Introduction
Premature ovarian insufficiency (POI) is a disorder
affecting ~1% of women under the age of 40 years (1) and is caused by two major mechanisms,
namely follicle dysfunction and follicle depletion (2), while the aetiology remains
undetermined in most cases (3).
In addition to complete loss-of-function mutations, partial
molecular defects may be the underlying cause of unexpected partial
phenotypes (4). Of note, certain
women with POI experience an unpredictable resumption of ovarian
function (1,5), with 3–10% conceiving spontaneously
after diagnosis (3,5). Genetic causes of non-syndromic POI
account for 10–15% of patients (2). Chromosomal abnormalities or rare
gene defects have been described in association with POI (2,6).
In non-syndromic POI, among autosomal genes, only rare mutations of
gonadotropin receptors or their ligands (7), of growth factors [bone morphogenetic
protein (BMP15), growth differentiation factor 9) or transcription
factors involved in follicular growth (splicing factor 1), of genes
involved in the establishment of the follicular pool and initial
recruitment (POF1B, folliculogenesis specific bHLH transcription
factor, NOBOX), and recently in meiosis (cohesin: Stromal antigen
3) have been described (4,6,8–12).
This strongly suggests that additional genetic factors remain to be
identified.
Gap junction intercellular communication (GJIC) has
a key role in mammalian ovarian follicle development (13). Gap junctions are channel clusters
formed by connexins, essential for oocyte-granulosa cell
communications. In mice, connexin 37 (Cx37) invalidation results in
ovarian folliculogenesis arrest at the preantral stage and female
infertility (14,15). Therefore, the present study
assessed the involvement of Cx37 in POI.
Subjects and methods
Patients and controls
The review boards of the institutions involved
approved this study and informed consent was obtained from all
patients. A total of 58 patients with POI from a national cohort
described in previous studies by our group were assessed (3,5).
These included 12 patients with primary and 46 with secondary
amenorrhea (including 21 with resumptive ovarian function). A
population of 142 Caucasian controls (from the Centre National de
Genotypage) was also studied.
DNA sequencing of the GJA4 gene
DNA was extracted from the peripheral leukocytes of
the patients. Automated direct genomic sequencing of the full
coding region of the gene encoding Cx37 (GJA4) was performed. Two
pairs of primers were used for polymerase chain reaction (PCR) to
amplify the coding regions of GJA4: Forward (F)1, 5'-GGA CGG AGG
CCG GGA GCC AT-3' and reverse (R)1, 5'-AGT GGC GCC TGT ACG GCT
GG-3'; F2, 5'-GGC AGG CTT CCT CTA TGG CC-3' and R2, 5'-TCC TGA GAA
GTC TGG CTG CCT GGG-3'.
PCR was performed using TaqDNA polymerase and PCR
mixture (cat. no. 10342-020; Invitrogen; Thermo Fisher Scientific,
Inc., Illkirch, France) under the following conditions: 94°C for 5
min, 30 cycles of 94°C for 30 sec, 62°C for 45 sec, 72°C for 1 min
and 60°C for 1 min, followed by a final elongation for 8 min at
72°C. A total of 10 primers were used for double-strand direct
genomic sequencing of GJA4 F1, 5'-GGA CGG AGG CCG GGA GCC AT-3' and
R1, 5'-GCA GAA GGA GGG GGA GCT-3'; F2, 5'-GAG CAG TCA GAT TTC GAG
TG-3' and R2, 5'-AGT GGC GCC TGT ACG GCT GG-3'; F3, 5'-CCT GTC TCG
GCG AGA AGA G-3' and R3, 5'-CTG CCT CAG CCG GGG GAT GA-3'; F4,
5'-GGC AGG CTT CCT CTA TGG CC-3' and R4, 5'-CAG AAC TGG GCC AAC CTG
A-3'; F5, 5'-GCT GTG TCG CTG CCT CAG-3' and R5, 5'-TCC TGA GAA GTC
TGG CTG CCT GGG-3'. Automated sequencing was performed with an ABI
310 Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA).
The c.860C>T heterozygous GJA4 mutation encoding
Cx37 p.Pro258Ser and introducing a HinfI restriction site
was also verified by restriction fragment length polymorphism
analysis. The c.1034G>A heterozygous GJA4 mutation encoding Cx37
p.Gly316Ser was verified in the control population by double-strand
direct sequencing.
Generation of GFP-Cx37-wild-type (WT) and
GFP-Cx37-mutant probes
Mutagenesis was assessed as follows: An OmicsLink™
Expression Clone [cytomegalovirus promoter; pReceiver-M29 (a,x,y)
Expression Clone; GeneCopoeia, Rockville, MD, USA] encoding the
wild-type Cx37 was applied. This vector encodes a fusion protein
with green fluorescent protein (GFP) at its N-terminus. The
QuikChange® II XL Site-Directed Mutagenesis kit (cat.
no. 200521; Stratagene, Cedar Creek, TX, USA). Mutant constructs
were engineered by oligonucleotide-mediated mutagenesis on this
vector by PCR. The c.749C>T (p.Pro258Ser) substitution was
engineered with two mutagenic primers: F,
5'-GGCACCTCCTCAGACTCTTACACGGACCAGGTCT-3' and R,
5'-AGACCTGGTCCGTGTAAGAGTCTGAGGAGGTGCC-3', binding at positions 757
and 790 of the complementary cDNA sequence, respectively, with
position +1 corresponding to the first nucleotide of the initiation
codon (ATG). The mutated bases are underlined. The c.946G>A
(p.Gly316Ser) mutation was engineered with two mutagenic primers:
F, 5'-GACCCACCCCCTCAGAATAGCCAAAAACCCCCAAGTC-3' and R,
5'-CTTGGGGGTTTTTGGCTATTCTGAGGGGGTGGGTC-3', binding at positions 928
and 964 of the cDNA sequence, respectively, with position +1
corresponding to the first nucleotide of the initiation codon
(ATG). The mutated bases are underlined. Mutants were verified by
direct sequencing with the following primer,
5'-TTAACCTGCTGGAGTTGGTG-3'.
Chemicals and antibodies
Calcein-red AM was purchased from Invitrogen (Thermo
Fisher Scientific, Inc., Illkirch, France), while rabbit
anti-giantin (1:1,000 dilution; ab80864), rabbit anti-early
endosome antigen 1 (EEA1; 1:1,000 dilution; ab2900) and mouse
anti-lysosome-associated membrane protein 2 (LAMP2; 1:100 dilution;
ab25631) (all from Abcam, Cambridge, UK) antibodies were purchased
from Covance (Princeton, NJ, USA), Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA) and BD Pharmingen (San Jose, CA, USA),
respectively, and goat anti-rabbit and anti-mouse secondary
antibodies conjugated to Alexa Fluor 647 (cat. nos. 115-605-146 and
111-605-144) were obtained from Jackson ImmunoResearch (West Grove,
PA, USA). GFP-Rab5 probe was a gift from M. Zerial Laboratory
(Dresden, Germany).
Cell culture and transfection
HeLa cells (ATCC, Manassas, VA, USA) were used for
functional studies, as they do not express functional connexins and
gap junctions. They were cultured in Dulbecco's modified Eagle's
medium (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal calf serum (Gibco, Waltham, MA, USA) and 1%
penicillin/streptomycin at 37°C in a humidified atmosphere
containing 5% CO2. The cells seeded into 96-well plates
at a density of 15,000 cells/well and were transfected in the
presence of Lipofectamine (Invitrogen; Thermo Fisher Scientific,
Inc.) as previously described (16). For each milliliter of transfection
mixture, 8 µl Lipofectamine was mixed with 200 µl
OptiMem (Invitrogen; Thermo Fisher Scientific, Inc.) and
simultaneously, 0.75 µg of each GFP-Cx37 (WT and/or mutant)
probe was mixed with 200 µl OptiMem in a separate vessel.
The two mixtures were incubated at room temperature in the dark for
45 min. The mixtures containing the probes were then individually
added to those containing Lipofectamine. The combination was mixed
smoothly and incubated for another 15 min at room temperature in
the dark. A total of 600 µl DMEM without serum was then
added dropwise to each well. The transfection efficiency was
assessed after 24 h by determining the proportion of total cells
transfected by GFP fluorescence. No significant difference in
transfection efficiency was observed between mutant and WT
expression vectors used separately [Cx37-P258S (47.12±1.65%) and
Cx37-G316S (46.81±3.46%) vs. Cx37-WT (50.46±2.07%); P=0.2771 and
0.4180, respectively, n=3].
Immunofluorescence imaging and
colocalization analysis
HeLa cells transfected for 24 h with expression
vectors encoding WT green fluorescence protein (GFP)-Cx37-WT, or
mutated GFP-Cx37-P258S or GFP-Cx37-G316S connexins, were fixed with
4% paraformaldehyde. The cells were permeabilized by incubation for
5 min in 0.1% saponin in phosphate-buffered saline (PBS), and then
blocked by incubation in 3% bovine serum albumin (Sigma-Aldrich,
St. Louis, MO, USA) and 0.01% saponin in PBS for 30 min at room
temperature. The cells were incubated at room temperature with
primary antibodies in blocking solution for 2 h, and then with
secondary antibodies conjugated with Alexa Fluor 647 and
4',6-diamidino-2-phenylindole (DAPI) for the labeling of nuclei for
1 h. Images were acquired with a Zeiss LSM512 confocal microscope
(Zeiss AG, Jena, Germany). The number of colocalized objects
(structures or vesicles positive for staining) was analyzed, as
previously described (17), with
ImageJ software (National Institutes of Health, Bethesda, MD, USA;
release 1.48e 19 October 2013). For each image, a colocalization
mask was first generated using the colocalization plugin for ImageJ
software (Colocalization Highlighter; Pierre Bourdoncle, Institut
Jacques Monod, Service Imagerie, Paris, France). The number of
objects in each channel (green, red and colocalized) was then
estimated with the particle counter of ImageJ software. Finally,
the number of colocalized particles was determined by dividing the
number of colocalized objects by the total number of particles.
Gap-fluorescence recovery after
photobleaching (FRAP) analysis
HeLa cells, either untransfected or transfected with
GFP-Cx37-WT, alone or together with GFP-Cx37-G316S or
GFP-Cx37-P258S expression vectors for 24 h, were loaded for 30 min
at 37°C with 5 µM calcein-red AM (Invitrogen; Thermo Fisher
Scientific, Inc.) in PBS. Dye transfer was then monitored with a
Zeiss LSM 510 confocal microscope, as previously described
(18). Randomly selected cells
were bleached with a 543-nm laser pulse and the recovery of
fluorescence intensity was monitored by analyzing data obtained
prior to bleaching, just after bleaching and 30 min after
photobleaching using Zeiss LSM software (version 2.01). The
percentage of fluorescence recovery was determined by normalizing
all values to the intensities prior to photo-bleaching set as 100%
and subtracting the normalized value for cells just after
photobleaching from the normalized value obtained at 30 min after
photobleaching. At least 20 cells were analyzed for each set of
experimental conditions and at least three separate experiments
were performed.
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. Student's t-test or analysis of variance followed by
Bonferroni's post hoc comparison was used for statistical analysis
(GraphPad, Prism, version 5.03). P<0.05 was considered to
indicate a statistically significant difference.
Results
A common African variant of human Cx37
may be associated with Caucasian primary ovarian insufficiency
Direct genomic sequencing of the GJA4 coding region
in 58 POI patients identified a single specific variant in two
Caucasian patients, which was absent in the ethnically matched
Caucasian controls. An identical heterozygous substitution [c.946
G>A, (NM_002060)] in these two patients led to the production of
a p.Gly316Ser (G316S) Cx37 mutant protein (Fig. 1A).
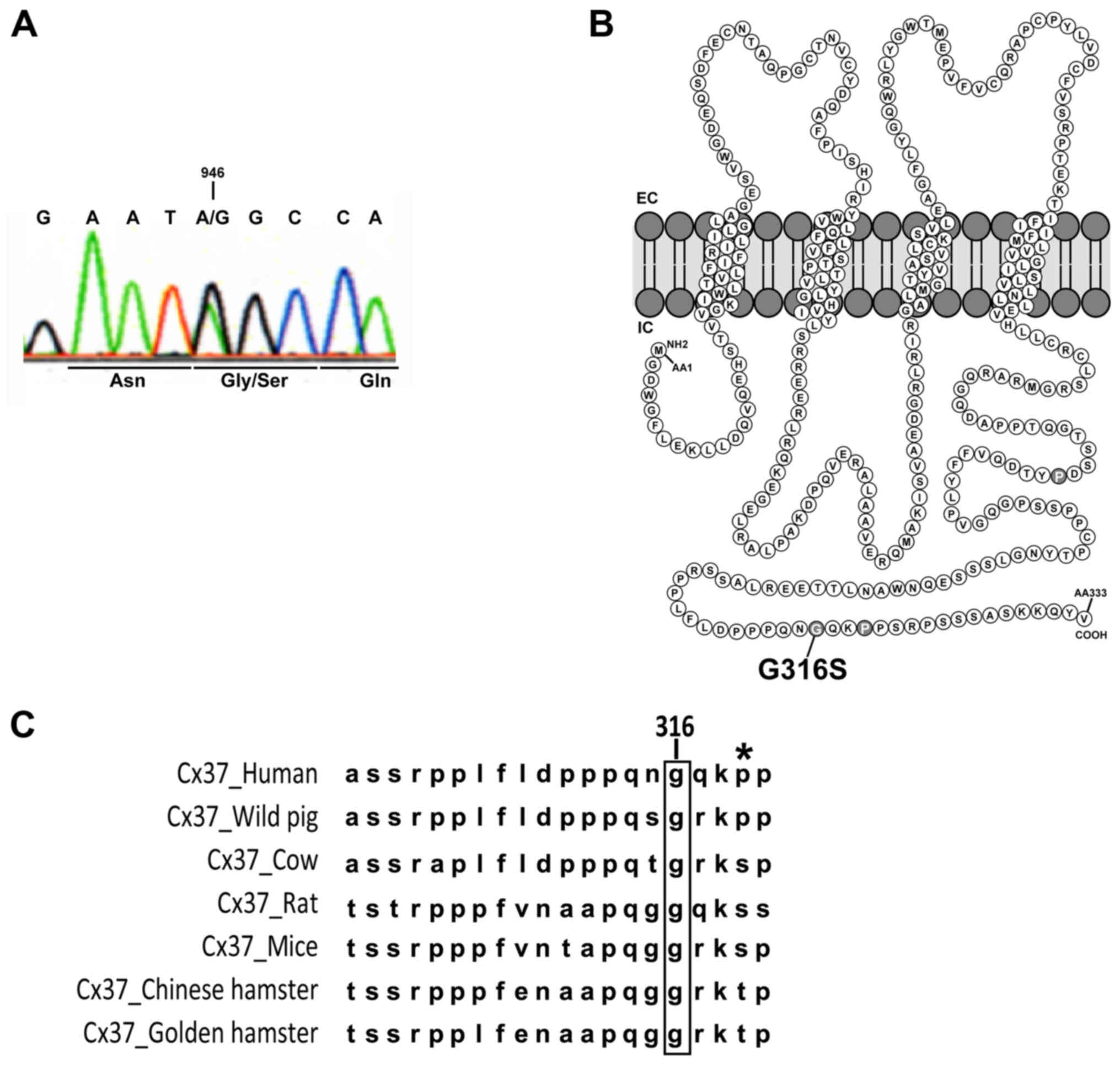 | Figure 1Gly316Ser mutation in human Cx37
identified in two patients with POI. (A) Direct genomic sequencing
of GJA4 for one POI patient with the c.946G>A variant. The
changed base and the corresponding codon are indicated. (B)
Schematic representation of Cx37 and localization of the G316S
residue in the C-terminal part of the intracellular domain. (C)
Alignment of Cx37 sequences in different species (human, Homo
sapiens; wild pig, Sus scrofa; cow, Bos taurus;
rat, Rattus norvegicus; mouse, Mus musculus; Chinese
hamster, Cricetulus griseus; golden hamster, Mesocricetus
auratus). The high level of conservation of the glycine 316
residue (in brackets) was apparent among the species. The asterisk
indicates the well-described P319S polymorphism of Cx37 present in
all populations studied in databases (note that this residue has
not been conserved during evolution). Cx, connexin; POI, primary
ovarian insufficiency; EC/IC, extra/intracellular space. |
The affected residue, at the Cx37 COOH-terminus
(Fig. 1B), is highly conserved
across species (Fig. 1C). Various
databases [National Center for Biotechnology Information, Ensembl,
1000 Genomes (1000G), exome variant server (ESV)] were searched for
the variants identified in the present study. The rs140949366
variant corresponding to c.946G>A encoding p.G316S was neither
detected in European, non-African North or South American,
Caribbean Puerto Rican and Asian populations according to the 1000G
database (0/1,688 chromosomes) nor detected in European Americans
according to the ESV database (0/8,600 chromosomes). However, the
rs140949366 variant was included in the 1000G and ESV databases,
but exclusively occurred in African populations. It was detected 12
times in a sample population of 1,088 individuals (or 2,176
chromosomes) from populations of African origin from Nigeria,
Kenya, Gambia and African Americans according to the 1,000G
database. This variant was also listed in the ESV database
exclusively in African Americans in a sample population of 12,885
chromosomes (121/4285). In the Exome Aggregation Consortium (ExAC)
database, this variant was listed only 9 times in 72,500 Caucasian
chromosomes, but no information was available regarding the gender
and phenotype of the relevant individuals and the pathogenicity of
the variant.
Two Pro258Ser and Pro319Ser polymorphisms
(rs28739284 and rs1764391) present in the POI patients were also
identified to be present in all ethnic populations of different
databases and also in the control population of the present
study.
In conclusion, an association between a Cx37
variant, which was absent in ethnically matched Caucasian
populations, but present in 2 Caucasian patients with POI was
identified. This variant is highly conserved across species,
suggesting a possible functional role.
Case presentations
The two patients with a Cx37 mutation
identified are described below
Case 1
The first patient was a 25-year-old Caucasian woman
who was referred to the Department of Endocrinology and
Reproductive Medicine, Pitie Salpetriere Hospital (Paris, France)
for secondary amenorrhea and primary infertility. She had a normal
pubertal development and regular menstrual cycles since the age of
11. She displayed secondary amenorrhea and experienced hot flushes
at the age of 23 years. No hormonal evaluation had been performed
at this age. Various sequential progestin tests resulted in vaginal
bleeding between the ages of 23 and 25 years. At the age of 25
years, the patient underwent hormonal and ultrasound evaluations at
the same department leading to the diagnosis of POI (Table I). Androgen levels [testosterone,
androstenedione and dehydroepiandrosterone sulfate (DHEA-S)] were
within normal ranges. Metabolic evaluation (glycaemia, cholesterol
and triglyceride levels) had no abnormalities. Ovarian
ultrasonography indicated the presence of a 34×20-mm cyst and two
5-mm follicles on the left ovary and of a normal-sized 30×14-mm
right ovary with a 5-mm follicle. The menstrual cycle resumed
spontaneously at 2 months after this evaluation, lasting 28 to 40
days. The patient revisited the center at the age of 28. She
continued to have regular menstrual cycles of 40 days and
considered pregnancy at this time. The patient's mother had 6
spontaneous pregnancies between the ages of 24 and 30 years,
resulting in 3 spontaneous abortions and 3 children, two of which
were male and had no reported fertility problems and case 1 of the
present study. The patient's mother presented with premature
ovarian failure at the age of 38 years and was unavailable for
further analysis.
 | Table IClinical, hormonal and US
characterisation of patients with connexin 37 mutation. |
Table I
Clinical, hormonal and US
characterisation of patients with connexin 37 mutation.
| Case | Type of amenorrhea
(age at onset) | Age at evaluation
(years) | Day of the
menstrual cycle | FSH UI/l | LH UI/l | E2
pg/ml | Inhibin B
pg/ml | AMH ng/ml | T ng/ml | P ng/ml | Ovarian surface
(right/left) cm2 | Presence of
follicles at US (size of the largest, mm) | Resumption of
ovarian function |
|---|
| Patient 1 | Secondary (23
years) | 25 | Amenorrhea | 40 | 10.8 | 20 | 15 | <0.05 | 0.3 | 0.3 | 3.4/5.9 | Yes (14) | Yes (resumption of
menstrual cycles) |
| 28 | 3 109 | | 51 | 46 | ND | ND | ND | 0.4 | ND | Yes (8) | |
| 28 | 8 106 | | 51 | 43 | ND | ND | ND | 0.4 | ND | ND | |
| Patient 2 | Secondary (25
years) | 31 | Amenorrhea | 73 | 36 | <10 | <10 | <0.05 | 0.2 | ND | ND | No | No |
Normal baseline
values
|
|---|
| Phase | FSH (U/l) | LH (UI/l) | E2
(pg/ml) | T (ng/ml) | P (ng/ml) | Inhibin B
(pg/ml) |
|---|
| Follicular | 3.5–12 | 1.5–8 | 20–85 | 0.1–0.48 | 0.2–1.5 | 10–276 |
| Ovulatory | 4.7–21.5 | 9.6–80 | 82–287 | 0.1–0.48 | 0.8–3.0 | |
| Luteal | 1.7–7.7 | 0.2–6.5 | 8–33 | 0.1–0.48 | 1.7–27.0 | |
| Menopause | 26–135 | 8–33 | <35 | 0.1–0.4 | 0.1–0.8 | <10 |
Case 2
The second patient was a 31-year-old Caucasian woman
born to consanguineous parents with congenital familial deafness of
unknown aetiology. Her brother also suffered from deafness. She had
a normal puberty and menses from the age of 13 onward. The
diagnosis of premature ovarian insufficiency was performed when she
was 25 and had developed secondary amenorrhea (Table I). At the age of 31 (October,
2007), hormonal measurements revealed the following:
Follicle-stimulatory hormone (FSH) N:3.5-12), 73 UI/l; lutenizing
hormone (LH), 36 U/l (N:1.5-8); estradiol, <10 pg/ml (N:20-85 in
the follicular phase); anti-mullerian hormone, <0.05 ng/ml
(N:2,2-6.8) and inhibin B, <10 pg/ml (N:10-276). Androgen levels
(testosterone, androstenedione and DHEA-S) were within the normal
range (Table I). Ovarian
ultrasonography at the age of 31 revealed two hypotrophic ovaries
without follicles. No further evaluation was performed since the
patient did not return to our center. Exploration of her deafness
in another reference center excluded a mutation of Cx26.
Functional studies of WT and mutated
connexons
Reduced gap-junction functionality in
cells expressing the Gly316Ser Cx37 variant
A Gap-FRAP analysis was performed on connexin-free
HeLa cells (18), which were
loaded with calcein-AM and photobleached. The fluorescence recovery
was then monitored for 30 min (Fig.
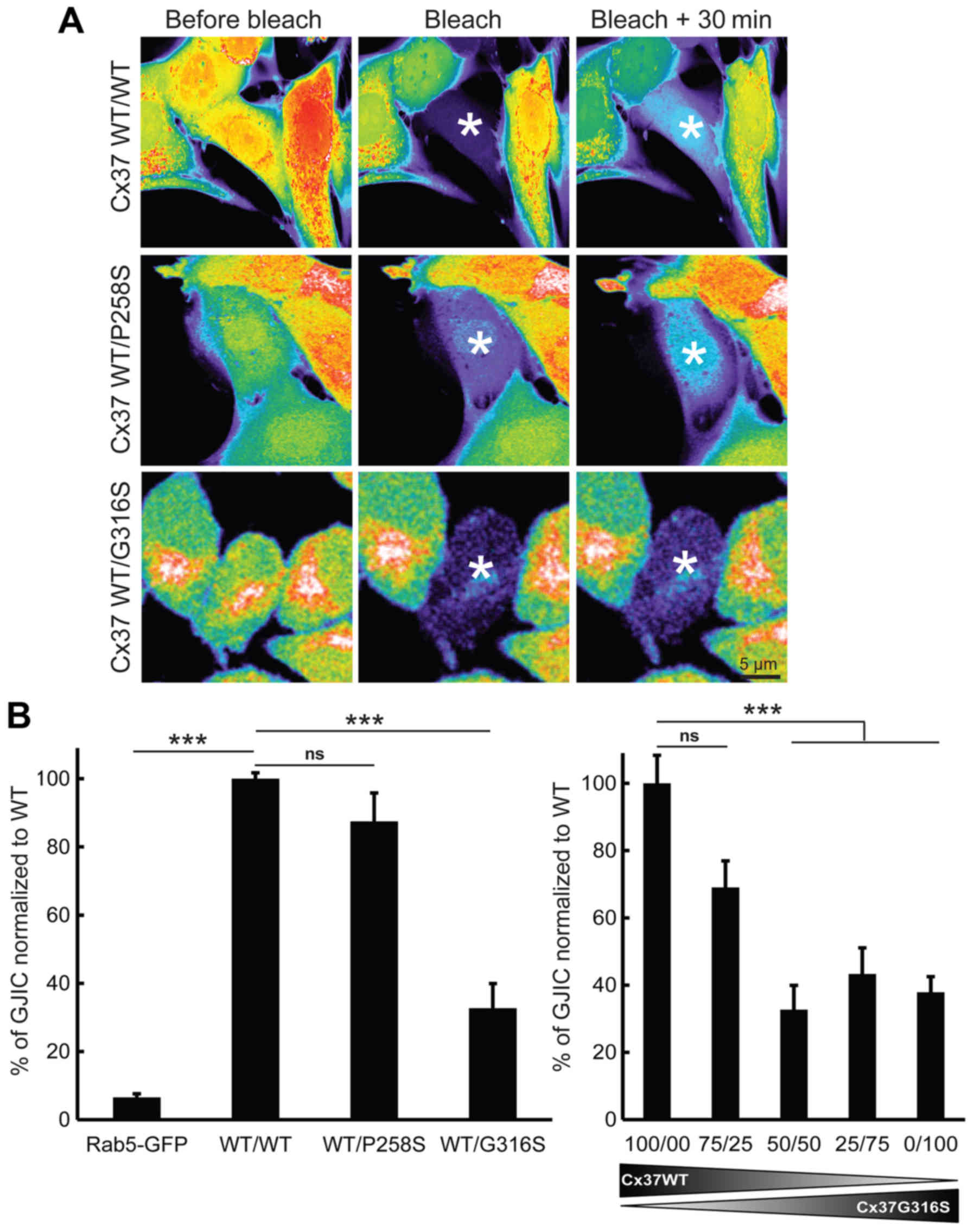
2A). Fluorescence recovery was almost undetectable when a
GFP-Rab5 expression vector was used as a negative control (Fig. 2B, left panel) (n=3). First, the
effect of the P258S variant was assessed. GJIC functionality in
cells expressing only Cx37-WT did not differ significantly from
that in cells co-expressing Cx37-WT and Cx37-P258S (P=0.2617; n=3)
(Fig. 2A, top and middle panels
and Fig. 2B, left panel). This
value was significantly higher than that of control cells
transfected with GFP-Rab5 (with a 20-fold increase; P=0.0001; n=3)
(Fig. 2B). The G316S variant,
which was identified in certain Caucasian POI patients but absent
in the controls of the present study and in Caucasian populations
of databases was then studied. In contrast to the observation for
the P258S variant, the G316S variant was less functional in
vitro. The functionality of GJIC was ~67.2±7.17% lower
(P=0.0005, n=5) when cells were co-transfected with Cx37-WT and
Cx37-G316S (50:50) expression vectors (Fig. 2A, bottom and Fig. 2B, left panel). The use of smaller
amounts of Cx37-G316S vector (75:25) rescued GJIC functionality
(only ~30.9±7.89% lower; P=0.0542; n=3) (Fig. 2B, right panel). Increasing the
ratio of Cx37-G316S to Cx37-WT (25:75 or 0/100) did not further
decrease GJIC functionality [56.7±7.77% (P=0.0076; n=3) and
62.1±4.62% (P=0.0029; n=3), respectively]. It was therefore
concluded that the G316S variant has a partial dominant-negative
effect on WT Cx37 in vitro.
Decreased expression of cell
surface-associated gap junctions in cells expressing the G316S-Cx37
variant
The presence of cell-surface gap junction plaque was
then assessed (Fig. 3A, arrows).
As expected, no difference was identified between cells transfected
with GFP-Cx37-WT and GFP-Cx37-P258S expression vectors (P=0.204;
n=3) (Fig. 3A). By contrast, much
weaker expression was observed in cells co-expressing GFP-Cx37-WT
and GFP-Cx37-G316S (47.73±8.59%; P=0.0052; n=3) (Fig. 3A). Furthermore, more
connexin-loaded large vesicles accumulated within cells transfected
with the G316S variant (Fig. 3A,
arrowheads). It was concluded that the G316S variant, but not the
P258S variant, decrease cell-surface gap junction plaque
expression.
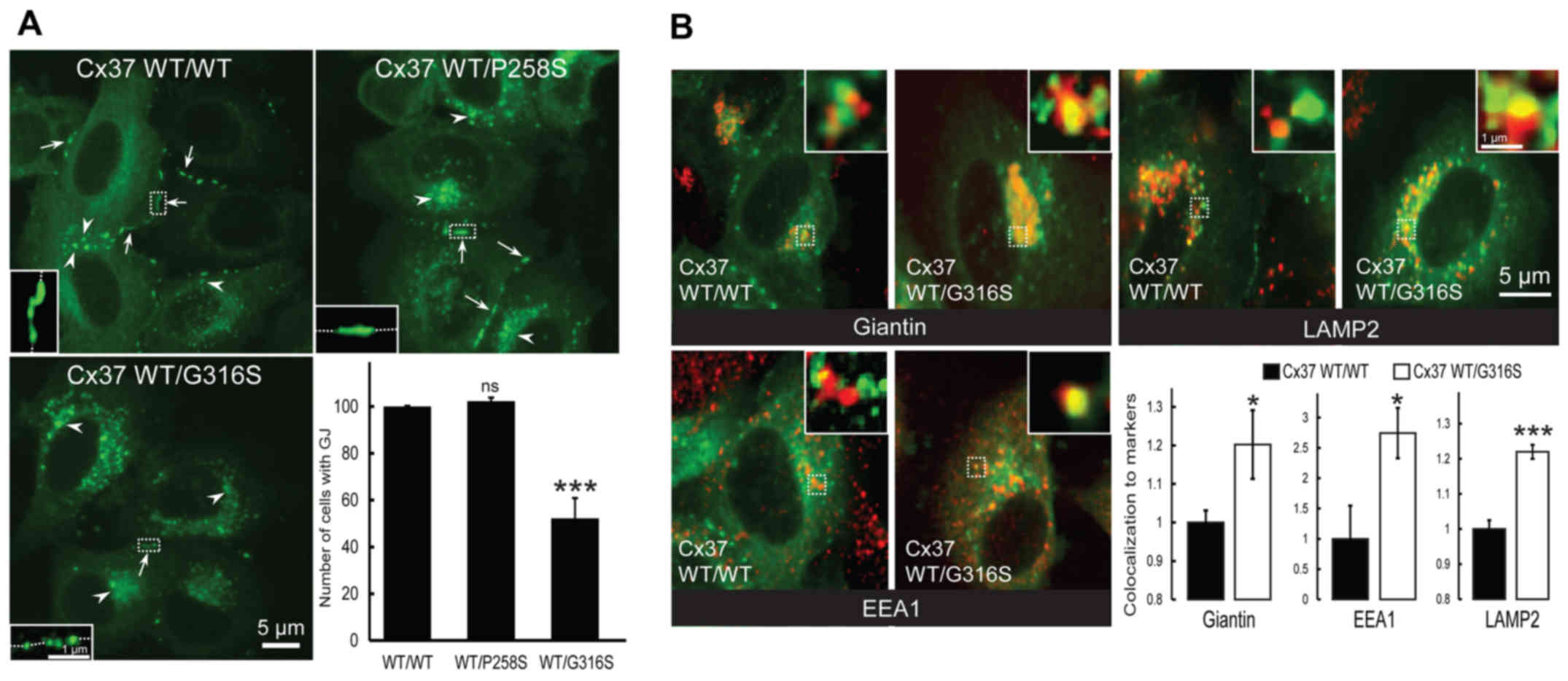 | Figure 3The Cx37-G316S variant markedly
decreases the number of gap junction plaques by decreasing Cx37
exocytosis and increasing Cx37 endocytosis and degradation. (A)
Representative images of HeLa cells transfected with GFP-Cx37-WT
alone or together with GFP-Cx37-P258S or GFP-Cx37-G316S expression
vectors. The number of gap junction plaques (arrows) was
significantly lower in cells expressing GFP-Cx37-G316S, which also
contained a larger number of cytoplasmic vesicles (arrowheads)
(scale bar, 5 µm). For each images, a higher magnification
was provided as inset and localized by dash-line squares to
visualized more easily gap junction plaques at the cell-cell
contacts (represented by dash-line in the insets). (B)
Representative images of HeLa cells transfected with GFP-Cx37-WT
alone or together with GFP-Cx37-G316S (green) and labeled (red) for
giantin (Golgi marker), EEA1 (early endosomes) or LAMP2
(lysosomes). For each image, a higher magnification of 2x2
µm was provided as insets and localized by dash-line squares
to visualized more easily co-localization (yellow color). The
bottom right panel displays the quantification of Cx37
colocalization with the various markers used. *P<0.05
and ***P<0.01. ns, not significant; Cx, connexin; GJ,
gap junction; WT, wild-type; GFP, green fluorescence protein; EEA1,
early endosome antigen 1; LAMP2, lysosome-associated membrane
protein 2. |
Altered trafficking of mutated G316S
connexons in HeLa cells
Co-localization experiments with intracellular
markers of Cx37-WT homomeric or Cx37-WT/Cx37-G316S heteromeric
connexons were then performed. Co-localization with Giantin (a
Golgi compartment marker) was significantly increased by the
variant Cx37-G316S, by 20.2±8.9% (P=0.0369; n=6) (Fig. 3B, top left and bottom right
panels), whereas that with EEA1, a specific endosomal marker, was
markedly increased, by 174.5±41.6% (P=0.0295; n=6) (Fig. 3B, bottom left and right panels),
and that with LAMP2, a specific lysosomal marker, was increased by
21.9±1.98% (P=0.0001; n=6) (Fig.
3B, top right and bottom right panels). The G316S-Cx37 variant
thus appeared to decrease gap junction plaque formation at the cell
surface, probably by increasing gap junction plaque endocytosis and
lysosomal degradation. By contrast, no deleterious functional
effect was detected with the P258S variant.
Discussion
The present study reported on an association between
a single Cx37 variant and POI in two of 58 Caucasian patients from
our national cohort (3), which
was absent in the Caucasian control population, corresponding to
two of the 58 patients with POI and 46 patients with secondary
amenorrhea.
A search of various databases indicated that the
G316S mutation has not been reported in European or Caucasian North
and South American, Caribbean and Asian populations. However, it
has been reported in African populations according to the 1000G and
ESV databases, strongly suggesting an ethnicity-specific
interaction with environmental and/or genetic factors. In the ExAC
database, in which a large number of chromosomes have been
included, the G316S variant is rare in Caucasian populations, with
9/72,500 alleles (0.01241%) vs. 285/10,196 alleles (2.795%)
detected in African populations. However, ExAC is unable to provide
any information on the gender and the clinical status of variant
carriers and it cannot be excluded that Caucasian female carriers
are affected or will develop POI prior to the age of 40 years.
However, it is unlikely that this variant is responsible for POI in
African populations. Ethnicity-specific effects on genetic variants
have been previously described (19–21). An important number of autosomal
dominant disorders are characterized by incomplete penetrance
(22), which may occur in
specific populations. The asymptomatic state appears to be more
common than the clinically overt state in several dominant
disorders of haemostasis and thrombosis. Reduced penetrance in
dominantly inherited diseases is likely to be the consequence of a
combination of different genetic and environmental factors, which
may vary among ethnic groups. Additional coding or non-coding
variants acting in cis or trans may influence the expression of a
potentially pathogenic allele or modulate its pathogenicity. In the
genetic background of the patients of the present study, changes in
conformation due to folding motif disruption or in protein-protein
interactions critical for Cx37 trafficking may lead to protein
retention within the cell and impaired GJIC function (23). In certain autosomal dominant
conditions, the two alleles of a disease-associated gene are not
expressed at the same level. This phenomenon drives a stronger
expression of either the mutant or the WT allele, which may
influence the clinical penetrance of the disorder. This is the case
for retinis pigmentosa type 11, erythropoietic protoporphyria or
Hirschsprung disease, in which penetrance depends not only on the
nature of the mutation but also on the allele gene dosage (22). Modifier genes may also influence
disease penetrance. The possibility that another mutation in an
unknown gene is necessary for the expression of the phenotype in
the Caucasian patients in a digenic mode of inheritance (24) or conversely that the African
populations are protected by a common allele cannot be ruled out. A
positive selection of the p.G316S variant in African populations
may have also occurred, as this has been reported for the genes
encoding globins, or in the case of glucose-6-phosphate
dehydrogenase deficiency, leading to hematologic disorders when
homozygous (sickle cell anemia or thalassemia) but protecting from
malaria in a heterozygous state (25). An extreme case of positive
selection across a wider evolutionary span has been reported for
BMP15. Indeed, the substitution Y235C, which was the first mutation
identified to be associated with POI (26), has invaded entire species and has
been fixed as the WT allele in the hominids Pan troglodites
and Pan paniscus (27).
Obviously, the genetic background of these hominids has compensated
any potential deleterious effects. Although not demonstrated in the
present study, a similar compensatory effect may be present in
African populations, which may not be unlikely given their higher
genetic diversity and extensive population substructure compared
with those of non-African populations. Africans also possess a
number of genetic adaptations that have evolved in response to
diverse climates and diets, as well as exposure to infectious
disease (28). Recent genome-wide
association studies on large populations have strongly associated a
non-synonymous single nucleotide popymorphism in the
mini-chromosome maintenance 8 homologous recombination repair
factor (MCM8) gene involved in DNA repair with the age of menopause
in Caucasian populations (29),
but not in African populations (30). As mutations of MCM8 are
responsible for POI (31), it is
likely that the variant associated with the age of menopause in
Caucasian populations is functional in those populations but not in
African populations. It is also possible that Africans may have
another mechanism to compensate the function of the MCM8
variant.
Several results provide additional support for the
possible involvement of this G316S variant in disease pathogenesis
in the Caucasian patients of the present study: i) The strong
evolutionary conservation of the mutated residue supporting an
important functional role; ii) the demonstrated functional
consequences of the Cx37-G316S variant in vitro, while the
P258S polymorphism present in different ethnic populations does not
have any functional effect; and iii) the correlation between the
phenotype of the patients with secondary amenorrhea and the partial
dominant-negative effect of this heterozygous variant on the WT
allele. Most of the connexin mutations implicated in human disease
are dominant-negative (32).
Cx37-deficient mice are infertile (14,15) due to arrested preantral follicle
development. A study on mice with chimeric ovaries containing null
mutant cells have indicated that GJIC between granulosa cells
involves Cx43, whereas GJIC between oocytes and granulosa cells is
constituted by Cx37 heterotypic channels (33).
The patients of the present study harbouring the
Gly316Ser variant had a less severe ovarian phenotype than
GJA4-knockout mice, as case 1 exhibited antral follicle development
and estrogen secretion and case 2 presented with secondary
amenorrhea. Indeed, the G316S variant has a partial
dominant-negative effect on the WT Cx37 in vitro. The
Cx37-G316S variant greatly decreased the number of gap junctions at
the cell surface (by 47.73±8.59%) and gap junction functionality
(by 67.2±7.17%). However, this difference was not statistically
significant (P=0.0838), indicating that poorer functionality
resulted mostly from changes in the gap junction number at the
cell-cell boundary. The G316 variant affects the Cx37 COOH-terminal
domain, which is important for protein-protein interactions and
trafficking (23,34). Connexin trafficking is regulated
by phosphorylation (34). The
Ser316 residue was predicted to be phosphorylated (NetPhos 2.0
freeware; Technical University of Denmark, Kongens Lyngby,
Denmark). This variant may therefore affect Cx37 trafficking and/or
gap junction stabilization at the cell surface.
In mice with streptozotocin-induced diabetes,
follicle maturation is delayed, with cumulus oocyte complex
expansion failure and anovulation or dysovulation (35,36). A 60% decrease in GJIC and a 36%
decrease in Cx37 protein levels (35) have been observed in this model,
close to the values determined by the present study for the
Cx37-G316S variant in vitro. These mice are subfertile,
rather than infertile, with low-frequency ovulation, and even rare
pregnancies. This phenotype is similar to that presented by case 1
of the present study, with resumption of ovarian function. Case 2
developed a secondary ovarian insufficiency, but the possibility of
an additional genetic defect cannot be excluded. The latter may
have impacted the phenotypic expression of the GJA4 mutation.
Alternatively, variable phenotypic expression of a single molecular
defect has been frequently documented in human reproductive
disorders. Indeed, intra- and inter-familiar phenotypic variability
has been documented for the same forkhead box L2 mutation, inducing
blepharophymosis syndrome type 1 (associated with ovarian failure)
occurring twice in the same family. This may reflect the
combination of the effect of modifier genes or of a different
genetic background, and possibly of the environment (37).
The resumption of ovarian function in the presence
of constantly high gonadotropin concentrations in case 1 suggests a
compensatory role of gonadotropins. FSH and LH regulate follicular
development and oocyte maturation, and high LH levels may
contribute to meiosis resumption (38,39). Connexin phosphorylation may also
be physiologically enhanced by gonadotropins (13,38). FSH increases Cx43 synthesis and
plasma membrane translocation (40,41). Similarly, FSH may also increase
Cx37 synthesis and connexon targeting to the cell surface up to the
threshold required for complete follicular function. Cx43 has been
demonstrated to be present in human cumulus cells in vitro
and may form heterotypic connexons with Cx37 in the oocyte
(42). Cx43 has been demonstrated
to compensate for defective Cx37 function (43). An analogous compensatory effect of
an FSH-induced Cx43 increase cannot be excluded in the patients of
the present study. Cx37 appears to be indispensable for complete
oocyte growth and meiotic competence in mice (15). Oocytes from Cx37-deficient mice
cannot enter the M phase (initial meiotic maturation) (15). However, the G316S mutation in the
patients of the present study led to the partial trapping of
certain G316S connexons within the cell, whereas the rest was
targeted to the cell surface and was demonstrated to be functional
in vitro. Partial oocyte maturation may thus be possible and
this, together with prolonged high gonadotropin concentrations in
the blood, may lead to the coordinate completion of follicular and
oocyte maturation and resumption of ovarian function in certain
patients.
The present study demonstrated an association
between a Cx37 variant absent in Caucasian populations but present
in 2 Caucasian POI patients, displaying a dominant-negative effect
in vitro. These results suggested that in the genetic
context of the Caucasian population, this variant may contribute to
the POI phenotype in vivo. Exome studies may be performed in
the future to identify further causative mutations associated with
the development of POI in the patients of the present study.
Further experiments in physiologically relevant models are required
to incriminate this Cx37 variant as having a causative role in the
pathology of POI alone or in association with another causative
genes, as observed in other reproductive diseases (24).
Acknowledgments
The authors would like to thank Dr Georges Pointis
(INSERM U1065, F-06000 Nice, France) and Dr Emmanuel Génin (INSERM
U1078, Brest, France) for their helpful discussions, as well as
Dominique Segretain (Paris Descartes University, 75005 Paris,
France) and Laurent Combettes (INSERM U757, 91400 Orsay, France)
for their helpful discussions and for providing access to their
laboratory facilities and materials.
References
|
1
|
European Society for Human Reproduction
and Embryology (ESHRE) Guideline Group on POI; Webber L, Davies M,
Anderson R, Bartlett J, Braat D, Cartwright B, Cifkova R, de Muinck
Keizer-Schrama S, Hogervorst E, Janse F, et al: ESHRE Guideline:
management of women with premature ovarian insufficiency. Hum
Reprod. 31:926–937. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nelson LM: Clinical practice. Primary
ovarian insufficiency. N Engl J Med. 360:606–614. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bachelot A, Rouxel A, Massin N, Dulon J,
Courtillot C, Matuchansky C, Badachi Y, Fortin A, Paniel B, Lecuru
F, et al: POF-GIS Study Group: Phenotyping and genetic studies of
357 consecutive patients presenting with premature ovarian failure.
Eur J Endocrinol. 161:179–187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beau I, Touraine P, Meduri G, Gougeon A,
Desroches A, Matuchansky C, Milgrom E, Kuttenn F and Misrahi M: A
novel phenotype related to partial loss of function mutations of
the follicle stimulating hormone receptor. J Clin Invest.
102:1352–1359. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bidet M, Bachelot A, Bissauge E, Golmard
JL, Gricourt S, Dulon J, Coussieu C, Badachi Y and Touraine P:
Resumption of ovarian function and pregnancies in 358 patients with
premature ovarian failure. J Clin Endocrinol Metab. 96:3864–3872.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tucker EJ, Grover SR, Bachelot A, Touraine
P and Sinclair AH: Premature ovarian insufficiency: New
perspectives on genetic cause and phenotypic spectrum. Endocr Rev.
37:609–635. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meduri G, Bachelot A, Cocca MP, Vasseur C,
Rodien P, Kuttenn F, Touraine P and Misrahi M: Molecular pathology
of the FSH receptor: New insights into FSH physiology. Mol Cell
Endocrinol. 282:130–142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang J, Zhang W, Jiang H and Wu BL:
Primary Ovarian Insufficiency Collaboration: Mutations in HFM1 in
recessive primary ovarian insufficiency. N Engl J Med. 370:972–974.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Caburet S, Arboleda VA, Llano E, Overbeek
PA, Barbero JL, Oka K, Harrison W, Vaiman D, Ben-Neriah Z,
García-Tuñó I, et al: Mutant cohesin in premature ovarian failure.
N Engl J Med. 370:943–949. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bouilly J, Bachelot A, Broutin I, Touraine
P and Binart N: Novel NOBOX loss-of-function mutations account for
62% of cases in a large primary ovarian insufficiency cohort. Hum
Mutat. 32:1108–1113. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wood-Trageser MA, Gurbuz F, Yatsenko SA,
Jeffries EP, Kotan LD, Surti U, Ketterer DM, Matic J, Chipkin J,
Jiang H, et al: MCM9 mutations are associated with ovarian failure,
short stature, and chromosomal instability. Am J Hum Genet.
95:754–762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
AlAsiri S, Basit S, Wood-Trageser MA,
Yatsenko SA, Jeffries EP, Surti U, Ketterer DM, Afzal S, Ramzan K,
Faiyaz-Ul Haque M, et al: Exome sequencing reveals MCM8 mutation
underlies ovarian failure and chromosomal instability. J Clin
Invest. 125:258–262. 2015. View
Article : Google Scholar :
|
|
13
|
Gershon E, Plaks V and Dekel N: Gap
junctions in the ovary: Expression, localization and function. Mol
Cell Endocrinol. 282:18–25. 2008. View Article : Google Scholar
|
|
14
|
Simon AM, Goodenough DA, Li E and Paul DL:
Female infertility in mice lacking connexin 37. Nature.
385:525–529. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carabatsos MJ, Sellitto C, Goodenough DA
and Albertini DF: Oocyte-granulosa cell heterologous gap junctions
are required for the coordination of nuclear and cytoplasmic
meiotic competence. Dev Biol. 226:167–179. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gilleron J, Fiorini C, Carette D, Avondet
C, Falk MM, Segretain D and Pointis G: Molecular reorganization of
Cx43, Zo-1 and Src complexes during the endocytosis of gap junction
plaques in response to a non-genomic carcinogen. J Cell Sci.
121:4069–4078. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gilleron J, Carette D, Fiorini C,
Dompierre J, Macia E, Denizot JP, Segretain D and Pointis G: The
large GTPase dynamin2: A new player in connexin 43 gap junction
endocytosis, recycling and degradation. Int J Biochem Cell Biol.
43:1208–1217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Carette D, Gilleron J, Decrouy X, Fiorini
C, Diry M, Segretain D and Pointis G: Connexin 33 impairs gap
junction functionality by accelerating connexin 43 gap junction
plaque endocytosis. Traffic. 10:1272–1285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tishkoff SA and Williams SM: Genetic
analysis of African populations: Human evolution and complex
disease. Nat Rev Genet. 3:611–621. 2002.PubMed/NCBI
|
|
20
|
Bentley-Lewis R, Powe C, Ankers E, Wenger
J, Ecker J and Thadhani R: Effect of race/ethnicity on hypertension
risk subsequent to gestational diabetes mellitus. Am J Cardiol.
113:1364–1370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jing L, Su L and Ring BZ: Ethnic
background and genetic variation in the evaluation of cancer risk:
A systematic review. PLoS On. 9:e975222014. View Article : Google Scholar
|
|
22
|
Cooper DN, Krawczak M, Polychronakos C,
Tyler-Smith C and Kehrer-Sawatzki H: Where genotype is not
predictive of phenotype: Towards an understanding of the molecular
basis of reduced penetrance in human inherited disease. Hum Genet.
132:1077–1130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hervé JC, Derangeon M, Sarrouilhe D,
Giepmans BN and Bourmeyster N: Gap junctional channels are parts of
multiprotein complexes. Biochim Biophys Acta. 1818:1844–1865. 2012.
View Article : Google Scholar
|
|
24
|
Pitteloud N, Durrani S, Raivio T and
Sykiotis GP: Complex genetics in idiopathic hypogonadotropic
hypogonadism. Front Horm Res. 39:142–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kwiatkowski DP: How malaria has affected
the human genome and what human genetics can teach us about
malaria. Am J Hum Genet. 77:171–192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Di Pasquale E, Beck-Peccoz P and Persani
L: Hypergonadotropic ovarian failure associated with an inherited
mutation of human bone morphogenetic protein-15 (BMP15) gene. Am J
Hum Genet. 75:106–111. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Auclair S, Rossetti R, Meslin C, Monestier
O, Di Pasquale E, Pascal G, Persani L and Fabre S: Positive
selection in bone morphogenetic protein 15 targets a natural
mutation associated with primary ovarian insufficiency in human.
PLoS On. 8:e781992013. View Article : Google Scholar
|
|
28
|
Campbell MC and Tishkoff SA: African
genetic diversity: Implications for human demographic history,
modern human origins, and complex disease mapping. Annu Rev
Genomics Hum Genet. 9:403–433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen CT, Liu CT, Chen GK, Andrews JS,
Arnold AM, Dreyfus J, Franceschini N, Garcia ME, Kerr KF, Li G, et
al: Meta-analysis of loci associated with age at natural menopause
in African-American women. Hum Mol Genet. 23:3327–3342. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Spencer KL, Malinowski J, Carty CL,
Franceschini N, Fernández-Rhodes L, Young A, Cheng I, Ritchie MD,
Haiman CA, Wilkens L, et al: Genetic variation and reproductive
timing: African American women from the Population Architecture
using Genomics and Epidemiology (PAGE) Study. PLoS On.
8:e552582013. View Article : Google Scholar
|
|
31
|
Yatsenko SA and Rajkovic A: Reproductive
aging and MCM8/9. Oncotarget. 6:15750–15751. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pando MJ, Gardiner CM, Gleimer M, McQueen
KL and Parham P: The protein made from a common allele of KIR3DL1
(3DL1*004) is poorly expressed at cell surfaces due to substitution
at positions 86 in Ig domain 0 and 182 in Ig domain 1. J Immunol.
171:6640–6649. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gittens JE and Kidder GM: Differential
contributions of connexin37 and connexin43 to oogenesis revealed in
chimeric reaggregated mouse ovaries. J Cell Sci. 118:5071–5078.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Johnson KE, Mitra S, Katoch P, Kelsey LS,
Johnson KR and Mehta PP: Phosphorylation on Ser-279 and Ser-282 of
connexin43 regulates endocytosis and gap junction assembly in
pancreatic cancer cells. Mol Biol Cell. 24:715–733. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chang AS, Dale AN and Moley KH: Maternal
diabetes adversely affects preovulatory oocyte maturation,
development, and granulosa cell apoptosis. Endocrinology.
146:2445–2453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ratchford AM, Esguerra CR and Moley KH:
Decreased oocyte-granulosa cell gap junction communication and
connexin expression in a type 1 diabetic mouse model. Mol
Endocrinol. 22:2643–2654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Baere E, Beysen D, Oley C, Lorenz B,
Cocquet J, De Sutter P, Devriendt K, Dixon M, Fellous M, Fryns JP,
et al: FOXL2 and BPES: Mutational hotspots, phenotypic variability,
and revision of the genotype-phenotype correlation. Am J Hum Genet.
72:478–487. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Norris RP, Freudzon M, Mehlmann LM, Cowan
AE, Simon AM, Paul DL, Lampe PD and Jaffe LA: Luteinizing hormone
causes MAP kinase-dependent phosphorylation and closure of connexin
43 gap junctions in mouse ovarian follicles: One of two paths to
meiotic resumption. Development. 135:3229–3238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang M, Ouyang H and Xia G: The signal
pathway of gonad-otrophins-induced mammalian oocyte meiotic
resumption. Mol Hum Reprod. 15:399–409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang HX, Gillio-Meina C, Chen S, Gong XQ,
Li TY, Bai D and Kidder GM: The canonical WNT2 pathway and FSH
interact to regulate gap junction assembly in mouse granulosa
cells. Biol Reprod. 89:392013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gilleron J, Carette D, Carpentier F,
Segretain D and Pointis G: Three-dimensional analysis of connexin
43 gap junction in the ex vivo rat seminiferous tubules: Short-term
effects of hormonal effectors. Microsc Res Tech. 72:845–855. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang HX, Tong D, El-Gehani F, Tekpetey FR
and Kidder GM: Connexin expression and gap junctional coupling in
human cumulus cells: Contribution to embryo quality. J Cell Mol
Med. 13:972–984. 2009. View Article : Google Scholar
|
|
43
|
Li TY, Colley D, Barr KJ, Yee SP and
Kidder GM: Rescue of oogenesis in Cx37-null mutant mice by
oocyte-specific replacement with Cx43. J Cell Sci. 120:4117–4125.
2007. View Article : Google Scholar : PubMed/NCBI
|