Introduction
The pathogenesis of inflammatory bowel disease (IBD)
in humans remains incompletely understood; however, considerable
evidence indicates that IBD results from an interaction between
genetic, immune and environmental factors (1,2).
IBD, including ulcerative colitis (UC) and Crohn's disease (CD), is
a prominent intestinal disease. The pathogenesis of IBD is
predominantly associated with intestinal immunodeficiency; this
dysregulated immune response results in chronic gut inflammation
and the presence of activated immune cells, which produce copious
amounts of interferon (IFN)-γ, tumor necrosis factor (TNF)-α,
interleukin (IL)-2 and IL-17, thereby promoting the production of
large amounts of inflammatory cytokines, and reactive oxygen and
nitrogen metabolites, which ultimately lead to organ damage
(3–5). Consistent with these findings,
restoration of the balance between inflammatory and
anti-inflammatory factors has reported as a useful treatment
strategy, which has been successfully tested in patients with IBD
and in experimental animal models of colitis (6–9).
Previous studies have reported that the aryl
hydrocarbon receptor (AhR) is a transcription factor, which serves
a protective role in IBD (10–12). AhR is extensively expressed in
vertebrate cells and is a member of the basic
helix-loop-helix/Per-Arnt-Sim homology superfamily (13). In the cytosol, AhR is present in
an inactive form that binds to numerous co-chaperones (14). Following ligand binding, AhR
dissociates from its chaperones and dimerises with AhR nuclear
translocator (ARNT). The AhR/ARNT complex then activates the
transcription of target genes, including cytochrome P450 1A1
(CYP1A1) (15). AhR has also been
revealed to be indirectly activated by endogenous AhR ligands,
including 6-formylindolo(3,2-b)carbazole (FICZ) (16). FICZ may activate AhR and
significantly inhibit the inflammatory response (17). A previous study in AhR-knockout
(KO) mice illustrated the role of AhR in the function and
development of various organs; skin defects and a spectrum of
hepatic abnormalities, as well as haematopoietic and vascular
abnormalities, were observed in AhR-KO mice (18). Therefore, reduced AhR expression
may lead to immunodeficiency and cause the immune system to secrete
a mass of inflammatory factors. Although numerous studies have
demonstrated a role for AhR in the inhibition of inflammation, the
detailed underlying mechanism remains unclear.
It has previously been reported that treatment of
tristetraprolin (TTP)-KO mice with dextran sulphate sodium (DSS)
leads to increased susceptibility to colitis, decreased colon
length and increased production of proinflammatory cytokines
(19). These findings suggest
that TTP deficiency may result in a defect in the resolution of
intestinal inflammation and in exacerbated IBD symptoms. TTP is the
prototypic member of the TIS11 family of RNA-binding proteins,
which has an important role in regulating the expression of
adenylate-uridylate-rich element (ARE)-containing mRNAs (20). RNA-binding proteins, including TTP
and human antigen R (HuR), recognise and bind to AREs in the
3′-untranslated regions (3′-UTRs) of their target mRNAs (20,21). Binding of TTP to its target mRNAs
promotes the degradation of numerous inflammatory factors,
including TNF-α, IFN-γ, IL-1β, IL-6, IL-8 and cyclooxygenase
(COX)-2 (22–24). Furthermore, TTP-KO mice display
severe autoimmune dysfunction, inflammatory arthritis and
ulcerative colitis, thus suggesting that TTP has an important role
in limiting the inflammatory response (25).
A previous study indicated that the expression of
TTP is downregulated in AhR-/- cells (26). Furthermore, AhR is able to
suppress the expression of COX-2, which is mediated by HuR, another
RNA-binding protein from the same family as TTP (27). Based on the results of previous
studies, the present study hypothesised that AhR may exert
protective effects against experimental colitis through the
regulation of TTP expression.
Materials and methods
Materials
FICZ was purchased from Enzo Life Sciences, Inc.
(Farmingdale, NY, USA). Lipopolysaccharide (LPS) was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Foetal bovine serum
(FBS) and Dulbecco's modified Eagle's medium (DMEM) were purchased
from HyClone (GE Healthcare Life Sciences, Logan, UT, USA).
Antibodies against AhR (cat. no. ab2770) were purchased from Abcam,
Inc. (Cambridge, MA, USA). Antibodies against CYP1A1 (cat. no.
13241-1-AP) and mitogen-activated protein kinase-activated protein
kinase 2 (MK2; cat. no. 13949-1-AP) were purchased from Wuhan
Sanying Biotechnology (Wuhan, China). Antibodies against TTP (cat.
no. sc-14030) were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Antibodies against phosphorylated (p)-MK2 were
purchased from Cell Signalling Technology, Inc. (Danvers, MA, USA).
Antibodies against GAPDH (cat. no. 10494-1-AP) were purchased from
Wuhan Sanying Biotechnology. Goat anti-mouse (cat. no. A0216) and
goat anti-rabbit (cat. no. A0208) horseradish peroxidase
(HRP)-conjugated secondary antibodies, Cy3-conjugated goat
anti-mouse immunoglobulin (Ig)G (cat. no. P0193) and fluorescein
isothiocyanate-conjugated goat anti-rabbit IgG (cat. no. P0186)
were purchased from Beyotime Institute of Biotechnology (Shanghai,
China). All primers used in the present study were synthesised by
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cell cultures
LoVo human intestinal epithelial cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA) and maintained in DMEM supplemented with 15% FBS, 100 U/ml
streptomycin and 100 u/ml penicillin at 37°C in a 5% CO2
atmosphere. The medium was refreshed every 2 days. The cells were
subcultured successively when they reached ~80% confluence.
Cell treatments
Cells were grown in 6-well plates and were incubated
with LPS (10 µg/ml) and/or FICZ (100 nM) for 8 h at 37°C.
Immunofluorescence and western blotting were used to detect target
protein expression, and reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) was used to determine mRNA
expression.
Small interfering (si)RNA
transfection
For transient knockdown of AhR expression, cells
were grown in DMEM with no antibiotics until they reached 80–100%
confluence. Transient transfection was performed using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After a
48-h transfection period, the cells were harvested. The sequences
of the AhR-targeting siRNA were: Sense,
5′-GGAACACCUACAUCUAGAAdTdT-3′ and antisense, 3′-dTdT
CCUUGUGGAUGUAGAUCUU-5′. The sequences of the negative control siRNA
were: Sense, 5′-GGGCAAAUCCCAAGAGGAAdTdT-3′ and antisense, 3′-dTdT
CCCGUUUAGGGUUCUCCUU-5′.
DSS-induced colitis
The present study was approved by the Laboratory
Animal Welfare and Ethic Committee of the Third Military Medical
university (Chongqing, China). Male C57BL/6J wild-type mice (n=60;
age, 6-8 weeks; weight, 18-22 g) and male C57BL/6J AhR-KO mice
(n=15; age, 6-8 weeks; weight, 18-22 g) were purchased from the
Experimental Animal Center at Daping Hospital of the Third Military
Medical University (Chongqing, China). Animals were bred and
maintained under specific pathogen-free conditions in a
temperature-controlled room (20±2°C) with circadian light-dark
cycles and free access to standard rodent chow and water. Mice were
randomly divided into the following three groups: Sham (n=10), DSS
(n=10), DSS + FICZ (n=10) group. Colitis was induced by
administration of 3% DSS (Sigma-Aldrich, Inc.; Merck KGaA)
dissolved in distilled water for 7 days. Mice in Sham group were
administered distilled water. In addition, FICZ (1 µg/mouse)
was administered daily, by intraperitoneal injection, beginning 2
days after the start of DSS administration. Changes in body weight
were recorded daily. Following DSS administration for 7 days, the
mice were sacrificed and intestinal mucosa samples were collected
for histology, and the determination of target mRNA and protein
expression levels.
Histological examination
Colon segments were washed three times in PBS and
were fixed in 4% paraformaldehyde overnight at 4°C. Ethanol was
used to dehydrate the tissues, which were embedded in paraffin. The
resulting tissue sections (4-µm thick) were stained with
haematoxylin for 5 min and eosin for 3 min at room temperature. The
sections were observed using a confocal microscope (LSM 5 PASCAL;
Carl Zeiss, Oberkochen, Germany).
Immunofluorescence staining
Cells were fixed with 4% parafor-maldehyde for 15
min at room temperature, and permeabilized with 0.2% Triton X-100
for 10 min at room temperature. Cells were then blocked in 5%
bovine serum albumin (BSA) (Sigma-Aldrich, Inc.; Merck KGaA) for 2
h at 37°C and incubated with mouse monoclonal anti-AhR (1:50) and
rabbit polyclonal anti-TTP (1:20) antibodies overnight at 4°C.
Subsequently, Cy3-conjugated goat anti-mouse immunoglobulin (Ig)G
(1:200) and fluorescein isothiocyanate-conjugated goat anti-rabbit
IgG (1:200) were used to stain the cells and sections,
respectively. Finally, 1 mg/ml DAPI (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to stain the nuclei for total cell
counting. The fluorescent signals were analysed by confocal laser
scanning microscopy (Leica LAS AF Lite, version 2.8.0; Leica,
Wetzlar, Germany), after the recording and merging of
single-stained images.
RNA purification and RT-qPCR
Total RNA from intestinal mucosa and LoVo cells was
extracted using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Spectrophotometry was used to determine the concentration of total
RNA. For RT-qPCR, 1 µg total RNA was converted to cDNA using
a RT kit (Takara Bio, Inc., Otsu, Japan) in a 20 µl volume
according to the following temperature protocol: 37°C for 15 min,
85°C for 5 sec, followed by 5 min at 4°C. Subsequently,
SYBR-Green-based qPCR was used to measure the mRNA expression
levels of CYP1A1, TTP, IFN-γ, TNF-α, IL-1β, IL-6, β-actin and
GAPDH. The average quantification cycle (Cq) value of triplicate
wells with each primer set was calculated as the amount of gene
product present in the sample. The relative mRNA expression levels
were determined according to the ratio between the Cq value of the
target gene and β-actin or GAPDH (28). The cycling conditions were as
following: 94°C for 10 min, 40 cycles at 94°C for 30 sec, 60°C for
1 min and 68°C for 30 sec, followed by 7 min at 68°C. The primer
sequences were designed using online tools (https://www.ncbi.nlm.nih.gov/tools/primer-blast/)
and are presented in Table I.
 | Table ISequences of primers used in reverse
transcription-quantitative polymerase chain reaction
experiments. |
Table I
Sequences of primers used in reverse
transcription-quantitative polymerase chain reaction
experiments.
| Species | Gene name | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| Human | | | |
| IL-1β |
CTTCGACACATGGGATAACG |
ATATCCTGTCCCTGGAGGTG |
| IL-6 G |
ACAGCCACTCACCTCTTCA |
CCTCTTTGCTGCTTTCACAC |
| CYP1A1 |
CCATGTCGGCCACGGAGTT |
ACAGTGCCAGGTGCGGCTT |
| TTP |
TTTAAGGGAGGCAATGAACC |
CAGGAGACACTGGAACCTCA |
| β-actin |
CCACGAAACTACCTTCAACTCC |
CGTGATCTCCTTCTGCATCCTG |
| Mouse | | | |
| IL-1β G |
AAATGCCACCTTTTGACAGTG |
TGGATGCTCTCATCAGGACAG |
| IL-6 |
CTTCCAGCCAGTTGCCTTCTTG |
GGTCTGTTGTGGGTGGTATCCTC |
| IFN-γ G |
CCACGGCACAGTCATTGA |
TGCTGATGGCCTGATTGTCTT |
| TNF-α |
CCACCACGCTCTTCTGTCTACTG |
GGGCTACGGGCTTGTCACTC |
| CYP1A1 |
CAATGAGTTTGGGGAGGTTACTG |
CCCTTCTCAAATGTCCTGTAGTG |
| TTP |
CTCGGAGGACTTTGGAACAT |
TGCAGTAGGCGAAGTAGGTG |
| GAPDH |
TGAAGGTCGGTGTGAACGGATTTGG |
ACGACATACTCAGCACCAGCATCAC |
Western blot analysis
LoVo cells were washed three times with PBS and were
lysed in cell lysis buffer (Intron Biotechnology, Inc., Seongnam,
Korea) for 20 min on ice. The lysed cells were then centrifuged at
12,000 × g for 10 min at 4°C, and the supernatant was collected.
The proteins were extracted from the intestinal mucosa using lysis
buffer, followed by homogenization through sonication and the
extraction mixture was centrifuged at 12,000 × g for 15 min at 4°C.
The protein concentrations were measured using a bicinchoninic acid
assay kit (Beyotime Institute of Biotechnology). Protein samples
(25 µg) were separated by 10% SDS-PAGE and the proteins were
transferred to polyvinylidene fluoride membranes (Amersham; GE
Healthcare, Chicago, IL, USA). The membranes were blocked in TBS
containing 0.05% Tween-20 (TBS-T) and 5% bovine serum albumin (BSA;
Sigma-Aldrich, Inc.; Merck KGaA) for 2 h at 37°C and were then
incubated with the following primary antibodies diluted in TBS-T
containing 5% BSA overnight at 4°C: Mouse anti-AhR (1:500), rabbit
anti-CYP1A1 (1:1,000), rabbit anti-TTP (1:200), rabbit anti-MK2
(1:1,000), rabbit anti-p-MK2 (1:1,000) and rabbit anti-GAPDH
(1:1,000). The membranes were washed three times in TBS-T, and were
then incubated with goat anti-mouse and goat anti-rabbit
horseradish peroxidase (HRP)-conjugated secondary antibodies
(1:5,000), after which they were washed a further three times in
TBS-T. Enhanced chemiluminescence (Millipore, Billerica, MA, USA)
was used to detect the binding of HRP-conjugated antibodies
according to the manufacturer's protocol.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Differences among groups were assessed by analysis of variance
using SPSS statistical software package (version 13.0; SPSS Inc.,
Chicago, IL, USA). The groups were compared using one-way analysis
of variance (ANOVA) and Q-test. Comparisons between groups were
conducted using paired t-tests. P≤0.05 was considered to indicate a
statistically significant difference.
Results
AhR deficiency results in increased
colitis severity
Initially, the present study observed that
administration of DSS produces more severe colitis in AhR-KO mice
compared with in C57BL/6J wild-type (WT) mice, as evidenced by
histological examination of colonic tissue (Fig. 1A). Furthermore, mice in the AhR-KO
group produced more inflammatory factors compared with those in the
DSS-treated WT group. The mRNA expression levels of IL-1β, IL-6,
IFN-γ and TNF-α were significantly increased in the AhR-KO + DSS
group compared with in the DSS-treated WT group (Fig. 1B). These findings suggested that a
deficiency in AhR may be associated with DSS-induced colitis.
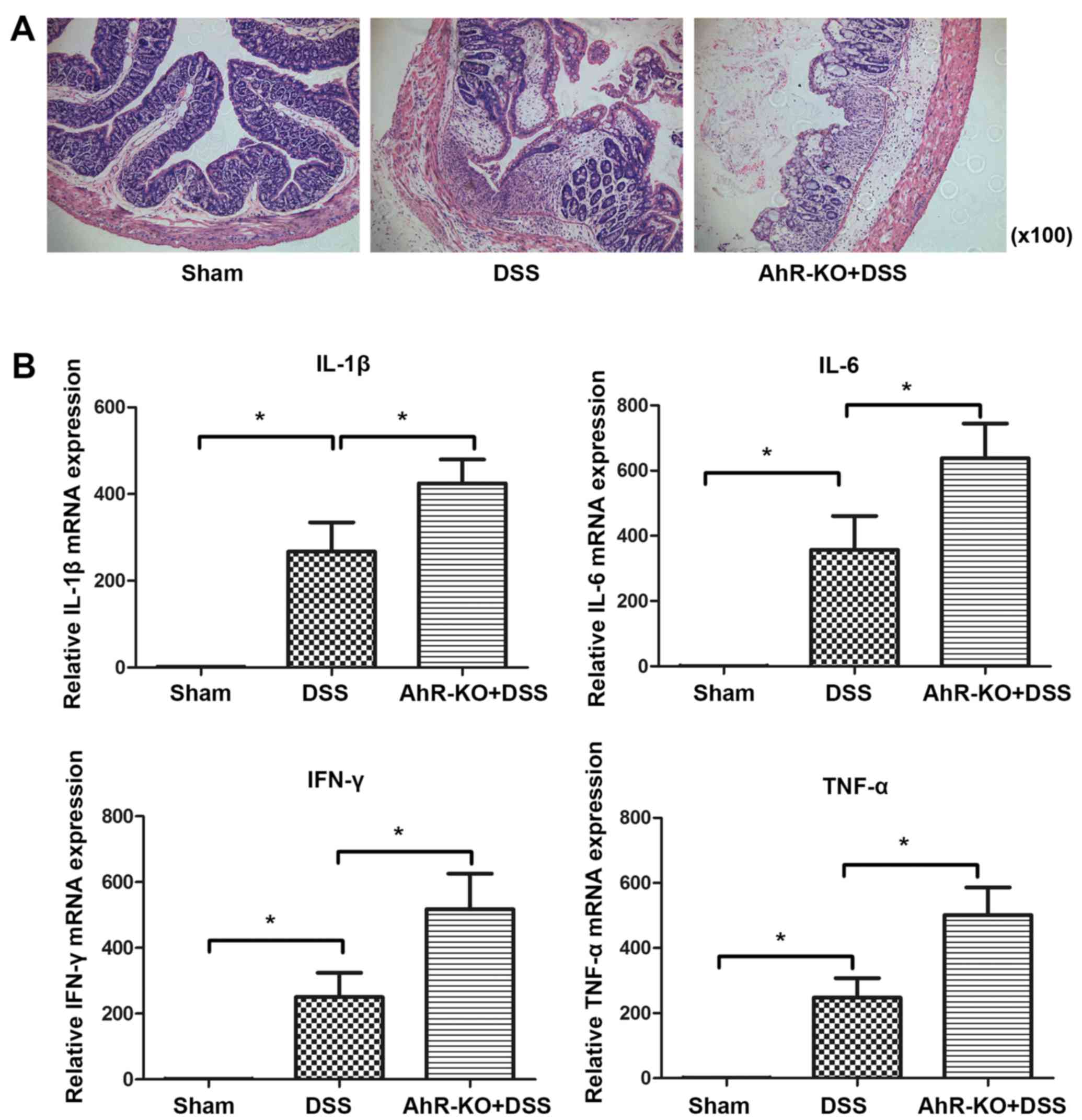 | Figure 1AhR deficiency results in increased
colitis severity. AhR-KO mice and C57BL/6J WT mice were treated
with 3% DSS for 7 days. (A) Representative haematoxylin and
eosin-stained colonic sections from DSS-treated C57BL/6J and AhR-KO
mice (magnification, ×100). (B) mRNA expression levels of IL-1β,
IL-6, IFN-γ and TNF-α in colonic samples from mice treated as
indicated in (A). mRNA expression was analysed by quantitative
polymerase chain reaction. Data are presented as the mean ±
standard deviation of three experiments, in which samples from 3
mice/group were analysed. *P<0.05. AhR, aryl
hydrocarbon receptor; DSS, dextran sulphate sodium; IFN-γ,
interferon-γ; IL, interleukin; KO, knockout; TNF-α, tumour necrosis
factor-α; WT, wild-type. |
AhR activation ameliorates DSS-induced
colitis
To investigate whether AhR activation could
attenuate experimental colitis, mice were administered FICZ for 5
days, beginning 2 days after the start of DSS administration
(Fig. 2). DSS-treated mice
exhibited significant weight loss 3-4 days after DSS treatment
(Fig. 2B). On day 8, mortality
rate in the DSS group was ~30% (Fig.
2C). In addition, colon length was decreased in DSS-treated
mice (Fig. 2A). Conversely, DSS +
FICZ-treated mice exhibited reduced weight loss and mortality
(~16.67%), and colon length was increased compared with in the
DSS-treated mice (Fig. 2A–C). In
addition, histological examination of colonic tissues from mice in
the various groups revealed that mice treated with FICZ developed
less severe colitis (Fig. 2E).
The expression levels of IL-1β, IL-6, IFN-γ and TNF-α were
significantly increased in colon samples from mice in the
DSS-induced colitis group compared with in the vehicle-treated
group (Fig. 2D). However, in mice
treated with FICZ the mRNA expression levels of IFN-γ, IL-1β, IL-6
and TNF-α were significantly reduced (Fig. 2D). These findings suggested that
administration of FICZ may attenuate inflammation associated with
DSS-induced colitis in mice, and that AhR has an important role in
this attenuation.
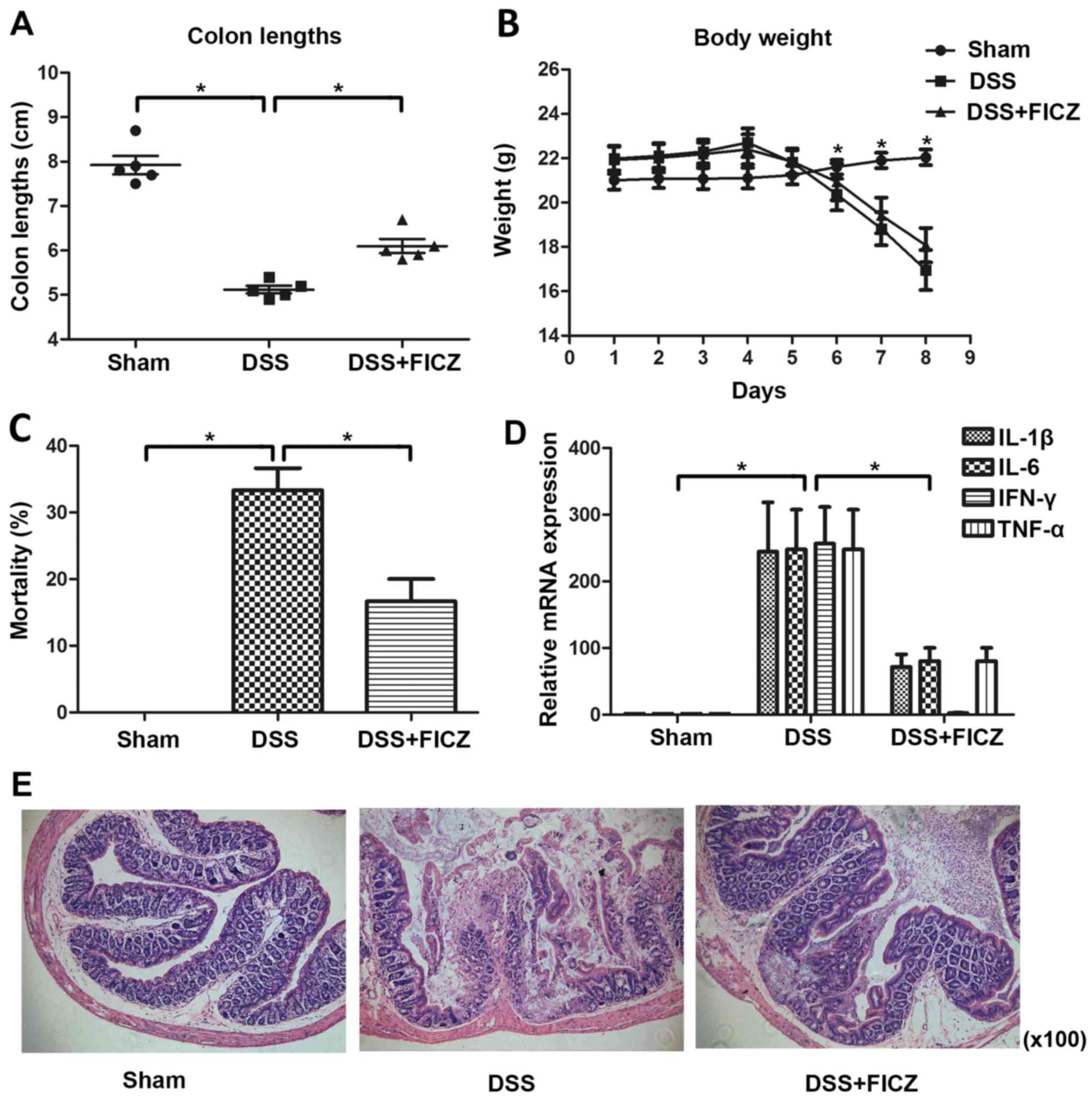 | Figure 2AhR activation ameliorates
DSS-induced colitis. (A) FICZ was intraperitoneally administered to
mice for 5 days, beginning 2 days after the start of DSS
administration. Colon lengths of mice in the sham, DSS and DSS +
FICZ groups were measured on day 8. Data are presented as the mean
± standard deviation of five experiments in which samples from 10
mice/group were analysed. (B) Changes in body weight were recorded
daily. Weight data are presented as the cumulative mean ± standard
deviation obtained from five separate experiments. In each
experiment, each group consisted of ≥10 mice. (C) Percentage of
mortality in the sham, DSS and DSS + FICZ groups was noted on day
8. Data are presented as the mean ± standard deviation. (D) mRNA
expression levels of IL-1β, IL-6, IFN-γ and TNF-α in colonic
samples from mice treated as indicated in (A). mRNA expression was
analysed by quantitative polymerase chain reaction. Data are
presented as the mean ± standard deviation of five experiments in
which samples from 10 mice/group were analysed. (E) Representative
haematoxylin and eosin-stained colonic sections of mice in the
sham, DSS and DSS + FICZ groups on day 8 (magnification, ×100).
*P<0.05 (n=10/group). AhR, aryl hydrocarbon receptor;
DSS, dextran sulphate sodium; FICZ, 6-formylindolo(3,2-b)carbazole;
IFN-γ, interferon-γ; IL, interleukin; KO, knockout; TNF-α, tumour
necrosis factor-α; WT, wild-type. |
FICZ upregulates expression of the
RNA-binding protein TTP in mice with DSS-induced colitis
Recent studies have reported that TTP serves an
important role in limiting inflammatory responses (29,30). In addition, treatment of TTP-KO
mice with DSS results in increased susceptibility to colitis
(19). Furthermore, it has been
demonstrated that TTP expression is decreased in AhR-/-
cells (26). To explore whether
TTP has a role in alleviating DSS-induced colitis, and to determine
the association between TTP and AhR in this process, the expression
levels of TTP were detected. The results indicated that the
expression levels of AhR, CYP1A1 and TTP were downregulated in
AhR-KO mice (Fig. 3A and B).
Similarly, the expression levels of AhR and TTP were significantly
decreased in mice with DSS-induced colitis (Fig. 3C and D).
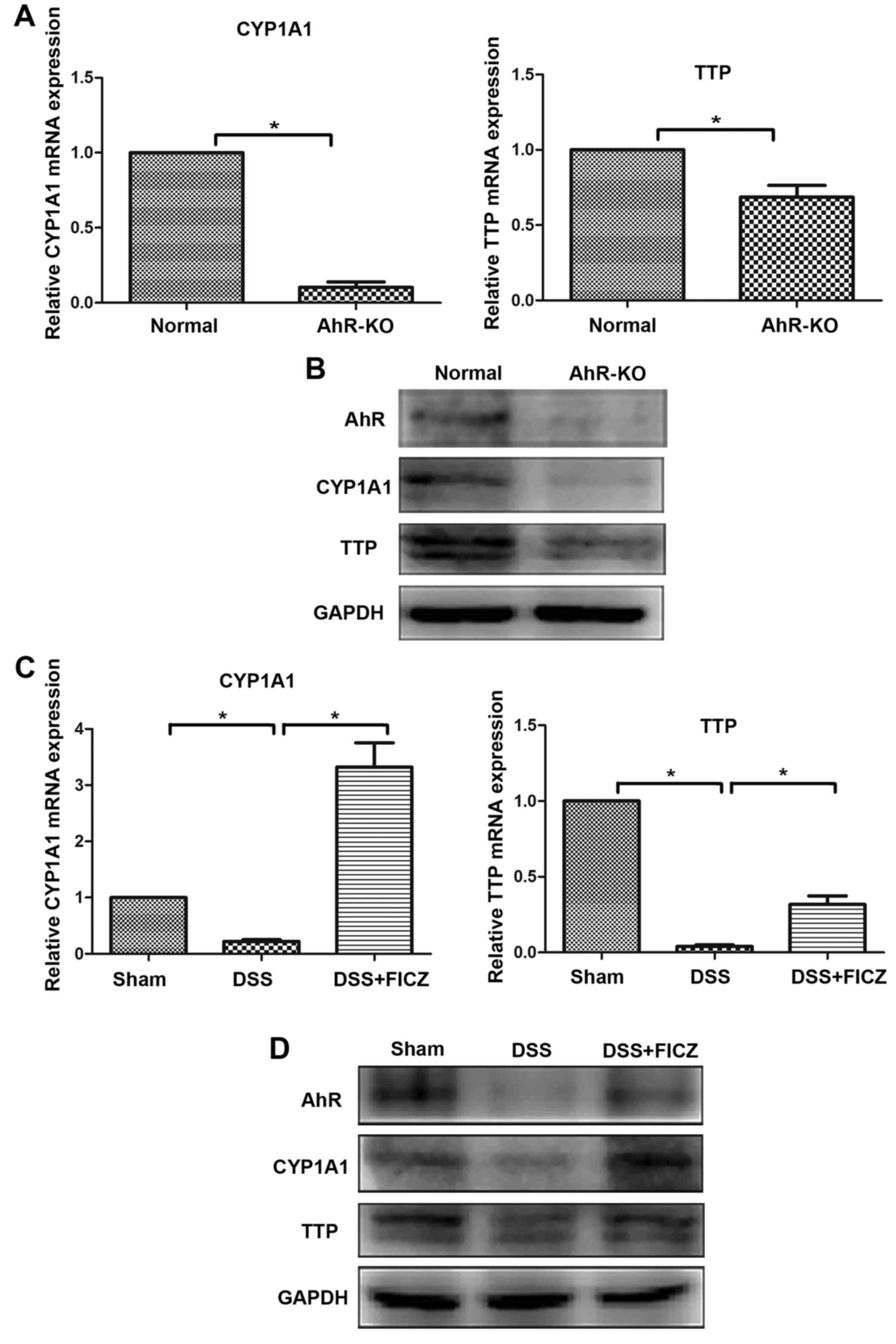 | Figure 3FICZ upregulates expression of the
RNA-binding protein TTP in mice with DSS-induced colitis. (A) mRNA
expression levels of CYP1A1 and TTP in colonic samples of WT and
aryl hydrocarbon receptor (AhR) KO mice were determined by RT-qPCR.
(B) Protein expression levels of AhR, CYP1A1 and TTP in colonic
samples from WT and AhR-KO mice, as detected by western blotting.
(C) FICZ was intraperitoneally administered to mice for 5 days,
beginning 2 days after the start of DSS administration. The mRNA
expression levels of CYP1A1 and TTP in colonic samples from the
sham, DSS and DSS + FICZ groups were determined by RT-qPCR. (D)
Mice were treated as indicated in (C), and the protein expression
levels of AhR, CYP1A1 and TTP from colonic samples was detected by
western blotting. Data are presented as the mean ± standard
deviation of five experiments in which samples from 10 mice/group
were analysed. *P<0.05. AhR, aryl hydrocarbon
receptor; CYP1A1, cytochrome P450 1A1; DSS, dextran sulphate
sodium; FICZ, 6-formylindolo(3,2-b)carbazole; KO, knockout;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; TTP, tristetraprolin; WT, wild-type. |
The present study demonstrated that TTP expression
was downregulated in AhR-KO mice. Therefore, in order to explore
whether activation of AhR could upregulate the expression of TTP,
mice were intraperitoneally injected with FICZ daily, beginning 2
days after the start of DSS administration. The expression levels
of AhR, CYP1A1 and TTP were significantly increased following
administration of FICZ to mice with DSS-induced colitis (Fig. 3C and D). Increased AhR, CYP1A1 and
TTP expression was confirmed by western blotting (Fig. 3D). Furthermore, alterations in
mRNA expression levels, as quantified by RT-qPCR, correlated with
these protein alterations (Fig.
3C).
FICZ upregulates the RNA-binding protein
TTP expression in an in vitro model
To further investigate the role of AhR and TTP in an
in vitro model, LoVo human intestinal epithelial cells were
treated with LPS (10 µg/ml) in the presence or absence of
FICZ (100 nM) for 8 h. The results demonstrated that IL-1β and IL-6
were significantly increased following treatment of the cells with
LPS. However, the mRNA expression levels of IL-1β and IL-6 were
significantly downregulated in the LPS + FICZ group compared with
in the LPS group (Fig. 4A). This
finding suggested that FICZ may inhibit LPS-induced inflammation in
an in vitro model. Subsequently, the present study aimed to
determine whether FICZ modulates the RNA-binding protein TTP. As
illustrated in Fig. 4B and C, the
protein and mRNA expression levels of TTP were significantly
increased in the LPS + FICZ group compared with in the LPS group.
These results are in accordance with those obtained in the animal
model. Unexpectedly, the protein expression levels of AhR were
decreased following treatment with FICZ. This may be due to AhR
degradation following FICZ treatment (31,32). Immunofluorescence staining
revealed that confluent LoVo cells treated with LPS and FICZ
displayed an increased intensity of TTP staining compared with the
LPS-treated group. In addition, the intensity of AhR staining was
decreased in the LPS and FICZ-treated groups compared to the
LPS-treated groups (Fig. 4D).
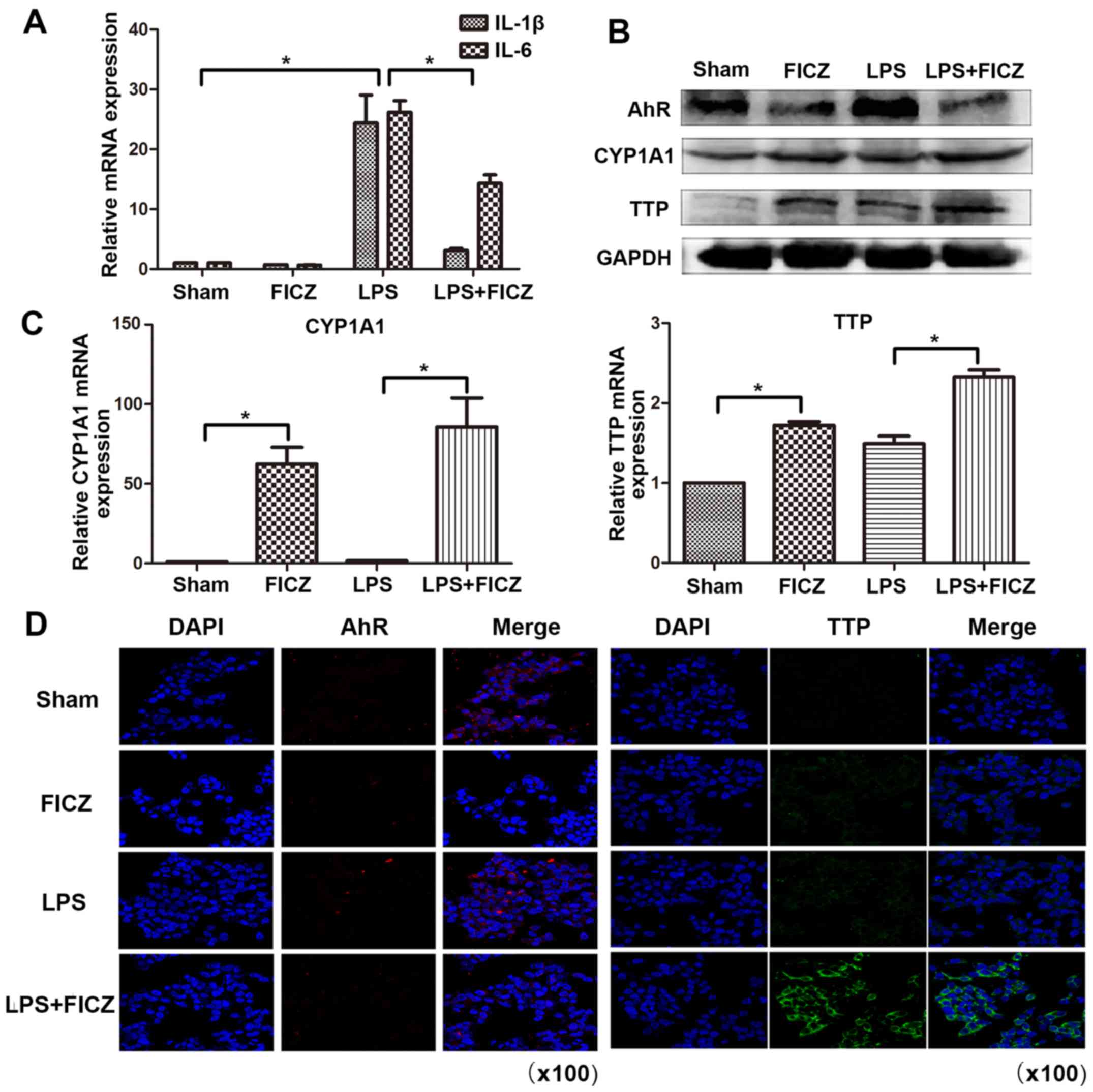 | Figure 4FICZ upregulates the RNA-binding
protein TTP expression in an in vitro model. (A-D) LoVo
human intestinal epithelial cells were treated with or without LPS
(10 µg/ml) and FICZ (100 nM) for 8 h. The mRNA expression
levels of (A) IL-1β, IL-6, (C) CYP1A1 and TTP were determined by
reverse transcription-quantitative polymerase chain reaction. (B)
Protein expression levels of AhR, CYP1A1 and TTP were
semi-quantified by western blotting. (D) Immunofluorescence was
used to detect AhR and TTP expression. Red and green signals
represent AhR and TTP, respectively (magnification, ×100) Data are
presented as the mean ± standard deviation. *P<0.05.
AhR, aryl hydrocarbon receptor; CYP1A1, cytochrome P450 1A1; FICZ,
6-formylindolo(3,2-b) carbazole; IL, interleukin; TTP,
tristetraprolin. |
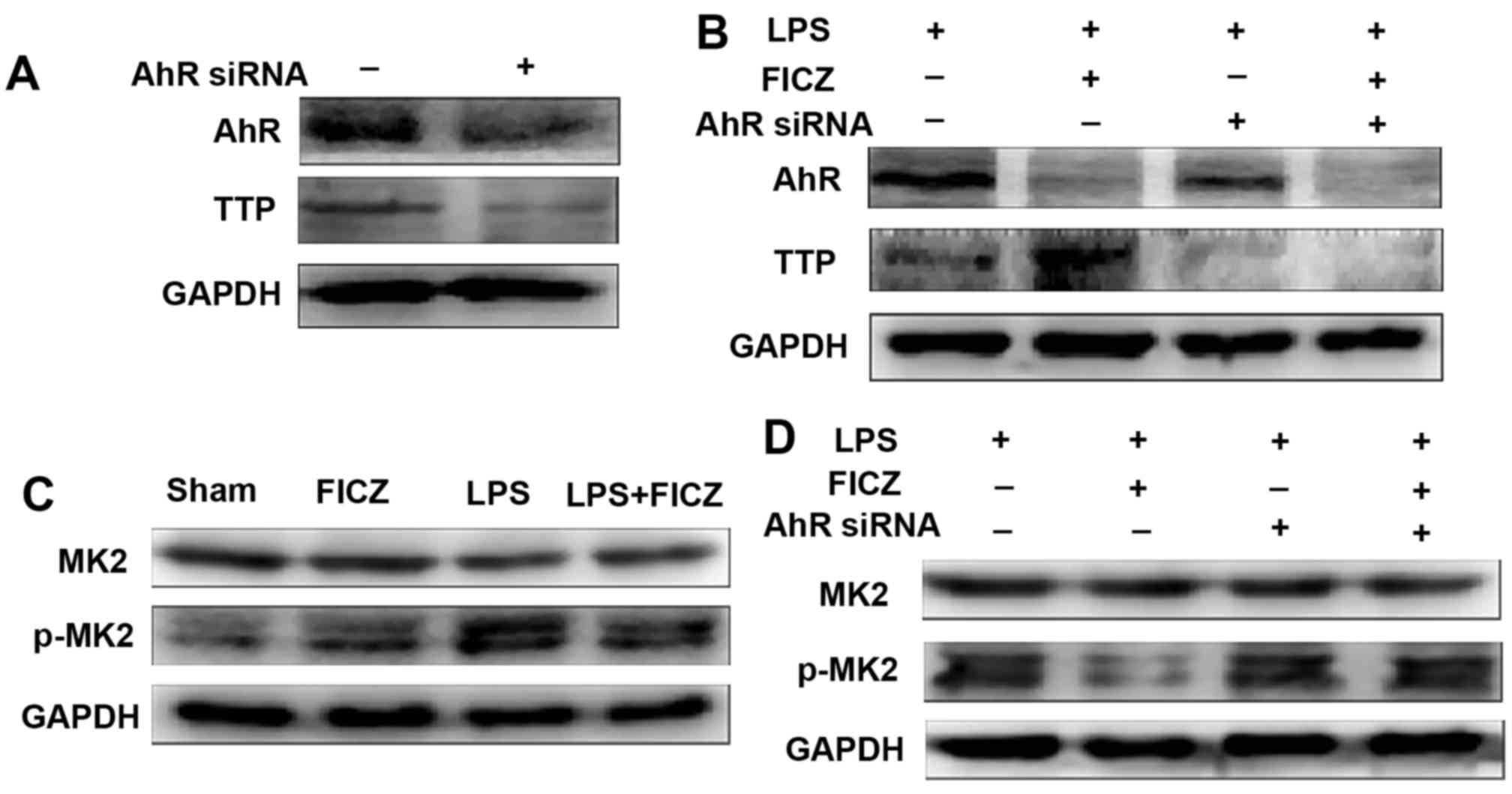
AhR regulates TTP expression through the
MK2/p-MK2 pathway
In order to explore the mechanism through which AhR
regulates TTP, AhR expression was silenced in LoVo cells, after
which the cells were treated with LPS and FICZ for 8 h. As
expected, the protein expression levels of AhR and TTP were
decreased in cells in which AhR expression was silenced (Fig. 5A). Concurrently, TTP expression in
AhR siRNA-transfected cells treated with LPS and FICZ was lower
than that in normal cells treated with LPS and FICZ (Fig. 5B). These findings indicated that
FICZ may upregulate TTP via AhR. In addition, LoVo cells were
treated with LPS and FICZ, and the expression levels of MK2 and
p-MK2 were measured, which are the classical upstream kinases of
the TTP pathway, in order to determine whether AhR mediates TTP
expression through the MK2/p-MK2 pathway. Notably, MK2 expression
was not significantly altered; however, p-MK2 expression was
reduced in the LPS + FICZ-treated group compared with in the
LPS-treated group (Fig. 5C).
Consistent with the expected results, these findings indicated that
AhR may mediate TTP expression via the MK2/p-MK2 pathway. It should
be noted that after AhR expression was silenced, MK2 expression was
still not significantly altered; however, FICZ no longer
downregulated p-MK2 expression in the LPS + FICZ-treated group
(Fig. 5D). These findings
suggested that AhR may serve a key role in the FICZ-mediated
downregulation of p-MK2 expression.
Discussion
The present study demonstrated that mice deficient
in AhR displayed increased susceptibility to colitis, and indicated
that activation of AhR by FICZ could ameliorate DSS-induced
colitis. In addition, AhR could inhibit the inflammation that
occurs in DSS-induced colitis via the MK2/p-MK2/TTP pathway.
It has previously been reported that AhR serves an
important role in IBD. AhR can inhibit inflammation and colitis in
mice via the upregulation of IL-22 (11), and AhR activation significantly
inhibited the production of IL-1β, IL-6 and TNF-α in LPS-treated
dendritic cells (17), and
attenuated LPS-induced inflammatory responses in SW480 human colon
carcinoma cells (10).
Furthermore, AhR can be activated by
2,3,7,8-tetra-chlorodibenzo-p-dioxin and attenuates inflammation
associated with CD (33). In
addition, β-naphthoflavone, a nontoxic agonist of AhR, has been
reported to suppress the pathogenesis of DSS-induced colitis and
attenuate DSS-induced colitis (10). These findings suggested that AhR
expression in intestinal epithelial cells is involved in the
prevention of colitis.
The present study examined the expression and
functional role of AhR in a mouse model of DSS-induced colitis. The
results indicated that DSS induces more severe colitis in AhR-KO
mice compared with in wild-type mice. This finding suggested that a
deficiency in AhR may result in a decrease in resistance to
inflammation. In addition, the present study assessed whether
activation of AhR by FICZ could attenuate DSS-induced colitis.
Treatment of mice with FICZ for 5 days, starting 2 days after the
initiation of DSS administration, significantly inhibited the
expression of inflammatory factors. Based on these findings, it may
be suggested that AhR activation attenuates DSS-induced
colitis.
The main aetiology of IBD is thought to be
associated with disordered immune function, in which the organism
releases a mass of inflammatory factors, resulting in an imbalance
between pro- and anti-inflammatory factors, which in turn disrupts
intestinal barrier function. In recent years, the mechanisms
through which factors that mediate the stability of mRNAs coding
inflammatory factors exert their anti-inflammatory effects have
attracted more attention. RNA-binding proteins, which may bind to
AREs located in the 3′-UTRs of target mRNAs and direct them to
exosomes for rapid degradation (34,35), can reduce the expression of
various inflammatory factors. RNA-binding proteins, including TTP
and HuR, are found in numerous protein families. It has previously
been reported that TTP-KO mice display more severe autoimmune
dysfunction, inflammatory arthritis and UC compared with their WT
counterparts (20). Furthermore,
in AhR-/- cells, the expression of TTP is downregulated
(26). AhR also inhibits
inflammation via the downregulation of another RNA-binding protein,
HuR (27). These findings
suggested that AhR activation may regulate activity of the
RNA-binding protein TTP.
The present study assessed whether AhR could
regulate TTP expression in the gut and investigated the role of TTP
in IBD. Initially, the results demonstrated that TTP expression was
decreased in DSS-treated mice compared with in vehicle-treated
animals. In addition, consistent with results reported in the
literature (26), the expression
levels of AhR and TTP were downregulated in AhR-KO mice. When
DSS-treated mice were treated with FICZ, TTP expression was
upregulated. These findings suggested that TTP expression may be
associated with IBD and that FICZ, which activates the AhR pathway,
may exert its anti-inflammatory effects by upregulating the
RNA-binding protein TTP.
Although AhR mediates the expression of TTP, the
mechanism underlying this effect remains unknown. It has been
suggested that phosphorylation of MK2, which occurs upstream of
TTP, modulates the activity and expression of TTP. MK2 can
phosphorylate the TTP protein (36.37). In addition, the stability,
expression and function of TTP are reported to be regulated by the
MK2/p-MK2 pathway (36,38,39). Activation of the MK2/p-MK2 pathway
has been reported to abolish TTP-mediated suppression of IL-6
3′-UTR reporter activity (40).
In addition, it has been demonstrated that the MK2/p-MK2 cascade is
involved in regulating the stability of ARE-containing mRNAs
(41). Recent studies have
clearly indicated that the MK2/p-MK2 signalling cascade can
regulate TTP-mediated mRNA stability of IL-6 and TNF-α (42).
In the present study, following treatment with FICZ,
MK2 expression was not significantly altered; however, the
expression of p-MK2 was downregulated. Conversely, when AhR
expression was silenced, FICZ no longer downregulated p-MK2
expression. These findings suggested that FICZ upregulates the
expression of TTP by downregulating p-MK2 expression.
In conclusion, the present study demonstrated that
AhR serves an important role in attenuating DSS-induced colitis.
AhR-mediated protection operates in part by inducing TTP via the
MK2/p-MK2 pathway. The present study is the first, to the best of
our knowledge, to demonstrate a dependence on TTP for AhR-mediated
protection against DSS-induced colitis. Since AhR may serve
important roles in protection against DSS-induced colitis, the
AhR-TTP pathway may be considered an attractive candidate for the
treatment of UC.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. NSFC
81330013 and NSFC 81272078 to H.Y., and NSFC81300275 to L.S.) and
the Program of Changjiang Scholars and Innovative Research (grant
no. IRT 13050 to H.Y.).
References
|
1
|
Bamias G, Martin C III, Mishina M, Ross
WG, Rivera-Nieves J, Marini M and Cominelli F: Proinflammatory
effects of TH2 cytokines in a murine model of chronic small
intestinal inflammation. Gastroenterology. 128:654–666. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fiocchi C: Inflammatory bowel disease:
etiology and pathogenesis. Gastroenterology. 115:182–205. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Strober W, Fuss IJ and Blumberg RS: The
immunology of mucosal models of inflammation. Annu Rev Immunol.
20:495–549. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Uhlig HH and Powrie F: The role of mucosal
T lymphocytes in regulating intestinal inflammation. Springer Semin
Immunopathol. 27:167–180. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaser A, Zeissig S and Blumberg RS:
Inflammatory bowel disease. Annu Rev Immunol. 28:573–621. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Targan SR: Hanauer Sb, Vandeventer SH,
Lloydmayer and Present D: A short-term study of chimeric monoclonal
antibody CA2 to tumor necrosis factor a for Crohn's disease. N Engl
J Med. 337:1029–1036. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baumgart DC and Sandborn WJ: Inflammatory
bowel disease: Clinical aspects and established and evolving
therapies. Lancet. 369:1641–1657. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Neurath MF, Fuss I, Kelsall BL, Stüber E
and Strober W: Antibodies to interleukin 12 abrogate established
experimental colitis in mice. J Exp Med. 182:1281–1290. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ito H, Takazoe M, Fukuda Y, Hibi T,
Kusugami K, Andoh A, Matsumoto T, Yamamura T, Azuma J and Nishimoto
N: A pilot randomized trial of a human anti-interleukin-6 receptor
monoclonal antibody in active Crohn's disease. Gastroenterology.
126:989–996; discussion 947. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Furumatsu K, Nishiumi S, Kawano Y, Ooi M,
Yoshie T, Shiomi Y, Kutsumi H, Ashida H, Fujii-Kuriyama Y, Azuma T,
et al: A role of the aryl hydrocarbon receptor in attenuation of
colitis. Dig Dis Sci. 56:2532–2544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Monteleone I, Rizzo A, Sarra M, Sica G,
Sileri P, Biancone L, MacDonald TT, Pallone F and Monteleone G:
Aryl hydrocarbon receptor-induced signals upregulate IL-22
production and inhibit inflammation in the gastrointestinal tract.
Gastroenterology. 141:237–248.e1. 2011. View Article : Google Scholar
|
|
12
|
Ji T, Xu C, Sun L, Yu M, Peng K, Qiu Y,
Xiao W and Yang H: Aryl hydrocarbon receptor activation
downregulates IL-7 and reduces inflammation in a mouse model of
DSS-induced colitis. Dig Dis Sci. 60:1958–1966. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Denis M, Cuthill S, Wikström AC,
Poellinger L and Gustafsson JA: Association of the dioxin receptor
with the Mr 90,000 heat shock protein: A structural kinship with
the glucocorticoid receptor. Biochem Biophys Res Commun.
155:801–807. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kewley RJ, Whitelaw ML and Chapman-Smith
A: The mammalian basic helix-loop-helix/PAS family of
transcriptional regulators. Int J Biochem Cell Biol. 36:189–204.
2004. View Article : Google Scholar
|
|
15
|
Sogawa K and Fujii-Kuriyama Y: Ah
receptor, a novel ligand-activated transcription factor. J Biochem.
122:1075–1079. 1997. View Article : Google Scholar
|
|
16
|
Lee YH, Lin CH, Hsu PC, Sun YY, Huang YJ,
Zhuo JH, Wang CY, Gan YL, Hung CC, Kuan CY, et al: Aryl hydrocarbon
receptor mediates both proinflammatory and anti-inflammatory
effects in lipopolysaccharide-activated microglia. Glia.
63:1138–1154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang C, Ye Z, Kijlstra A, Zhou Y and Yang
P: Activation of the aryl hydrocarbon receptor affects activation
and function of human monocyte-derived dendritic cells. Clin Exp
Immunol. 177:521–530. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barouki R, Coumoul X and
Fernandez-Salguero PM: The aryl hydrocarbon receptor, more than a
xenobiotic-interacting protein. FEBS Lett. 581:3608–3615. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Joe Y, Uddin MJ, Zheng M, Kim HJ, Chen Y,
Yoon NA, Cho GJ, Park JW and Chung HT: Tristetraprolin mediates
anti-inflammatory effect of carbon monoxide against DSS-induced
colitis. PLoS One. 9:e887762014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sanduja S, Blanco FF, Young Le, Kaza V and
Dixon DA: The role of tristetraprolin in cancer and inflammation.
Front Biosci (Landmark Ed). 17:174–188. 2012. View Article : Google Scholar
|
|
21
|
Fan XC and Steitz JA: Overexpression of
HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo
stability of ARE-containing mRNAs. EMBO J. 17:3448–3460. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carballo E, Lai WS and Blackshear PJ:
Feedback inhibition of macrophage tumor necrosis factor-alpha
production by tristetraprolin. Science. 281:1001–1005. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kratochvill F, Machacek C, Vogl C, Ebner
F, Sedlyarov V, Gruber AR, Hartweger H, Vielnascher R, Karaghiosoff
M, Rülicke T, et al: Tristetraprolin-driven regulatory circuit
controls quality and timing of mRNA decay in inflammation. Mol Syst
Biol. 7:5602011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Molle C, Zhang T, Ysebrant de Lendonck L,
Gueydan C, Andrianne M, Sherer F, Van Simaeys G, Blackshear PJ, Leo
O and Goriely S: Tristetraprolin regulation of interleukin 23 mRNA
stability prevents a spontaneous inflammatory disease. J Exp Med.
210:1675–1684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taylor GA, Carballo E, Lee DM, Lai WS,
Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE,
Haynes BF, et al: A pathogenetic role for TNF alpha in the syndrome
of cachexia, arthritis, and autoimmunity resulting from
tristetraprolin (TTP) deficiency. Immunity. 4:445–454. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang X, Fan Y, Karyala S, Schwemberger S,
Tomlinson CR, Sartor MA and Puga A: Ligand-independent regulation
of transforming growth factor beta1 expression and cell cycle
progression by the aryl hydrocarbon receptor. Mol Cell Biol.
27:6127–6139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zago M, Sheridan JA, Nair P, Rico de Souza
A, Gallouzi IE, Rousseau S, Di Marco S, Hamid Q, Eidelman DH and
Baglole CJ: Aryl hydrocarbon receptor-dependent retention of
nuclear HuR suppresses cigarette smoke-induced cyclooxygenase-2
expression independent of DNA-binding. PLoS One. 8:e749532013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Kratochvill F, Gratz N, Qualls JE, Van De
Velde LA, Chi H, Kovarik P and Murray PJ: Tristetraprolin limits
inflammatory cytokine production in tumor-associated macrophages in
an mRNA decay-independent manner. Cancer Res. 75:3054–3064. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ross EA, Smallie T, Ding Q, O'Neil JD,
Cunliffe HE, Tang T, Rosner DR, Klevernic I, Morrice NA, Monaco C,
et al: Dominant suppression of inflammation via targeted mutation
of the mRNA destabilizing protein tristetraprolin. J Immunol.
195:265–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song Z and Pollenz RS: Ligand-dependent
and independent modulation of aryl hydrocarbon receptor
localization, degradation, and gene regulation. Mol Pharmacol.
62:806–816. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pollenz RS: The mechanism of AH receptor
protein down-regulation (degradation) and its impact on AH
receptor-mediated gene regulation. Chem Biol Interact. 141:41–61.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Benson JM and Shepherd DM: Aryl
hydrocarbon receptor activation by TCDD reduces inflammation
associated with Crohn's disease. Toxicol Sci. 120:68–78. 2011.
View Article : Google Scholar :
|
|
34
|
Lai WS, Carballo E, Strum JR, Kennington
EA, Phillips RS and Blackshear PJ: evidence that tristetraprolin
binds to AU-rich elements and promotes the deadenylation and
destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol.
19:4311–4323. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen CY, Gherzi R, Ong SE, Chan EL,
Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M and Karin M:
AU binding proteins recruit the exosome to degrade ARE-containing
mRNAs. Cell. 107:451–464. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mahtani KR, Brook M, Dean JLE, Sully G,
Saklatvala J and Clark AR: Mitogen-activated protein kinase p38
controls the expression and posttranslational modification of
tristetraprolin, a regulator of tumor necrosis factor alpha mRNA
stability. Mol Cell Biol. 21:6461–6469. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chrestensen CA, Schroeder MJ, Shabanowitz
J, Hunt DF, Pelo JW, Worthington MT and Sturgill TW: MAPKAP kinase
2 phosphorylates tristetraprolin on in vivo sites including Ser178,
a site required for 14-3-3 binding. J Biol Chem. 279:10176–10184.
2004. View Article : Google Scholar
|
|
38
|
Brook M, Tchen CR, Santalucia T, McIlrath
J, Arthur JS, Saklatvala J and Clark AR: Posttranslational
regulation of tristetraprolin subcellular localization and protein
stability by p38 mitogen-activated protein kinase and extracellular
signal-regulated kinase pathways. Mol Cell Biol. 26:2408–2418.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hitti E, Iakovleva T, Brook M, Deppenmeier
S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A and
Gaestel M: Mitogen-activated protein kinase-activated protein
kinase 2 regulates tumor necrosis factor mRNA stability and
translation mainly by altering tristetraprolin expression,
stability, and binding to adenine/uridine-rich element. Mol Cell
Biol. 26:2399–2407. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao W, Liu M, D'Silva NJ and Kirkwood KL:
Tristetraprolin regulates interleukin-6 expression through p38
MAPK-dependent affinity changes with mRNA 3′ untranslated region. J
Interferon Cytokine Res. 31:629–637. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun L, Stoecklin G, Van Way S,
Hinkovska-Galcheva V, Guo RF, Anderson P and Shanley TP:
Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from
dephosphorylation by protein phosphatase 2a and stabilizes tumor
necrosis factor-alpha mRNA. J Biol Chem. 282:3766–3777. 2007.
View Article : Google Scholar
|
|
42
|
O'Dea KP, Dokpesi JO, Tatham KC, Wilson MR
and Takata M: Regulation of monocyte subset proinflammatory
responses within the lung microvasculature by the p38 MAPK/MK2
pathway. Am J Physiol Lung Cell Mol Physiol. 301:L812–L821. 2011.
View Article : Google Scholar : PubMed/NCBI
|