Introduction
Ischemic heart disease is a leading cause of
morbidity and mortality (1,2).
Reperfusion of coronary arteries in response to thrombolytic
treatment or percutaneous coronary intervention is crucial for
reducing ischemia-induced heart damage (3–5).
However, reperfusion may induce additional myocardial injury,
including cardiomyocyte death and loss of cardiac function; this is
known as myocardial ischemia/reperfusion (I/R) injury (6–8).
The cellular mechanisms underlying I/R injury remain to be
completely elucidated. Increasing evidence has suggested that
ischemia initiates myocardial apoptosis, which is amplified by
reperfusion, thus contributing to cardiac cell death (9,10).
Conversely, suppressing apoptotic processes can minimize
I/R-induced cardiac damage (11).
Transient receptor potential vanilloid 1 (TRPV1) is
a ligand-gated nonselective cation channel, which is primarily
expressed in sensory nerves that innervate cardiovascular tissues,
including the heart and blood vessels (12,13). TRPV1 has been considered to act as
a molecular integrator of numerous chemical and physical mediators,
including noxious heat, low pH, capsaicin and lipid metabolites
(14–16). TRPV1 expressed in the cardiac
sensory nerves, which conduct angina pain signals (17,18), may function as a molecular sensor
for the detection of tissue ischemia and the modulation of cardiac
function (12). Pharmacological
studies have suggested that activation of TRPV1 with exogenous
agonists protects the heart from I/R injury (19,20), whereas TRPV1 gene deletion may
impair cardiac recovery following I/R (21). Furthermore, activation of TRPV1
inhibits hypoxia/reoxygenation-induced apoptosis in rat hippocampal
neurons via activation of the phosphatidylinositol 3-kinase/protein
kinase B (PI3K/Akt) and extracellular signal-regulated protein
kinase 1/2 (ERK1/2) signaling pathways (22), which are key regulators of cell
growth and survival (23,24). During I/R, both signaling pathways
are activated and confer cardioprotective effects through the
recruitment of downstream anti-apoptotic molecules (25); however, the role of TRPV1 in
myocardial apoptosis in response to I/R injury remains to be fully
characterized. Furthermore, it is currently unknown whether the
PI3K/Akt and ERK1/2 signaling pathways are involved in
TRPV1-mediated myocardial apoptosis in I/R. The present study aimed
to determine the effects of TRPV1 activation on myocardial
apoptosis in response to I/R injury and explored the downstream
signaling mechanism of TRPV1 activation.
Materials and methods
Animals and reagents
Male TRPV1 knockout (TRPV1−/−) and
wild-type (WT) C57BL/6J mice (n=54 each; weight, 25–30 g; age,
10–12 weeks) were provided by the Experimental Animal Center of
Chongqing Medical University (Chongqing, China) and were maintained
under specific pathogen-free conditions (temperature, 22°C;
humidity, 60%), with a 12-h light/dark cycle and with free access
to food and water. All surgical procedures performed on mice were
conducted under sodium pentobarbital anesthesia [50 mg/kg,
intraperitoneal (IP) injection], and all efforts were made to
minimize their suffering. The present study was approved by the
Ethics Committee of Chongqing Medical University. The mice were
treated in accordance with the recommendations listed in the Guide
for the Care and Use of Laboratory Animals of the National
Institutes of Health (NIH) (26).
LY294002 (a PI3K inhibitor) and 2,3,5-triphenyl
tetrazolium chloride (TTC) were purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany); antibodies against phosphorylated
(p)-Akt (Ser473; cat. no. 9271), Akt (cat. no. 9272), p-ERK1/2
(Thr202/Thr204; cat. no. 4370), ERK1/2 (cat. no. 9102), B-cell
lymphoma-2 (Bcl-2; cat. no. 2870) and Bcl-2-associated X protein
(Bax; cat. no. 14796) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Anti-GAPDH antibody (cat. no.
AG019) and the bicinchoninic acid (BCA) protein assay kit were
purchased from Beyotime Institute of Biotechnology (Haimen, China).
The terminal deoxynucleotidyl transferase-mediated dUTP nick-end
labeling (TUNEL) reaction mixture was purchased from Roche
Diagnostics (Laval, QC, Canada).
Langendorff heart preparation
Mice were treated with heparin (500 U/kg, IP) and
anesthetized with pentobarbital sodium (50 mg/kg, IP) prior to
thoracotomy. The hearts were rapidly excised, placed into ice-cold
Krebs-Henseleit (K-H) buffer (composition in mM: NaCl 118, KCl 4.7,
MgSO4 1.2, KH2PO4 1.2,
CaCl2 2.5, NaHCO3 25, Na-EDTA 0.5 and glucose
11) and perfused in a Langendorff apparatus within 2 min under a
constant pressure of 80 mmHg. The perfusion fluid was oxygenated
with a mixture of 95% O2 and 5% CO2, and was
maintained at pH 7.4. In addition, temperature of the K-H buffer
was maintained at 37°C throughout the experiment. A fluid-filled
balloon connected to a pressure transducer was inserted into the
left ventricle (LV) via the mitral valve to monitor LV pressure.
The volume of the balloon was adjusted to maintain a stable LV
end-diastolic pressure of 5–8 mmHg during initial
equilibration.
Experimental protocol
The isolated mouse hearts were randomly divided into
the following six groups (n=9/group): i) WT Sham group; ii)
TRPV1−/− Sham group; iii) WT I/R group; iv)
TRPV1−/− I/R group; v) WT I/R + LY294002 group; and vi)
TRPV1−/− I/R + LY294002 group. To induce I/R, the
perfused hearts were stabilized for 30 min and subjected to global
normothermic (37°C) ischemia (no flow) for 30 min, followed by 60
min of reperfusion. Conversely, hearts in the sham groups were
perfused with K-H solution continuously until the end of the
experiment. Hearts in the LY294002 treatment group were perfused
for 10 min with LY294002 (50 µM) in K-H buffer prior to induction
of global ischemia, the hearts were then subjected to global
ischemia followed by reperfusion without LY294002. LY294002 was
initially dissolved in dimethyl sulfoxide and then in K-H buffer to
reach a final concentration of 50 µM. The concentration of LY294002
used in the present study has been reported to specifically abolish
PI3K activity and inhibit Akt phosphorylation, but not the
phosphorylation of other protein kinases, including
phosphatidylinositol 4-kinase, protein kinase C, mitogen-activated
protein kinase or c-Src (27).
The experimental protocol is presented in Fig. 1.
Determination of myocardial infarct
size
At the end of reperfusion, the hearts were frozen at
−20°C for 15 min and cut into five pieces along the longitudinal
heart axis. Heart sections were incubated for 10 min in 1% TTC at
37°C in the dark. Subsequently, the sections were soaked in 4%
paraformaldehyde in phosphate buffer overnight at 4°C to enhance
the contrast of the stain. Viable myocardium exhibited red
staining, whereas the infarcted area exhibited white staining. Each
image was digitally photographed and the infarct size was analyzed
using ImageJ software version 1.49v (NIH, Bethesda, MD, USA). The
infarct size was expressed as a percentage of the total area of the
heart.
Measurement of myocardial cell
apoptosis
For tissue TUNEL staining, the heart samples were
fixed in 4% paraformaldehyde at 25°C for 24 h, embedded in paraffin
and cut into transverse sections (5 µm). Myocardial
apoptosis was assessed using a TUNEL staining kit according to the
manufacturer's protocol. The numbers of TUNEL-positive myocyte
nuclei and total myocyte nuclei were counted in 10 different fields
for each stained section at high magnification (objective, ×400).
The number of TUNEL-positive nuclei (brown staining) was calculated
using ImageJ software (NIH) according to the following formula:
Percentage of TUNEL-positive myocyte nuclei = TUNEL-positive
myocyte nuclei/total myocyte nuclei × 100%.
Western blot analysis
Total proteins were extracted from mouse heart
tissues and protein concentrations were assessed using a BCA
protein assay kit. The mouse heart tissues were homogenized using
radioimmunoprecipitation lysis buffer (RIPA; Beyotime Institue of
Biotechnology) and then centrifuged at 4°C. Equal amounts of
protein (4 µg/µl) were separated by 10–12% SDS-PAGE
and were transferred onto polyvinylidene fluoride (PVDF) membranes
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The PVDF membranes
were blocked in 5% non-fat milk in Tris-buffered saline containing
0.05% Tween-20 for 2 h at room temperature, and were then incubated
overnight at 4°C with primary antibodies against GAPDH (1:1,000
dilution), p-Akt (Ser473) (1:1,000 dilution), Akt (1:1,000
dilution), p-ERK1/2 (Thr202/Thr204) (1:2,000 dilution), ERK1/2
(1:1,000 dilution), Bcl-2 (1:1,000 dilution) and Bax (1:1,000
dilution). Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies (1:5,000
dilution; ZB-5305; Beijing Zhongshan Golden Bridge Biotechnology
Co., Beijing, China) for 2 h at room temperature. The
immunoreactive proteins were visualized using an enhanced
chemiluminescence detection system (Pierce; Thermo Fisher
Scientific Inc., Waltham, MA, USA). The band density was analyzed
by ImageJ software (NIH).
Statistical analysis
SPSS 17.0 statistical software (SPSS, Inc., Chicago,
IL, USA) was used for all statistical analyses. Data are expressed
as the means ± standard deviation. Significance was determined
using either an unpaired Student's t-test (differences in
TUNEL-positive cells and infarct size in TRPV1−/− hearts
compared with in WT hearts) or one-way analysis of variance
followed by the Tukey-Kramer multiple comparison test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Myocardial apoptosis is increased in
TRPV1−/− hearts following I/R
To investigate the role of TRPV1 in apoptosis, WT
and TRPV1−/− hearts were subjected to I/R, and
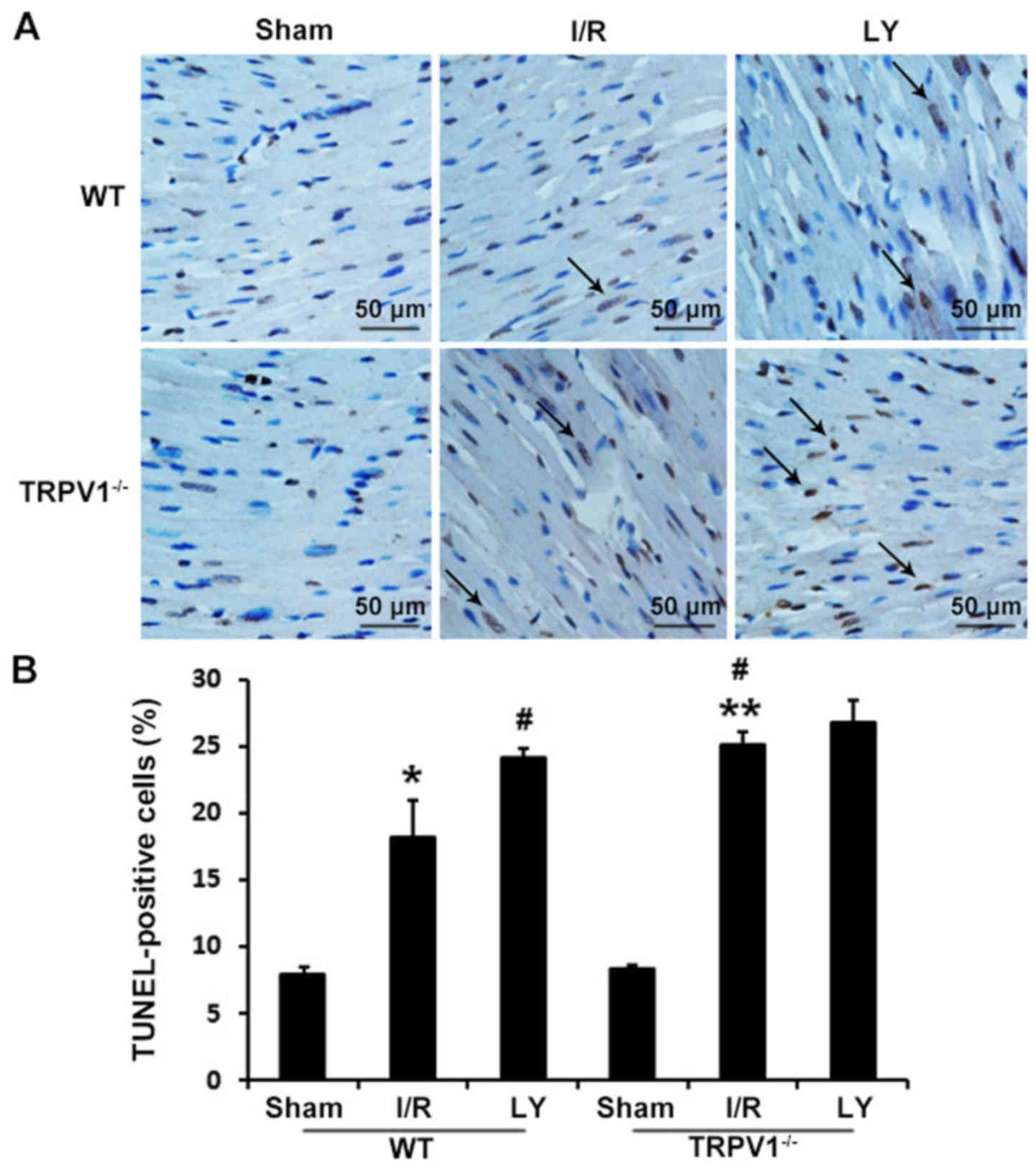
myocardial apoptosis was detected using TUNEL staining (Fig. 2). The percentage of TUNEL-positive
cardiomyocytes was markedly increased in the TRPV1−/−
and WT hearts following I/R compared with in the respective sham
control groups (P<0.01). Furthermore, the percentage of
TUNEL-positive cardiomyocytes in TRPV1−/− hearts
subjected to I/R was significantly greater than in WT hearts
(25.10+1.03 vs. 18.20+2.79%; P<0.01). In addition, treatment
with the PI3K inhibitor LY294002 prior to I/R, increased myocardial
apoptosis in WT (P<0.01) but not in TRPV1−/− hearts
(P>0.05) (Fig. 2).
Infarct size is larger in
TRPV1−/− hearts following I/R
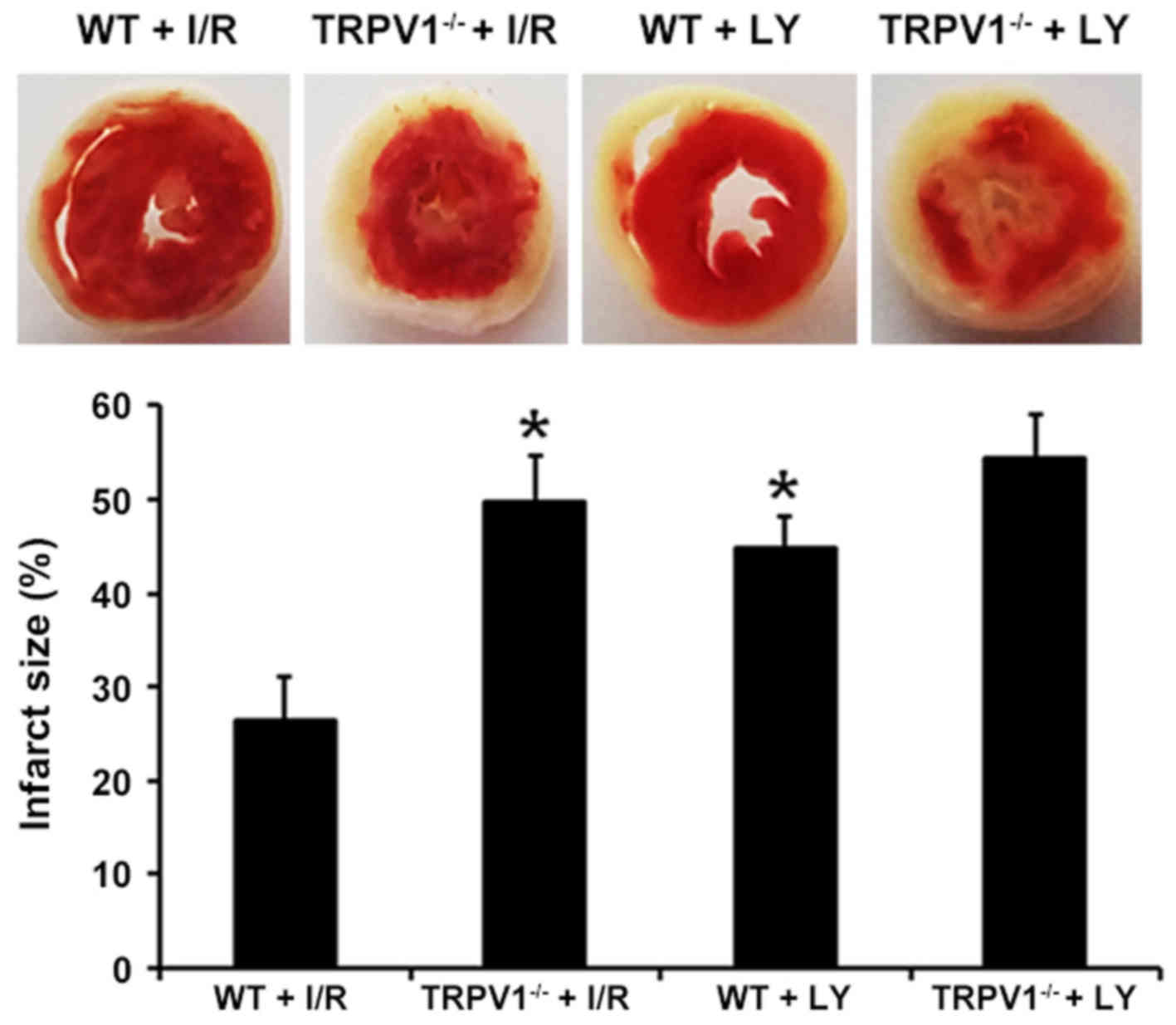
Myocardial infarct size was assessed using TTC
staining (Fig. 3). Myocardial
infarct size was markedly increased in the TRPV1−/− and
WT groups following I/R compared with in the corresponding sham
groups (P<0.01) (data not shown). Notably, infarct size in
TRPV1−/− hearts was markedly increased compared with in
the WT hearts (49.58+4.83 vs. 26.32+4.57%; P<0.01). In addition,
treatment with LY294002 significantly increased infarct size in WT
(P<0.01) but not in TRPV1−/− hearts (P>0.05)
(Fig. 3).
PI3K/Akt signaling is involved in the
anti-apoptotic effects of TRPV1
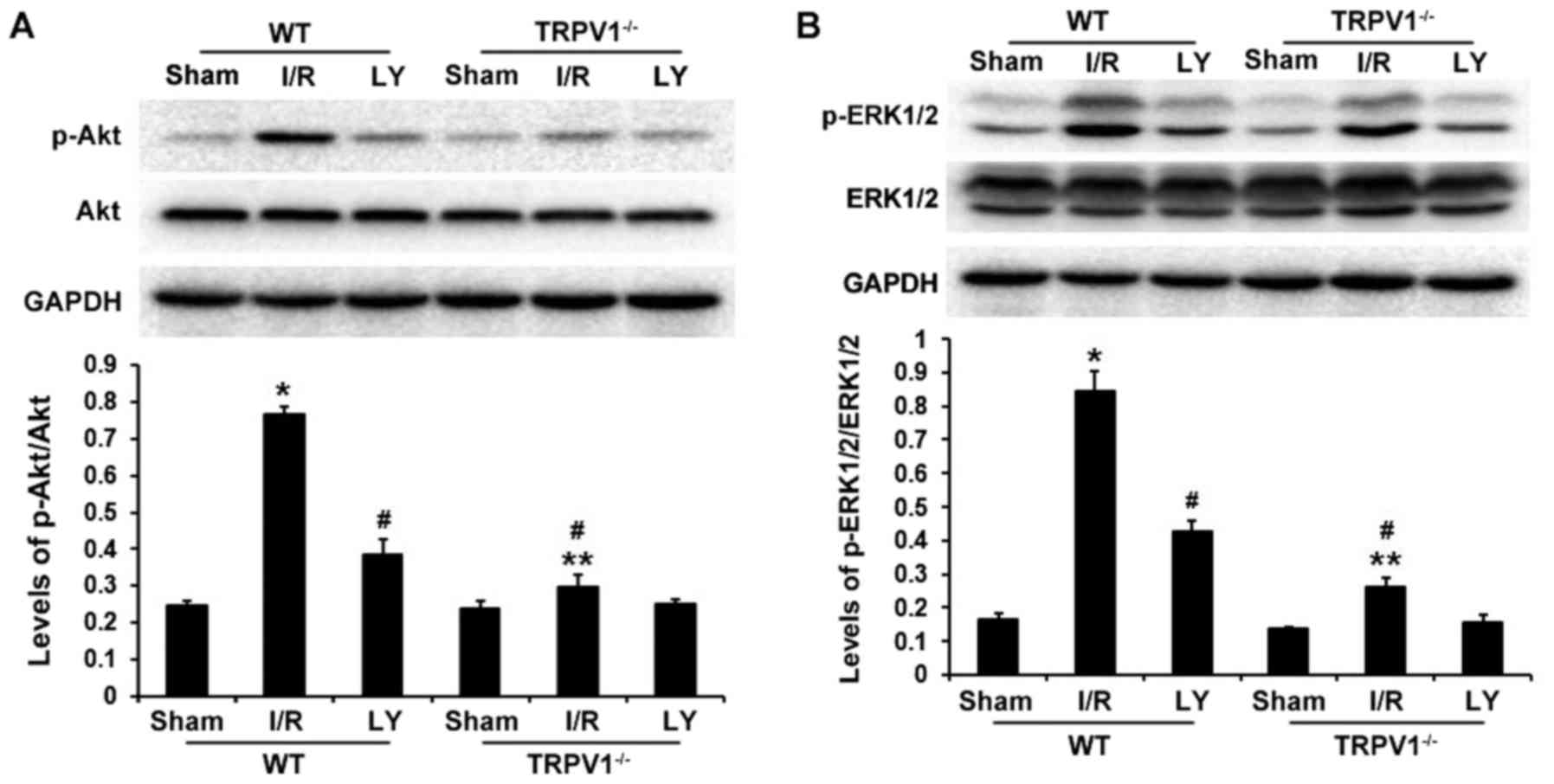
The PI3K/Akt and ERK1/2 survival signaling pathways
serve key roles in protecting cardiomyocytes from apoptosis during
I/R injury. To determine the downstream signaling pathway
associated with the effects of TRPV1, Akt and ERK1/2
phosphorylation was detected in heart samples exposed to I/R
(Fig. 4). Myocardial I/R
significantly increased p-Akt and p-ERK1/2 expression in
TRPV1−/− and WT hearts (P<0.05), without any
significant changes in total ERK1/2 and Akt protein levels. The
ratios of p-AKT/AKT and p-ERK1/2/ERK1/2 were significantly higher
following I/R in WT hearts compared with in TRPV1−/−
hearts (P<0.01); however, these ratios were decreased following
LY294002 treatment (P<0.01) (Fig.
4). These results suggested that the PI3K/Akt signaling pathway
may be involved in the beneficial effects of TRPV1 during I/R
injury.
TRPV1 activation increases Bcl-2/Bax
ratio by activating the PI3K-Akt signaling pathway
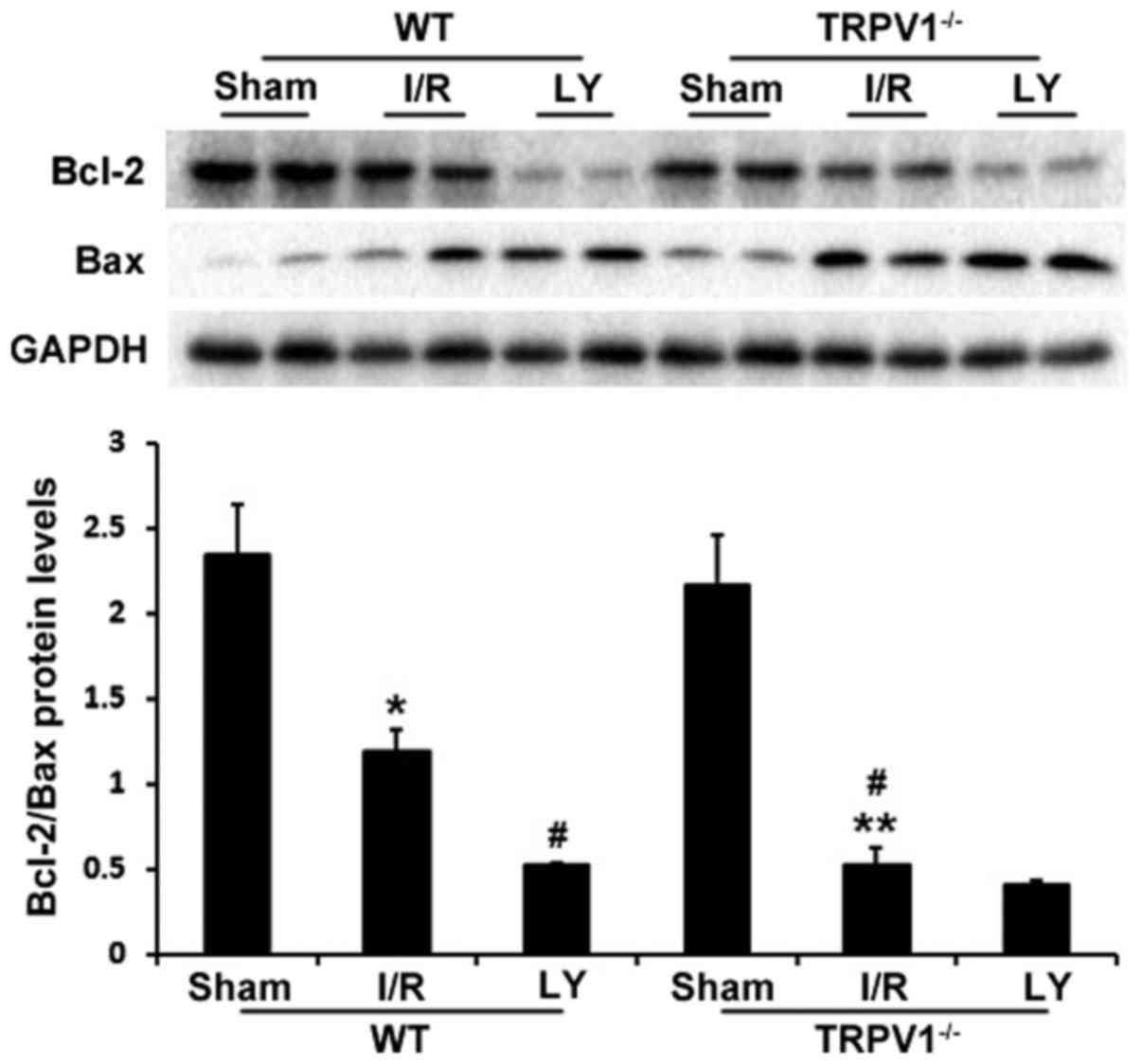
To further examine the mechanism of apoptosis, the
protein expression levels of Bax and Bcl-2 were determined
(Fig. 5). Bcl-2 and Bax have
major roles in determining cell survival or death in response to
apoptotic stimuli (28,29). The present study demonstrated that
I/R resulted in a significant decrease in Bcl-2/Bax protein ratio
compared with in the sham group in TRPV1−/− and WT
hearts (P<0.05). In addition, the ratio of Bcl-2/Bax was lower
in the of TRPV1−/− hearts compared with in WT hearts
following I/R (P<0.01). Furthermore, treatment with LY294002
markedly decreased Bcl-2/Bax ratio in WT hearts (P<0.01) but not
in TRPV1−/− hearts (Fig.
5).
Discussion
The present study demonstrated that TRPV1 gene
knockdown significantly increased myocardial apoptosis and
infarction during I/R. In addition, treatment with the PI3K
inhibitor LY294002 increased infarct size and number of
TUNEL-positive cardiomyocytes in WT but not in TRPV1−/−
hearts. These results indicated that TRPV1 may protect the heart
against I/R injury, possibly through its anti-apoptotic effects via
activating the PI3K/Akt signaling pathway.
The importance of the TRPV1 channel in regulating
heart function has recently been highlighted. TRPV1 can be
activated by numerous metabolites that are accumulated during
myocardial ischemia (12). Prior
induction of TRPV1 may confer a benefit to the myocardium against
further severe damage. This concept is supported by evidence that
suggests that short episodes of sub-lethal ischemia may induce
ischemic preconditioning, and that TRPV1 knockout abrogates the
effects of ischemic preconditioning (30). In addition, TRPV1 activation
promotes recovery of cardiac systolic/diastolic function during I/R
(21). Apoptosis is a type of
programmed cell death, which significantly contributes to
myocardial I/R injury (9,10); therefore, inhibition of myocardial
cell apoptosis may prevent cell loss and attenuate cardiac injury
during myocardial I/R (11).
Apoptosis-associated proteins, including Bcl-2 and Bax serve
pivotal roles in apoptosis (28,29). In particular, apoptosis is
regulated by the ratio of Bcl-2 and Bax protein expression
(31,32). Previous studies have indicated
that TRPV1 is involved in the regulation of apoptosis (22,33–35). Capsaicin has been reported to
significantly reduce reperfusion-induced liver injury by reducing
apoptosis due to activation of TRPV1 (34). Therefore, the present study
investigated the role of TRPV1 in myocardial apoptosis during I/R.
The results demonstrated that the Bcl-2/Bax ratio was significantly
reduced in TRPV1−/− compared with WT hearts following
I/R. Furthermore, I/R increased the percentage of TUNEL-positive
cells and infarct size in TRPV1−/− hearts compared with
in WT hearts. These findings suggested that TRPV1 activation
protects cardiomyocytes from I/R injury by suppressing myocardial
apoptosis. Although cardiac function was not assessed in the
present study, our previous study demonstrated that TRPV1
deficiency results in increased mortality, aggravated inflammatory
response, enhanced cardiac fibrosis and exaggerated progression of
LV remodeling 7 days after myocardial infarction (36). However, further studies are
required to investigate the role of TRPV1 in regulating cardiac
function following I/R in vivo.
Previous studies have revealed that capsaicin
activates the PI3K/Akt and ERK1/2 pathways in dorsal root ganglion
neurons (37) and human HepG2
cells (38) through activation of
the capsaicin receptor TRPV1. The PI3K/Akt and ERK1/2 signaling
pathways, when specifically activated at the time of myocardial
reperfusion, may inhibit cardiomyocyte apoptosis and attenuate I/R
injury through targeting downstream molecules, including Bcl-2 and
Bax (39,40). Therefore, the present study
investigated the role of TRPV1 in phosphorylation of Akt and ERK1/2
in hearts subjected to I/R. The results indicated that the ratios
of p-AKT/AKT and p-ERK1/2/ERK1/2 were upregulated in
TRPV1−/− and WT hearts following I/R compared with in
the sham groups. In addition, the ratios of p-AKT/AKT and
p-ERK1/2/ERK1/2 were lower in TRPV1−/− hearts compared
with in WT hearts. Notably, treatment with LY294002 decreased
Bcl-2/Bax ratio, and increased infarct size and TUNEL-positive
cardiomyocytes in WT but not in TRPV1−/− hearts. These
results suggested that TRPV1 may inhibit I/R-induced cardiomyocyte
apoptosis via PI3K/Akt activation. In addition, treatment with
LY294002 significantly inhibited p-ERK1/2 levels in WT but not
TRPV1−/− hearts following I/R. Previous evidence has
suggested that PI3K inhibition suppresses ERK1/2 activation induced
by capsaicin and nerve growth factor in primary sensory dorsal root
ganglion neurons (41), which is
supported by the present study. However, in a previous study, PI3K
has been reported to inhibit, rather than increase, ERK1/2
activation (42). It has been
suggested that the ability of PI3K inhibitors to suppress ERK1/2
activation depends on cell type, the type of stimuli and the
strength of signals (43,44). In the present study, PI3K
inhibition decreased I/R-induced ERK1/2 activation in WT mice, thus
suggesting that ERK1/2 activation is PI3K-dependent. Further
studies are required to explore the precise role of the ERK1/2
signaling pathway in TRPV1-induced cardiac protection.
As an ex vivo model, the Langendorff
preparation has its limitations. For example, the isolated and
perfused heart is denervated and its performance is not regulated
by neurohumoral factors. In addition, crystalloid-perfused hearts
are prone to tissue edema, which has a negative impact on cardiac
function, particularly in I/R study protocols >2 h, which is not
the case in the present study. However, the preparation is simple,
reproducible and enables the study of the heart without other organ
systems, which may confound physiological assessment. Due to these
advantages, the Langendorff model has been used for >100 years
to generate insightful data.
In conclusion, the present study demonstrated that
TRPV1 may exert anti-apoptotic effects against myocardial I/R
injury via PI3K/Akt signaling activation in isolated mouse hearts.
These data suggested that TRPV1 may be considered a potential
target for pharmacological intervention to reduce cardiac damage
and improve clinical outcomes following cardiac I/R.
Glossary
Abbreviations
Abbreviations:
|
Bax
|
B-cell lymphoma 2-associated X
protein
|
|
ERK1/2
|
extracellular signal-regulated protein
kinase 1/2
|
|
PI3K/Akt
|
phosphatidylinositol 3-kinase/protein
kinase B
|
|
TRPV1
|
transient receptor potential vanilloid
1
|
|
TTC
|
2,3,5-triphenyl tetrazolium
chloride
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling
|
Acknowledgments
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81170188 and 30971212), the
Natural Science Foundation of Chongqing (grant no. CSCT2009BB5069)
and the National Key Subject Construction Project (grant no.
2011170).
References
|
1
|
Bolli R, Becker L, Gross G, Mentzer R Jr,
Balshaw D and Lathrop DA; NHLBI Working Group on the Translation of
Therapies for Protecting the Heart from Ischemia: Myocardial
protection at a crossroads: The need for translation into clinical
therapy. Circ Res. 95:125–134. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanada S, Komuro I and Kitakaze M:
Pathophysiology of myocardial reperfusion injury: Preconditioning,
postconditioning, and translational aspects of protective measures.
Am J Physiol Heart Circ Physiol. 301:H1723–H1741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Silber S, Albertsson P, Avilés FF, Camici
PG, Colombo A, Hamm C, Jørgensen E, Marco J, Nordrehaug JE, Ruzyllo
W, et al Guidelines for percutaneous coronary interventions: The
Task Force for Percutaneous Coronary Interventions of the European
Society of Cardiology. Eur Heart J. 26:804–847. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Steg PG, James SK, Atar D, Badano LP,
Blömstrom-Lundqvist C, Borger MA, Di Mario C, Dickstein K, Ducrocq
G, Fernandez-Aviles F, et al Task Force on the management of
ST-segment elevation acute myocardial infarction of the European
Society of Cardiology (ESC): ESC Guidelines for the management of
acute myocardial infarction in patients presenting with ST-segment
elevation. Eur Heart J. 33:2569–2619. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ibáñez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y and Ren J: Targeting autophagy for
the therapeutic application of histone deacetylase inhibitors in
ischemia/reperfusion heart injury. Circulation. 129:1088–1091.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tao J, Zhu W, Li Y, Xin P, Li J, Liu M, Li
J, Redington AN and Wei M: Apelin-13 protects the heart against
ischemia-reperfusion injury through inhibition of ER-dependent
apoptotic pathways in a time-dependent fashion. Am J Physiol Heart
Circ Physiol. 301:H1471–H1486. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Konstantinidis K, Whelan RS and Kitsis RN:
Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc
Biol. 32:1552–1562. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song JQ, Teng X, Cai Y, Tang CS and Qi YF:
Activation of Akt/GSK-3beta signaling pathway is involved in
intermedin(1–53) protection against myocardial apoptosis induced by
ischemia/reperfusion. Apoptosis. 14:1061–1069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pan HL and Chen SR: Sensing tissue
ischemia: Another new function for capsaicin receptors?
Circulation. 110:1826–1831. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szallasi A and Blumberg PM: Vanilloid
(Capsaicin) receptors and mechanisms. Pharmacol Rev. 51:159–212.
1999.PubMed/NCBI
|
|
14
|
Caterina MJ, Schumacher MA, Tominaga M,
Rosen TA, Levine JD and Julius D: The capsaicin receptor: A
heat-activated ion channel in the pain pathway. Nature.
389:816–824. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Caterina MJ, Leffler A, Malmberg AB,
Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI
and Julius D: Impaired nociception and pain sensation in mice
lacking the capsaicin receptor. Science. 288:306–313. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Davis JB, Gray J, Gunthorpe MJ, Hatcher
JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson
K, et al: Vanilloid receptor-1 is essential for inflammatory
thermal hyperalgesia. Nature. 405:183–187. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zahner MR, Li DP, Chen SR and Pan HL:
Cardiac vanilloid receptor 1-expressing afferent nerves and their
role in the cardiogenic sympathetic reflex in rats. J Physiol.
551:515–523. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bolli RandLatif A: No pain, no gain: the
useful function of angina. Circulation. 112:3541–3543. 2005.
View Article : Google Scholar
|
|
19
|
Sexton A, McDonald M, Cayla C, Thiemermann
C and Ahluwalia A: 12-Lipoxygenase-derived eicosanoids protect
against myocardial ischemia/reperfusion injury via activation of
neuronal TRPV1. FASEB J. 21:2695–2703. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rang WQ, Du YH, Hu CP, Ye F, Xu KP, Peng
J, Deng HW and Li YJ: Protective effects of evodiamine on
myocardial ischemia-reperfusion injury in rats. Planta Med.
70:1140–1143. 2004. View Article : Google Scholar
|
|
21
|
Wang L and Wang DH: TRPV1 gene knockout
impairs postischemic recovery in isolated perfused heart in mice.
Circulation. 112:3617–3623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dai Z, Xiao J, Liu SY, Cui L, Hu GY and
Jiang DJ: Rutaecarpine inhibits hypoxia/reoxygenation-induced
apoptosis in rat hippocampal neurons. Neuropharmacology.
55:1307–1312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hausenloy DJ and Yellon DM: Reperfusion
injury salvage kinase signalling: Taking a RISK for
cardioprotection. Heart Fail Rev. 12:217–234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bucciarelli LG, Ananthakrishnan R, Hwang
YC, Kaneko M, Song F, Sell DR, Strauch C, Monnier VM, Yan SF,
Schmidt AM, et al: RAGE and modulation of ischemic injury in the
diabetic myocardium. Diabetes. 57:1941–1951. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cross TG, Scheel-Toellner D, Henriquez NV,
Deacon E, Salmon M and Lord JM: Serine/threonine protein kinases
and apoptosis. Exp Cell Res. 256:34–41. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Council N: Guide for the care and use of
laboratory animals: Eighth edition. Guide for the Care & Use of
Laboratory Animals. 327:963–965. 2011.
|
|
27
|
Rahman S, Li J, Bopassa JC, Umar S, Iorga
A, Partownavid P and Eghbali M: Phosphorylation of GSK-3β mediates
intralipid-induced cardioprotection against ischemia/reperfusion
injury. Anesthesiology. 115:242–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Childs AC, Phaneuf SL, Dirks AJ, Phillips
T and Leeuwenburgh C: Doxorubicin treatment in vivo causes
cytochrome C release and cardiomyocyte apoptosis, as well as
increased mitochondrial efficiency, superoxide dismutase activity,
and Bcl-2:Bax ratio. Cancer Res. 62:4592–4598. 2002.PubMed/NCBI
|
|
29
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhong B and Wang DH: TRPV1 gene knockout
impairs preconditioning protection against myocardial injury in
isolated perfused hearts in mice. Am J Physiol Heart Circ Physiol.
293:H1791–H1798. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maulik N, Engelman RM, Rousou JA, Flack JE
III, Deaton D and Das DK: Ischemic preconditioning reduces
apoptosis by upregulating anti-death gene Bcl-2. Circulation.
100(Suppl 19): II369–II375. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Patel JR and Brewer GJ: Age-related
differences in NFkappaB translocation and Bcl-2/Bax ratio caused by
TNFalpha and Abeta42 promote survival in middle-age neurons and
death in old neurons. Exp Neurol. 213:93–100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Costa MA, Fonseca BM, Keating E, Teixeira
NA and Correia-da-Silva G: Transient receptor potential vanilloid 1
is expressed in human cytotrophoblasts: Induction of cell apoptosis
and impairment of syncytialization. Int J Biochem Cell Biol.
57:177–185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harada N, Okajima K, Kurihara H and
Nakagata N: Stimulation of sensory neurons by capsaicin increases
tissue levels of IGF-I, thereby reducing reperfusion-induced
apoptosis in mice. Neuropharmacology. 52:1303–1311. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun Z, Han J, Zhao W, Zhang Y, Wang S, Ye
L, Liu T and Zheng L: TRPV1 activation exacerbates
hypoxia/reoxygenation-induced apoptosis in H9C2 cells via calcium
overload and mitochondrial dysfunction. Int J Mol Sci.
15:18362–18380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang W, Rubinstein J, Prieto AR, Thang LV
and Wang DH: Transient receptor potential vanilloid gene deletion
exacerbates inflammation and atypical cardiac remodeling after
myocardial infarction. Hypertension. 53:243–250. 2009. View Article : Google Scholar
|
|
37
|
Tang HB and Nakata Y: The activation of
transient receptor potential vanilloid receptor subtype 1 by
capsaicin without extracellular Ca2+ is involved in the
mechanism of distinct substance P release in cultured rat dorsal
root ganglion neurons. Naunyn Schmiedebergs Arch Pharmacol.
377:325–332. 2008. View Article : Google Scholar
|
|
38
|
Joung EJ, Li MH, Lee HG, Somparn N, Jung
YS, Na HK, Kim SH, Cha YN and Surh YJ: Capsaicin induces heme
oxygenase-1 expression in HepG2 cells via activation of PI3K-Nrf2
signaling: NAD(P)H:quinone oxidoreductase as a potential target.
Antioxid Redox Signal. 9:2087–2098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen S, Liu J, Liu X, Fu Y, Zhang M, Lin
Q, Zhu J, Mai L, Shan Z, Yu X, et al: Panax notoginseng saponins
inhibit ischemia-induced apoptosis by activating PI3K/Akt pathway
in cardiomyocytes. J Ethnopharmacol. 137:263–270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weston CR, Balmanno K, Chalmers C,
Hadfield K, Molton SA, Ley R, Wagner EF and Cook SJ: Activation of
ERK1/2 by deltaRaf-1:ER* represses Bim expression
independently of the JNK or PI3K pathways. Oncogene. 22:1281–1293.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhuang ZY, Xu H, Clapham DE and Ji RR:
Phosphatidylinositol 3-kinase activates ERK in primary sensory
neurons and mediates inflammatory heat hyperalgesia through TRPV1
sensitization. J Neurosci. 24:8300–8309. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rommel C, Clarke BA, Zimmermann S, Nuñez
L, Rossman R, Reid K, Moelling K, Yancopoulos GD and Glass DJ:
Differentiation stage-specific inhibition of the Raf-MEK-ERK
pathway by Akt. Science. 286:1738–1741. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Duckworth BC and Cantley LC: Conditional
inhibition of the mitogen-activated protein kinase cascade by
wortmannin. Dependence on signal strength. J Biol Chem.
272:27665–27670. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wennström S and Downward J: Role of
phosphoinositide 3-kinase in activation of ras and
mitogen-activated protein kinase by epidermal growth factor. Mol
Cell Biol. 19:4279–4288. 1999. View Article : Google Scholar : PubMed/NCBI
|