Introduction
Alzheimer's disease (AD) is a progressive disorder
of the brain and nervous system thought to be caused by the buildup
of amyloid plaques and loss of pyramidal neurons in the brain
(1). AD disrupts the electrical
signals between neurons that are responsible for the formation of
thoughts and memories (2). It is
considered that the formation of plaques, caused by extracellular
deposits of amyloid β (Aβ), is responsible for the pathology of AD
(3). Therefore, early removal of
plaque in the brain may be critical to facilitate the effective
treatment of AD. Genetic models of AD, particularly amyloid
precursor protein/presenilin 1 (APP/PS) mice, have been widely used
to elucidate the pathogenesis of AD and develop appropriate
treatments (4,5). APP/PS mice exhibit typical AD
phenotypes, including the impairment of memory and hippocampal
long-term potentiation (LTP), loss of pyramidal cells and
accumulation of Aβ; however, these symptoms usually appear in mice
at a late stage (>6 months of age) (4). Aβ injections induce neurotoxicity
and this may be a novel method of inducing AD in mice, enabling
further research to be performed; cell death in mice with a typical
AD phenotype has been observed in a short period of time following
injection of Aβ (6,7).
MicroRNAs (miRNAs) are endogenous non-coding small
RNA molecules that are 21–25 nucleotides long, and regulate the
degradation and translation of target proteins (8). The function of miRNAs has been well
documented in the development and treatment of various types of
cancer (9–11). miRNAs serve critical roles in
neurodegeneration and neuroprotection (12,13) and it has been reported that miRNAs
regulate spinogenesis in the central nervous system (14). Furthermore, previous studies have
suggested that miRNAs are involved in the pathogenesis of AD
(15,16).
It has been demonstrated that miR-107 expression is
downregulated in the temporal cortical grey matter during the early
phases of AD (17,18). Furthermore, accumulation of Aβ in
the brain serves an important role in the pathogenesis of AD
(6). It has been reported that
miRNAs regulate multiple aspects of AD development and progression,
indicating that targeting miRNAs may be a novel strategy of
treating AD (15,16). Furthermore, it has been
demonstrated that osthole decreases Aβ levels in AD by upregulating
miR-107 (19). However, it
remains unknown whether miR-107 is critical in the synthesis of
plaques and whether it serves a specific role in AD. The present
study used an Aβ1-42-induced neurotoxicity rat model to investigate
the function of miR-107 in AD and elucidate the potential
mechanisms involved. The results indicate that miR-107 may be a
potential method of treating AD.
Materials and methods
Animals and AD modeling
A total of 60 male C57 mice (6 months old, weighing
25–30 g) were purchased from the Shanghai Laboratory Animal Center
(Shanghai, China). Mice were housed in a room maintained at a
temperature of 23±2°C, a relative humidity of 45–65% and
experienced a 12 h light/dark cycle. Animals had ad libitum
access to food and water. All experimental procedures were approved
by the Ethics Committee of Henan University of Science and
Technology (Luoyang, China).
Animals were randomly divided into five groups
(each, n=12): A control group, an Aβ model group, a scramble
control (SC)+Aβ model group, a miR-107 mimic + Aβ model group and
an miR-107 mimic + control group. Single intracerebroventricular
(icv) injections of Aβ aggregates were used to induce AD symptoms,
as previously described (20).
Aβ1-42 (2 μg/mouse; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) was dissolved and diluted in distilled water, and
incubated to induce aggregation at 37°C for 4 days. Rats received
one injection of aggregated Aβ1-42 (1 μl) directly into the
third ventricle of the mouse brain (specific parameters:
Anteroposterior, −2.5 mm; mediolateral, 0 mm; dorsoventral, −3.0 mm
relative to bregma). A vehicle (1 μl saline) was injected
(icv) as a control.
Treating mice with the miR-107 mimic
miR-107 mimic and SC were provided by Shanghai
GenePharma Co., Ltd. (Shanghai, China) and administered to the mice
icv 30 min prior to icv injection of 1 μl Aβ1-42. A total of
7 days after injection with Aβ1-42, a Morris water maze test was
conducted to test spatial memory. A total of 6 mice in each group
were then sacrificed to isolate hippocampal slices for
electrophysiological experiments. Additional mice underwent Nissl
staining, as well as biochemical or molecular experiments.
Morris water maze
A total of 7 days after Aβ1-42 injections, the
spatial learning and memory ability of mice was assessed using a
Morris water maze test (Panlab, Barcelona, Spain). A training
protocol lasting for 5 consecutive days was used that involved
conducting 4 trials/day in a water maze. The maximum time allowed
for each mouse to find the platform was 90 sec. Each trial had a
different starting point and if mice failed to find the platform in
90 sec, they would be guided to the platform manually and kept at
the platform for 10 sec. The time spent by each mouse to reach the
platform, known as the escape latency period, was counted as 90
sec.
The spatial probe test was conducted on day 6 and
lasted for 90 sec. The platform was removed and each mouse was
released opposite the target quadrant, facing the wall of the pool.
In the probe test, the time that mice spent in the target quadrant
was recorded to assess their spatial memory.
Electrophysiological experiments
Mice were anesthetized with isoflurane (1% in
oxygen) via inhalation. Following anesthesia, mice used in
electrophysiological experiments were decapitated, brains were
quickly isolated and placed on ice. Subsequently, brains were
immersed in pre-cooled cutting solution (124 NaCl, 26
NaHCO3, 10 D-glucose, 3 KCl, 1.25
KH2PO4, 5 MgSO4 and 3.4
CaCl2) and hippocampal slices (400 μm) were
prepared in cutting solution, as previously described (21). Slices were transferred to an
interface recording chamber consisting of a warm, humidified
atmosphere of 95% O2/5% CO2 and were
continuously perfused with oxygenated and preheated (32±0.5°C)
artificial cerebrospinal fluid (aCSF; 110 mM NaCl, 5 mM KCl, 2.5 mM
CaCl2, 1.5 mM MgSO4, 1.24 mM
KH2PO4, 10 mM D-glucose, 27.4 mM
NaHCO3) at a speed of 1.5 ml/min. The field excitatory
postsynaptic potential was elicited by stimulating the Schaffer
collateral pathway with twisted nichrome wires. The input-output
and LTP induced by θ-burst stimulation (TBS; 10 bursts of 4 pulses
at 100 Hz delivered at 5-Hz intervals) were measured.
ELISA
Hippocampal Aβ1-42 was measured using ELISA.
Hippocampi from different groups were homogenized in homogenization
buffer (5 M guanidine HCl/50 mM Tris-HCl) and centrifuged at 11,587
× g for 10 min at 4°C. Protein concentrations were determined using
a bicinchoninic acid (BCA) assay kit (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Supernatant fractions were analyzed using
an Aβ1-42 ELISA kit (cat. no. KHB3441; Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's protocol. Absorbance
was measured at 450 nm using a microplate reader.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the hippocampus using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). RT was
then performed using the Moloney murine leukemia virus reverse
transcriptase (cat. no. M1705; Promega, Madison, WI, USA) and dNTPs
(Thermo Fisher Scientific, Inc.) in First-Strand Buffer (cat. no.
M1705; Promega) for 50 min at 37°C following the manufacturer's
protocol. qPCR was performed to quantify miR-107 expression in the
hippocampus using the mmu-mir-107 RT-PCR Detection and U6
Calibration kit (cat. no. abx096666; Abbexa Ltd., Cambridge, UK) on
a quantitative thermal cycler (Mastercycler® ep
realplex; Eppendorf, Hamburg, Germany). The thermocycling
conditions were as follows: 95°C denaturation for 45 sec, 58°C
annealing for 60 sec and 72°C extension 60 sec for 35 cycles. The
following primers were used in qPCR (5′-3′): miR-107 forward,
5′-GCCAAGCCCACTCAGCTGCCAGCC′-3 and reverse,
5′-GGCTGGCAGCTGAGTGGGCTTGGC-3′; U6, forward,
5′-CTCGCTTCGGCAGCACA-3′ and reverse, 5′-AACGCTTCACGAATTTGCGT-3′.
The relative expression of mature miR-107 was calculated against U6
RNA (the internal control) using the 2ΔΔCq method
(22).
Western blotting
Hippocampi homogenates were obtained and lysed using
radioimmunoprecipitation assay lysis buffer (Thermo Fisher
Scientific, Inc.). Protein concentrations were measured using the
BCA protein assay kit (Thermo Fisher Scientific, Inc.). Equivalent
amounts of proteins (25 μg/lane) were underwent SDS-PAGE
(12% gel) and transferred onto nitrocellulose membranes. Membranes
were blocked in 5% skim milk for 2 h at room temperature and
incubated with primary antibodies at 4°C overnight. The primary
antibodies used were for brain-derived neurotrophic factor (BDNF;
dilution 1:1,000, cat. no. AB1534; EMD Millipore, Billerica, MA,
USA), phosphorylated (p)-Tau (dilution 1:3,000, cat.no. 12885,),
Tau (dilution 1:3,000, cat. no. 4019p-tropomyosin receptor kinase
(TrkB; dilution 1:3,000, cat. no. 4619), TrkB (dilution 1:3,000,
cat. no. 4607), p-AKT (dilution 1:3,000, cat. no. 4060), AKT
(dilution 1:3,000, cat. no. 2920) (all from CST Biological
Reagents, Co., Ltd.) and GAPDH (1:10,000, cat. no. AB2302; EMD
Millipore). Following three washes with phosphate-buffered saline
(PBS), membranes were labeled with specific horseradish peroxidase
(HRP)-coupled secondary antibodies [anti-mouse immunoglobulin (Ig)
G HRP or anti-rabbit IgG HRP; cat. no. A16104SAMPLE, Thermo Fisher
Scientific, Inc.) for 2 h at room temperature. Protein bands were
visualized by staining with a chemiluminescent substrate detection
reagent (Thermo Fisher Scientific, Inc.). Grayscale analysis of
target bands was performed using ImageJ software version 7.0
(National Institutes of Health, Bethesda, MD, USA).
Statistical analyses
Data are presented as the mean ± standard error of
the mean. All statistical analyses were performed using GraphPad
Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA). One-way
analysis of variance followed by Bonferroni correction was
performed to compare differences between groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-107 mimic prevents the Aβ-induced
reduction of miR-107 in the hippocampus
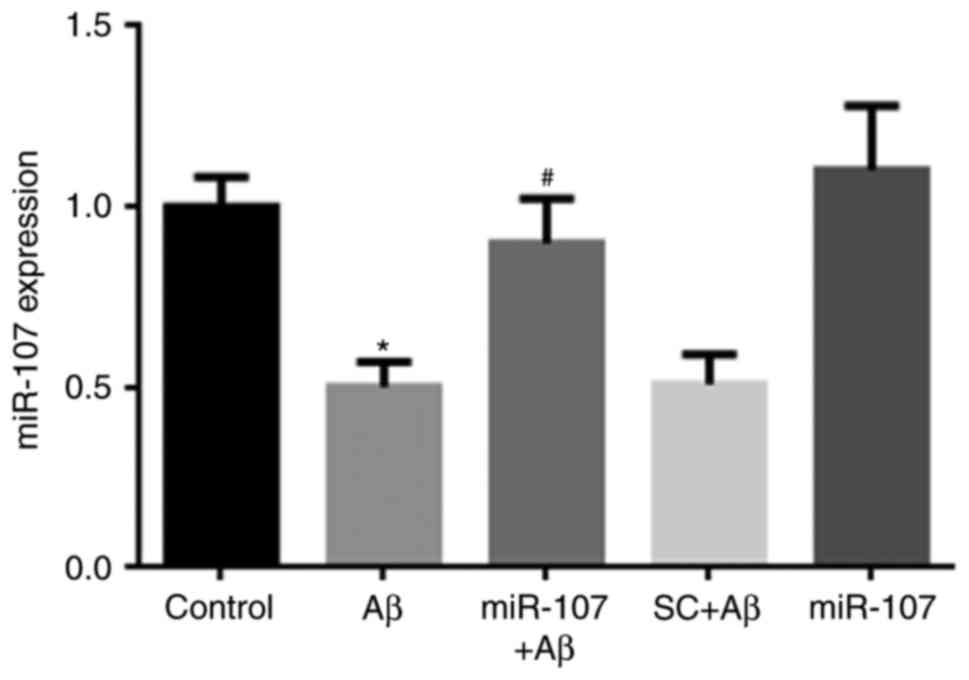
miR-107 expression in the hippocampus was measured 7
days following AD induction or miR-107 treatment. Following Aβ
injection, miR-107 levels were significantly decreased compared
with the control (P<0.05) (Fig.
1). The miR-107 mimic significantly prevented the reduction of
miR-107 compared with the Aβ group (P<0.05); however, the
scramble control did not affect miR-107 levels. Additionally,
application of the miR-107 mimic in control mice did not affect
miR-107 expression.
miR-107 mimic prevents Aβ-induced cell
death in the CA1 region
Cell loss is a major pathological characteristic of
the AD phenotype; therefore the present study speculated whether
miR-107 would prevent cell loss in the Aβ model. Nissl staining was
used to measure cell numbers in the CA1 region 7 days following AD
induction or miR-107 treatment (Fig.
2A). Significant cell loss was observed in the CA1 region of
Aβ-injected mice compared with control mice (P<0.05) (Fig. 2B and C). However, application of
the miR-107 mimic significantly prevented the cell loss induced by
Aβ (P<0.05). The SC did not prevent the cell loss induced by Aβ.
Furthermore, the administration of miR-107 to normal mice did not
affect cell numbers.
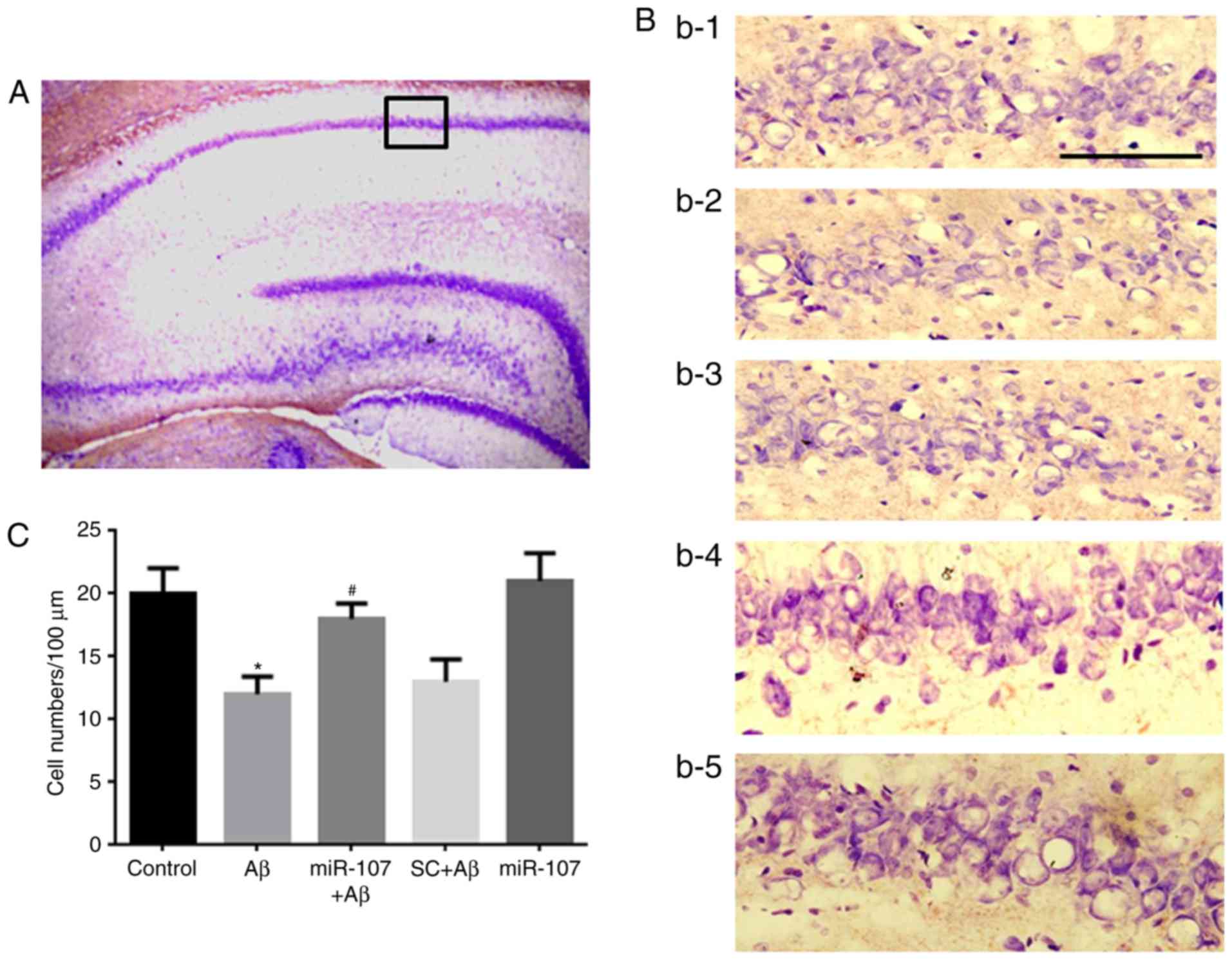 | Figure 2miR-107 mimic prevents Aβ-induced
cell loss in the CA1 region. (A) Image of hippocampus following
Nissl staining (magnification, ×100). The black square indicates
the CA1 region, amplified (magnification, ×400). (B) Images of the
CA1 region. B1, B2, B3, B4 and B5 represent the control, Aβ, SC+Aβ,
miR-107+Aβ and miR-107 groups, respectively. Purple staining
indicates pyramidal neurons. Scale bar, 100. (C) Quantification of
cell numbers in the CA1 region in the different groups. Data are
presented as the mean ± standard error of the mean, as determined
by one-way analysis of variance. n=12. *P<0.05 vs.
control; #P<0.05 vs. SC+Aβ. μm. SC, scramble
control; Aβ, amyloid β; miR, microRNA. |
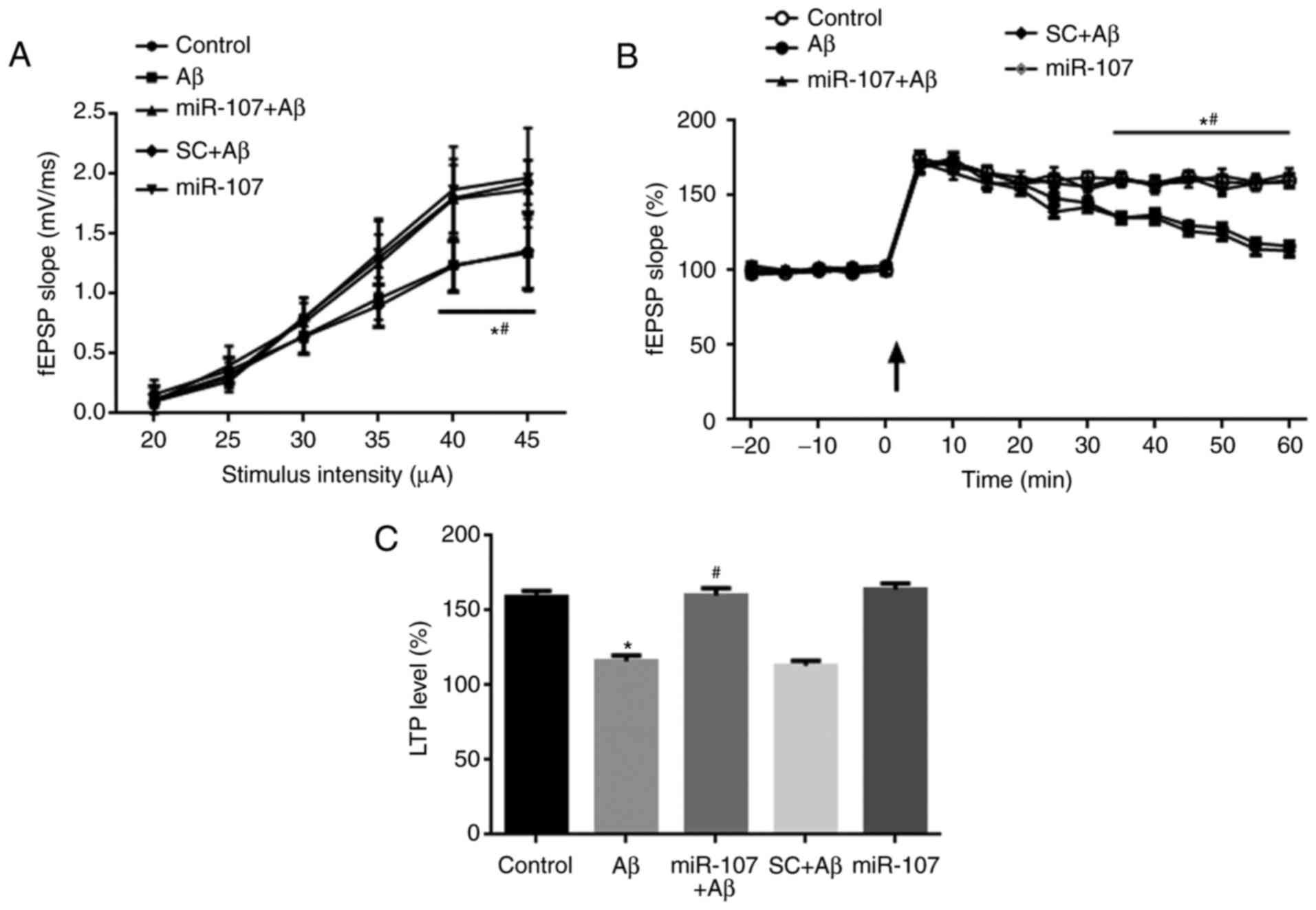
miR-107 mimic prevents the Aβ-induced
impairment of basal synaptic transmission and LTP
Electrophysiological experiments were used to detect
synaptic transmission and LTP in Schaffer collateral-CA1 synapses 7
days following AD induction or miR-107 treatment. The input-output
of synaptic transmission was significantly reduced when delivering
a 40- and 50-μA stimulus in the Aβ group compared with the
control (P<0.05) (Fig. 3A).
Furthermore, application of the miR-107 mimic but not SC
significantly prevented the impairment of input-output compared
with the Aβ group (P<0.05). The SC had no effect on synaptic
transmission following AD induction. LTP was induced in the
hippocampi of control mice but was significantly impaired in Aβ
model mice (P<0.050 (Fig. 3B).
However, the miR-107 mimic significantly reversed LTP impairment
(P<0.05). SC had no effect on the LTP compared with the Aβ
group. Quantification of the LTP data at 1 h demonstrated that LTP
was significantly reduced in the Aβ group compared with the control
(P<0.05) (Fig. 3C). This
reduction was reversed by the miR-107 mimic (P<0.05), but not by
the SC. The application of miR-107 in normal controls did not
affect the LTP.
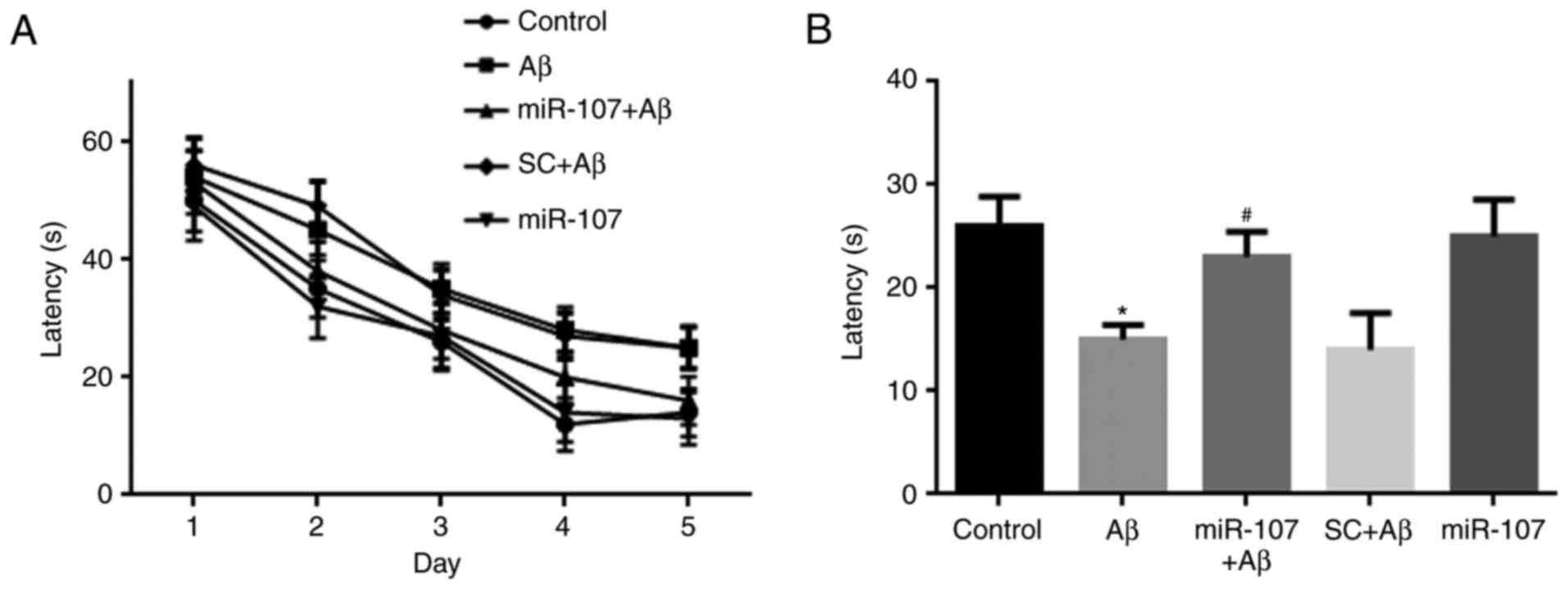
miR-107 mimic prevents the Aβ-induced
impairment of spatial memory
Spatial memory was examined using the Morris water
maze 7 days following AD induction or miR-107 treatment. Mice that
were administered Aβ exhibited reduced learning during the training
period (Fig. 4A), although this
reduction was not significant. During the test period, the time
spent in the platform quadrant by the Aβ group was significantly
lower than that of the control group (P<0.05). However, miR-107
prevented memory impairment in Aβ-injected mice (P<0.05)
(Fig. 4B) although it did not
promote memory in control mice.
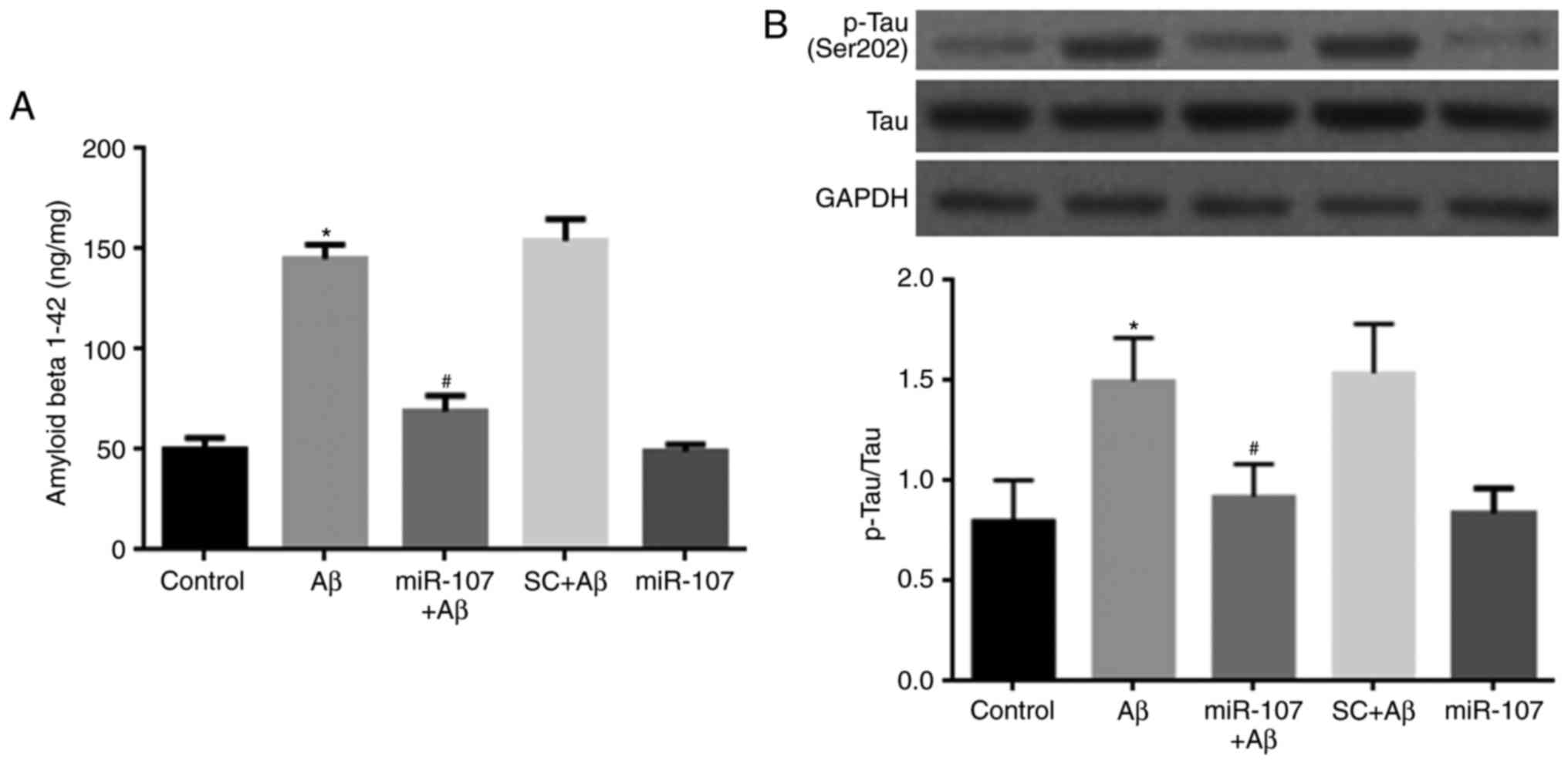
miR-107 mimic prevents the Aβ-induced
increase of Aβ 1-42 and p-Tau
AD proteins were also detected 7 days following AD
induction or miR-107 treatment. Levels of endogenous Aβ 1-42 were
significantly increased in Aβ-injected mice compared with controls
(P<0.05) (Fig. 5A). miR-107
significantly reduced the levels of Aβ 1-42 in Aβ-injected mice
(P<0.05) but not in normal mice. The level of Tau
phosphorylation was also measured. Aβ significantly increased p-Tau
levels compared with the control (P<0.05) (Fig. 5B), however, this was reversed
following the administration of miR-107 (P<0.05). miR-107
administration did not affect Tau phosphorylation in healthy
control mice.
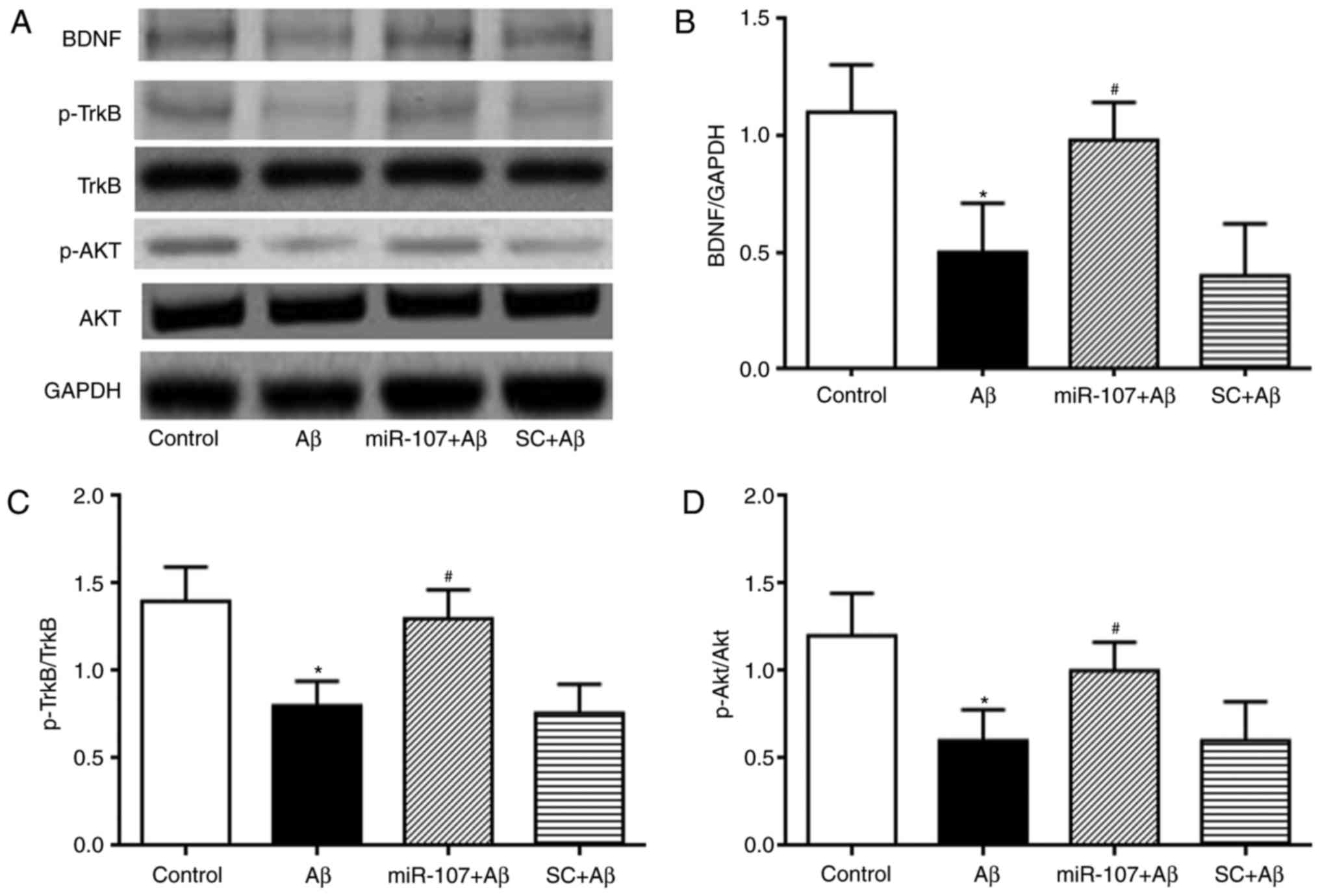
miR-107 mimic prevents the Aβ-induced
depression of the BDNF-TrkB and AKT pathways
The mechanisms involved behind the effects of
miR-107 in the Aβ model of AD were determined. The expression of
p-AKT, p-TrkB and BDNF were measured and quantified. Levels of
mature BDNF were significantly reduced in Aβ-injected mice compared
with the control (P<0.05) (Fig. 6A
and B). This reduction was attenuated by the miR-107 mimic
(P<0.05). miR-107 did not affect the expression of mature BDNF
in normal control mice. The expression of p-TrkB was also
determined in the different groups. Aβ reduced the expression of
p-TrkB compared with the control (P<0.05); however, treatment
with miR-107 significantly increased TrkB phosphorylation in the Aβ
group (P<0.05) (Fig. 6A and
C). Finally, AKT phosphorylation was also quantified. Aβ
reduced the expression of p-AKT compared with the control
(P<0.05) (Fig. 6A and D),
whereas treatment with miR-107 reversed this reduction
(P<0.05).
Discussion
The neuroprotective action of miR-107 has been
previously reported (23,24); therefore the present study
explored the potential function of miR-107 in the treatment of AD.
The results demonstrated that miR-107 expression in the
hippo-campus was downregulated in the Aβ-induced mouse model and
that treatment with an miR-107 mimic prevented the reduction of
miR-107 expression in the hippocampus induced by Aβ injection.
Importantly, the miR-107 mimic prevented Aβ-induced behavioral and
electrophysiological abnormalities. In addition, it was
demonstrated that the miR-107 mimic prevented the production of
amyloid plaques and the loss of pyramidal neurons in the CA1
region.
Although the functions of miRNAs have been
extensively investigated in cancer (25,26), the potential role of these
structures in neurodegenerative diseases has also attracted
attention (27). The results of
previous studies indicated that miRNAs are involved in a variety of
biological processes associated with the onset or treatment of AD
(27–29). It has been demonstrated that
soluble Aβ (sAβ) oligomers contribute to the pathogenesis of AD;
the sAβ-induced expression of miR-134, miR-145 and miR-210 is fully
reversed by two selective N-methyl-d-aspartate (NMDA) receptor
inhibitors (30). In the same
study, insoluble Aβ fibrils, which constitute the extracellular
plaques, did not induce changes in the miRNA profile (30). Prefibrillar sAβ are more toxic
than their insoluble fibrillar counterparts and sAβ oligomers may
represent an early trigger of synaptic damage and cognitive
impairment in AD (31).
Clinically, it has been reported that miR-107 levels are reduced in
the temporal cortical gray matter during the early stages of AD
progression (18). Furthermore,
the overexpression of miR-107 in the hippocampus exhibits
antidepressant-like effects (32). Consistent with the results of a
previous study (17), the present
study demonstrated that miR-107 expression was reduced in an AD
model induced by administration of Aβ. miR-107 mimics not only
promoted miR-107 expression in the hippocampus, but also reversed
the impairment of spatial memory in a model of Aβ.
It has been proposed that LTP is the primary
cellular model for memory (33,34). The impairment of hippocampal LTP
is typical of AD model mice (4).
The present study reported that TBS-induced LTP in Schaffer
collateral-CA1 synapses was impaired in AD model mice. It was also
demonstrated that input-output of the basal synaptic transmission
was reduced in an AD model, consistent with the results of previous
studies (4,5). The miR-107 mimic reversed the
impairment of basal synaptic transmission. As reported previously,
the reduction in excitatory synaptic transmission could also
contribute to the LTP impairment observed in AD (35). In addition, inhibition of the
BDNF-TrkB pathway, observed following Aβ injection, may account for
the impairment of LTP (36,37).
Pyramidal neuron death has also been proposed as a
pathological factor underlying the AD phenotype (38–40). The present study determined the
number of pyramidal neurons in the CA1 area. Significant cell loss
in the CA1 region following Aβ injection was observed; the miR-107
mimic attenuated the cell loss caused by Aβ injection. Apoptosis is
defined as programmed cell death and is responsible for the
majority of cell death that occurs in neurodegenerative diseases
(41). Aβ treatment induces
apoptosis in hippocampal neurons, most likely via suppression of
the AKT signaling pathway (42,43). The AKT pathway is an important
cell survival pathway (44);
therefore elimination of p-AKT also induces the loss of pyramidal
neurons following Aβ injection. Although the exact mechanisms of
action that contribute to neuronal loss were not elucidated in the
present study, the reduction of BDNF-TrkB and AKT activity may
contribute to cell loss. The results also indicated that Aβ
injection reduced the input-output of basal synaptic transmission.
The loss of pyramidal neurons was consistent with the
electrophysiological results.
It has been demonstrated that miR-107 directly
downregulates Dicer-1, a gene that encodes an enzyme essential for
processing miRNA precursors (45). This results in the inhibition of
vascular endothelial growth factor translation and a decrease in
its expression, leading to angiogenesis following stroke (45). Novel bioinformatics predictions,
in situ hybridization experiments and biochemical
validations indicate that miR-107 may be involved in the
progression of AD by increasing β-secretase (18). Although the present study
contained no direct evidence indicating that the miR-107 mimic
ameliorates spatial memory in the AD model via the BDNF-TrkB
pathway, the results indicate that BDNF expression is downregulated
in AD and that the miR-107 mimic attenuates the reduction in BDNF
expression induced by Aβ administration. Furthermore, it has been
reported that miR-107 may regulate BDNF expression (46). The phosphorylation of TrkB
exhibited a similar trend to BDNF expression; the BDNF-TrkB pathway
is known to regulate neuronal function, most likely by modulating
AKT phosphorylation (47). The
present study demonstrated that AKT phosphorylation was decreased
in the AD model, but was reversed by treatment with miR-107 mimic.
The reductions in AKT phosphorylation and suppression of the
BDNF-TrkB pathway may account for the impairment of hippocampal LTP
and cell loss observed in AD.
In conclusion, the results of the present study
indicate that miR-107 levels are reduced in mice with AD phenotypes
and that treatment with an miR-107 mimic increases miR-107
expression and ameliorates some of the AD phenotypes. Additionally,
it was identified that the BDNF-TrkB signaling pathway may be
responsible for the effects of the miR-107 mimic on Aβ-induced
neurotoxicity. miR-107 may therefore serve as a candidate
therapeutic target for the treatment of AD.
References
|
1
|
Selkoe DJ and Hardy J: The amyloid
hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med.
8:595–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gong NJ, Chan CC, Leung LM, Wong CS, Dibb
R and Liu C: Differential microstructural and morphological
abnormalities in mild cognitive impairment and Alzheimer's disease:
Evidence from cortical and deep gray matter. Hum Brain Map.
38:2495–2508. 2017. View Article : Google Scholar
|
|
3
|
Luo J, Wärmländer SK, Gräslund A and
Abrahams JP: Cross-interactions between the Alzheimer disease
amyloid-beta peptide and other amyloid proteins. A FURTHER ASPECT
OF THE AMYLOID CASCADE HYPOTHESIS. J Biol Chem. 292:20462017.
View Article : Google Scholar
|
|
4
|
Hong X, Liu J, Zhu G, Zhuang Y, Suo H,
Wang P, Huang D, Xu J, Huang Y, Yu M, et al: Parkin overexpression
ameliorates hippocampal long-term potentiation and beta-amyloid
load in an Alzheimer's disease mouse model. Hum Mol Genet.
23:1056–1072. 2014. View Article : Google Scholar
|
|
5
|
Li F, Han G and Wu K: Tanshinone IIA
alleviates the AD phenotypes in APP and PS1 transgenic mice. BioMed
Res Int. 2016:76318012016.PubMed/NCBI
|
|
6
|
Wang P, Wu Q, Wu W, Li H, Guo Y, Yu P, Gao
G, Shi Z, Zhao B and Chang YZ: Mitochondrial ferritin deletion
exacerbates β-amyloid-induced neurotoxicity in mice. Oxid Med Cell
Longev. 2017:10203572017. View Article : Google Scholar
|
|
7
|
Zhu J, Liao S, Zhou L and Wan L:
Tanshinone IIA attenuates Aβ25-35 -induced spatial memory
impairment via upregulating receptors for activated C kinase1 and
inhibiting autophagy in hippocampus. J Pharm Pharmacol. 69:191–201.
2017. View Article : Google Scholar
|
|
8
|
Quinlan S, Kenny A, Medina M, Engel T and
Jimenez-Mateos EM: MicroRNAs in Neurodegenerative Diseases. Int Rev
Cell Mol Biol. 334:309–343. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rupaimoole R and Slack FJ: MicroRNA
therapeutics: Towards a new era for the management of cancer and
other diseases. Nat Rev Drug Discov. 16:203–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reddy KB: MicroRNA (miRNA) in cancer.
Cancer Cell Int. 15:382015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hayes J, Peruzzi PP and Lawler S:
MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol
Med. 20:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Recasens A, Perier C and Sue CM: Role of
microRNAs in the regulation of α-Synuclein expression: A systematic
review. Front Mol Neurosci. 9:1282016. View Article : Google Scholar
|
|
13
|
Molasy M, Walczak A, Szaflik J, Szaflik JP
and Majsterek I: MicroRNAs in glaucoma and neurodegenerative
diseases. J Hum Genet. 62:105–112. 2017. View Article : Google Scholar
|
|
14
|
Impey S, Davare M, Lesiak A, Fortin D,
Ando H, Varlamova O, Obrietan K, Soderling TR, Goodman RH and
Wayman GA: An activity-induced microRNA controls dendritic spine
formation by regulating Rac1-PAK signaling. Mol Cell Neurosci.
43:146–156. 2010. View Article : Google Scholar :
|
|
15
|
Nagaraj S, Laskowska-Kaszub K, Dębski KJ,
Wojsiat J, Dąbrowski M, Gabryelewicz T, Kuźnicki J and Wojda U:
Profile of 6 microRNA in blood plasma distinguish early stage
Alzheimer's disease patients from non-demented subjects.
Oncotarget. 8:16122–16143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reddy PH, Tonk S, Kumar S, Vijayan M,
Kandimalla R, Kuruva CS and Reddy AP: A critical evaluation of
neuropro-tective and neurodegenerative MicroRNAs in Alzheimer's
disease. Biochem Biophys Res Commun. 483:1156–1165. 2017.
View Article : Google Scholar
|
|
17
|
Nelson PT and Wang WX: MiR-107 is reduced
in Alzheimer's disease brain neocortex: Validation study. J
Alzheimer's Dis. 21:75–79. 2010. View Article : Google Scholar
|
|
18
|
Wang WX, Rajeev BW, Stromberg AJ, Ren N,
Tang G, Huang Q, Rigoutsos I and Nelson PT: The expression of
microRNA miR-107 decreases early in Alzheimer's disease and may
accelerate disease progression through regulation of beta-site
amyloid precursor protein-cleaving enzyme 1. J Neurosci.
28:1213–1223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiao Y, Kong L, Yao Y, Li S, Tao Z, Yan Y
and Yang J: Osthole decreases beta amyloid levels through
up-regulation of miR-107 in Alzheimer's disease. Neuropharmacology.
108:332–344. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim TI, Lee YK, Park SG, Choi IS, Ban JO,
Park HK, Nam SY, Yun YW, Han SB, Oh KW and Hong JT: l-Theanine, an
amino acid in green tea, attenuates beta-amyloid-induced cognitive
dysfunction and neurotoxicity: Reduction in oxidative damage and
inactivation of ERK/p38 kinase and NF-kappaB pathways. Free Radic
Biol Med. 47:1601–1610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Chen H, Wu S, Cheng Y, Li Q, Wang J
and Zhu G: MPP+ inhibits mGluR1/5-mediated long-term depression in
mouse hippocampus by calpain activation. Eur J Pharmacol.
795:22–27. 2017. View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Yang ZB, Zhang Z, Li TB, Lou Z, Li SY,
Yang H, Yang J, Luo XJ and Peng J: Up-regulation of brain-enriched
miR-107 promotes excitatory neurotoxicity through down-regulation
of glutamate transporter-1 expression following ischaemic stroke.
Clin Sci (Lond). 127:679–689. 2014. View Article : Google Scholar
|
|
24
|
Wang WX, Wilfred BR, Madathil SK, Tang G,
Hu Y, Dimayuga J, Stromberg AJ, Huang Q, Saatman KE and Nelson PT:
miR-107 regulates granulin/progranulin with implications for
traumatic brain injury and neurodegenerative disease. Am J Pathol.
177:334–345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang RK, Li X, Mu N, Hrydziuszko O,
Garcia-Majano B, Larsson C and Lui WO: MicroRNA expression profiles
in nonepithelial ovarian tumors. Int J Oncol. 52:55–66. 2017.
|
|
26
|
Cao K, Li J, Chen J, Qian L, Wang A, Chen
X, Xiong W, Tang J, Tang S, Chen Y, et al: microRNA-33a-5p
increases radiosen-sitivity by inhibiting glycolysis in melanoma.
Oncotarget. 8:83660–83672. 2017.PubMed/NCBI
|
|
27
|
Müller M, Kuiperij HB, Claassen JA,
Küsters B and Verbeek MM: MicroRNAs in Alzheimer's disease:
Differential expression in hippocampus and cell-free cerebrospinal
fluid. Neurobiol Aging. 35:152–158. 2014. View Article : Google Scholar
|
|
28
|
Liu W, Cai H, Lin M, Zhu L, Gao L, Zhong
R, Bi S, Xue Y and Shang X: MicroRNA-107 prevents amyloid-beta
induced blood-brain barrier disruption and endothelial cell
dysfunction by targeting Endophilin-1. Exp Cell Res. 343:248–257.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yao J, Hennessey T, Flynt A, Lai E, Beal
MF and Lin MT: MicroRNA-related cofilin abnormality in Alzheimer's
disease. PLoS One. 5:e155462010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li JJ, Dolios G, Wang R and Liao FF:
Soluble beta-amyloid peptides, but not insoluble fibrils, have
specific effect on neuronal microRNA expression. PLoS One.
9:e907702014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shrestha BR, Vitolo OV, Joshi P,
Lordkipanidze T, Shelanski M and Dunaevsky A: Amyloid beta peptide
adversely affects spine number and motility in hippocampal neurons.
Mol Cell Neurosci. 33:274–282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang YF, Yang CH, Huang CC and Hsu KS:
Vascular endothelial growth factor-dependent spinogenesis underlies
antidepressant-like effects of enriched environment. J Biol Chem.
287:40938–40955. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cooke SF and Bliss TV: Long-term
potentiation and cognitive drug discovery. Curr Opin Investig
Drugs. 6:25–34. 2005.PubMed/NCBI
|
|
34
|
Bliss TV, Collingridge GL and Morris RG:
Introduction = Long-term potentiation and structure of the issue.
Philos Trans R Soc Lond B, Biol Sci. 358:607–611. 2003. View Article : Google Scholar
|
|
35
|
Zhu G, Liu Y, Wang Y, Bi X and Baudry M:
Different patterns of electrical activity lead to long-term
potentiation by activating different intracellular pathways. J
Neurosci. 35:621–633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Borba EM, Duarte JA, Bristot G, Scotton E,
Camozzato AL and Chaves ML: Brain-derived neurotrophic factor serum
levels and hippocampal volume in mild cognitive impairment and
dementia due to Alzheimer disease. Dement Geriatr Cogn Dis Extra.
6:559–567. 2016. View Article : Google Scholar
|
|
37
|
Nie J, Tian Y, Zhang Y, Lu YL, Li LS and
Shi JS: Dendrobium alkaloids prevent Abeta25-35-induced neuronal
and synaptic loss via promoting neurotrophic factors expression in
mice. PeerJ. 4:e27392016. View Article : Google Scholar
|
|
38
|
Braak H and Braak E: Ratio of pyramidal
cells versus non-pyramidal cells in the human frontal isocortex and
changes in ratio with ageing and Alzheimer's disease. Prog Brain
Res. 70:185–212. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ditter SM and Mirra SS: Neuropathologic
and clinical features of Parkinson's disease in Alzheimer's disease
patients. Neurology. 37:754–760. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Maingret V, Barthet G, Deforges S, Jiang
N, Mulle C and Amédée T: PGE2-EP3 signaling pathway impairs
hippocampal presynaptic long-term plasticity in a mouse model of
Alzheimer's disease. Neurobiology Aging. 50:13–24. 2017. View Article : Google Scholar
|
|
41
|
Saleem S and Biswas SC: Tribbles
Pseudokinase 3 induces both apoptosis and autophagy in
amyloid-β-induced neuronal death. J Biol Chem. 292:2571–2585. 2017.
View Article : Google Scholar
|
|
42
|
Xiao H, Zhang Q, Peng Y, Tang G, Liao Y,
Zhuang X, Ye WC, Wang Y and Shi L:
7-(4-Hydroxy-3-methoxyphenyl)-1-phenyl-4E- hepten-3-one alleviates
Abeta1-42 induced cytotoxicity through PI3K-mTOR pathways. Biochem
Biophys Res Commun. 484:365–371. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen L, Ou S, Zhou L, Tang H, Xu J and Guo
K: Formononetin attenuates Aβ25-35-induced cytotoxicity in HT22
cells via PI3K/Akt signaling and non-amyloidogenic cleavage of APP.
Neurosci Lett. 639:36–42. 2017. View Article : Google Scholar
|
|
44
|
Zhu G, Wang X, Wu S and Li Q: Involvement
of activation of PI3K/Akt pathway in the protective effects of
puerarin against MPP+-induced human neuroblastoma SH-SY5Y cell
death. Neurochem Int. 60:400–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Y, Mao L, Gao Y, Baral S, Zhou Y and Hu
B: MicroRNA-107 contributes to post-stroke angiogenesis by
targeting Dicer-1. Sci Rep. 5:133162015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xia H, Li Y and Lv X: MicroRNA-107
inhibits tumor growth and metastasis by targeting the BDNF-mediated
PI3K/AKT pathway in human non-small lung cancer. Int J Oncol.
49:1325–1333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qian Q, Liu Q, Zhou D, Pan H, Liu Z, He F,
Ji S, Wang D, Bao W, Liu X, et al: Brain-specific ablation of Efr3a
promotes adult hippocampal neurogenesis via the brain-derived
neurotrophic factor pathway. FASEB J. 31:2104–2113. 2017.
View Article : Google Scholar : PubMed/NCBI
|