Introduction
Severe sepsis, characterized by an overwhelming
systemic immune response to pathogen-associated molecules, such as
lipopolysaccharides, may lead to multiple organ failure, shock and
even death (1–3). Cardiac impairment, characterized by
a reduced ejection fraction and collapsed circulation, is the major
consequence of septic shock, and it has been demonstrated to be the
greatest risk factor for severe sepsis-associated mortality
(4). Severe septic myocardium
launches a cascade of overwhelming inflammatory responses, with
excessive circulating myocardial depressant factors, as well as
overactive neutrophil and macrophage infiltration (5). In addition, increasing evidence
indicates that myocardium apoptosis and the inflammatory response
lead to lipopolysaccharide-induced cardiac malfunction (6,7),
suggesting that anti-apoptosis measures may ameliorate cardiac
impairment.
Toll-like receptors (TLRs) are critical
pattern-recognition receptors that mediate the innate immune
response, and activate signaling networks to promote the
inflammatory cascade (8).
However, inappropriate and unregulated production of
pro-inflammatory cytokines and mediators results in reduced cardiac
output, cardiovascular collapse and mortality (9,10).
There are two major intracellular adaptor proteins
that mediate TLR signaling: Myeloid differentiation factor 88
(MyD88) and Toll or interleukin-1 receptor-domain-containing
adaptor-inducing interferon-β (IFN-β) (TRIF) (11). All TLRs, except TLR3, are
activated via a MyD88-dependent signaling pathway, and subsequently
activate cytosolic nuclear factor-κB (NF-κB), which serves a
critical role in host defense and tissue inflammation, and is
involved in the pathogenesis of cardiac dysfunction during severe
sepsis (12). MyD88 deficiency
confers powerful protection against lipopolysaccharide-induced
cardiac dysfunction and mortality risk by alleviating
cytokine-induced inflammation and reducing neutrophil and
macrophage infiltration (13).
TRIF has been demonstrated to be involved in TLR4
and TLR3 signaling, type I IFN regulatory factor-3 (IRF-3)
induction, IFN-β activation and slower NF-κB activation. Thus, it
regulates the production of various cytokines, including
inflammatory cytokines and apoptosis-related inducing factors
(14). Systematic deletion of the
TRIF gene has been demonstrated to effectively protect mice from
endotoxic shock and ischemia-reperfusion injury, with improved
cardiac function and survival (15). However, few studies have
investigated the role and mechanism of TRIF signaling in severe
septic myocardium (15–17). Furthermore, the relative
contributions of MyD88 and TRIF signaling to the development of
cardiac dysfunction in a model of polymicrobial severe sepsis are
unknown.
The present study investigated the effect of MyD88-
and TRIF-dependent pathways in the TLR-mediated cardiac
inflammatory response in a murine model of cecum ligation and
puncture (CLP)-induced severe sepsis. The importance of each
pathway in influencing myocardial function was also examined. The
present study may shed light on a potential useful therapy for
sepsis-induced cardiac dysfunction via MyD88 and TRIF blockade.
Materials and methods
Animal model
A total of 64 male C57/BL6 mice weighing 20–25 g and
aged 6–8 weeks were purchased from the Model Animal Research Center
of Nanjing University (Nanjing, China) (stock no. J003752). The
mice were maintained in a specific pathogen-free environment, with
a humidity of 50%, constant temperature of 26°C, and a 12-h
light/dark cycle. The mice had access to food and water ad libitum.
All animal care and experimental procedures conformed strictly to
the regulations of the Institutional Animal Care and Use Committee
of Central South University (Changsha, China). The study protocol
was reviewed and approved by the Medical Ethical Committee of the
Second Xiangya Hospital of Central South University (Changsha,
China). A total of 64 mice were divided randomly into four groups
(n=16/group; 8 for observation of survival rate, 8 for heart sample
analysis): The wild-type (WT)-sham, WT-CLP, anti-MyD88-CLP and
anti-TRIF-CLP groups.
Severe sepsis was generated by CLP. Briefly,
following anesthetization with 1.5% pentobarbital sodium (40 mg/kg;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) injected
intraperitoneally, a 1.0-cm long incision was made on the abdomen
of each mouse. The cecum was exposed, ligated below the ileocecal
valve with a silk 4–0 suture, and punctured twice with a 20-gauge
needle. Anti-MyD88 (5 µl/g; ab2068) and anti-TRIF antibodies
(5 µl/g; ab13810) (both from Abcam, Cambridge, UK) were
administered to the respective mice through the tail veins 2 h
before CLP. The sham group underwent laparotomy without CLP.
Following surgery, pre-warmed normal saline (37°C; 0.1 ml/g body
weight) was administered subcutaneously to maintain fluid
resuscitation and avoid iatrogenic hypothermia. The survival rate
of each group was recorded from 12–24 h after sham operation or
CLP.
Echocardiographic assessment of left
ventricular (LV) contraction and function
Echocardiography was performed at 6 and 12 h after
CLP under light anesthesia by pentobarbital sodium [40 mg/kg,
intraperitoneal (IP)] using an ultrasound scanner (Acuson S3000)
coupled with an 18.0-MHz linear transducer (both from Siemens
Healthineers, Erlangen, Germany). A single experienced operator who
was blinded to the experimental design acquired all images. LV
systolic function was assessed by fractional shortening (FS) using
the M-mode method at the mid-papillary level in the parasternal
short-axis view. LV internal diameters at end diastole and end
systole were measured. Strain was measured using a speckle tracking
approach for the middle of the posterior wall on short-axis views
during at least three consecutive heartbeats, and analyzed online
using Velocity Vector Imaging software v. 3.5 (Siemens
Healthineers). This approach enables rapid and accurate
quantitative assessment of global and regional myocardial function
(18). Strain was defined as
relative deformation of the myocardium (19), which represents myocardial
contraction sensitivity, and was expressed as a percentage.
Serum cardiac troponin I (cTnI)
measurement
Under anesthesia with pentobarbital sodium (40
mg/kg, IP), blood samples were collected via puncture from the
inferior vena cava by opening the abdomen in all groups 12 h after
CLP. Mice were then sacrificed via cervical dislocation. Blood
samples were centrifuged at 589 × g/min for 10 min at 4°C. The
serum cTnI level was measured using an ELISA Quantikine mouse kit
(M6000B, MTA00B and MLB00C; R&D Systems Europe, Ltd., Abingdon,
UK) according to the manufacturer's recommendations.
Quantification of mRNA in heart
tissue
Following euthanasia, the hearts were harvested and
stored at −80°C after being quickly immersed in liquid nitrogen for
reverse transcription-polymerase chain reaction (RT-PCR) assays.
Total RNA was purified from heart tissue using TRIzol reagent
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according
to the manufacturer's protocol. RT-PCR was performed to amplify
mouse interleukin-1 (IL-1), IL-6, tumor necrosis factor-α (TNF-α)
and β-actin mRNA. Using 2 µl reverse transcriptase (Promega
Corp., Madison, WI, USA), reactions were performed in a final
volume of 20 µl with the gene-specific primers listed in
Table I. The expression of the
genes examined was normalized to that of β-actin as an internal
control. Amplified PCR products were evaluated by 1.7% agarose gel
electrophoresis and ethidium bromide staining. The integral optical
density (IOD) of the electrophoretic bands was quantified. The data
presented in the figures was the ratio of the IOD of the target
gene to the IOD of the reference gene. Results were interpreted
using Image-Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
 | Table IPrimers used for reverse
transcription-polymerase chain reaction. |
Table I
Primers used for reverse
transcription-polymerase chain reaction.
| Gene | Primer | Sequence (5′–3′) |
|---|
| IL-1 | Sense |
GCACTCTGCTTGCTCACCTTTA |
| Antisense |
GGTCTCTAAGGCCAGAGGTGGA |
| IL-6 | Sense |
AGTGCGACCTGGACATCCG |
| Antisense |
TGGCTCTAACAGTCCGCCTAG |
| Tumor necrosis
factor-α | Sense |
AGTGCGACCTGGACATCCG |
| Antisense |
TGGCTCTAACAGTCCGCCTAG |
| β-actin | Sense |
AGTGCGACCTGGACATCCG |
| Antisense |
TGGCTCTAACAGTCCGCCTAG |
| Vascular cell
adhesion molecule-1 | Sense |
CGGTCATGGTCAAGTGTTTG |
| Antisense |
GAGATCCAGGGGAATTCA |
| Intercellular
adhesion molecule-1 | Sense |
TATCGGGATGGTGAAGTCT |
| Antisense |
GGCGGTAATAGGTGTAAATG |
| β-actin | Sense |
AGTGCGACCTGGACATCCG |
| Antisense |
TGGCTCTAACAGTCCGCCTAG |
| Interferon | Sense |
GGAGCATGGATGTGATCAAG |
| Antisense |
GAGTTCACTGATGGCTTTGC |
| Myeloid
differentiation factor 88 | Sense |
GGAACAGACCAACTATCGGC |
| Antisense |
GAGACAACCACTACCATCCG |
| Toll or
interleukin-1 receptor-domain-containing | Sense |
CACCTTCTGCGAGGATTTC |
| adaptor-inducing
interferon-β | Antisense |
GCTGCTCATCAGAGACTGGT |
| Interferon
regulatory factor-3 | Sense |
TCTGCCCTCAACCGCAAAGAAG |
| Antisense |
TACTGCCTCCACCATTGGTGTC |
| Nuclear
factor-κB | Sense |
GGGACTACGACCTGAATGCT |
| Antisense |
GGGCACGGTTGTCAAAGAT |
| β-actin | Sense |
TCTGGCACCACACCTTCT |
| Antisense |
GATCTGGGTCATCTTCTCAC |
Histopathological examination
Samples of hearts were dissected and fixed in 10%
buffered formalin at room temperature for >48 h, and
subsequently embedded in paraffin, cut into 5-µm sections,
and stained with hematoxylin and eosin at 37°C for 15 min.
Formalin-fixed, paraffin-embedded heart tissue was deparaffinized
and blocked with 2% bovine serum albumin (Sigma-Aldrich; Merck
KGaA) for 30 min at room temperature. To assess neutrophil
accumulation in heart tissue, the sections were incubated with
rabbit polyclonal anti-Gr-1 antibody (1:50; ab25377; Abcam).
Macrophages in the myocardium were examined with rabbit polyclonal
anti-cluster of differentiation (CD)45 antibody (1:200; ab3638;
Abcam). These antibodies were applied for 12 h at 4°C. Following
this, the secondary antibody (horseradish peroxidase goat
anti-rabbit immunoglobulin G; 1:200; G23303; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) was applied for 50 min at
room temperature. Subsequently, the slides were treated with a
3,3′-diaminobenzidine staining system (Dako; Agilent Technologies,
Inc., Santa Clara, CA, USA) according to the manufacturer's
protocol. Hematoxylin was used to counterstain the nuclei for 3 min
at room temperature. The slides were observed under a light
microscope (Carl Zeiss AG, Oberkochen, Germany) at magnifications
of ×200 and ×400.
Myocardial myeloperoxidase (MPO)
assay
Tissue MPO activity was determined in the heart.
Heart samples were harvested, excised, homogenized, rinsed and
dissolved in 60 ml phosphate-buffered saline (pH 6). Subsequently,
0.9 ml tissue homogenate was mixed well with 0.1 ml myocardial MPO
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and
activity was assayed using a MPO Colorimetric activity assay kit
(Nanjing Jiancheng Bioengineering Institute). After resting at 37°C
in a water bath for 15 min, the mixture was added to color carrier
and H2O2. Following this, the mixture was
incubated at 60°C for 10 min in a water bath, and the rate of
change in absorbance at 460 nm was measured using a
spectrophotometer (CE 9000; Cecil Instruments, Ltd., Cambridge,
UK). Myocardial MPO activity was expressed as U/l protein in wet
tissue.
In situ apoptosis assay
Apoptosis assays, including those conducted to
detect caspase-3 and Fas/Fas ligand (FasL) activity, were performed
using the RT-PCR method described above. The primers used are
listed in Table II.
| Table IIPrimers used for myocardial apoptosis
assays. |
Table II
Primers used for myocardial apoptosis
assays.
| Gene | Primer | Sequence
(5′–3′) |
|---|
| Fas | Sense |
CCGCAGGCTGCCCACACAGG |
| Antisense |
CTACCTTAGTGACCTTTTCA |
| Fas ligand | Sense |
AGAACTCCGTGAGCCAAC |
| Antisense |
TGTGTCTTCCCATTCCAG |
| Caspase-3 | Sense |
GGGATGAAAGCGCAGTGTCCTGC |
| Antisense |
AGACTCCGGCAGTAGTCGCCTC |
| β-actin | Sense |
TCTGGCACCACACCTTCT |
| Antisense |
GATCTGGGTCATCTTCTCAC |
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
All data were presented as the mean ± standard deviation.
Comparison among groups was performed using one-way analysis of
variance with Bonferroni post hoc tests for statistical
significance. Survival rates were estimated using the log-rank
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
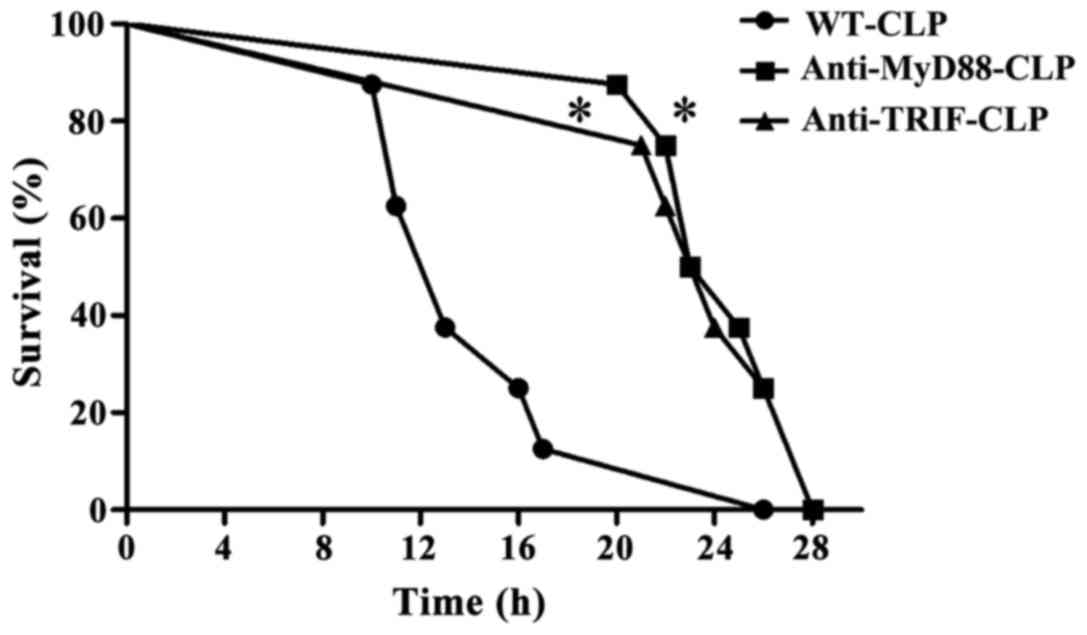
Survival rate during severe sepsis is
lower in WT-CLP mice than in anti-MyD88 and anti-TRIF mice
A total of 12 h after CLP, WT-CLP mice displayed
septic symptoms, including ruffled hair, slow physical actions,
shivering and low temperature. The survival rate at 16 h was ~20%.
Mice in the anti-MyD88-CLP and anti-TRIF-CLP groups presented signs
of moderate health, such as shiny hair, and normal physical actions
and temperature throughout the observation period. The survival
rates at 16 h were 100% and at 24 h in these groups were 50 and
37%, respectively, which was significantly higher than that in the
WT-CLP group (P<0.05). These results indicate that MyD88 and
TRIF signaling contributed to the severe sepsis-linked high
mortality rate observed in WT-CLP mice (Fig. 1).
Cardiac function is better in mice in the
anti-MyD88 and anti-TRIF groups than in mice in the WT group
following CLP
M-mode studies indicated that baseline LV
contractile function was similar in the four experimental groups.
CLP resulted in significantly deteriorated LV function, as
evidenced by 20 and 40% reductions in strain at 6 and 12 h after
CLP, respectively, compared with the baseline measurements (WT-CLP
vs. WT-sham: 6 h post-CLP, 16.6 vs. 20.3%, P<0.05; 12 h
post-CLP, 12.5 vs. 19.3%, P<0.01) (Table III). In addition, compared with
the baseline, the WT-CLP group demonstrated reductions in FS of 21%
at 12 h after CLP (41 vs. 52%, P<0.05) and 50% at 24 h after CLP
(27 vs. 52%, P<0.01) (Table
III). The anti-MyD88- and anti-TRIF-CLP groups conserved LV
contractile function, as demonstrated by significantly higher
global longitudinal strain compared with that in the WT-CLP group
at each time-point (all P<0.05) (Table III), as well as improved FS at
24 h after CLP (anti-MyD88- and anti-TRIF-CLP vs. WT-CLP: 38 and 42
vs. 27%, P<0.05) and smaller LV internal diameters at end
systole at 24 h (anti-MyD88-and anti-TRIF-CLP vs. WT-CLP: 1.8 and
1.73 vs. 2.3 mm, P<0.05) (Table
III).
| Table IIISerial echocardiographic measurements
before and after CLP in the different experimental groups. |
Table III
Serial echocardiographic measurements
before and after CLP in the different experimental groups.
| Measurement in each
group | Time-point
|
|---|
| Baseline | 6 h | 12 h | 24 h |
|---|
| Heart rate
(bpm) | | | | |
| WT-sham | 611.0±9.0 | 612.0±12.0 | 622.0±13.0 | 588.0±12.0 |
| WT-CLP | 601.0±14.0 | 562.0±19.0 | 493.0±25.0 | 432.0±30.0 |
|
Anti-MyD88-CLP | 598.0±11.0 | 611.0±14.0 | 558.0±15.0 | 500.0±24.0 |
| Anti-TRIF-CLP | 604.0±10.0 | 610.0±14.0 | 562.0±19.0 | 502.0±26.0 |
| Blood pressure
(mmHg) | | | | |
| WT-sham | 84.0±2.0 | 84.0±2.0 | 83.7±3.0 | 83.0±3.2 |
| WT-CLP | 94.0±3.2 | 72.0±4.3a | 55.0±6.8b | 37.0±6.0b |
|
Anti-MyD88-CLP | 88.0±2.2 | 80.3±3.0a | 71.0±4.5a, c | 60.0±6.1b, c |
| Anti-TRIF-CLP | 89.0±2.3 | 84.0±2.7 | 73.0±3.6a, c | 62.0±6.8b, c |
| LVIDd (mm) | | | | |
| WT-sham | 3.0±0.1 | 3.0±0.0 | 2.9±0.1 | 3.0±0.1 |
| WT-CLP | 3.2±0.1 | 3.3±0.1 | 3.5±0.1a | 3.8±0.1a |
|
Anti-MyD88-CLP | 3.0±0.1 | 3.1±0.1 | 3.1±0.2c | 3.5±0.1a, c |
| Anti-TRIF-CLP | 3.1±0.1 | 3.2±0.1 | 3.2±0.2 | 3.4±0.1 |
| LVIDs (mm) | | | | |
| WT-sham | 1.5±0.0 | 1.6±0.1 | 1.5±0.0 | 1.5±0.1 |
| WT-CLP | 1.5±0.1 | 1.5±0.1a | 1.9±0.1a | 2.3±0.1b |
|
Anti-MyD88-CLP | 1.4±0.1 | 1.4±0.1 | 1.6±0.1 | 1.8±0.1a, c |
| Anti-TRIF-CLP | 1.4±0.1 | 1.4±0.1 | 1.5±0.1 | 1.7±0.1a, c |
| Fractional
shortening (%) | | | | |
| WT-sham | 50.0±2.1 | 47.0±1.3 | 49.0±1.6 | 50.0±2.0 |
| WT-CLP | 52.0±3.0 | 51.0±1.6 | 41.0±1.9a | 27.0±2.2b |
|
Anti-MyD88-CLP | 53.0±2.2 | 53.0±1.8 | 50.0±2.0c | 38.0±2.2a, c |
| Anti-TRIF-CLP | 53.0±1.7 | 52.0±2.0 | 50.0±1.8c | 42.0±1.9a, c |
| Strain | | | | |
| WT-sham | 19.6±1.7 | 20.3±2.0 | 19.3±2.4 | 19.1±2.1 |
| WT-CLP | 20.3±2.2 | 16.6±1.7a,d | 12.5±2.0b,e | 10.4±2.0b |
|
Anti-MyD88-CLP | 20.4±2.2 | 18.8±2.1c | 17.2±2.4a, c | 15.3±1.4a, c |
| Anti-TRIF-CLP | 19.8±2.4 | 18.7±2.2c | 17.6±2.5a, c | 15.6±1.8a, c |
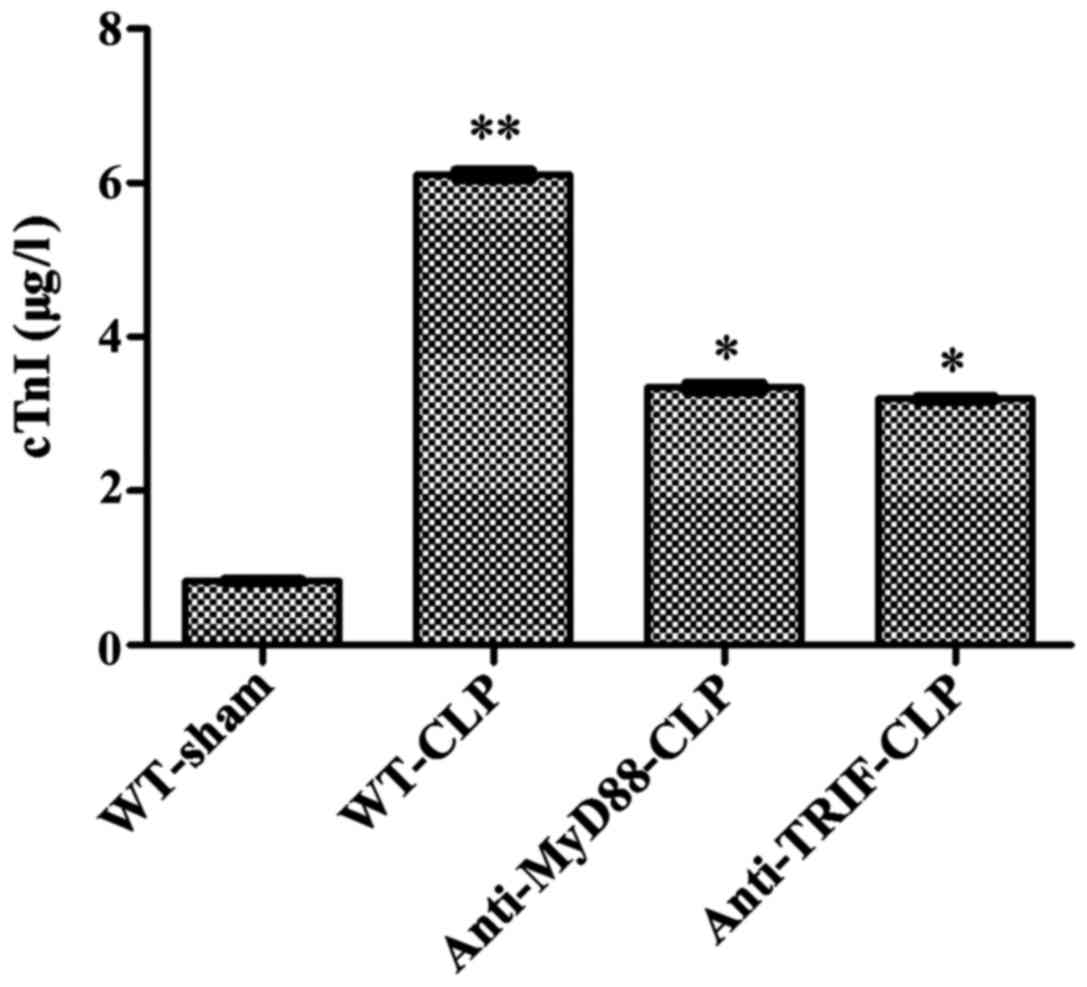
The circulating level of cTnI in WT-CLP mice was six
times higher than that in WT-sham mice (6.12±0.22 vs. 0.84±0.06
µg/l, respectively, P<0.05), and approximately two times
higher than those in anti-MyD88-CLP and anti-TRIF-CLP mice
(6.12±0.22 vs. 3.34±0.11 and 3.19±0.14 µg/l, respectively,
P<0.05), with no significant difference between the latter two
groups. These results further demonstrate that mice treated with
anti-MyD88 and anti-TRIF antibodies exhibited less severe
sepsis-induced cardiac injury than mice in the WT-CLP group
(Fig. 2).
Anti-MyD88 and anti-TRIF antibodies have
critical effects on the inhibition of myocardial inflammatory
cytokines following CLP
After 24 h, CLP induced a significant increase in
myocardial IL-1, IL-6 and TNF-α mRNA production compared with the
levels observed in the WT-sham group (P<0.05) (Fig. 3). These levels were similar in the
anti-MyD88-CLP, anti-TRIF-CLP and WT-sham groups, and significantly
lower than the levels in the WT-CLP group (P<0.05). Production
was slightly, but not significantly, higher in the anti-MyD88 CLP
group than in the anti-TRIF CLP group.
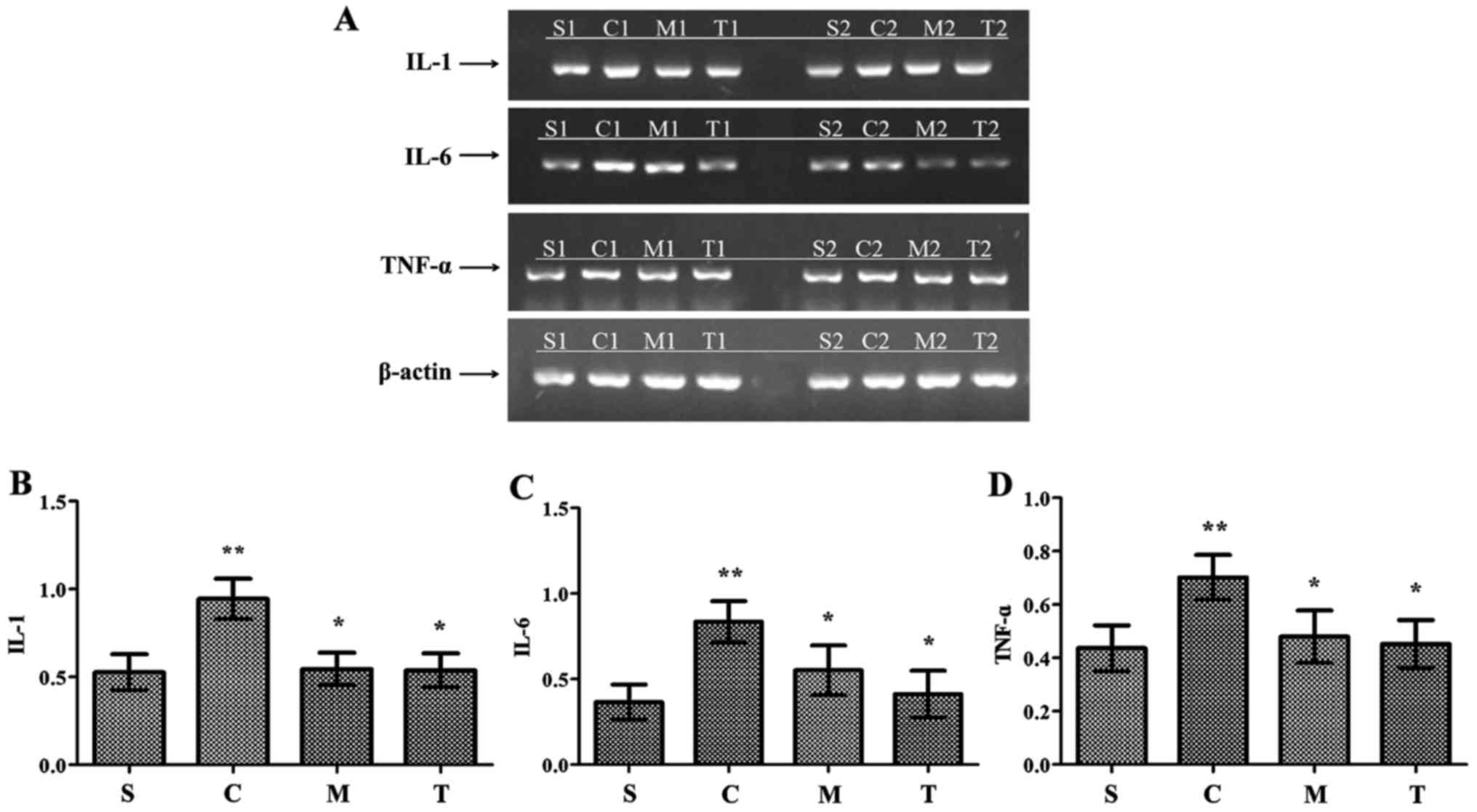 | Figure 3Myocardial inflammatory cytokines are
reduced by anti-MyD88 and anti-TRIF antibody treatments following
CLP. (A) Reverse transcription-polymerase reaction results for
inflammatory cytokine expression levels in each group. Expression
levels of (B) IL-1, (C) IL-6 and (D) TNF-α were decreased in the
anti-MyD88- and anti-TRIF-CLP groups compared with the levels in
the WT-CLP group. n=8/group. **P<0.05 vs. S;
*P<0.05 vs. C. S, WT-sham group; C, WT-CLP group; M,
anti-MyD88-CLP group; T, anti-TRIF-CLP group; MyD88, myeloid
differentiation factor 88; TRIF, Toll or interleukin-1
receptor-domain-containing adaptor-inducing interferon-β; CLP,
cecum ligation and puncture; WT, wild-type; IL, interleukin; TNF-α,
tumor necrosis factor-α. |
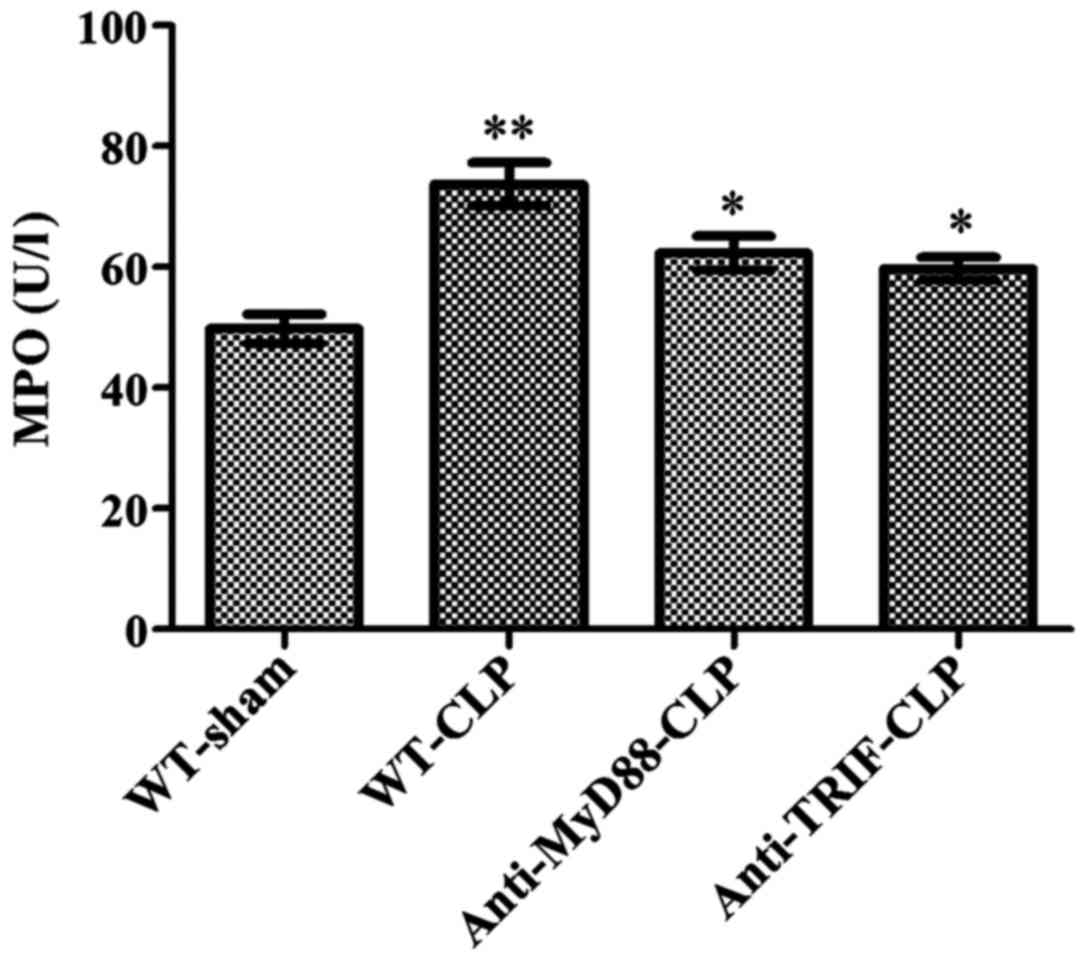
MyD88 and TRIF have equivalent effects on
neutrophil infiltration following CLP
Following CLP, the myocardial MPO activity level was
significantly higher in mice of the WT-CLP group than the level in
WT-sham mice (73 vs. 49 µ/l, respectively, P<0.05)
(Fig. 4). In contrast, MPO
activity levels were significantly reduced in the anti-MyD88-CLP
and anti-TRIF-CLP groups compared to the level in the WT-CLP group
(62 and 59 vs. 73 µ/l, respectively, P<0.05) (Fig. 4). No significant difference was
observed between the anti-MyD88 and anti-TRIF antibody groups,
indicating that these antibodies had equivalent effects on
alleviating neutrophil infiltration.
Increased expression of adherent molecules in heart
tissue, including vascular cell adhesion molecule-1 (VCAM-1) and
intracellular adhesion molecule-1 (ICAM-1), was observed in the
WT-CLP group compared with the WT-sham group (P<0.05) (Fig. 5). The levels of VCAM-1 and ICAM-1
mRNA were significantly lower in the anti-MyD88-CLP and
anti-TRIF-CLP groups than the levels in the WT-CLP group
(P<0.05) (Fig. 5).
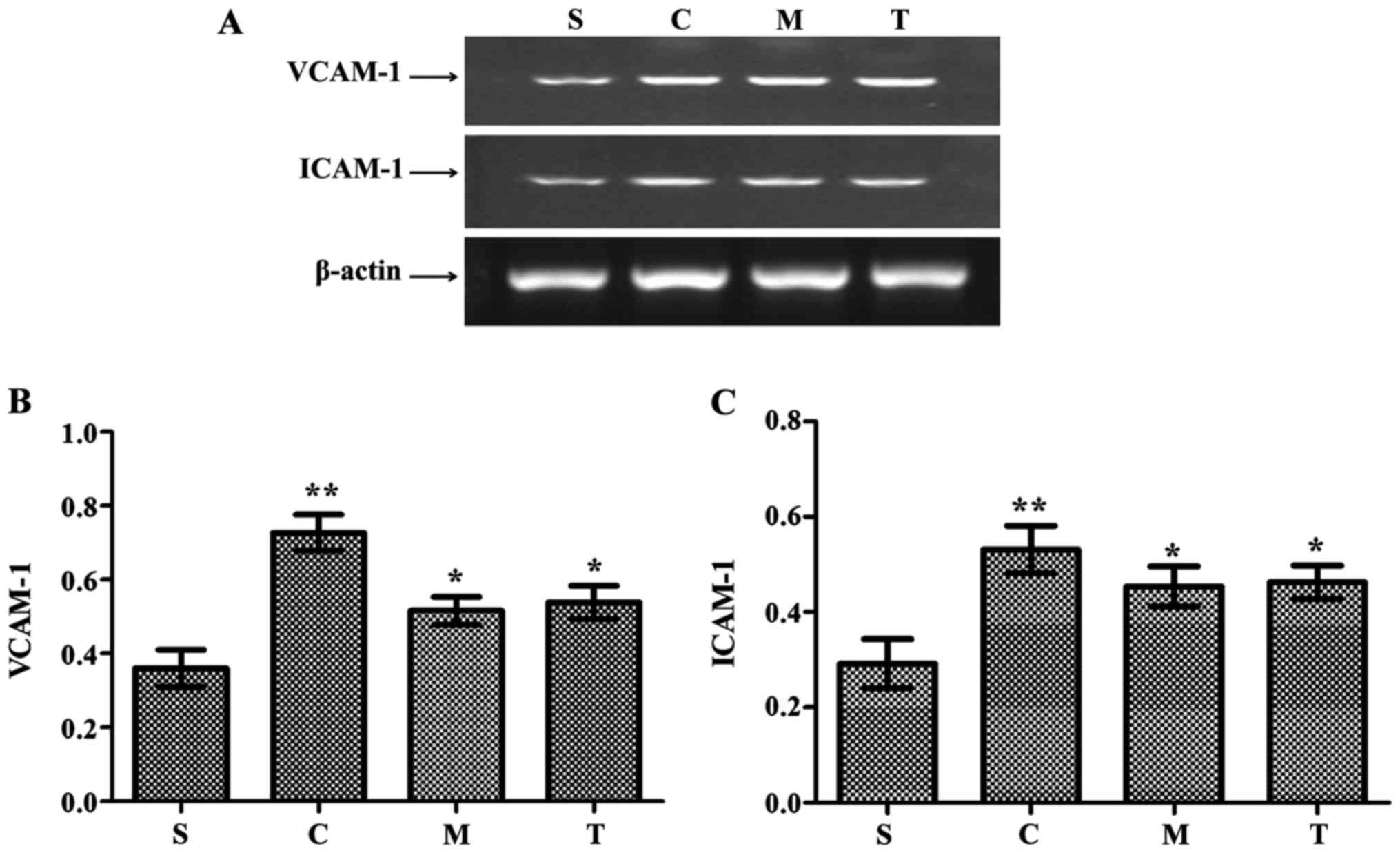 | Figure 5Anti-MyD88 and anti-TRIF antibody
treatments inhibit the expression of VCAM-1 and ICAM-1. (A) Reverse
transcription-polymerase chain reaction results and
semi-quantitative analysis of (B) VCAM-1 and (C) ICAM-1 mRNA
expression levels. n=8/group. **P<0.05 vs. S;
*P<0.05 vs. C. S, WT-sham group; C, WT-CLP group; M,
anti-MyD88-CLP group; T, anti-TRIF-CLP group; MyD88, myeloid
differentiation factor 88; TRIF, Toll or interleukin-1
receptor-domain-containing adaptor-inducing interferon-β; CLP,
cecum ligation and puncture; WT, wild-type; VCAM-1, vascular cell
adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1. |
Anti-MyD88 and anti-TRIF treatments
improve myocardial structure and reduce neutrophil and macrophage
infiltration during severe sepsis
In the WT-sham group, myocardial fibers were
arranged regularly, with distinct striation and no apparent
degeneration or necrosis. In the WT-CLP group, edema, local
swelling and some myocardial necrosis with disorganized myocardial
fibers were evident in the center of reperfusion (Fig. 6).
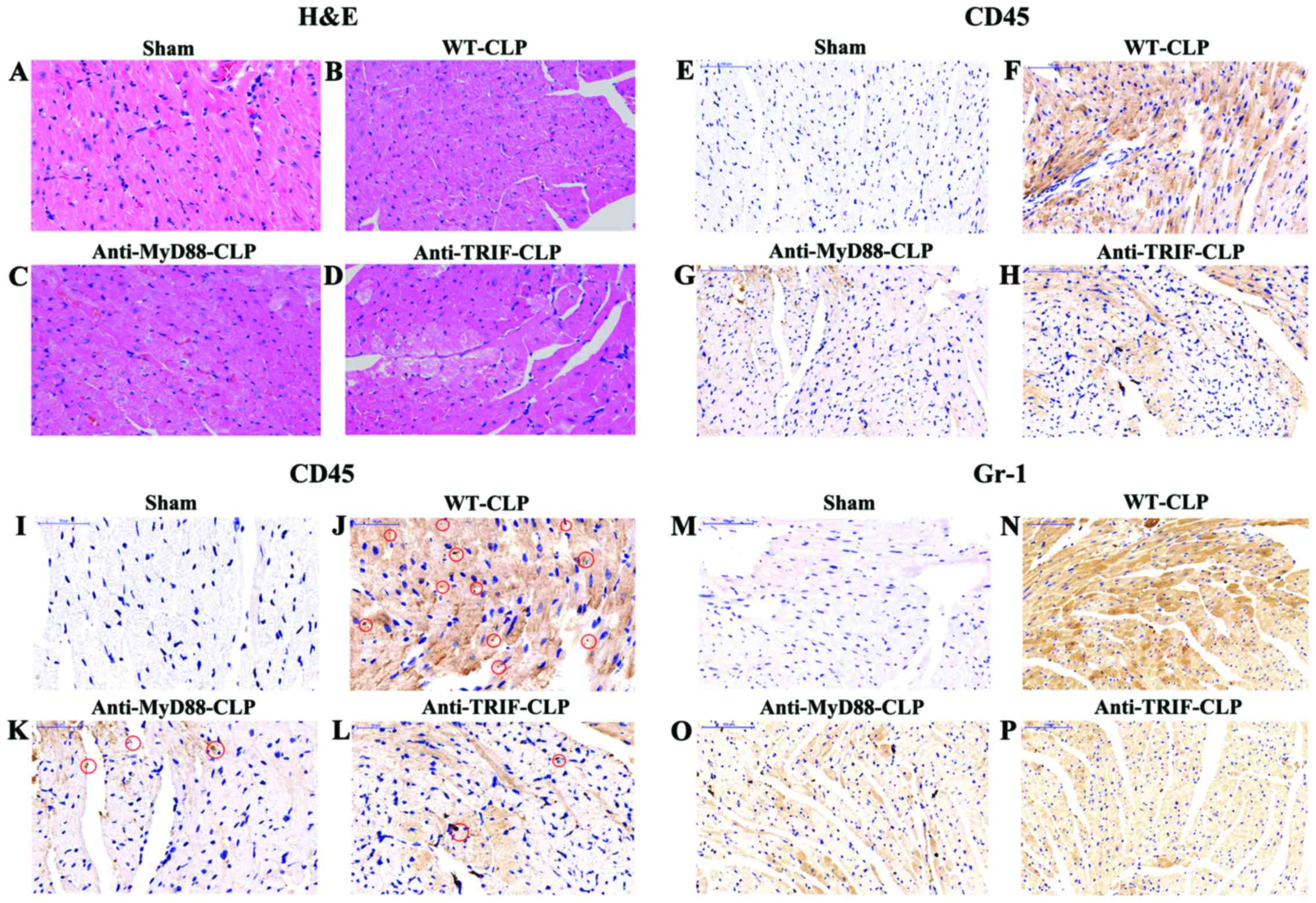 | Figure 6Anti-MyD88 and anti-TRIF treatments
reduce myocardial injury and neutrophil infiltration in mice with
severe sepsis after 24 h. (A-D) H&E staining demonstrated
moderate edema, local swelling, and some myocardial necrosis along
with disorganized myocardial fibers in mice in the WT-CLP group
(magnification, ×200). (E-L) Incubation with anti-CD45 antibody
indicated that there were increased neutrophils (brown staining) in
the hearts of mice in the WT-CLP group compared to the levels in
the hearts of mice in the anti-MyD88- and anti-TRIF-CLP groups
[(E-H) magnification, ×200; (I-L) magnification, ×400)]. Red
circles indicate CD45-positive cardiomyocytes, stained in brown.
(M-P) Incubation with anti-rabbit polyclonal anti-Gr-1 antibody
indicated that there were increased macrophages (brown staining) in
the hearts of mice in the WT-CLP group compared to the levels in
the hearts of mice in the anti-MyD88- and anti-TRIF-CLP groups
[(M-P) magnification, ×200]. MyD88, myeloid differentiation factor
88; TRIF, Toll or interleukin-1 receptor-domain-containing
adaptor-inducing interferon-β; CLP, cecum ligation and puncture;
WT, wild-type; H&E, hematoxylin and eosin; CD45, cluster of
differentiation 45. |
Neutrophil and macrophage infiltration, demonstrated
by a pervasive brown-yellow color in cardiac muscle cells, was
observed in myocardial tissue samples from the WT-CLP,
anti-MyD88-CLP and anti-TRIF-CLP groups. Consistent with the
myocardial MPO results, the areas of neutrophil and macrophage
infiltration in myocardial tissue samples from mice in the WT-CLP
group were larger than in those from WT-sham mice. This expression
in the anti-MyD88-CLP and anti-TRIF-CLP groups did not differ from
that in the WT-sham group, and was notably lower than that in the
WT-CLP group (Fig. 6).
Anti-MyD88 and anti-TRIF antibodies
attenuate myocardial apoptosis in severe sepsis
Fas/FasL mRNA levels were significantly increased in
hearts from mice of the WT-CLP group compared with the levels in
the WT-sham group (P<0.05). Furthermore, compared to WT-CLP
mice, the levels were significantly lower in the hearts of mice in
the anti-TRIF-and anti-MyD88-CLP groups (P<0.05) (Fig. 7). Notably, the level of caspase-3
activity was slightly, yet significantly, higher in WT-CLP mice
than in mice of the anti-TRIF- and anti-MyD88-CLP groups
(P<0.05) (Fig. 7).
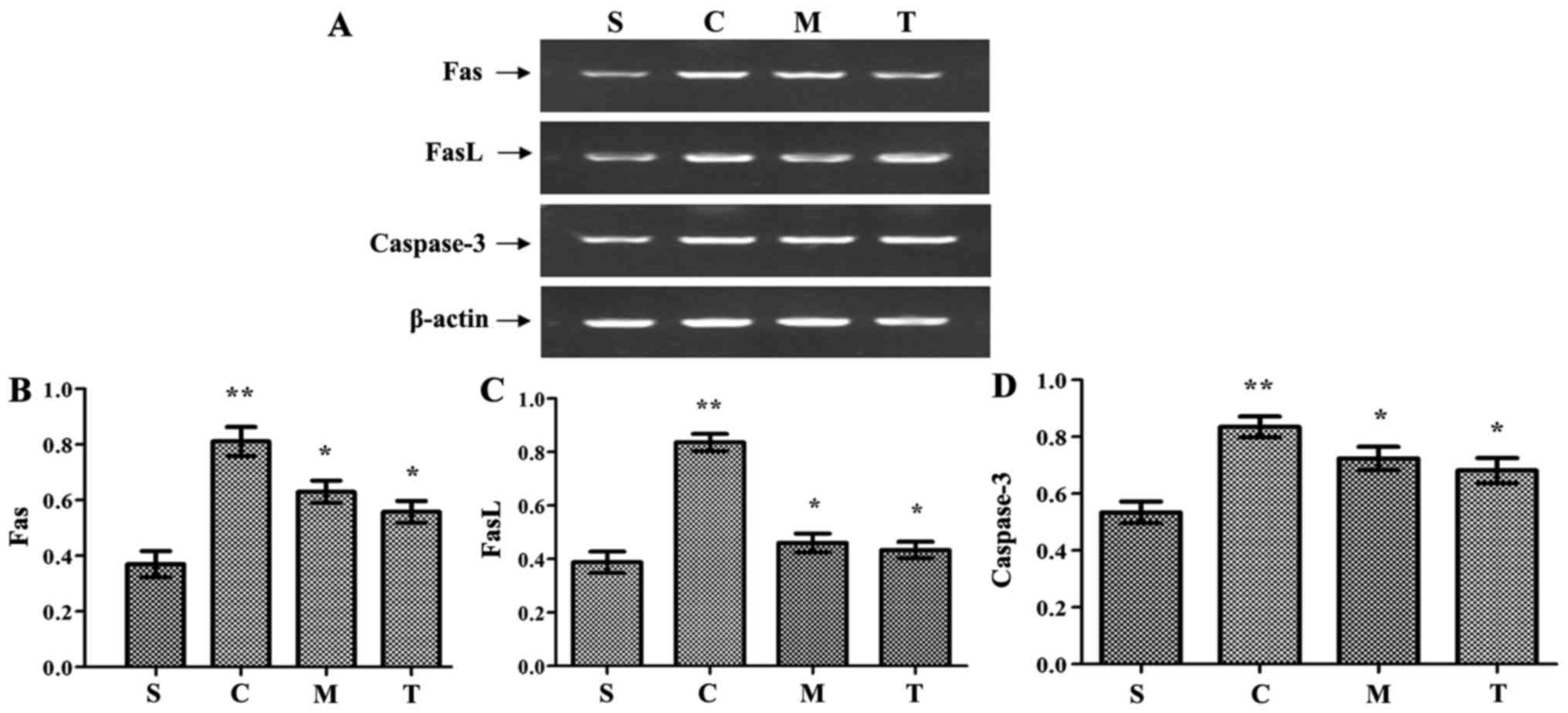 | Figure 7Inhibition of MyD88 and TRIF reduces
the expression of apoptosis cytokines, including Fas, FasL and
caspase-3, following CLP. (A) FAS, FASL and caspase-3 expression
was detected by reverse transcription-polymerase chain reaction.
Semi-quantitative analysis of (B) FAS, (C) FASL and (D) caspase-3
expression. n=8/group. **P<0.05 vs. S;
*P<0.05 vs. C. S, WT-sham group; C, WT-CLP group; M,
anti-MyD88-CLP group; T, anti-TRIF-CLP group; MyD88, myeloid
differentiation factor 88; TRIF, Toll or interleukin-1
receptor-domain-containing adaptor-inducing interferon-β; CLP,
cecum ligation and puncture; WT, wild-type; FasL, Fas ligand. |
MyD88, TRIF, NF-κB and IRF-3 expression was
significantly higher in the WT-CLP group than in the WT-sham group
(P<0.05) (Fig. 8). These
levels were also significantly higher in the WT-CLP group than in
the anti-MyD88- and anti-TRIF-CLP groups (P<0.05) (Fig. 8). These results suggest that: i)
MyD88 and TRIF promote cardiac injury during severe sepsis; ii)
MyD88 is a key adaptor protein in the TLR-mediated MyD88-dependent
NF-κB activation pathway, and TRIF is a key adaptor protein in the
TLR-mediated MyD88-independent pathway; and iii) TLR domain
alignment of the MyD88-dependent and TRIF-dependent pathways serves
an essential role in impairment of cardiac function during severe
sepsis.
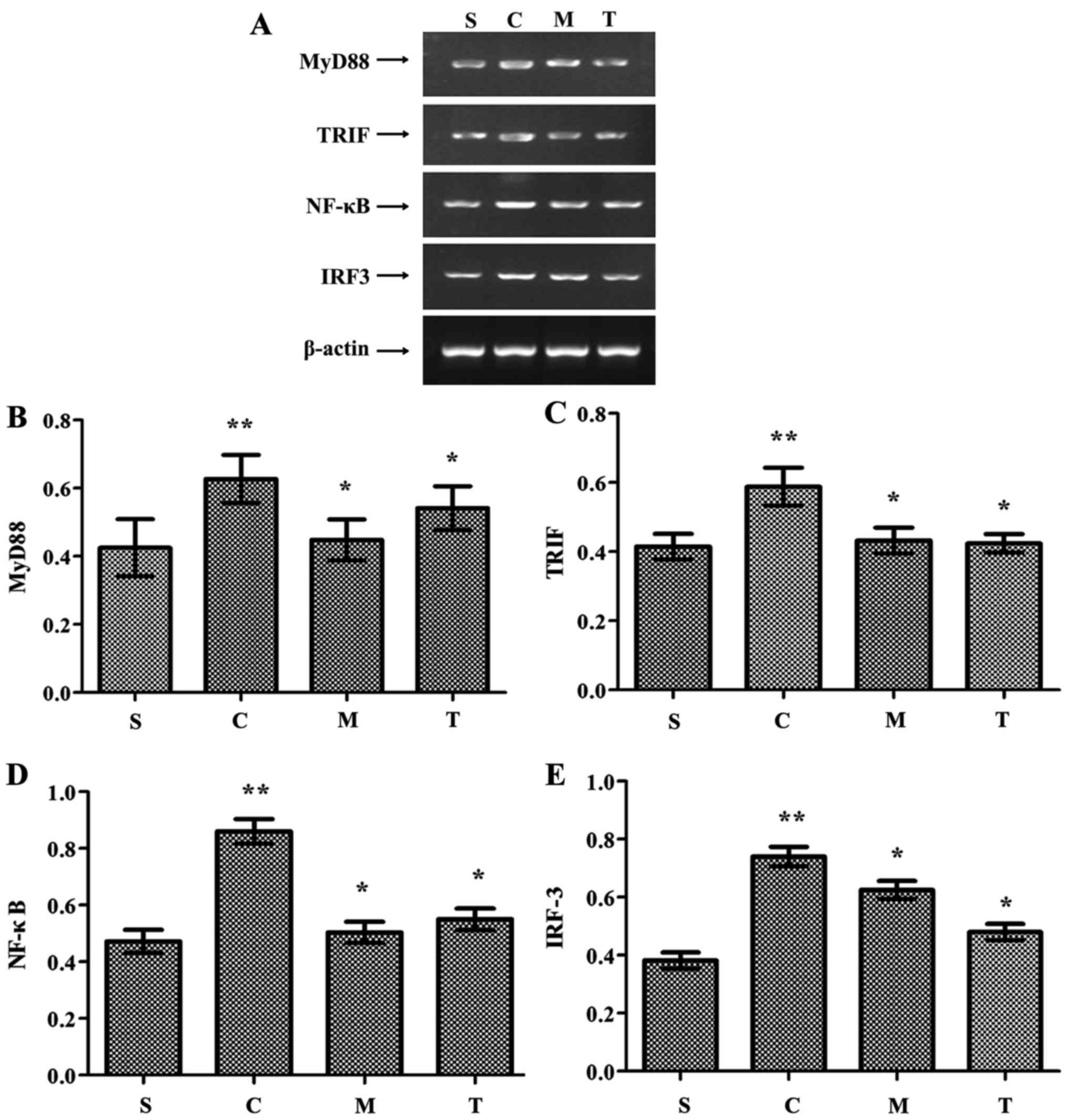 | Figure 8Inhibition of MyD88 and TRIF reduces
the expression of MyD88, TRIF, NF-κB and IRF-3 in the myocardium
following CLP. (A) MyD88, TRIF, NF-κB and IRF-3 expression detected
by reverse transcription polymerase chain reaction.
Semi-quantitative analysis of (B) MyD88, (C) TRIF, (D) NF-κB and
(E) IRF-3 expression. **P<0.05 vs. S;
*P<0.05 vs. C. S, WT-sham group; C, WT-CLP group; M,
anti-MyD88-CLP group; T, anti-TRIF-CLP group; MyD88, myeloid
differentiation factor 88; TRIF, Toll or interleukin-1
receptor-domain-containing adaptor-inducing interferon-β; CLP,
cecum ligation and puncture; WT, wild-type; NF-κB, nuclear
factor-κB; IRF-3, interferon regulatory factor-3. |
Discussion
Clinical and experimental studies have demonstrated
that inflammatory cardiomyopathy, frequently due to
infection-triggered myocarditis, is an early and fatal complication
of septic shock (20). The
pathogenesis of myocardial injury is predominantly due to
uncontrolled inflammatory responses, including excessive secretion
of inflammatory cytokines and robust neutrophil and macrophage
infiltration (20,21). Activated neutrophils react with
inflammatory cytokines, disrupt the blood vessel endothelium,
promote platelet aggregation and adhesion to endothelium, and block
blood flow, consequently leading to cardiac dysfunction (21).
The present study demonstrated that the two major
downstream signaling pathways of TLRs, the MyD88- and
TRIF-dependent pathways, serve important roles in sepsis-induced
cardiac injury following CLP. The present study also indicated that
MyD88 and TRIF inhibition have a protective effect on cardiac
function during sepsis. TLRs have been demonstrated to be important
in these pathological processes (10). MyD88 is one of the adaptor
proteins of TLRs, which holds a key position in the cascade of
inflammatory promotion. MyD88 blockade could decrease IL-1β, IL-6β
and TNF-α mRNA levels and modulate circulating neutrophil and
macrophage recruitment to protect myocardial dysfunction (22,23). ICAM-1 and VCAM-1 are known to be
adhesion factors, inducing neutrophil and macrophage infiltration
of the myocardium during septic shock (24). The present study indicated
significantly decreased myocardial MPO levels, and ICAM and VCAM
expression, as well as reduced neutrophil and macrophage
accumulation in mice in the anti-MyD88-CLP group. These results
suggest that MyD88 promotes neutrophil and macrophage recruitment,
adhesion to the endothelium, and blockage of blood flow, resulting
in myocardial injury. These observations indicate that MyD88
signaling serves a critical role in the impairment of cardiac
function.
The TRIF-dependent signaling pathway leads to the
activation of IRF-3 and induction of IFN-β, activating NF-κB and
inducing the synthesis and release of inflammatory cytokines,
representing the major host immune mechanism (14). It has been reported by Feng et
al (25) that TRIF signaling
was implicated in the aggravation of the inflammatory response and
high mortality caused by endotoxic shock; however, lack of TRIF did
not change cardiac impairment in polymicrobial peritoneal sepsis.
We speculate that this difference may be attributed to the
different animal models used in the study. Another study
demonstrated that TRIF signaling has a critical role in protection
against ischemia-reperfusion-induced myocarditis by maintaining LV
function and reducing myocardial ischemia size (14). However, the authors argued that
the mechanism by which TRIF signaling mediates cardiac impairment
was independent of the inflammatory response, as deletion of TRIF
did not activate neutrophil recruitment or cardiac cytokine
secretion (15). The present data
indicated that TRIF blockade alleviates inflammatory cytokine
secretion, and neutrophil and macrophage recruitment, thereby
maintaining myocardial function. Consistently, TRIF-deficient mice
had a limited capacity to establish an immune response, accompanied
by decreased neutrophil infiltration and minimized requirement for
a systemic inflammatory response during viral myocar-ditis and
bacterial sepsis (26). We infer
that the difference between TRIF signaling activation in
ischemia-reperfusion and the TLR4-TRIF pathway in the present study
was due to the different pathophysiology mechanism between
ischemia-reperfusion and severe sepsis. Though innate immunity
serves a critical role in myocardial injury in response to severe
sepsis and ischemia-reperfusion, the cellular and molecular
mechanisms in myocardial injury in ischemia-reperfusion is
complicated, involving energy metabolism disorder and free radical
generation (27).
Another mechanism of myocardial sepsis is apoptosis.
Growing evidence suggests that apoptotic pathways in the myocardium
are activated during sepsis (28). Previous studies have suggested
that TLR3-TRIF signaling mediates apoptosis in pancreatic
infection, myocardial ischemia-reperfusion injury, and
sepsis-induced myocardial damage (15,29). TLR3-TRIF activation has been
demonstrated to stimulate the extrinsic and intrinsic apoptotic
pathways by upregulating TNF-related apoptosis-inducing ligand and
its receptors, and down-regulating the anti-apoptotic protein,
B-cell lymphoma 2 (30). MyD88
signaling in myocardial apoptosis is linked closely to NF-κB
expression; however, the relationship between cardiac NF-κB
activity and apoptosis in injury remains unclear. NF-κB has proven
to be detrimental in some model systems, and protective in others
(13), for the secondary relation
to the NF-κB pathway promotes the transcription of pro- and
anti-apoptotic proteins (31).
Thus, it was concluded that, in attenuating myocardial
deterioration, the inhibition of MyD88 signaling predominantly
focused on reducing the inflammatory reaction. High levels of
apoptotic regulatory factors, such as Fas/FasL and caspase-3, were
observed in hearts from mice who received CLP in the present study.
The inhibition of MyD88 and TRIF signals decreased the levels of
these regulatory factors. Fas/FasL and caspase-3 levels were
slightly higher in the anti-MyD88-CLP than in the anti-TRIF-CLP
group; however, this difference was not significant. We speculate
that the near equivalence of apoptosis regulatory factors between
the anti-MyD88-CLP and anti-TRIF-CLP groups in the present study
indicate that MyD88 and TRIF are both essential for severe
sepsis-induced cardiac dysfunction, thus may be used as targets for
treatment.
Some limitations of the present study should be
noted. Firstly, the murine model of CLP induces a severe systemic
inflammatory response that is often observed in the early stage of
sepsis. It does not create immune suppression, which is often
exhibited in patients who succumb to the chronic phase of sepsis.
Therefore, the role of MyD88 and TRIF signaling in cardiac
dysfunction under those conditions remains to be investigated.
Secondly, the administration of anti-MyD88 and anti-TRIF antibodies
in the present model did not permit the delineation of the specific
roles of their upstream signaling, including TLR3 and TLR4, in
mediating cardiac dysfunction. Therefore, the effect of specific
TLRs in sepsis-induced cardiac dysfunction requires further
research.
In conclusion, the present study demonstrated that
inhibition of MyD88 and TRIF improves survival and cardiac function
in a murine model of CLP-induced severe sepsis. MyD88 and TRIF
inhibition have an equivalent effect on cardiac function. MyD88 and
TRIF signaling pathways may be promising targets for the treatment
of severe sepsis-linked myocardial dysfunction in clinical
practice.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81201096 and
81503445) and Hunan Province Science and Technology program (grant
nos. 2013SK3041 and 2016JJ2095).
References
|
1
|
Schefold JC, Bierbrauer J and
Weber-Carstens S: Intensive care unit-acquired weakness (ICUAW) and
muscle wasting in critically ill patients with severe sepsis and
septic shock. J Cachexia Sarcopenia Muscle. 1:147–157. 2010.
View Article : Google Scholar
|
|
2
|
No authors listed. American College of
Chest Physicians/Society of Critical Care Medicine Consensus
Conference: Definitions for sepsis and organ failure and guidelines
for the use of innovative therapies in sepsis. Crit Care Med.
20:864–874. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Angus DC, Linde-Zwirble WT, Lidicker J,
Clermont G, Carcillo J and Pinsky MR: Epidemiology of severe sepsis
in the United States: Analysis of incidence, outcome, and
associated costs of care. Crit Care Med. 29:1303–1310. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parrillo JE, Parker MM, Natanson C,
Suffredini AF, Danner RL, Cunnion RE and Ognibene FP: Septic shock
in humans. Advances in the understanding of pathogenesis,
cardiovascular dysfunction, and therapy. Ann Intern Med.
113:227–242. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rudiger A and Singer M: Mechanisms of
sepsis-induced cardiac dysfunction. Crit Care Med. 35:1599–1608.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Niu J, Azfer A and Kolattukudy PE:
Protection against lipopolysaccharide-induced myocardial
dysfunction in mice by cardiac-specific expression of soluble Fas.
J Mol Cell Cardiol. 44:160–169. 2008. View Article : Google Scholar
|
|
7
|
Fauvel H, Marchetti P, Chopin C,
Formstecher P and Nevière R: Differential effects of caspase
inhibitors on endotoxin-induced myocardial dysfunction and heart
apoptosis. Am J Physiol Heart Circ Physiol. 280:H1608–H1614. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Neill LA, Golenbock D and Bowie AG: The
history of Toll-like receptors - redefining innate immunity. Nat
Rev Immunol. 13:453–460. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mann DL: The emerging role of innate
immunity in the heart and vascular system: For whom the cell tolls.
Circ Res. 108:1133–1145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chao W: Toll-like receptor signaling: A
critical modulator of cell survival and ischemic injury in the
heart. Am J Physiol Heart Circ Physiol. 296:H1–H12. 2009.
View Article : Google Scholar :
|
|
11
|
Iwasaki A and Medzhitov R: Toll-like
receptor control of the adaptive immune responses. Nat Immunol.
5:987–995. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weighardt H, Kaiser-Moore S, Vabulas RM,
Kirschning CJ, Wagner H and Holzmann B: Cutting edge: Myeloid
differentiation factor 88 deficiency improves resistance against
sepsis caused by polymicrobial infection. J Immunol. 169:2823–2827.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guidotti LG and Chisari FV: Noncytolytic
control of viral infections by the innate and adaptive immune
response. Annu Rev Immunol. 19:65–91. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen C, Feng Y, Zou L, Wang L, Chen HH,
Cai JY, Xu JM, Sosnovik DE and Chao W: Role of extracellular RNA
and TLR3-Trif signaling in myocardial ischemia-reperfusion injury.
J Am Heart Assoc. 3:e0006832014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaczorowski DJ, Nakao A, Vallabhaneni R,
Mollen KP, Sugimoto R, Kohmoto J, Zuckerbraun BS, McCurry KR and
Billiar TR: Mechanisms of Toll-like receptor 4 (TLR4)-mediated
inflammation after cold ischemia/reperfusion in the heart.
Transplantation. 87:1455–1463. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Si R, Feng Y, Chen HH, Zou L, Wang
E, Zhang M, Warren HS, Sosnovik DE and Chao W: Myocardial ischemia
activates an injurious innate immune signaling via cardiac heat
shock protein 60 and Toll-like receptor 4. J Biol Chem.
286:31308–31319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Urheim S, Edvardsen T, Torp H, Angelsen B
and Smiseth OA: Myocardial strain by Doppler echocardiography.
Validation of a new method to quantify regional myocardial
function. Circulation. 102:1158–1164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Heimdal A, Støylen A, Torp H and Skjaerpe
T: Real-time strain rate imaging of the left ventricle by
ultrasound. J Am Soc Echocardiogr. 11:1013–1019. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cimolai MC, Alvarez S, Bode C and Bugger
H: Mitochondrial mechanisms in septic cardiomyopathy. Int J Mol
Sci. 16:17763–17778. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vinten-Johansen J: Involvement of
neutrophils in the pathogenesis of lethal myocardial reperfusion
injury. Cardiovasc Res. 61:481–497. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Castoldi A, Braga TT, Correa-Costa M,
Aguiar CF, Bassi EJ, Correa-Silva R, Elias RM, Salvador F,
Moraes-Vieira PM, Cenedeze MA, et al: TLR2, TLR4 and the MYD88
signaling pathway are crucial for neutrophil migration in acute
kidney injury induced by sepsis. PLoS One. 7:e375842012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu X, Zhao H, Graveline AR, Buys ES,
Schmidt U, Bloch KD, Rosenzweig A and Chao W: MyD88 and NOS2 are
essential for Toll-like receptor 4-mediated survival effect in
cardiomyocytes. Am J Physiol Heart Circ Physiol. 291:H1900–H1909.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alves-Filho JC, de Freitas A, Spiller F,
Souto FO and Cunha FQ: The role of neutrophils in severe sepsis.
Shock. 30(Suppl 1): 3–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feng Y, Zou L, Zhang M, Li Y, Chen C and
Chao W: MyD88 and Trif signaling play distinct roles in cardiac
dysfunction and mortality during endotoxin shock and polymicrobial
sepsis. Anesthesiology. 115:555–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zeng X, Moore TA, Newstead MW, Deng JC,
Kunkel SL, Luster AD and Standiford TJ: Interferon-inducible
protein 10, but not monokine induced by gamma interferon, promotes
protective type 1 immunity in murine Klebsiella pneumoniae
pneumonia. Infect Immun. 73:8226–8236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soraya H, Rameshrad M, Mokarizadeh A and
Garjani A: Metformin attenuates myocardial remodeling and
neutrophil recruitment after myocardial infarction in rat.
Bioimpacts. 5:3–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McDonald TE, Grinman MN, Carthy CM and
Walley KR: Endotoxin infusion in rats induces apoptotic and
survival pathways in hearts. Am J Physiol Heart Circ Physiol.
279:H2053–H2061. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cavassani KA, Ishii M, Wen H, Schaller MA,
Lincoln PM, Lukacs NW, Hogaboam CM and Kunkel SL: TLR3 is an
endogenous sensor of tissue necrosis during acute inflammatory
events. J Exp Med. 205:2609–2621. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang
W, He Q, Deng M, Liu X, Li G, et al: Toll-like receptor 3 (TLR3)
induces apoptosis via death receptors and mitochondria by
up-regulating the trans-activating p63 isoform alpha (TAP63alpha).
J Biol Chem. 286:15918–15928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Delhalle S, Blasius R, Dicato M and
Diederich M: A beginner's guide to NF-kappaB signaling pathways.
Ann NY Acad Sci. 1030:1–13. 2004. View Article : Google Scholar
|