Introduction
Gallstone disease, the most common biliary disorder
affecting 10–15% of the adult population, is a significant health
problem worldwide, with an incidence that has been increasing due
to the rising epidemic of obesity and metabolic syndrome (1). Bile contains cholesterol, bile salts
and phospholipids, and the precipitation of excessive cholesterol
in bile leads to gallstone formation (2). To overcome the extremely low
solubility of cholesterol in aqueous solutions, cholesterol in bile
exists as mixed micelles of bile salts and phospholipids. Excess
cholesterol, a lack of bile salts or phospholipids, or a
combination of these factors can lead to the supersaturation of
bile and the precipitation of cholesterol (2,3).
Therefore, cholesterol homeostasis is required to prevent gallstone
formation.
The balance between cholesterol intake/biosynthesis
and biliary cholesterol secretion is important for cholesterol
homeostasis (4). The master
regulator of cholesterol biosynthesis, sterol regulatory
element-binding protein-2 (SREBP-2), regulates the expression of
numerous genes involved in cholesterol homeostasis, including the
rate-limiting enzyme in cholesterol biosynthesis,
3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) (4). In addition, the cytochrome P450
genes, cytochrome P450 family 7 subfamily A member 1
(CYP7A1), CYP7B1, CYP27A1 and CYP8B1,
encode enzymes that catalyze various steps in cholesterol
catabolism and bile acid synthesis. Transcriptional regulation of
CYP7A1, which encodes cholesterol 7α-hydroxylase and
catalyzes the initial rate-limiting step in cholesterol catabolism
(5), has been relatively
well-established. The farnesoid X receptor (FXR) induces expression
of an atypical orphan nuclear receptor, small heterodimer partner
(SHP), which then inhibits liver-related homolog-1 transactivation,
thereby inhibiting Cyp7a1 transcription (5). In addition, SHP and the pregnane X
receptor (PXR) interact with hepatocyte nuclear factor 4α and block
its recruitment of peroxisome proliferator-activated receptor-γ
coactivator-1α to CYP7A1 chromatin, thereby inhibiting
CYP7A1 transcription (5).
Finally, the ATP-binding cassette (ABC) transporters ABCB4 [also
known as multidrug resistance protein 2 (Mdr2)], ABCB11 [also known
as bile salt export pump (Bsep)] and ABCG5/ABCG8 heterodimers serve
essential roles in the biliary secretion of phospholipids, bile
acids and cholesterol, respectively (6). Regulation of cholesterol metabolism
is a therapeutic target, and statins are reported to be effective
to treat gallstone disease (7)
and to reduce the risk of cholecystectomy (8). However, the efficacy of statin
treatment for gallstone diseases remains under debate (9). Ursodeoxycholic acid (UDCA) was Food
and Drug Administration (FDA) approved for cholesterol gallstone
dissolution through the increase in bile flow and the alteration of
the hydrophobicity index of the bile acid pool (10). However, UDCA use in neonatal and
infant cholestasis was reported to be ineffective and unsafe, and
it can also cause severe toxicities, including hepatitis, pruritus,
cholangitis, ascites, vanishing bile duct syndrome and liver
failure, in adults (10).
Therefore, novel drugs to treat gallstone disease are required to
apply to the range of patients.
The proteasome, which degrades ubiquitinated
proteins, has emerged as an attractive therapeutic target to treat
various cancer types (11).
Bortezomib (BT), the first FDA-approved prote asome inhibitor, has
been used clinically to treat patients with multiple myelomas and
mantle cell lymphoma (12).
Moreover, BT has been under investigation to expand its therapeutic
effects to various other conditions, including fatty liver
(13), hepatic injury by ductal
ligation (14), drug-induced
liver injury (15), pancreatic
cancer (16), breast cancer
(17), renal cell carcinoma
(18) and prostate cancer
(19). In addition, other
proteasome inhibitors, including carfilzomib (CF), CEP-18770,
PR-047 and NPI-0052, have also been developed and tested for the
treatment of a variety of diseases (20).
The present study investigated the effects and
mechanism of proteasome inhibition in gallstone formation using two
proteasome inhibitors, BT and CF by reducing cholesterol synthesis
and biliary secretion. Furthermore, it was found that reduced
transcription of Cyp7a1 by proteasome inhibition was
associated with c-Jun N-terminal protein kinase (JNK) activation
and that reduced ABCG5/ABCG8 levels resulted from reduced
phosphorylation of protein kinase A (PKA).
Materials and methods
Materials
The following materials were purchased: i) SP600125,
8-bromo-cAMP, Sp-cAMPS and anti-Cyp7a1 (catalog no. SAB4301212)
antibody (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany); ii)
PD98059 and anti-p38 (catalog no. 8690), anti-phosphorylated
(phospho)-p38 (Thr180/Tyr182; catalog no. 4511), anti-JNK (catalog
no. 9252), anti-p-JNK (Thr183/Tyr185; catalog no. 9255),
anti-extracellular signal-regulated kinase (ERK; catalog no. 4695),
anti-p-ERK (Thr202/Tyr204; 4370), anti-p-PKA (Thr197; catalog no.
5661) and anti-PKA (catalog no. 5842) antibodies (all Cell
Signaling Technology, Inc., Danvers, MA, USA); iii) anti-SREBP-2
(catalog no. NV100-74543) antibody (Novus Biologicals, Littleton,
CO, USA); iv) anti-Cyp27a1 antibody (catalog no. GTX103718;
GeneTex, San Antonio, TX, USA); v) anti-HMGCR antibody (catalog no.
ab174830) (Abcam, Cambridge, MA, USA); vi) anti-BSEP (catalog no.
sc-74500), anti-MDR2 (sc-58221), anti-ABCG5 (catalog no. sc-517207)
and anti-ABCG8 (catalog no. sc-30111) antibodies (all Santa Cruz
Biotechnology, Inc., Dallas, TX, USA); vii) anti-glyceraldehyde
3-phosp hate dehydrogenase (GAPDH) antibody (catalog no. MAB374)
(EMD Millipore, Billerica, MA, USA); viii) anti-mouse-horseradish
peroxidase (HRP; catalog no. 115-036-003) and anti-rabbit-HRP
(catalog no. 111-035-003) antibodies (both Jackson Laboratory, Ben
Harbor, ME, USA); and ix) BT and CF (both Biovision, Inc.,
Milpitas, CA, USA).
Animals and lithogenic diet
provision
Male C57BL/6J mice (n=30; 6 weeks old) were
purchased from Orient Bio, Inc. (Seoul, South Korea) and were
housed under special pathogen-free conditions. Experimental
procedures were approved by the Animal Ethics Committee at Lee Gil
Ya Cancer and Diabetes Institute of Gachon University, and all
animals were treated in accordance with the Animal Care Guidelines
of Lee Gil Ya Cancer and Diabetes Institute of Gachon University.
Mice were housed in a controlled environment at 21–23°C and 51–54%
humidity, with a 12 h light-dark cycle, and supplied with food and
water ad libitum. Mice were fed a lithogenic diet (D12336;
Research Diets Inc., New Brunswick, NJ, USA) and were injected with
BT (0.5 mg/kg/week) or with CF (0.5 mg/kg/week) for 12 weeks.
Control mice were injected with an equal amount of dimethyl
sulfoxide. Mice were euthanized with low flow (10–30% of total
volume) of CO2, and sera, livers and gall bladders were
collected and stored at −80°C until further analysis.
Cell culture and treatment
Hep3B cells were purchased from the American Type
Culture Collection (Rockville, MD, USA) and grown in Dulbecco's
modified Eagle's medium supplemented with 10% fetal bovine serum
and 1% penicillin/streptomycin (all Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA). The Hep3B cells were treated with 50 nM
BT, CF or epoxomicin (Epoxo) for 48 h along with 10 μM
SP600125 (JNK inhibitor), 10 μM PD98059 (ERK inhibitor), 10
μM 8-bromo-cAMP (PKA activator) and 10 μM Sp-cAMPS
(PKA activator).
Western blotting
Liver and Hep3B cells were lysed with RIPA buffer
[50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium
deoxycholate and 0.1% SDS] containing protease and phosphatase
inhibitors (Sigma-Aldrich; Merck KGaA) at 4°C for 1 h. Lysates were
centrifuged at 12,000 × g, at 4°C for 10 min, supernatants were
collected and the protein concentrations of the supernatants were
measured using Protein Assay Dye reagent (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Proteins (50 μg) were loaded and
separated on 8–13% SDS-polyacrylamide gels, and then transferred to
nitrocellulose membranes (Bio-Rad Laboratories, Inc.). Membranes
were blocked with 5% bovine serum albumin (Sigma-Aldrich; Merck
KGaA) in phosphate-buffered saline with 0.1% Tween-20 for 1 h at
4°C and then were incubated with primary antibodies (diluted
1:1,000) overnight at 4°C. Membranes were then incubated with
secondary antibodies (diluted 1:10,000) for 1 h at room
temperature. Protein bands were detected using Enhanced
Chemiluminescence Western Blotting Detection reagents (Amersham
Biosciences, Little Chalfont, UK) and a ChemiDoc MP imaging system
(Bio-Rad Laboratories, Inc.).
Cholesterol, phospholipid and bile acid
measurements
Cholesterol, phospholipid and bile acid levels in
sera, bile and livers were measured using the total cholesterol and
cholesteryl ester colorimetric/fluorometric assay kit (Biovision,
Inc.), the phospholipid assay kit (Sigma-Aldrich; Merck KGaA) and
the total bile acids assay kit (Biovision, Inc.), respectively,
according to the manufacturer's instructions.
Reverse transcription-quantitative
polymerase chain reaction (qPCR)
Total mRNAs from mouse livers were extracted with
the NucleoSpin RNA II kit (Macherey-Nagel, Duren, Germany), and
cDNAs were synthesized using a Verso cDNA Synthesis kit (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). qPCR was performed with
SYBR-Green (Thunderbird; Toyobo Co., Ltd., Osaka, Japan) using a
CFX Connect™ Real-Time PCR Detection system (Bio-Rad Laboratories).
The thermal cycling conditions were as follows: 95°C for 1 min,
followed by 40 cycles of 95°C for 15 sec, 60°C for 45 sec. Relative
gene expression was calculated using the 2−ΔΔCq method
(21). The primers that were used
are described in Table I. GAPDH
was used as the reference gene.
 | Table IPrimers used for quantitative
polymerase chain reaction. |
Table I
Primers used for quantitative
polymerase chain reaction.
| Genes | Primer
sequences |
|---|
| srebp-2
(mouse) | F:
5′-ACTGACCAGCACCCATACTC-3′ |
| R:
5′-CAGGAGGAGAGTTGGAACCA-3′ |
| hmgcr
(mouse) | F:
5′-ATCTCCTCTCCACAAAGCTT-3′ |
| R:
5′-CATTCTCACAGCAAGCTCCC-3′ |
| cyp7b1
(mouse) | F:
5′-GCATCATCCGAGAAGTGCAG-3′ |
| R:
5′-ATGAGTGGAGGAAAGAGGGC-3′ |
| cyp7a1
(mouse) | F:
5′-CTCCGGGCCTTCCTAAATCA-3′ |
| R:
5′-ACAGCGTTAGATATCCGGCT-3′ |
| cyp27a1
(mouse) | F:
5′-GAGAGTGAATCAGGGGACCA-3′ |
| R:
5′-TCAGGAATGGAGGGTTTCAG-3′ |
| cyp8b1
(mouse) | F:
5′-AGATTGCAGCGTCTCTTCCAT-3′ |
| R:
5′-CCTTGCTCCCTCAGAAACTG-3′ |
| bsep
(abcb11) (mouse) | F:
5′-GGGTTCTACAGGGGTTGGAA-3′ |
| R:
5′-GTGAACTTGGCCACACTCAG-3′ |
| mdr2
(abcb4) (mouse) | F:
5′-TCGCAGAGAACATCGCCTAT-3′ |
| R:
5′-TCTCGATGAAGGGGTGGATG-3′ |
| abcg5
(mouse) | F:
5′-AATTTTGGGGGAATTTCCAG-3′ |
| R:
5′-GTCCTGTGGTTGGCTCATCT-3′ |
| abcg8
(mouse) | F:
5′-CCTGATCCGTCGTCAGATTT-3′ |
| R:
5′-CCATGGCCGTAGTAAAGGAA-3′ |
| fxr
(nr1h4) (mouse) | F:
5′-TGGGTACCAGGGAGAGACTG-3′ |
| R:
5′-GTGAGCGCGTTGTAGTGGTA-3′ |
| pxr
(nr1i2) (mouse) | F:
5′-CCCATCAACGTAGAGGAGGA-3′ |
| R:
5′-TCTGAAAAACCCCTTGCATC-3′ |
| shp
(nr0b2) (mouse) | F:
5′-AGCTGGGTCCCAAGGAGTAT-3′ |
| R:
5′-GGTACCAGGGCTCCAAGACT-3′ |
| lxrα
(nr1h3) (mouse)' | F:
5′-TACGTCTCCATCAACCACCC-3′ |
| R:
5′-CTTGCTCTGAATGGACGCTG-3′ |
| lxrβ
(nr1h2) (mouse) | F:
5′-CAGACGCTACAACCACGAGA-3′ |
| R:
5′-ATGAATTCCACCTGCAAGCC-3′ |
|
gapdha
(mouse) | F:
5′-CGACTTCAACAGCAACTCCCACTCTTCC-3′ |
| R:
5′-TGGGTGGTCCAGGGTTTCTTACTCCTT-3′ |
Statistical analysis
All of the experiments were repeated at least three
independent times, and data are presented as the mean ± standard
error of the mean. Statistical significance was calculated using
analysis of variance followed by Tukey's post hoc test (GraphPad
Prism 6.0; GraphPad Software, San Diego, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
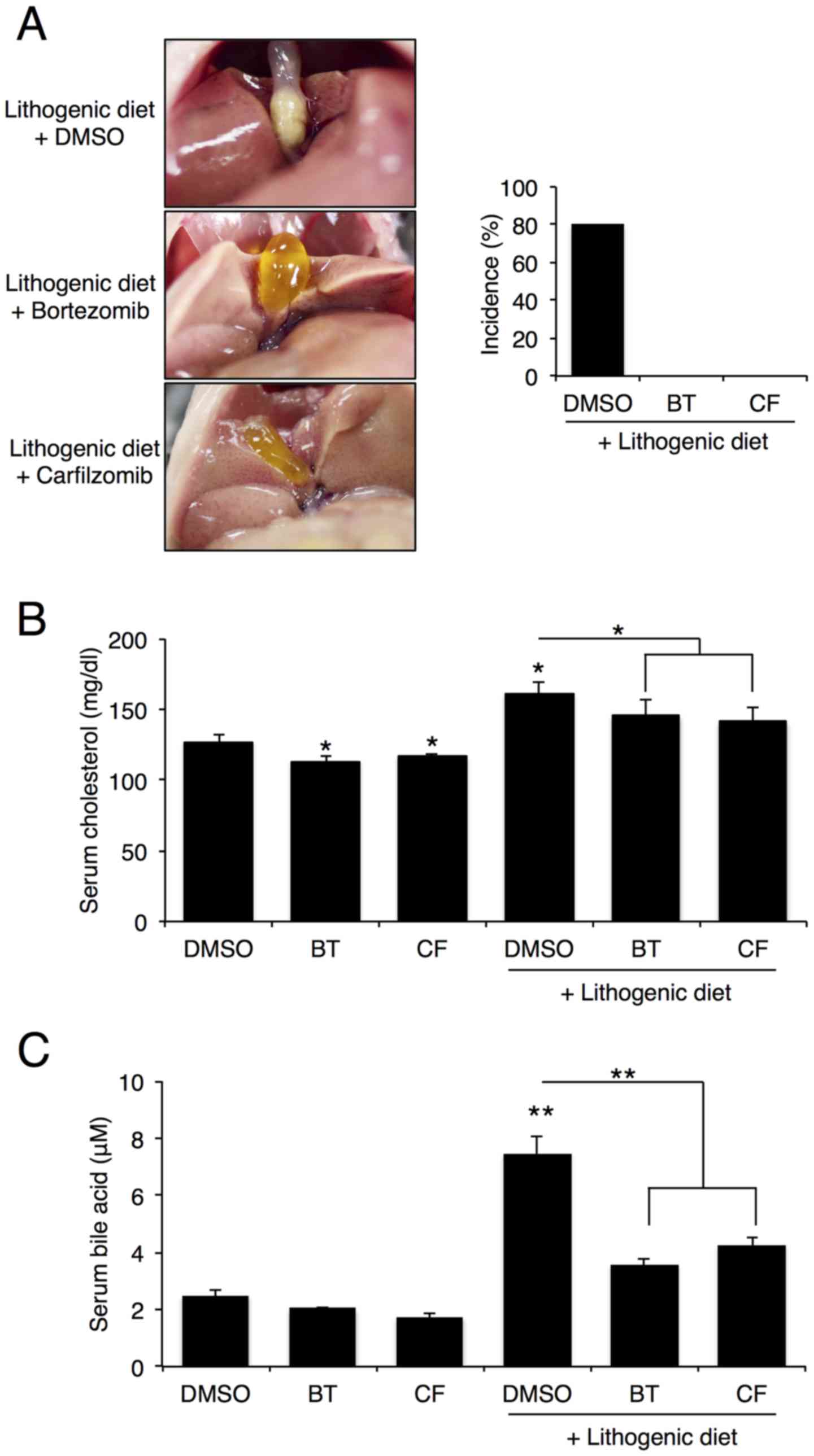
Proteasome inhibition reduces gallstone
formation in mice
To examine the role of proteasome inhibition on
gallstone formation, C57BL/6J mice were intraperitoneally injected
with BT or CF (0.5 mg/kg/week) and fed a lithogenic diet for 12
weeks. After 12 weeks, 8 out of the 10 mice that were fed a
lithogenic diet, exhibited gallstone formation, whereas none of the
mice that were fed a lithogenic diet and were treated with BT or CF
displayed gallstone formation (Fig.
1A). As expected, serum cholesterol and bile acid levels
increased after 12 weeks on a lithogenic diet, but proteasome
inhibition with BT or CF significantly reduced these levels
(Fig. 1B and C).
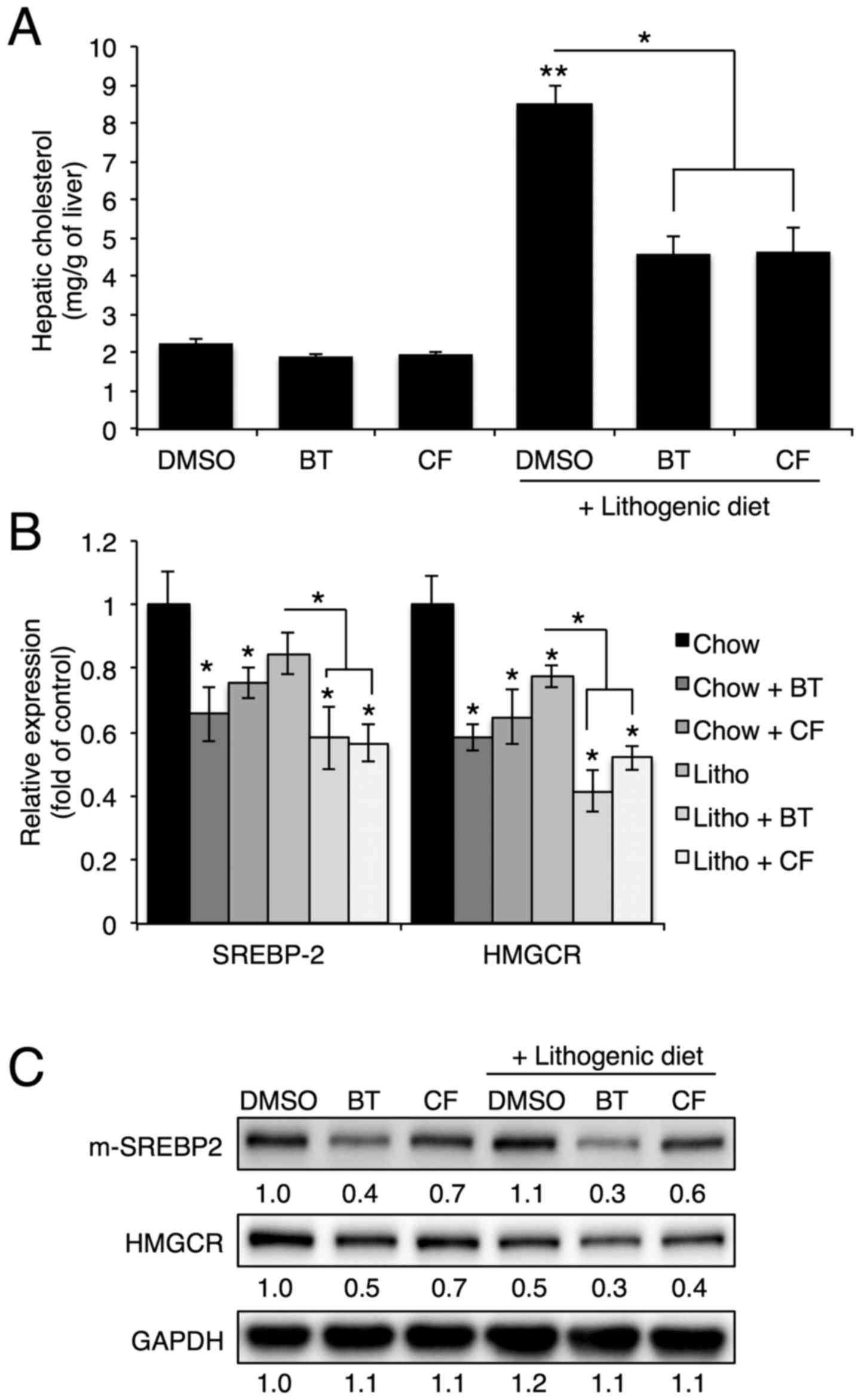
Proteasome inhibition reduces cholesterol
and bile acid synthesis in the liver
To investigate the protective mechanism of
proteasome inhibition on gallstone formation, the hepatic
cholesterol levels and expression of genes involved in cholesterol
synthesis were analyzed. In accordance with serum cholesterol
levels, hepatic cholesterol levels that were elevated with a
lithogenic diet were significantly reduced upon BT or CF
administration (Fig. 2A).
Proteasome inhibition reduced the mRNA and protein levels of
Srebp-2, the master regulator of cholesterol synthesis, and
Hmgcr, the rate-limiting enzyme of cholesterol synthesis
(Fig. 2B and C) (4).
Next, the hepatic bile acid levels and expression of
genes involved in bile acid synthesis were examined. Proteasome
inhibition reduced the bile acid levels in the liver that were
elevated by the lithogenic diet (Fig.
3A). Proteasome inhibition also reduced mRNA and protein
expression of hepatic genes involved in bile acid synthesis,
including Cyp7a1, Cyp7b1, Cyp27a1 and
Cyp8b1 (Fig. 3B and
C).
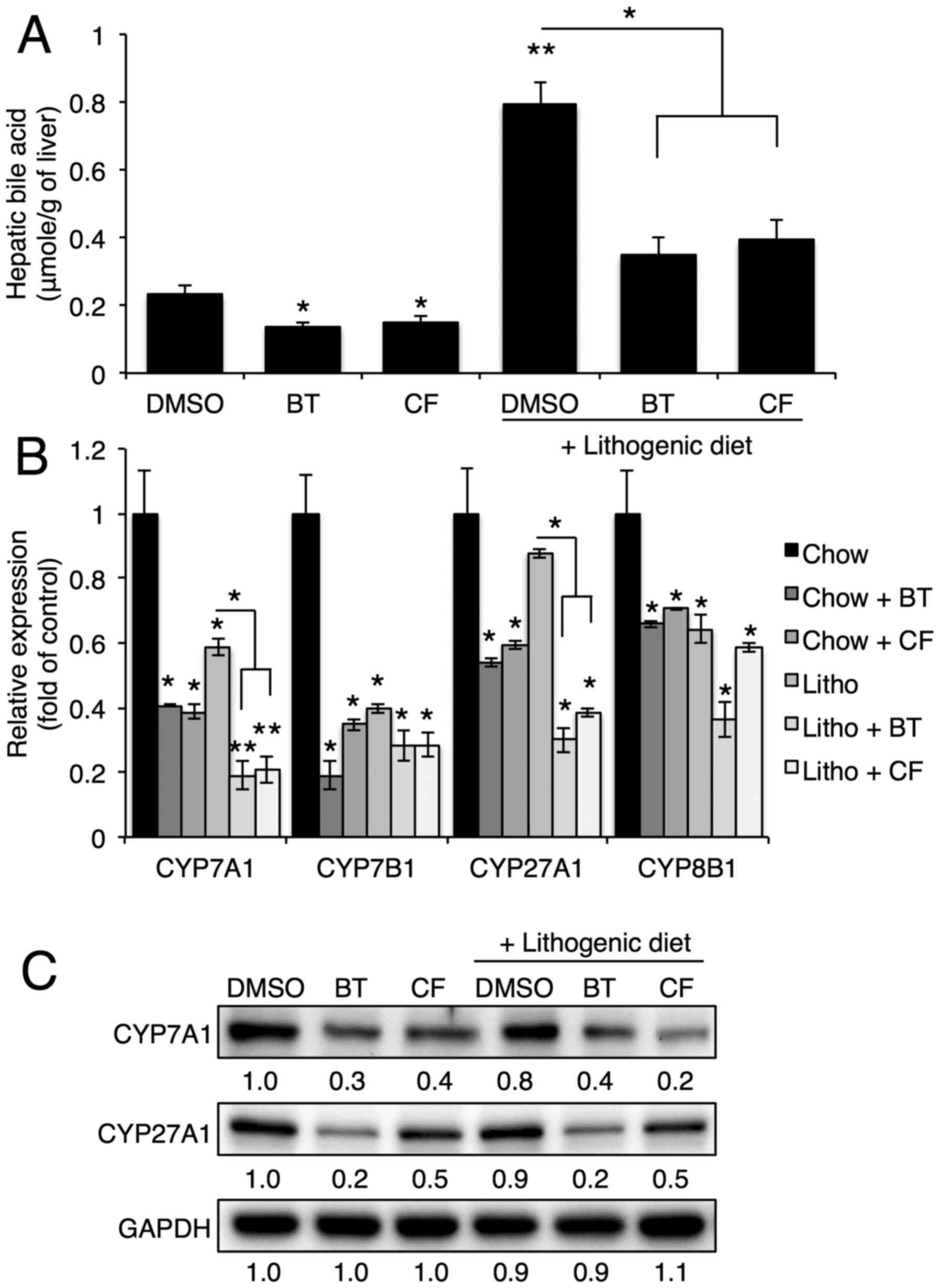 | Figure 3Proteasome inhibition reduces bile
acid synthesis by reducing Cyp7a1, Cyp7b1, Cyp27a1 and Cyp8b1
expression. (A) Hepatic bile acid levels upon BT or CF injection
were quantified with commercial kits (n=3). (B) mRNA levels of
Cyp7a1, Cyp7b1, Cyp27a1 and Cyp8b1 in the livers of mice injected
with BT or CF were measured by quantitative polymerase chain
reaction (n=3). (C) Representative western blot analyses of Cyp7a1
and Cyp27a1. Densitometric values for each band are provided below
the images. Data are expressed as the mean ± standard error of the
mean. *P<0.05 and **P<0.01. Three
independent experiments were performed. BT, bortezomib; CF,
carfilzomib; GAPDH, glyceraldehyde 3-phosp hate dehydrogenase;
DMSO, dimethyl sulfoxide; CYP7B1, oxysterol 7α-hydroxylase; CYP7A1,
cholesterol 7α-hydroxylase; CYP27A1, sterol 27-hydroxylase; CYP8B1,
sterol 12α-hydroxylase; Litho, lithogenic diet. |
Proteasome inhibition reduces biliary
cholesterol secretion
To investigate the effects of proteasome inhibition
on biliary secretion, bile composition was further analyzed using
commercial kits. Abcg5/Abcg8 heterodimers, Bsep and Mdr2 are
crucial for the biliary secretion of cholesterol, bile acids and
phospholipids, respectively (6).
Notably, BT and CF treatments reduced biliary cholesterol levels,
but not biliary phospholipid or bile acid levels (Fig. 4A), suggesting that proteasome
inhibition alters only the biliary secretion of cholesterol. In
accordance with reduced biliary cholesterol levels, Abcg5
and Abcg8 mRNA and protein levels were downregulated upon BT
or CF injection, whereas Bsep and Mdr2 levels were not
significantly affected (Fig. 4B and
C). These data indicate that proteasome inhibition of Abcg5 and
Abcg8 expression results in reduced biliary secretion of
cholesterol and prevents gallstone formation.
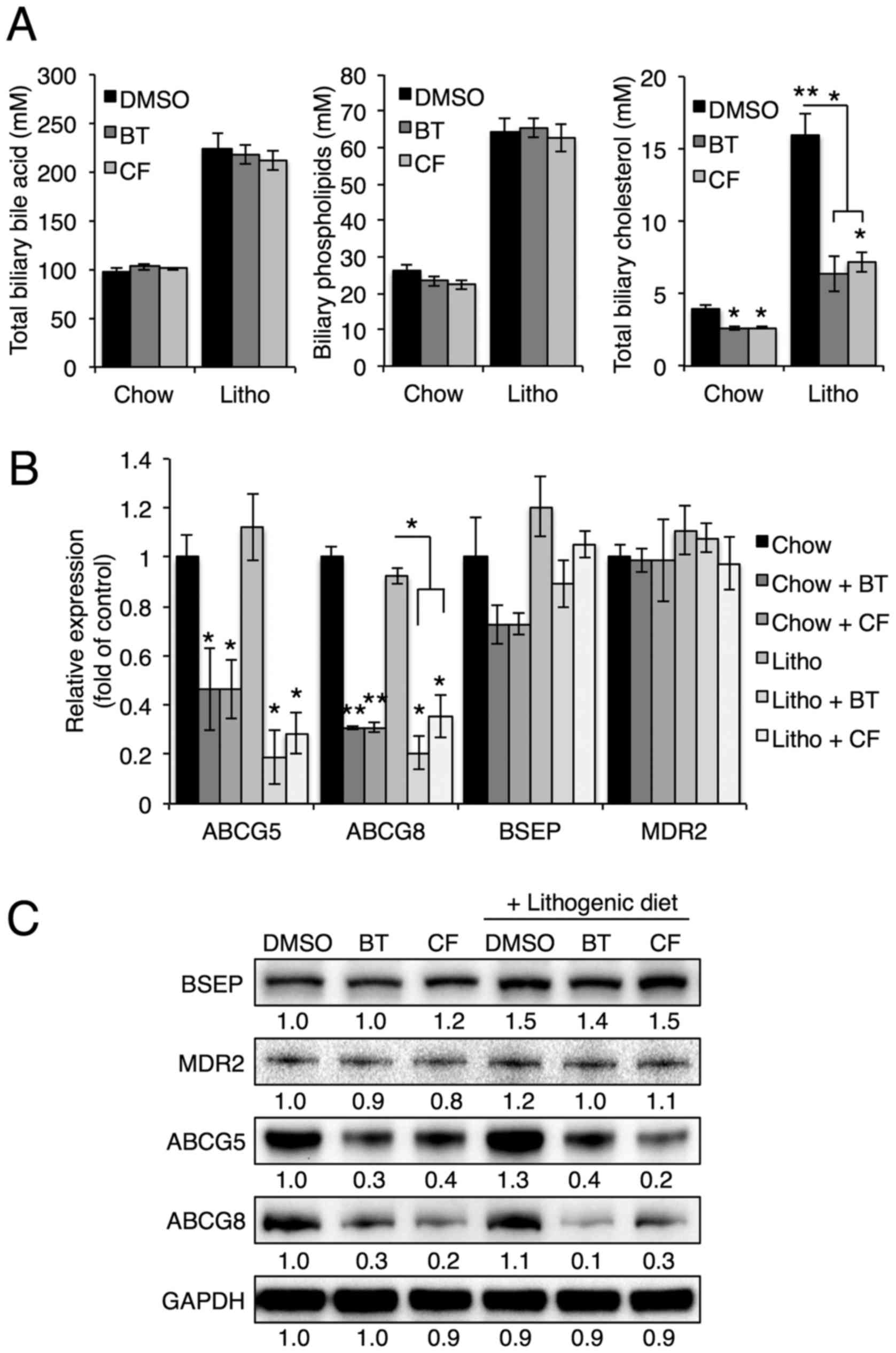 | Figure 4Proteasome inhibition reduces
cholesterol secretion by reducing expression of Abcg5 and Abcg8.
(A) Biliary cholesterol, bile acid and phospholipid levels were
measured in mice injected with BT or CF. (B) mRNA levels of ABCG5,
ABCG8, BSEP and MDR2 in the livers of mice injected with BT or CF
were measured by quantitative polymerase chain reaction (n=3). (C)
Representative western blot analyses of BSEP, MDR2, ABCG5 and
ABCG8. Densitometric values for each band are provided below the
images. Data are expressed as the mean ± standard error of the
mean. *P<0.05, **P<0.01 and
***P<0.001. Three independent experiments were
performed. BT, bortezomib; CF, carfilzomib; GAPDH, glyceraldehyde
3-phosp hate dehydrogenase; DMSO, dimethyl sulfoxide; Litho,
lithogenic diet; BSEP, bile salt export pump; MDR2, multidrug
resistance protein 2; ABCG, ATP-binding cassette transporter. |
Proteasome inhibition induces JNK
phosphorylation, which regulates Cyp7a1 expression
Cholesterol 7α-hydroxylase, which is encoded by
Cyp7a1, catalyzes the initial rate-limiting step in
cholesterol catabolism (5), and
Cyp7a1 transcription has been shown to be inhibited by the
transcription factors SHP, FXR and PXR (5). To examine whether these
transcription factors are involved in the reduction of Cyp7a1
levels as a result of proteasome inhibition, SHP, FXR and PXR
levels were examined upon treatment with proteasome inhibitors. As
they have been shown to inhibit Cyp7a1 transcription, SHP,
FXR and PXR levels were expected to increase upon proteasome
inhibition; however, SHP, FXR and PXR levels were reduced in livers
treated with proteasome inhibitors (Fig. 5A). Thus it was concluded that SHP,
FXR and PXR do not serve roles in the proteasome inhibition-induced
reduction of Cyp7a1.
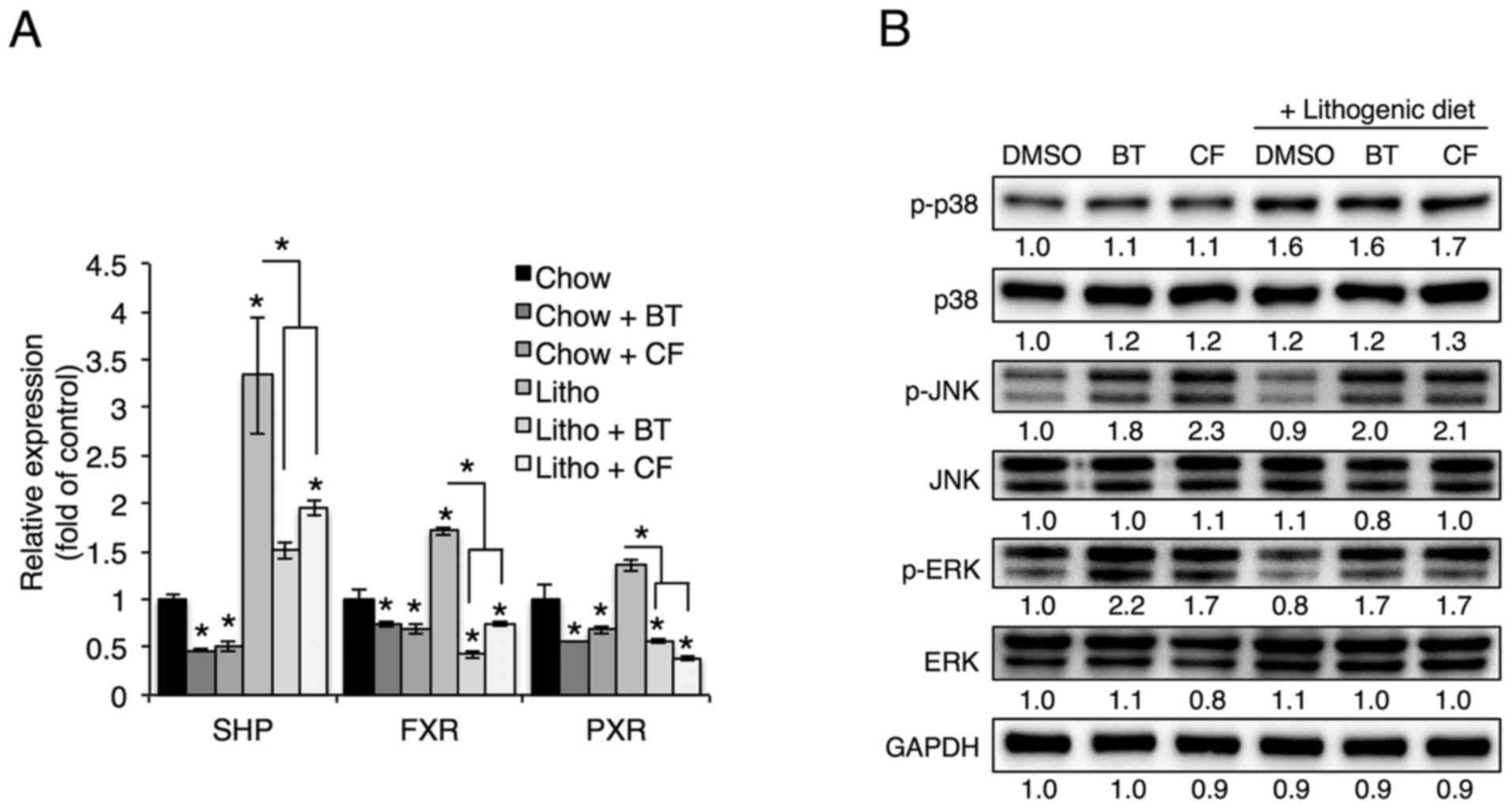 | Figure 5Proteasome inhibition increases
phosphorylation of JNK and ERK. (A) mRNA levels of SHP, FXR and PXR
in the livers of mice injected with BT or CF were measured by
quantitative polymerase chain reaction (n=3). (B) Representative
western blot analyses of proteins in the MAPK pathway.
Densitometric values for each band are provided below the images.
Data are expressed as the mean ± standard error of the mean.
*P<0.05. Three independent experiments were
performed. BT, bortezomib; CF, carfilzomib; GAPDH, glyceraldehyde
3-phosp hate dehydrogenase; DMSO, dimethyl sulfoxide; Litho,
lithogenic diet; JNK, c-Jun N-terminal protein kinase; ERK,
extracellular signal-regulated kinase; MAPK, mitogen-activated
protein kinase; SHP, small heterodimer partner; FXR, farnesoid X
receptor; PXR, pregnane X receptor. |
As mitogen-activated protein kinase (MAPK) has also
been shown to regulate Cyp7a1 transcription (22–24), the phosphorylation of p38, ERK and
JNK was examined in livers treated with proteasome inhibitors.
Hepatic phosphorylation of JNK and ERK, but not p38, was elevated
upon administration of BT and CF (Fig. 5B). To determine whether proteasome
inhibition-induced phosphorylation of JNK or of ERK led to the
reduced expression of cholesterol and bile acid synthesis genes,
the JNK inhibitor SP600125 and the ERK inhibitor PD98059 were used.
In accordance with in vivo data, Hep3B cells treated with
the proteasome inhibitors BT, CF and Epoxomicin displayed elevated
ERK and JNK phosphorylation accompanied by concomitant reductions
in mature form of (m)-SREBP2, HMGCR, CYP7A1 and CYP27A1 levels
(Fig. 6A). Inhibition of JNK, but
not ERK, reversed the downregulation of CYP7A1 and CYP27A1 that was
induced by proteasome inhibition, but the proteasome
inhibition-induced reduction of SREBP-2 and HMGCR was not reversed
with JNK or ERK inhibitors (Fig.
6B).
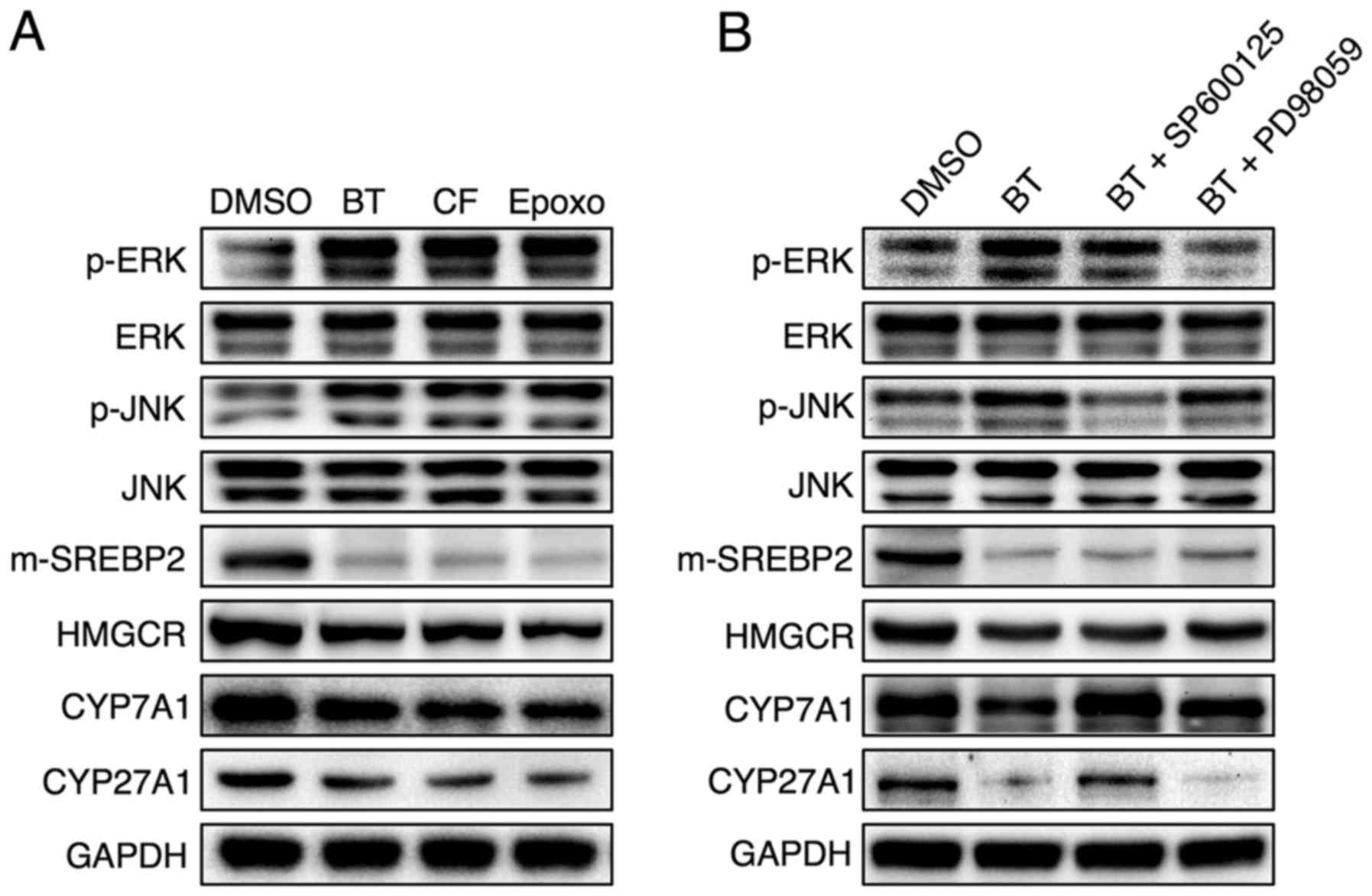 | Figure 6JNK phosphorylation as a result of
proteasome inhibition serves a critical role in the downregulation
of Cyp7a1 and Cyp27a1. Representative western blot analyses of (A)
Hep3B cells treated with 50 nM BT, 50 nM CF or 50 nM Epoxomicin
(Epoxo) and (B) Hep3B cells treated with 50 nM BT and 10 μM
SP600125 (JNK inhibitor) or 10 μM PD98059 (ERK inhibitor).
Three independent experiments were performed. BT, bortezomib; CF,
carfilzomib; Epoxo, epoxomicin; GAPDH, glyceraldehyde 3-phosp hate
dehydrogenase; DMSO, dimethyl sulfoxide; JNK, c-Jun N-terminal
protein kinase; ERK, extracellular signal-regulated kinase; CYP7A1,
cholesterol 7α-hydroxylase; CYP27A1, sterol 27-hydroxylase;
m-SREBP-2, mature form of sterol regulatory element-binding
protein-2; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase. |
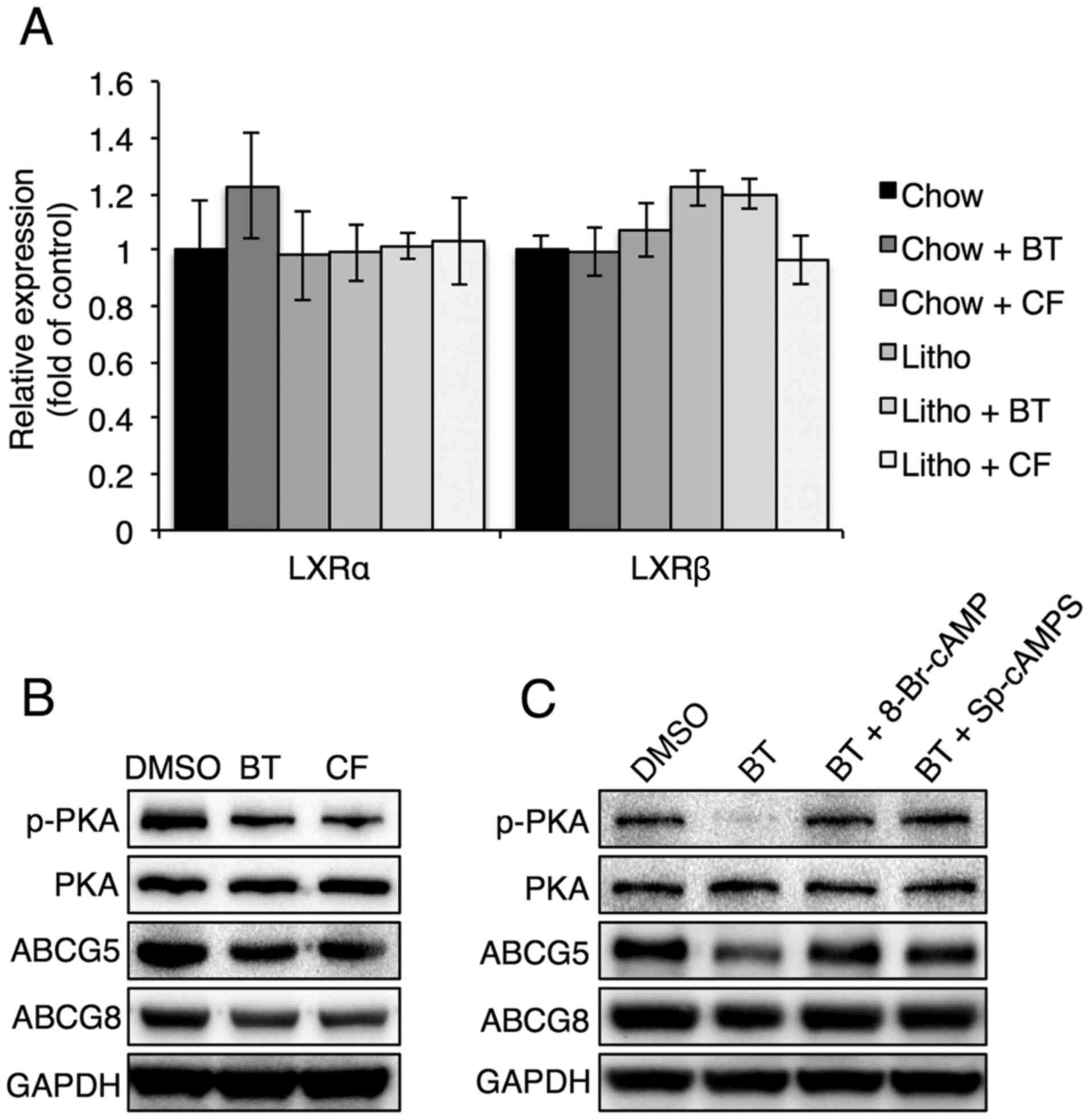
Proteasome inhibition reduces ABCG5 and
ABCG8 levels by reducing PKA phosphorylation
To evaluate how proteasome inhibitors regulate ABCG5
and ABCG8, the levels of liver X receptor (LXR)α and LXRβ, which
are known to regulate ABCG5 and ABCG8, were examined in mouse
livers in the presence of proteasome inhibitors (25). It was found that LXRα and LXRβ
levels were not affected by proteasome inhibitors, suggesting that
the proteasome inhibitor-induced downregulation of ABCG5 and ABCG8
was not caused by LXRα and LXRβ (Fig.
7A). Thus, the PKA signaling pathway, which has also been shown
to affect ABCG5 and ABCG8 expression, was analyzed (26). Reduced PKA phosphorylation was
found in Hep3B cells that were treated with proteasome inhibitors
(Fig. 7B). It was also found that
treatment with a PKA activator, 8-Br-cAMP or Sp-cAMPS, reversed the
proteasome inhibition-induced reduction of ABCG5 and ABCG8 levels
(Fig. 7C), confirming that PKA
phosphorylation serves a critical role in the regulation of ABCG5
and ABCG8 levels during proteasome inhibition.
Discussion
Gallstone disease is one of the most common
gastrointestinal diseases worldwide, and the cholesterol,
phospholipid and bile acid composition of bile serves an important
role in the pathogenesis of cholesterol gallstone formation
(2). For example, cholesterol
overcomes its low solubility in aqueous solutions by forming
micelles with phospholipids and bile acids (2,3).
Clinically, the main strategies to prevent gallstone disease have
been focused on reducing cholesterol synthesis, reducing
cholesterol secretion or inducing FXR, which increases bile acid
and phospholipid secretion by increasing BSEP and MDR2 levels
(27). In the present study,
proteasome inhibition with BT or CF prevented gallstone formation
by reducing cholesterol synthesis (via reductions in SREBP-2 and
HMGCR) and by reducing cholesterol secretion (via reductions in
ABCG5 and ABCG8). As excess cholesterol in bile can precipitate and
lead to gallstone formation, reducing biliary cholesterol by
proteasome inhibition is a strategy that can be utilized clinically
to prevent gallstone disease. Considering that BT is currently
utilized to treat hepatic conditions and certain cancer types, the
protective effect of BT on gallstone formation has clinical
importance.
Hepatic cholesterol levels, hepatic bile acid levels
and the expression of genes involved in bile acid synthesis were
significantly reduced upon proteasome inhibition. Reduced
degradation of cholesterol to bile acids may lead to a higher
concentration of free cholesterol that can be secreted leading to
cholesterol saturation and gallstone formation (28). However, in contrast to hepatic
bile acid levels, biliary bile acid levels were not affected by
proteasome inhibition, thus biliary bile acid levels do not appear
to affect gallstone formation. Rather, hepatic bile acid synthesis
may be reduced to compensate for the reduction in hepatic
cholesterol induced by proteasome inhibition.
In the present study, Cyp7a1 and Cyp27a1 protein
levels were regulated by JNK phosphorylation as a result of
proteasome inhibition. JNK phosphorylation has been shown to be
induced by inflammatory cytokines, including tumor necrosis
factor-α (TNF-α) and interleukin-1β (IL-1β) (29,30). However, TNF-α and IL-1β levels in
the liver and serum were not altered upon proteasome inhibition
(data not shown), suggesting that they are not involved in
proteasome inhibition-induced phosphorylation of JNK. Although
proteasome inhibition has been shown to activate MAPK (31,32), to the best of our knowledge, this
is the first study to demonstrate that JNK regulates genes involved
in bile acid synthesis upon proteasome inhibition. In contrast to
the regulation of Cyp7a1 and Cyp27a1, JNK or ERK inhibition was
unable to reverse the reduction in SREBP-2 and HMGCR levels induced
by proteasome inhibition. The mechanism of the reduction in SREBP-2
and HMGCR levels by proteasome inhibition remains to be
elucidated.
Proteasome inhibition also reduced the hepatic
levels of the cholesterol transporters ABCG5 and ABCG8, but did not
reduce the hepatic levels of the phospholipid and bile acid
transporters MDR2 and BSEP, respectively. These data indicate that
proteasome inhibition reduced hepatic cholesterol secretion to bile
and reduced hepatic cholesterol synthesis. Although LXR activation
has been shown to increase gallstone formation by regulating ABCG5
and ABCG8 levels (25,33), BT or CF injection did not affect
LXRα and LXRβ levels. PKA has also been shown to regulate ABCG5 and
ABCG8 expression (26), and the
present study showed that PKA phosphorylation was reduced by
proteasome inhibition. In addition, it was found that PKA
activation by 8-bromo-cAMP or Sp-cAMPS reversed the reduction of
ABCG5 and ABCG8 upon proteasome inhibition, confirming the role of
PKA in the proteasome inhibition-induced regulation of ABCG5 and
ABCG8.
Although proteolytic activity of the 26S proteasome
is ATP- and ubiquitin-dependent, c-AMP-induced PKA activation also
stimulates the proteolytic activity of the proteasome (34). The regulatory subunit of PKA has
been shown to be degraded via the proteasome (35), thus proteasome inhibition would
lead to accumulation of the regulatory subunit of PKA. As the
regulatory subunit of PKA functions as an inhibitor, PKA
phosphorylation would be reduced (36).
BT is currently used clinically as a first-line
chemotherapeutic agent for multiple myeloma, and high doses of BT
induce endoplasmic reticulum stress and cell death (12,15,37). In addition, high doses of BT often
lead to side effects, such as weight loss and painful neuropathy
(38). It has been shown that
administration of a low dose of BT (0.5 mg/kg/week) does not
increase ER stress (13).
Consistent with this, no signs of ER stress were observed with this
low BT dose in the present study (data not shown). Although several
studies have shown the protective effects of proteasome inhibition
for liver diseases, including drug-induced hepatotoxicity (15) and ethanol-induced steatosis
(13), to the best of our
knowledge, this study is the first to show that proteasome
inhibitors could be used to prevent gallstone formation.
In conclusion, in the present study, proteasome
inhibitors reduced hepatic cholesterol and bile acid synthesis, and
reduced biliary cholesterol secretion by reducing expression of the
cholesterol transporters ABCG5 and ABCG8. This finding may lead to
novel strategies for the treatment of gallstone disease.
Glossary
Abbreviations
Abbreviations:
|
ABCG
|
ATP-binding cassette transporter
|
|
BSEP
|
bile salt export pump
|
|
Cyp7a1
|
cholesterol 7α-hydroxylase
|
|
Cyp7b1
|
oxysterol 7α-hydroxylase
|
|
Cyp27a1
|
sterol 27-hydroxylase
|
|
Cyp8b1
|
sterol 12α-hydroxylase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FXR
|
farnesoid X receptor
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
HMGCR
|
3-hydroxy-3-methylglutaryl-CoA
reductase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
LXR
|
liver X receptor
|
|
MDR
|
multidrug resistance
|
|
PKA
|
protein kinase A
|
|
PXR
|
pregnane X receptor
|
|
SHP
|
small heterodimer partner
|
|
SREBP
|
sterol regulatory element-binding
protein
|
Acknowledgments
This study was supported by the Gachon University
Research Fund of 2013 (grant no. 2013-M050).
References
|
1
|
Stinton LM and Shaffer EA: Epidemiology of
gallbladder disease: cholelithiasis and cancer. Gut Liver.
6:172–187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kosters A, Jirsa M and Groen AK: Genetic
background of cholesterol gallstone disease. Biochim Biophys Acta.
1637:1–19. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Portincasa P, Moschetta A and Palasciano
G: Cholesterol gallstone disease. Lancet. 368:230–239. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tao R, Xiong X, DePinho RA, Deng CX and
Dong XC: Hepatic SREBP-2 and cholesterol biosynthesis are regulated
by FoxO3 and Sirt6. J Lipid Res. 54:2745–2753. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chiang JY: Bile acids: regulation of
synthesis. J Lipid Res. 50:1955–1966. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van Erpecum KJ: Pathogenesis of
cholesterol and pigment gallstones: an update. Clin Res Hepatol
Gastroenterol. 35:281–287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ahmed MH, Hamad MA, Routh C and Connolly
V: Statins as potential treatment for cholesterol gallstones: an
attempt to understand the underlying mechanism of actions. Expert
Opin Pharmacother. 12:2673–2681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Merzon E, Weiss NS, Lustman AJ, Elhayani
A, Dresner J and Vinker S: Statin administration and risk of
cholecystectomy: a population-based case-control study. Expert Opin
Drug Saf. 9:539–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cariati A and Piromalli E: Limits and
perspective of oral therapy with statins and aspirin for the
prevention of symptomatic cholesterol gallstone disease. Expert
Opin Pharmacother. 13:1223–1227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kotb MA: Molecular mechanisms of
ursodeoxycholic acid toxicity & side effects: ursodeoxycholic
acid freezes regeneration & induces hibernation mode. Int J Mol
Sci. 13:8882–8914. 2012. View Article : Google Scholar :
|
|
11
|
Adams J, Palombella VJ and Elliott PJ:
Proteasome inhibition: a new strategy in cancer treatment. Invest
New Drugs. 18:109–121. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Field-Smith A, Morgan GJ and Davies FE:
Bortezomib (Velca-detrade mark) in the treatment of multiple
myeloma. Ther Clin Risk Manag. 2:271–279. 2006. View Article : Google Scholar
|
|
13
|
Oliva J, French SW, Li J and Bardag-Gorce
F: Proteasome inhibitor treatment reduced fatty acid,
triacylglycerol and cholesterol synthesis. Exp Mol Pathol.
93:26–34. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anan A, Baskin-Bey ES, Isomoto H, Mott JL,
Bronk SF, Albrecht JH and Gores GJ: Proteasome inhibition
attenuates hepatic injury in the bile duct-ligated mouse. Am J
Physiol Gastrointest Liver Physiol. 291:G709–G716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park WJ, Kim SY, Kim YR and Park JW:
Bortezomib alleviates drug-induced liver injury by regulating
CYP2E1 gene transcription. Int J Mol Med. 37:613–622. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nawrocki ST, Carew JS, Pino MS, Highshaw
RA, Dunner K Jr, Huang P, Abbruzzese JL and McConkey DJ: Bortezomib
sensitizes pancreatic cancer cells to endoplasmic reticulum
stress-mediated apoptosis. Cancer Res. 65:11658–11666. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Codony-Servat J, Tapia MA, Bosch M, Oliva
C, Domingo-Domenech J, Mellado B, Rolfe M, Ross JS, Gascon P,
Rovira A, et al: Differential cellular and molecular effects of
bortezomib, a proteasome inhibitor, in human breast cancer cells.
Mol Cancer Ther. 5:665–675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kondagunta GV, Drucker B, Schwartz L,
Bacik J, Marion S, Russo P, Mazumdar M and Motzer RJ: Phase II
trial of bortezomib for patients with advanced renal cell
carcinoma. J Clin Oncol. 22:3720–3725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Papandreou CN, Daliani DD, Nix D, Yang H,
Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, et al:
Phase I trial of the proteasome inhibitor bortezomib in patients
with advanced solid tumors with observations in
androgen-independent prostate cancer. J Clin Oncol. 22:2108–2121.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dick LR and Fleming PE: Building on
bortezomib: second-generation proteasome inhibitors as anti-cancer
therapy. Drug Discov Today. 15:243–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Wooton-Kee CR, Coy DJ, Athippozhy AT, Zhao
T, Jones BR and Vore M: Mechanisms for increased expression of
cholesterol 7alpha-hydroxylase (Cyp7a1) in lactating rats.
Hepatology. 51:277–285. 2010. View Article : Google Scholar
|
|
23
|
Gupta S, Stravitz RT, Dent P and Hylemon
PB: Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene
expression by bile acids in primary rat hepatocytes is mediated by
the c-Jun N-terminal kinase pathway. J Biol Chem. 276:15816–15822.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stroup D: Kinase/phosphatase regulation of
CYP7A1. Front Biosci. 10:1678–1692. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Repa JJ, Berge KE, Pomajzl C, Richardson
JA, Hobbs H and Mangelsdorf DJ: Regulation of ATP-binding cassette
sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha
and beta. J Biol Chem. 277:18793–18800. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamazaki Y, Yasui K, Hashizume T, Suto A,
Mori A, Murata Y, Yamaguchi M, Ikari A and Sugatani J: Involvement
of a cyclic adenosine monophosphate-dependent signal in the
diet-induced canalicular trafficking of adenosine
triphosphate-binding cassette transporter g5/g8. Hepatology.
62:1215–1226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moschetta A, Bookout AL and Mangelsdorf
DJ: Prevention of cholesterol gallstone disease by FXR agonists in
a mouse model. Nat Med. 10:1352–1358. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bertolotti M, Gabbi C, Anzivino C, Carulli
L and Carulli N: Changes in bile acid synthesis in gallstone
disease: cause, consequence, or neither? Hepatology. 46:1664–1665.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li T, Jahan A and Chiang JY: Bile acids
and cytokines inhibit the human cholesterol 7 alpha-hydroxylase
gene via the JNK/c-jun pathway in human liver cells. Hepatology.
43:1202–1210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu X, Qi Y, Tian B, Chen D, Gao H, Xi C,
Xing Y and Yuan Z: Maternal protein restriction induces alterations
in hepatic tumor necrosis factor-α/CYP7A1 signaling and disorders
regulation of cholesterol metabolism in the adult rat offspring. J
Clin Biochem Nutr. 55:40–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Choi CH, Lee BH, Ahn SG and Oh SH:
Proteasome inhibition-induced p38 MAPK/ERK signaling regulates
autophagy and apoptosis through the dual phosphorylation of
glycogen synthase kinase 3β. Biochem Biophys Res Commun.
418:759–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang HQ, Liu BQ, Gao YY, Meng X, Guan Y,
Zhang HY and Du ZX: Inhibition of the JNK signalling pathway
enhances proteasome inhibitor-induced apoptosis of kidney cancer
cells by suppression of BAG3 expression. Br J Pharmacol.
158:1405–1412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uppal H, Zhai Y, Gangopadhyay A, Khadem S,
Ren S, Moser JA and Xie W: Activation of liver X receptor
sensitizes mice to gallbladder cholesterol crystallization.
Hepatology. 47:1331–1342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang F, Hu Y, Huang P, Toleman CA,
Paterson AJ and Kudlow JE: Proteasome function is regulated by
cyclic AMP-dependent protein kinase through phosphorylation of
Rpt6. J Biol Chem. 282:22460–22471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hegde AN, Goldberg AL and Schwartz JH:
Regulatory subunits of cAMP-dependent protein kinases are degraded
after conjugation to ubiquitin: a molecular mechanism underlying
long-term synaptic plasticity. Proc Natl Acad Sci USA.
90:7436–7440. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin JT, Chang WC, Chen HM, Lai HL, Chen
CY, Tao MH and Chern Y: Regulation of feedback between protein
kinase A and the proteasome system worsens Huntington's disease.
Mol Cell Biol. 33:1073–1084. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takenokuchi M, Miyamoto K, Saigo K and
Taniguchi T: Bortezomib causes ER stress-related death of acute
promyelocytic leukemia cells through excessive accumulation of
PML-RARA. Anticancer Res. 35:3307–3316. 2015.PubMed/NCBI
|
|
38
|
Meregalli C, Carozzi VA, Sala B, Chiorazzi
A, Canta A, Oggioni N, Rodriguez-Menendez V, Ballarini E, Ceresa C,
Nicolini G, et al: Bortezomib-induced peripheral neurotoxicity in
human multiple myeloma-bearing mice. J Biol Regul Homeost Agents.
29:115–124. 2015.PubMed/NCBI
|
|
39
|
Liu G, Friggeri A, Yang Y, Park Y-J,
Tsuruta Y and Abraham E: miR-147, a microRNA that is induced upon
Toll-like receptor stimulation, regulates murine macrophage
inflammatory responses. Proc Natl Acad Sci USA. 106:15819–15824.
2009. View Article : Google Scholar : PubMed/NCBI
|