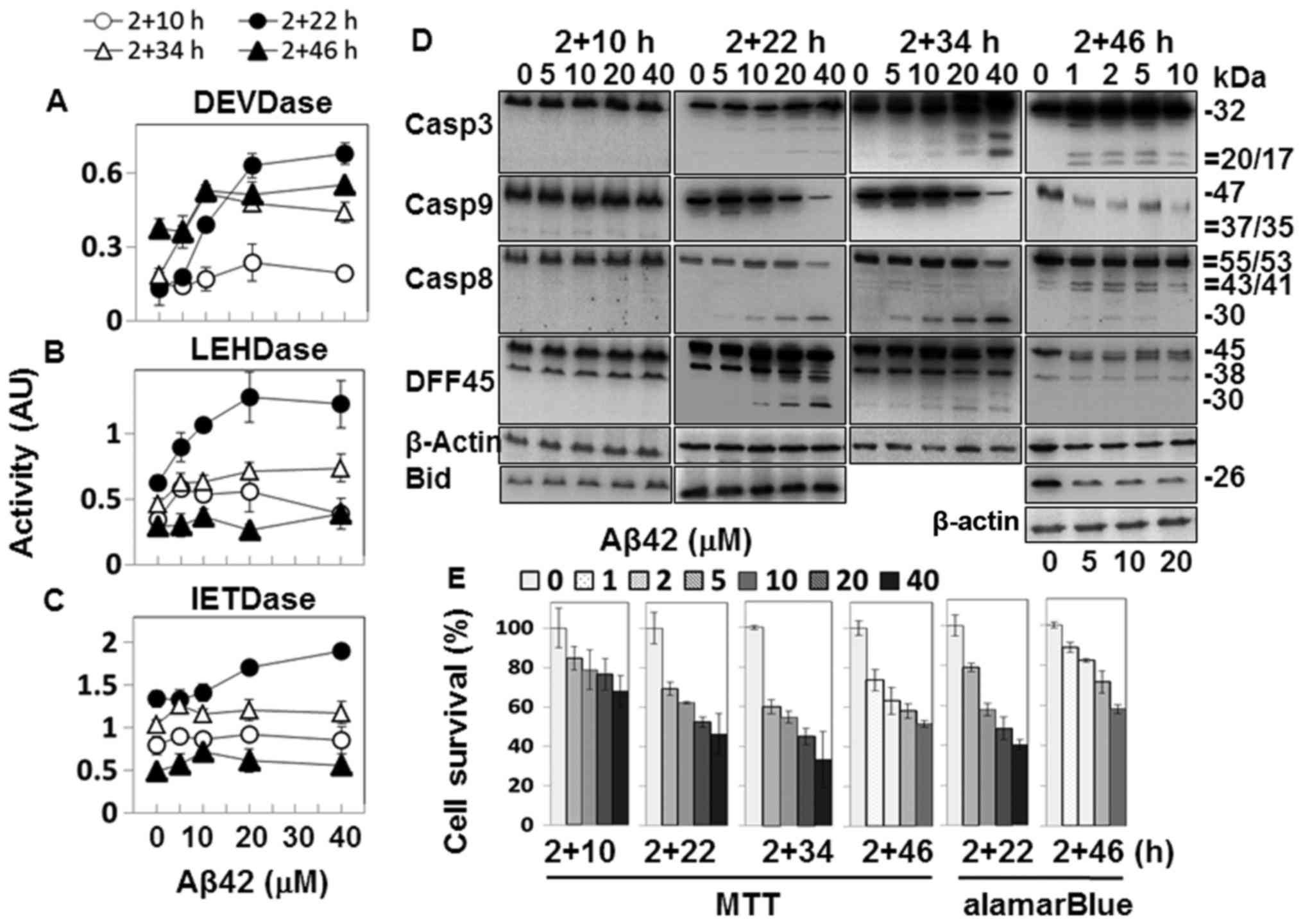
as this resulted in robust caspase activation (Fig. 2A–C).
The samples with 2+22 h and longer incubation
durations showed caspase-3-like DEVDase activity in an
Aβ42-dose-dependent manner (Fig.
2A). A processed fragment of ~20 kDa was detected in the 2+22 h
and 2+34 h samples treated with high concentrations of Aβ42, and
the fully processed ~17 kDa fragment was recognized in the 2+46 h
samples in addition (Fig. 2D).
Although the band intensity of the processed fragment was the
strongest at 2+34 h, and the additional ~17-kDa fragment was
present in the 2+46-h samples (Fig.
2D), DEVDase activities of these samples were similar to that
of the 2+22 h sample (Fig. 2A),
indicating that the activity and the level of observed fragment
processing were not closely correlated. A reduction in DFF45, a
substrate of caspase-3, was also observed (Fig. 2D), confirming the catalytic
activation of caspase-3.
The Aβ42-dose-dependent caspase-9-like LEHDase was
prominently activated in the 2+22 h sample, and to a lesser degree,
in the samples with longer incubation durations (Fig. 2B). The levels of procaspase-9 were
decreased in the 2+22 h sample and in samples with longer
incubation durations treated with high doses of Aβ42 (Fig. 2D). These data support the
activation of caspase-9. The processing of procaspase-3 (Fig. 2D), the substrate of caspase-9,
further supports catalytic activation of caspase-9. Cleaved
products of ~35/37 kDa, detected in the analysis of apoptosome
(Fig. 4C), were not detected in
the samples (Fig. 2D), possibly
due to insufficient intensity of the immunoblot signals.
Caspase-8-like IETDase activity was also the highest
in the 2+22 h sample, although the correlation between Aβ42-dose
and the activity was weak (Fig.
2C). Procaspase-8 was processed in the 2+22 h sample and
samples with longer incubation durations, but two different types
of fragments were detected depending on the incubation duration
(Fig. 2D). In the 2+22 h samples,
a fragment of ~30 kDa (p30) was largely detected, while the typical
41/43 kDa fragments (p41/43) were detected in the 2+46 h samples.
p30 is a fragment of procaspase-8 lacking the prodomain (37), while the p41/43 fragment contains
the prodomain and the large domain which becomes the large subunit
when caspase-8 is fully maturated. Thus, the cleavage sites to form
the two different fragments are distinct (37).
In order to determine the cytotoxicity of the Aβ42
peptide preparation, cell death was assessed in the double
treatment samples. All the samples tested showed
Aβ42-dose-dependent cell death (Fig.
2E). The levels of cell death evaluated by MTT-formazan
reduction were generally higher than those by alamarBlue assay
(2+22 h and 2+46 h samples of MTT vs. those of alamarBlue), as seen
in the single treatment samples (Fig.
1E). Reductions in the values were slightly higher in the
double treatment samples than in the equivalent single treatment
samples (i.e. 2+22 vs. 24 h). The single and double treatment
samples generally exhibited similar levels of cell death.
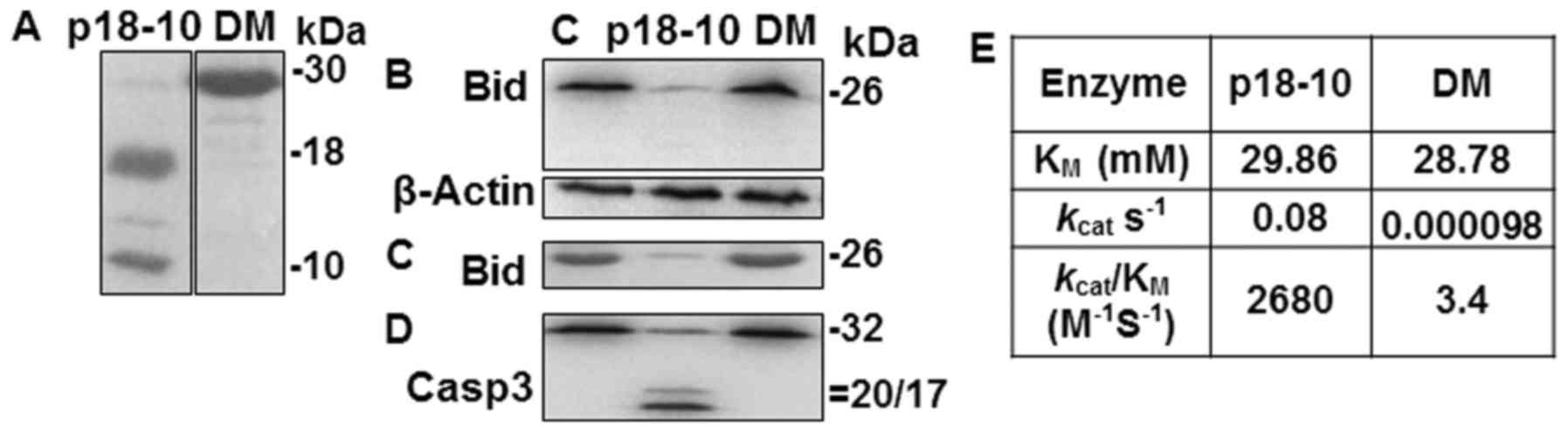
Catalytic activity of p30 and p41/43 on
Bid processing
Bid, a substrate of active caspase-8 (38), was not cleaved in cells
transfected with a plasmid encoding a p30-equivalent enzyme
(39) and the catalytic activity
of the purified p30-equivalent enzyme was weak (39–41). In the 2+22 h samples which
contained p30, the immunoblot signal intensity of Bid was similar
to that of control samples without Aβ42 treatment (Fig. 2D). Thus, it is reasonable to
assume that p30 cannot cleave Bid. To confirm this, we determined
whether p30 would process Bid, using a purified prodomainless
caspase-8 double mutant protein mimicking p30 (DM) (see Materials
and methods) (Fig. 3A) (42). The catalytic activity was compared
with that of the enzyme containing the prodomainless equivalent of
p41/43 (p18-10) (Fig. 3A), which
is known as the major enzyme for Bid processing (43). Purified p18-10 was able to process
procaspase-3 and Bid from crude cell extracts and purified Bid as
expected, while DM exhibited inefficient processing (Fig. 3B–D). The kcat of
DM on IETD-based synthetic substrate was ~800-fold lower than that
of p18-10 (Fig. 3E), indicating
that the lower activity of DM was due to the slower catalytic
velocity, consistent with the previous studies (39,40,42).
Bid reduction was observed in the cells incubated
for 2+46 h with Aβ42 at concentrations higher than 5 µM
(Fig. 2D). The timing of p41/43
generation correlated well with the reduction in Bid, indicating
that catalytic activation of caspase-8 was responsible for Bid
processing, as reported previously (38). It was noted that production of
p41/43 and Bid reduction were observed after activity of the
caspase-3 (substrate of caspases-8 and -9)-like DEVDase reached its
highest level (2+46 h samples) (Fig.
2A).
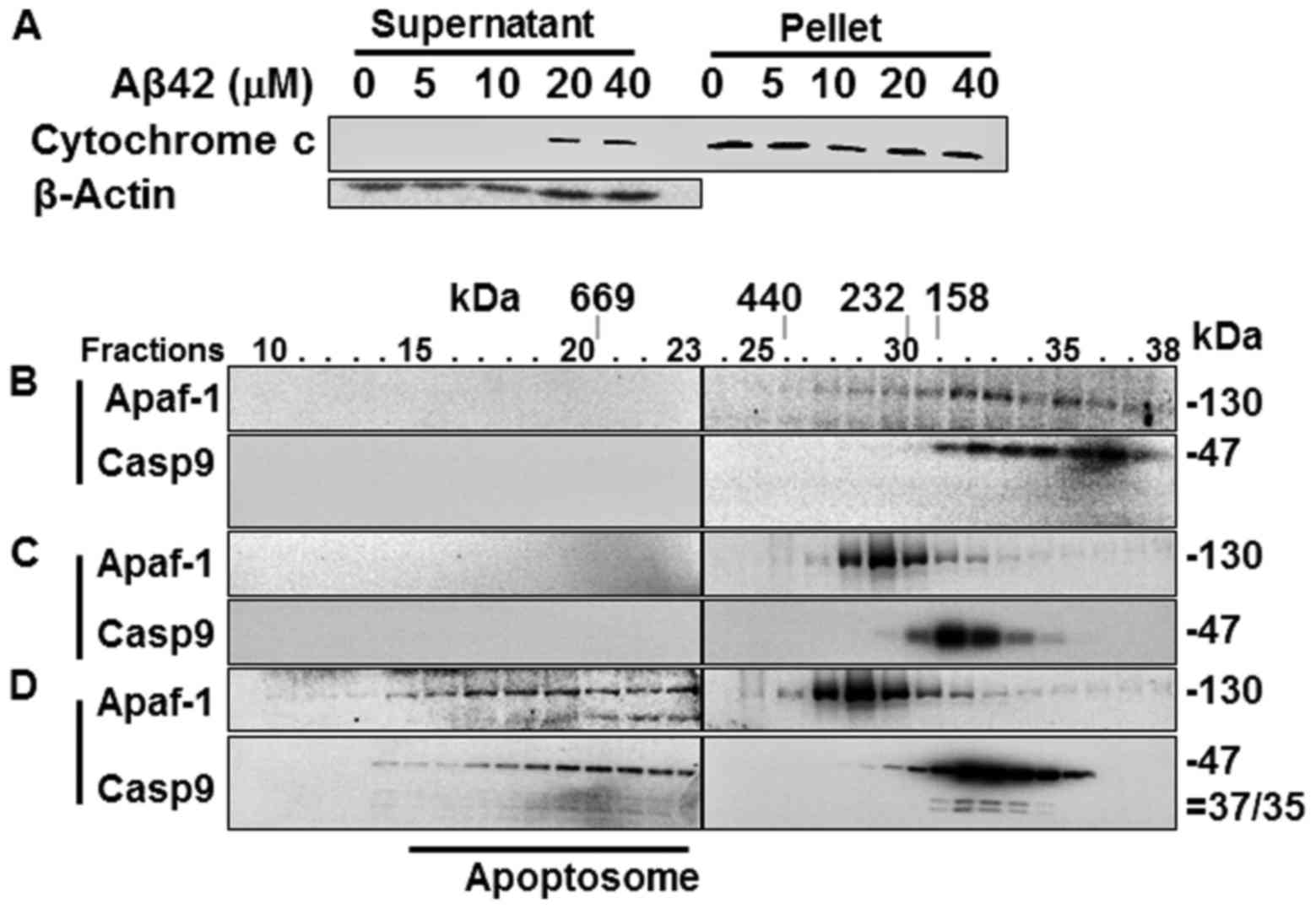
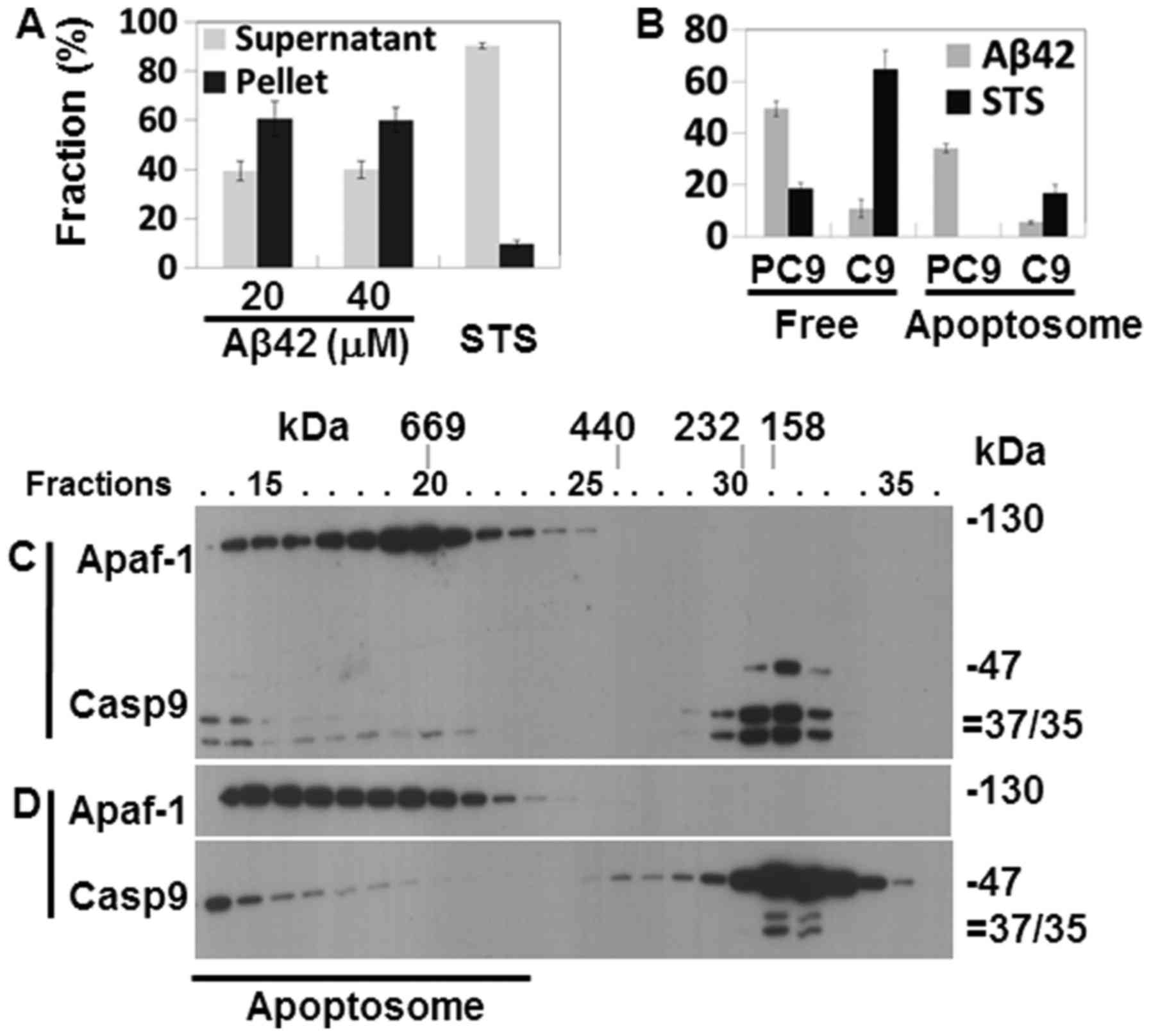
Cytochrome c release from the
mitochondria and the formation of the Apaf-1 apoptosome in the
Aβ42-doubly-treated cells
Catalytic activity and the processing of caspases-9
and -3 in the Aβ42-doubly-treated cells (Fig. 2A–D) suggest that the IAPW was
activated in the cells. To confirm the activation of the pathway,
cytochrome c release from the mitochondria was probed in the
2+22 h samples that showed potent activation and processing of
caspase-3, and reduction of procas-pase-9 (Fig. 2A–D). cytochrome c was
detected in the soluble fraction of cell extracts prepared from the
samples treated with 20 and 40 µM Aβ42, evidencing the
release of the protein in corresponding cells (Fig. 4A). Subsequently, the formation of
apoptosome, a hallmark of the IAPW, was examined in the 2+22 h
sample treated with 20 µM Aβ42. Apaf-1 and caspase-9 were
detected only in the later fractions (>25) of SEC in the control
sample prepared from Aβ42-non-treated cells (Fig. 4B) or cells treated once with Aβ42
for 24 h (Fig. 4C), whereas they
were detected in the earlier fractions (~15–23) in the sample
treated with 20 µM of Aβ42 for 2+22 h (Fig. 4C). These data clearly indicated
the formation of apoptosome in the 2+22 h sample. Thus, we
concluded that IAPW was activated in the cells. It was noted that
the 35/37 kDa fragments of caspase-9, which were not detected in
the total lysate (Fig. 2D), were
clearly detected in the western blot analysis following SEC
(Fig. 4C). They were detected in
this analysis, probably since the protein was concentrated during
chromatography.
The Aβ42-induced activation of the IAPW was compared
with the previous results obtained from the cells treated with STS,
which has been employed as an IAPW-inducing agent (27). The levels of cytochrome c
recovered in the supernatant fraction were low (~40% of total
cytochrome c protein) in the 2+22 h sample treated with 20
or 40 µM of Aβ42 (Fig.
4A), compared with that of STS-treated cells (~90%) (Fig. 5A). Cytochrome c is an
essential constituent of apoptosome. Thus, the reduced level of
released cytochrome c by Aβ42 compared to STS could
negatively influence robust activation of IAPW (see
Discussion).
The apoptosome formed in the sample treated for 2+22
h with 20 µM Aβ42 (Fig.
4D) also differed from that of the STS-treated cells in terms
of unprocessed and processed caspase-9 abundance in the apoptosome
complex. Unprocessed procaspase-9 (47 kDa) comprised the majority
(~34% of total caspase-9 protein) of caspase-9 (~40%) bound to the
protein complex formed in the 2+22 h Aβ42 treated sample (Fig. 5B), whereas only processed
caspase-9 (35/37 kDa) was detected in the complex of STS-treated
cells (17% of total caspase-9 protein) (Fig. 5B). Consistently, the level of
processed caspase-9 was much lower in the 2+22 h samples than in
STS-treated cells (11 vs. 65%) (Fig.
5B). Cell-free experiments were performed as an attempt to
understand the differential caspase-9 processing in the two samples
(STS-treated cells vs. 2+22 h sample treated with Aβ42). Apoptosome
formed in the cell lysate treated with dATP and cytochrome c
contained only processed 35/37 kDa caspase-9 as that formed in the
STS-treated cells (Fig. 5C),
whereas the apoptosome formed by dATP and cytochrome c in
the presence of 20 µM Aβ42 contained only unprocessed
procaspase-9 (Fig. 5D). These
data clearly indicate that Aβ42 inhibited procaspase-9 processing
in the apoptosome. It is noted that more caspase-9, processed or
unprocessed, was detected in apoptosome of the 2+22 h sample (~40%)
than in STS-treated cells (~17%), implying that suppressive effect
of Aβ42 on the formation of apoptosome by binding to procaspase-9
(27) was overcome. Taken
together, it seems that IAPW may not be fully activated by Aβ42 as
substantiated by inefficient catalytic processing of procaspase-9
(and reduced levels of released cytochrome c) which could be
the cause of the low response (such as caspase activation) of cells
to the peptide (Fig. 2).
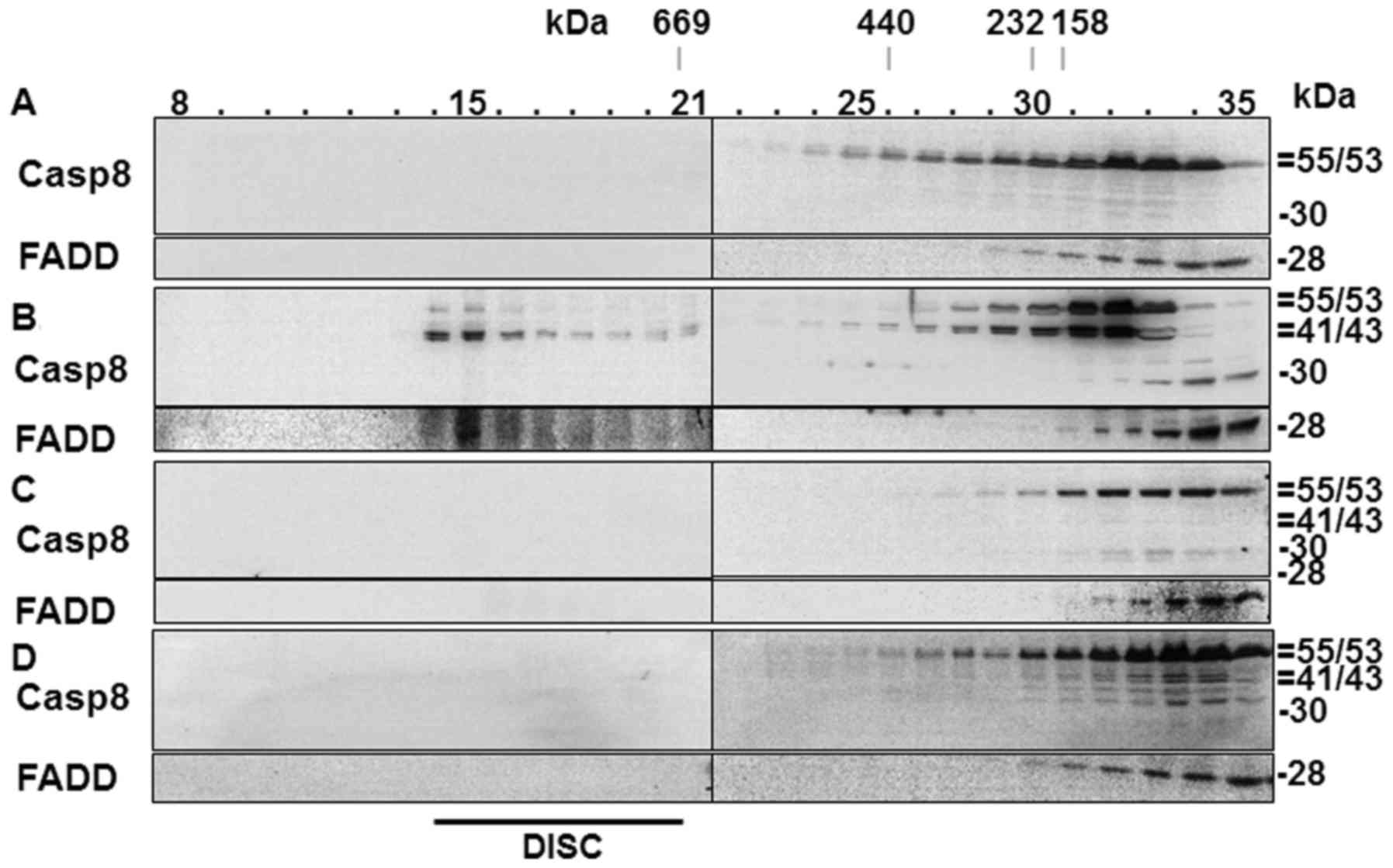
DISC is not formed in Aβ42-treated
cells
A number of studies have indicated that EAPW plays a
crucial role in the apoptotic process induced by Aβ42 (13,14,44). In the present study, we observed
activation of caspase-8 and Bid processing (Fig. 2D), which are hallmarks of EAPW
activation in the type II cells such as HeLa cells used in the
present study (45). Another
hallmark of the pathway is the formation of DISC (34,37), which has not been shown in
Aβ42-treated cells previously. We probed formation of DISC in the
Aβ42-doubly-treated cells in the present study to confirm its
involvement in the cells. In the SEC analysis of control cells
which were not treated with any cell death agent, procaspase-8 and
FADD proteins were detected only in the later fractions (>22)
(Fig. 6A), while fractions ~14–21
contained the two proteins in a positive control sample prepared
from cells treated with actinomycin-D followed by tumor necrosis
factor-α (TNF-α) (Fig. 6B). The
two proteins recovered in the early fractions (~14–21) are
components of DISC (13).
In the cells incubated for 2+22 h with 40 µM
Aβ42, which showed the highest levels of DEVDase, LEHDase and
IETDase activities, and the production of processed caspases
(Fig. 2A–D), procaspase-8 and
FADD were detected only in the later fractions (>22) (Fig. 6C), indicating that DISC was not
formed in those samples. DISC formation was further examined in the
cells incubated for 2+46 h with 10 µM Aβ42, in which the
generation of the p41/43 caspase-8 fragment, Bid reduction, and
IETDase activation were evident (Fig.
2A–D). DISC was also not detected in these cells (Fig. 6D). Thus, we hypothesized that the
formation of p41/43 and Bid processing in the cells incubated for
2+46 h with 10 µM Aβ42 were through a pathway other than
EAPW. Higher concentrations (>20 µM) of Aβ42 could not be
employed in the experiment with 2+46 h incubation to probe the
presence of DISC by SEC, as the cells were so severely damaged that
it was difficult to obtain a sufficient amount of protein from
these cells (data not shown). It was noted that p30 was present in
the 2+22 h sample (Fig. 6C),
consistent with that observed in Fig.
2D, while p41/43 was additionally detected in the 2+46 h sample
(Fig. 6D). These data confirm
that the formation of the p30 fragment of caspase-8 precedes that
of the p41/43 fragment.
Fragmentation of procaspase-8 into p30
and p41/43 in the cell-free apoptotic process
Since DISC was not formed in the cells doubly
treated with Aβ42 (Fig. 6C and D)
and that caspase-3 activation preceded caspase-8 activation and Bid
cleavage (Fig. 2A–D), we
speculated that caspases-3 and -6 activated in the IAPW (46–48) is responsible for the formation of
p30 and p41/43. Caspase-3 was activated in the 2+22 h sample with
20 µM Aβ42 as shown in Figs.
2A and D, and 6A and B.
Caspase-6 activation in the cells was verified by catalytic
activation with a VEID-based synthetic substrate and the production
of a ~23-kDa caspase-6 fragment (Fig.
7A and B).
To confirm and characterize the fragmentation of
procas-pase-8 in the IAPW, we employed a cell-free experimental
approach, in which cell extracts were incubated with 1 mM dATP and
10 µM cytochrome c, leading to the formation of the
Apaf-1 apoptosome (27) or with
purified caspases-3 and -6. As expected, procaspase-8 and its
substrate Bid were processed in the samples containing dATP and
cytochrome c, but only p41/43 was detected (Fig. 7C, second lane). The p30 fragment
was seen in the following experiments in which a lower dose (1
µM) of cytochrome c was used (Fig. 7C, lanes 3–7). In these samples,
procaspase-8 processing was incomplete, suggesting that p30 might
exist only in the early stage of apoptosis, consistent with the
results of Figs. 2D and 5C and D.
We added purified caspases-3 and -6 to the prepared
cell extract, and examined whether each fragment of caspase-8 was
generated, to determine which enzyme was responsible for the
formation of each caspase-8 fragment. Each purified enzyme was
added at a concentration lower than that which can cause complete
fragmentation of procaspase-8, to reproduce the results of the
cell-based experiments (Fig. 2D).
Purified caspase-3 could induce the formation of both p30 and
p41/43 fragments (Fig. 7D), while
caspase-6 induced only the formation of p41/43 fragment (Fig. 7E). The cell-free samples shown in
Fig. 7C–E seemed to reflect the
situation of the samples incubated for 2+22 h with 20–40 µM
Aβ42, as shown in Fig. 2D, in
consideration of the level of Bid and processed caspase-8. Taken
together, data from the cell-free experiments confirm that both p30
and p41/43 fragments could be formed through the IAPW, although
physiological implication of the differential activities of
caspases-3 and -6 on procaspase-8 processing remains to be
investigated.
In the present study, we demonstrated that the IAPW
was activated in Aβ42-doubly-treated cells. However, it was noted
that DEVDase and VEIDase activity, and processing of the two
caspases were less in the Aβ42-treated cells than those in
STS-treated cells by several fold (Fig. 7A and B). The limited activation of
caspases-3 and -6 in the Aβ42-treated cells seemed to be
responsible for the incomplete activation of caspase-8 (Figs. 2D and 6C–E). On the other hand, it is also
possible that Aβ42 could negatively affect the activity of
caspases-3 and -6 for the processing of procaspase-8. To test this
hypothesis, the effect of Aβ42 on the formation of p30 and p41/43
was explored in the cell-free samples with purified caspases-3 and
-6 proteins. Caspase-6 generated similar levels of p30 and p41/43
fragments independent of Aβ42 treatment, while caspase-3 appeared
to be only slightly affected by the peptide in producing the
fragments (Fig. 7F). Thus, it was
concluded that the lower levels of caspases-3 and -6 activation,
but not their activity, were responsible for the incomplete
activation of caspase-8 in the Aβ42-doubly-treated cells (Fig. 2A–C). This is consistent with our
previous study demonstrating that Aβ42 did not affect the
activities of caspases except on procas-pase-9 (27).
Discussion
Although caspase is not robustly activated in
Aβ-treated cells (Figs. 1 and
6A and B) (26), and Aβ42 can suppress IAPW by
interacting with procaspase-9 (27), accumulating data, including ours,
have implicated the apoptotic process as a mechanistic feature of
the cell death induced by Aβ42 (19,49). In the present study, we sought a
caspase-activating condition in Aβ42-treated cells and explored the
apoptosis pathways involved under the condition to assess cytotoxic
properties of Aβ42 and its mechanistic features. To achieve this,
we explored various culture systems and methods associated with
Aβ42 treatment in the experiments.
Caspase activity was detected in the cells treated
'once' with Aβ42 in the previous studies (9,12).
In the present study, the activity was also detected in the cells
singly treated with the peptide (Fig.
1A–C). However, Aβ42-dose dependency of the activity was not
observed (Fig. 1A–C) and
furthermore, the processed fragments of caspases were not detected
(Fig. 1D). In fact, detection of
processed fragments of caspases may not be necessary to prove the
activation of the enzyme, because an undetectable amount of
activated caspase can exhibit catalytic activity with the synthetic
substrates (data not shown). Thus, we think that a certain low
level of caspase activation could have occurred in the single
treatment samples. However, it is questionable whether the activity
was elicited solely by the addition of Aβ42. For example, it is
possible that serum deprivation of the cells before Aβ42 treatment
caused increased background activity. Thus, it is not clear whether
the activity was due to Aβ42 treatment or other unknown
factors.
The activation of caspase was observed in cells
'doubly' treated with Aβ42 in which Aβ42-dose dependency of the
activity was evident, and the processed fragments of caspases or
reduction of procaspase was detected (Fig. 2A–D). The Apaf-1 apoptosome was
also formed in the Aβ42-doubly-treated cells (Fig. 4B), suggesting that the IAPW was
activated in the cells. Polymerization of the peptide (50) likely provides a strong signal to
activate the apoptotic pathway and related caspases, although the
mechanism underlying the necessity of the double treatment of Aβ42
to induce the activation of caspases remains poorly understood. The
suppressive effect of Aβ42 on the formation of the apoptosome
(27) seemed to be overcome in
the cells. Relatively longer exposure (2+22 h in the present study
vs. 4+6 h) (27) of cells to Aβ42
might be an inducing factor for the formation of apoptosome.
However, Aβ42-induced IAPW was different from that
of induced by other agent (STS) in that less cytochrome c
was released from the mitochondria and the majority of procaspase-9
in the Apaf-1 complex was not processed in the peptide-treated
cells (Figs. 4D and 5). Previously we showed that Aβ42
inhibited activity of purified procaspase-9 (27), consistent with the present
observation. On the other hand, we doubt that the lower level of
cytochrome c in the 2+22 h sample (Fig. 5A) negatively affected the
processing of procaspase-9, because our previous data showed that
cytochrome c concentration variation to a certain extent did
not lead to differential processing of caspase-9 (27). It is speculated that processing
efficiency of caspase-9 was high in the STS-treated cells and the
lower affinity of processed caspase-9 for Aβ42 (27) led to the lower level of caspase-9
bound to apoptosome in the cells (Fig. 5B). We suggest that processing of
procaspase-9 in the apoptosome is the key difference in Aβ42 and
STS-induced IAPW than binding of the protein to the Apaf-1
apoptosome.
Some studies have indicated that EAPW is elicited
in Aβ-treated cells (12,44). This conclusion is usually based on
activation of caspase-8, cleavage of Bid, upregulation of related
proteins, and downregulation of EAPW-inhibiting proteins in the
cells. However, the formation of DISC, one of the essential
processes of the EAPW, has not been shown in these studies, to the
best of our knowledge. To confirm the involvement of EAPW in
Aβ-induced apoptosis, DISC formation was explored in selected
samples in the present study. However, we could not detect DISC
formation in the 2+22 h sample with 40 µM Aβ42, in which
caspase-3-like DEVDase and other caspase-related activities reached
their highest levels (Figs. 2A–D
and 5C). Furthermore, DISC
formation was not seen in the 2+46 h sample with 10 µM Aβ42,
in which p41/43 production and Bid cleavage, main processes that
DISC formation leads to, occurred (Figs. 2D and 5D). DISC may be formed in cells under
other treatment conditions, for example, with longer exposure of
cells to Aβ42, at higher concentrations. However, the role of the
hypothetical DISC formation and EAPW activation in Aβ42-induced
apoptosis are still questionable, because the apoptotic processes
could be completed in the cells without DISC, as evidenced in the
2+22 h samples with 40 µM Aβ42 (Figs. 2A–D and 5C). It was expected that DISC would be
formed before activation of caspase-3-related activity if EAPW
played a crucial role in the induced apoptosis. Taken together, the
formation of DISC and the activation of EAPW were not observed in
cells in which Aβ-induced apoptosis occurred to a significant
extent. Therefore, it is questionable whether they played active
roles in the Aβ-induced apoptotic process.
It is reasonable to assume that the p41/43
fragments of caspase-8 was generated by IAPW in the cells, because
DISC formation was absent in the selected samples (Fig. 6D). We confirmed that p41/43 was
produced in the cell-free experiments in which IAPW was
reconstituted by addition of dATP and cytochrome c to the
cell extracts, consistently with previous studies (33,48) (Fig.
7C). In IAPW, p41/43 is produced by catalytically activating
the cleavage of procaspase-8 by caspases-3 and -6 (46,51). The low levels of p41/43 in the
Aβ42-doubly-treated cells, therefore, appear to be attributable to
the lower activation of the caspases-3 and -6 (Fig. 7A and B), which could be a result
of the inhibitory effect of Aβ42 on the formation of the Apaf-1
apoptosome, as reported previously (27). One of the results of the
incomplete activation of caspase-8 is the lower level of Bid
processing, as shown in Aβ42-doubly-treated cells (Fig. 2D). The level of Bid processing may
not be sufficient to cause proper operation of the amplification
loop, which acts as a positive feedback to reinforce the apoptotic
death process in the type II cells such as HeLa cells used here
(45). The reduced activity of
caspases and lack of the amplification loop in the corresponding
cells imply limited activation of IAPW.
The p30 fragment of caspase-8 was also detected in
the Aβ42-treated cells (Fig. 2D).
The presence of the caspase-8 fragment p30 has rarely been
demonstrated in previous studies. It was generated in cells treated
with CD95 by formation of DISC and further processed into p18 and
p10 fragments through subsequent autocatalytic processes (37). Here, we showed for the first time
that p30 was formed in the cells doubly treated with Aβ42 without
formation of DISC (Fig. 6C and
D). It could be produced in the cell-free assay with
reconstituted IAPW (Fig. 7C–E),
suggesting a potential role of the pathway in generating the
fragment. p30 was catalytically inefficient in processing
procaspase-3 and Bid (Fig. 3B–D).
Thus, the physiological meaning of p30 formation in Aβ42-treated
cells presently remains unknown. It is hypothesized that p30 can
sensitize cells toward apoptosis as suggested previously (37), based on the observation that it
was produced at earlier times of the treatment prior to formation
of the p41/43 fragments (Fig.
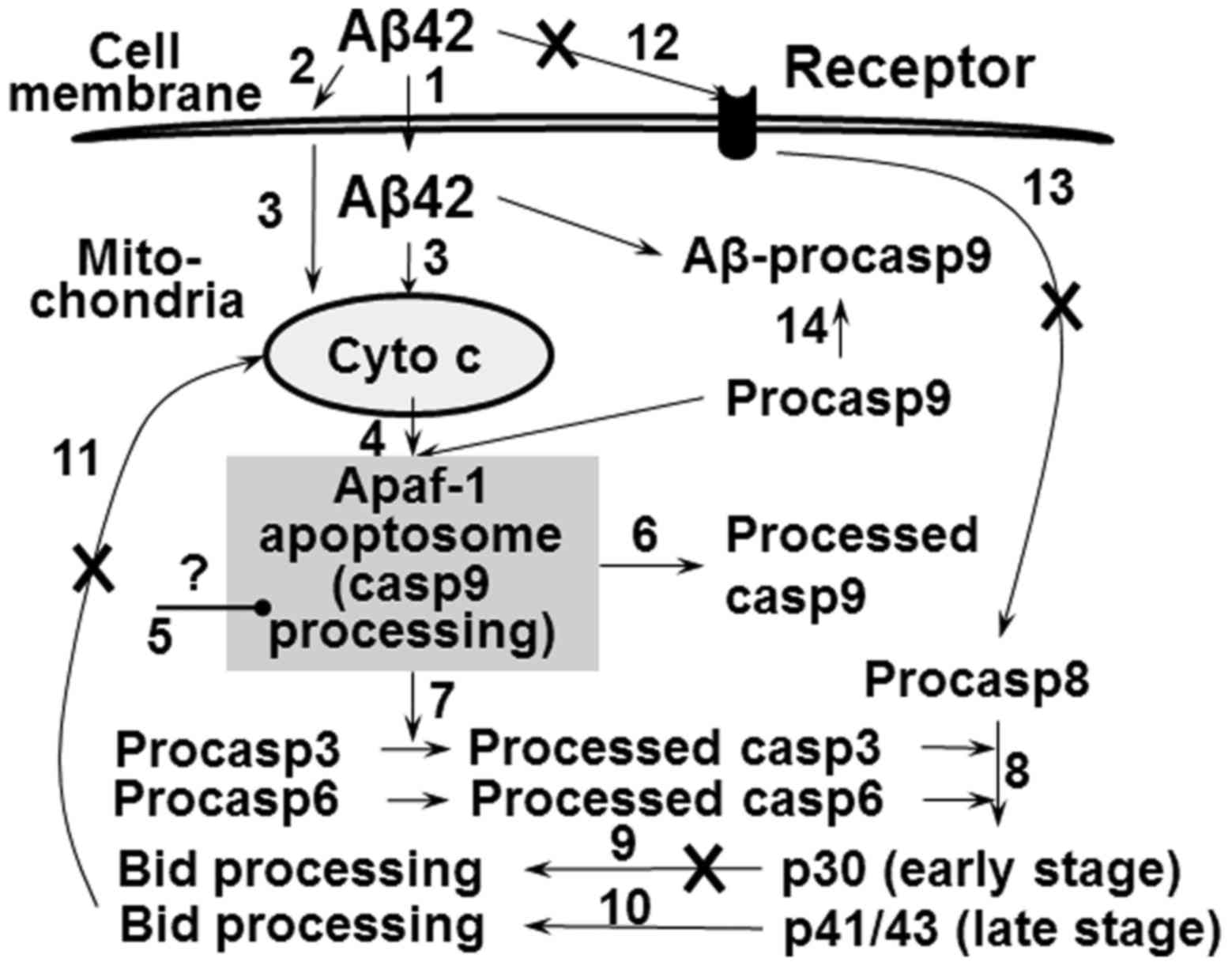
2D). Proposed model for the effect of Aβ42 on apoptosis pathway
was summarized in Fig. 8.
A previous study showed that the double treatment
of Aβ42 was necessary to elicit the cell death (31). Many studies, however, have
indicated that the single treatment was able to induce cell death.
In the present study, cell death evaluated by MTT and alamarBlue
assay occurred in a dose-dependent manner in the cells singly
treated with Aβ42 (Fig. 1E).
Similar levels of cell death were observed in cells doubly treated
with the peptide (Fig. 2E).
Remarkably, the apoptotic process accompanying caspase activation
was observed only in the Aβ42-doubly-treated cells (Fig. 2), but not in the singly treated
cells. These results imply that the cell death could occur via
distinct pathways in the two different samples. Study to elucidate
the differential death pathway for each case is ongoing. It is
difficult to imagine that neuronal cells are exposed to Aβ42 just
once in the affected patient. Thus, the experimental condition with
two or more treatments of the peptide to cells may reflect more
accurately the actual physiological conditions, and accordingly, it
can be suggested that IAPW plays a role in the Aβ-induced cell
death despite its limited activation. It is to be hoped that
comprehensive characterization of the nature of the multiple
treatments and related cell death pathway will provide novel
insight into Aβ-associated pathology and control of AD.
Abbreviations:
|
Aβ
|
amyloid-β
|
|
AD
|
Alzheimer's disease
|
|
DISC
|
death-inducing signaling-complex
|
|
EAPW
|
extrinsic apoptotic pathway
|
|
IAPW
|
intrinsic apoptotic pathway
|
|
MTT
|
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
PBS
|
phosphate- buffered saline
|
|
SDS-PAGE
|
sodium dodecylsulfate-polyacrylamide
gel electrophoresis
|
|
SEC
|
size-exclusion column
chromatography
|
|
STS
|
staurosporine
|
Acknowledgments
This study was partially supported by funding from
the Korea Research Foundation Grant from the Korean Government (no.
K201474026) and the Chosun University research fund in 2014. MII is
a member of Korean Government Scholarship Program-2010 (Graduate
stream).
References
|
1
|
Esler WP and Wolfe MS: A portrait of
Alzheimer secretases - new features and familiar faces. Science.
293:1449–1454. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mattson MP: Pathways towards and away from
Alzheimer's disease. Nature. 430:631–639. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kayed R, Head E, Thompson JL, McIntire TM,
Milton SC, Cotman CW and Glabe CG: Common structure of soluble
amyloid oligomers implies common mechanism of pathogenesis.
Science. 300:486–489. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neudecker P, Robustelli P, Cavalli A,
Walsh P, Lundström P, Zarrine-Afsar A, Sharpe S, Vendruscolo M and
Kay LE: Structure of an intermediate state in protein folding and
aggregation. Science. 336:362–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Selkoe DJ; American College of Physicians;
American Physiological Society: Alzheimer disease: Mechanistic
understanding predicts novel therapies. Ann Intern Med.
140:627–638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duyckaerts C, Delatour B and Potier MC:
Classification and basic pathology of Alzheimer disease. Acta
Neuropathol. 118:5–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Walsh DM, Minogue AM, Sala Frigerio C,
Fadeeva JV, Wasco W and Selkoe DJ: The APP family of proteins:
Similarities and differences. Biochem Soc Trans. 35:416–420. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chong YH, Shin YJ, Lee EO, Kayed R, Glabe
CG and Tenner AJ: ERK1/2 activation mediates Abeta oligomer-induced
neurotoxicity via caspase-3 activation and tau cleavage in rat
organotypic hippocampal slice cultures. J Biol Chem.
281:20315–20325. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ellerby LM and Orr HT: Neurodegenerative
disease: Cut to the chase. Nature. 442:641–642. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nikolaev A, McLaughlin T, O'Leary DD and
Tessier-Lavigne M: APP binds DR6 to trigger axon pruning and neuron
death via distinct caspases. Nature. 457:981–989. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Picone P, Carrotta R, Montana G, Nobile
MR, San Biagio PL and Di Carlo M: Abeta oligomers and fibrillar
aggregates induce different apoptotic pathways in LAN5
neuroblastoma cell cultures. Biophys J. 96:4200–4211. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fossati S, Ghiso J and Rostagno A: TRAIL
death receptors DR4 and DR5 mediate cerebral microvascular
endothelial cell apoptosis induced by oligomeric Alzheimer's Aβ.
Cell Death Dis. 3:e3212012. View Article : Google Scholar
|
|
14
|
Fossati S, Ghiso J and Rostagno A:
Insights into caspase-mediated apoptotic pathways induced by
amyloid-β in cerebral micro-vascular endothelial cells.
Neurodegener Dis. 10:324–328. 2012. View Article : Google Scholar
|
|
15
|
Boatright KM and Salvesen GS: Mechanisms
of caspase activation. Curr Opin Cell Biol. 15:725–731. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mace PD, Riedl SJ and Salvesen GS: Caspase
enzymology and activation mechanisms. Methods Enzymol. 544:161–178.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Troy CM, Friedman JE and Friedman WJ:
Mechanisms of p75-mediated death of hippocampal neurons. Role of
caspases. J Biol Chem. 277:34295–34302. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Truzzi F, Marconi A, Atzei P, Panza MC,
Lotti R, Dallaglio K, Tiberio R, Palazzo E, Vaschieri C and
Pincelli C: p75 neurotrophin receptor mediates apoptosis in
transit-amplifying cells and its overexpression restores cell death
in psoriatic keratinocytes. Cell Death Differ. 18:948–958. 2011.
View Article : Google Scholar :
|
|
19
|
Cha MY, Han SH, Son SM, Hong HS, Choi YJ,
Byun J and Mook-Jung I: Mitochondria-specific accumulation of
amyloid β induces mitochondrial dysfunction leading to apoptotic
cell death. PLoS One. 7:e349292012. View Article : Google Scholar
|
|
20
|
Murakami Y, Ohsawa I, Kasahara T and Ohta
S: Cytoprotective role of mitochondrial amyloid beta
peptide-binding alcohol dehydrogenase against a cytotoxic aldehyde.
Neurobiol Aging. 30:325–329. 2009. View Article : Google Scholar
|
|
21
|
Lee DY, Lee KS, Lee HJ, Kim DH, Noh YH, Yu
K, Jung HY, Lee SH, Lee JY, Youn YC, et al: Activation of PERK
signaling attenuates Abeta-mediated ER stress. PLoS One.
5:e104892010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Verdier Y and Penke B: Binding sites of
amyloid beta-peptide in cell plasma membrane and implications for
Alzheimer's disease. Curr Protein Pept Sci. 5:19–31. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Verdier Y, Zarándi M and Penke B: Amyloid
beta-peptide interactions with neuronal and glial cell plasma
membrane: Binding sites and implications for Alzheimer's disease. J
Pept Sci. 10:229–248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dahlgren KN, Manelli AM, Stine WB Jr,
Baker LK, Krafft GA and LaDu MJ: Oligomeric and fibrillar species
of amyloid-beta peptides differentially affect neuronal viability.
J Biol Chem. 277:32046–32053. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Resende R, Ferreiro E, Pereira C and
Resende de Oliveira C: Neurotoxic effect of oligomeric and
fibrillar species of amyloid-beta peptide 1-42: Involvement of
endoplasmic reticulum calcium release in oligomer-induced cell
death. Neuroscience. 155:725–737. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thapa A, Woo ER, Chi EY, Sharoar MG, Jin
HG, Shin SY and Park IS: Biflavonoids are superior to
monoflavonoids in inhibiting amyloid-β toxicity and fibrillogenesis
via accumulation of nontoxic oligomer-like structures.
Biochemistry. 50:2445–2455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharoar MG, Islam MI, Shahnawaz M, Shin SY
and Park IS: Amyloid β binds procaspase-9 to inhibit assembly of
Apaf-1 apoptosome and intrinsic apoptosis pathway. Biochim Biophys
Acta. 1843:685–693. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thapa A, Shahnawaz M, Karki P, Raj Dahal
G, Sharoar MG, Yub Shin S, Sup Lee J, Cho B and Park IS:
Purification of inclusion body-forming peptides and proteins in
soluble form by fusion to Escherichia coli thermostable proteins.
Biotechniques. 44:787–796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chromy BA, Nowak RJ, Lambert MP, Viola KL,
Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P,
et al: Self-assembly of Abeta(1–42) into globular neurotoxins.
Biochemistry. 42:12749–12760. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karki P, Dahal GR and Park IS: Both
dimerization and inter-domain processing are essential for
caspase-4 activation. Biochem Biophys Res Commun. 356:1056–1061.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wogulis M, Wright S, Cunningham D,
Chilcote T, Powell K and Rydel RE: Nucleation-dependent
polymerization is an essential component of amyloid-mediated
neuronal cell death. J Neurosci. 25:1071–1080. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Slaughter MR, Bugelski PJ and O'Brien PJ:
Evaluation of alamar blue reduction for the in vitro assay of
hepatocyte toxicity. Toxicol In Vitro. 13:567–569. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karki P, Seong C, Kim JE, Hur K, Shin SY,
Lee JS, Cho B and Park IS: Intracellular K(+) inhibits apoptosis by
suppressing the Apaf-1 apoptosome formation and subsequent
downstream pathways but not cytochrome c release. Cell Death
Differ. 14:2068–2075. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Majkut J, Sgobba M, Holohan C, Crawford N,
Logan AE, Kerr E, Higgins CA, Redmond KL, Riley JS, Stasik I, et
al: Differential affinity of FLIP and procaspase 8 for FADD's DED
binding surfaces regulates DISC assembly. Nat Commun. 5:33502014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Estus S, Tucker HM, van Rooyen C, Wright
S, Brigham EF, Wogulis M and Rydel RE: Aggregated amyloid-beta
protein induces cortical neuronal apoptosis and concomitant
'apoptotic' pattern of gene induction. J Neurosci. 17:7736–7745.
1997.PubMed/NCBI
|
|
36
|
Xue WF, Homans SW and Radford SE:
Systematic analysis of nucleation-dependent polymerization reveals
new insights into the mechanism of amyloid self-assembly. Proc Natl
Acad Sci USA. 105:8926–8931. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hoffmann JC, Pappa A, Krammer PH and
Lavrik IN: A new C-terminal cleavage product of procaspase-8, p30,
defines an alternative pathway of procaspase-8 activation. Mol Cell
Biol. 29:4431–4440. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oberst A, Pop C, Tremblay AG, Blais V,
Denault JB, Salvesen GS and Green DR: Inducible dimerization and
inducible cleavage reveal a requirement for both processes in
caspase-8 activation. J Biol Chem. 285:16632–16642. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pop C, Oberst A, Drag M, Van Raam BJ,
Riedl SJ, Green DR and Salvesen GS: FLIP(L) induces caspase 8
activity in the absence of interdomain caspase 8 cleavage and
alters substrate specificity. Biochem J. 433:447–457. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pop C, Fitzgerald P, Green DR and Salvesen
GS: Role of proteolysis in caspase-8 activation and stabilization.
Biochemistry. 46:4398–4407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Salvesen GS and Dixit VM: Caspase
activation: The induced-proximity model. Proc Natl Acad Sci USA.
96:10964–10967. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Beaudouin J, Liesche C, Aschenbrenner S,
Hörner M and Eils R: Caspase-8 cleaves its substrates from the
plasma membrane upon CD95-induced apoptosis. Cell Death Differ.
20:599–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qian MC, Liu J, Yao JS, Wang WM, Yang JH,
Wei LL, Shen YD and Chen W: Caspase-8 mediates amyloid-β-induced
apoptosis in differentiated PC12 cells. J Mol Neurosci. 56:491–499.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hippe D, Lytovchenko O, Schmitz I and
Lüder CG: Fas/CD95-mediated apoptosis of type II cells is blocked
by Toxoplasma gondii primarily via interference with the
mitochondrial amplification loop. Infect Immun. 76:2905–2912. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cowling V and Downward J: Caspase-6 is the
direct activator of caspase-8 in the cytochrome c-induced apoptosis
pathway: Absolute requirement for removal of caspase-6 prodomain.
Cell Death Differ. 9:1046–1056. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Monnier PP, D'Onofrio PM, Magharious M,
Hollander AC, Tassew N, Szydlowska K, Tymianski M and Koeberle PD:
Involvement of caspase-6 and caspase-8 in neuronal apoptosis and
the regenerative failure of injured retinal ganglion cells. J
Neurosci. 31:10494–10505. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Slee EA, Harte MT, Kluck RM, Wolf BB,
Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri
ES, et al: Ordering the cytochrome c-initiated caspase cascade:
Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a
caspase-9-dependent manner. J Cell Biol. 144:281–292. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Morishima Y, Gotoh Y, Zieg J, Barrett T,
Takano H, Flavell R, Davis RJ, Shirasaki Y and Greenberg ME:
Beta-amyloid induces neuronal apoptosis via a mechanism that
involves the c-Jun N-terminal kinase pathway and the induction of
Fas ligand. J Neurosci. 21:7551–7560. 2001.PubMed/NCBI
|
|
50
|
Jan A, Adolfsson O, Allaman I, Buccarello
AL, Magistretti PJ, Pfeifer A, Muhs A and Lashuel HA: Abeta42
neurotoxicity is mediated by ongoing nucleated polymerization
process rather than by discrete Abeta42 species. J Biol Chem.
286:8585–8596. 2011. View Article : Google Scholar
|
|
51
|
Wieder T, Essmann F, Prokop A, Schmelz K,
Schulze-Osthoff K, Beyaert R, Dörken B and Daniel PT: Activation of
caspase-8 in drug-induced apoptosis of B-lymphoid cells is
independent of CD95/Fas receptor-ligand interaction and occurs
downstream of caspase-3. Blood. 97:1378–1387. 2001. View Article : Google Scholar : PubMed/NCBI
|