Introduction
Atherosclerosis is a complex, chronic progressive
disease, and a major cause of mortality worldwide (1,2).
The atherosclerotic process comprises three steps: Fatty streak
formation, atheroma formation and atherosclerotic plaque formation
(3). Serious clinical
consequences of atherosclerosis, including myocardial infarction
and stroke, are caused by acute rupture of unstable plaques
(4). The proliferation and
migration of vascular smooth muscle cells are important in the
pathogenesis of atherosclerosis and in the rupture of unstable
plaques (5–7).
Metformin is a biguanide class drug, which is widely
used for the clinical treatment of type 2 diabetes mellitus
(8); its fundamental use is to
suppress gluconeogenesis and promote glucose metabolism (9). Previous studies have reported that
metformin exerts vascular protective effects beyond its role in
treating diabetes (10,11). A recent study indicated that
metformin is associated with the suppression of
diabetes-accelerated atherosclerosis (12). In addition, it has been suggested
that metformin may have a role in preventing and treating atheroma,
and protecting against plaque rupture (13). Furthermore, metformin may inhibit
smooth muscle cell proliferation and migration (14,15).
In response to metformin, ataxia
telangiectasia-mutated (ATM) protein is phosphorylated at Ser-1981
(16). It has been reported that
ATM activates p53 via phosphorylation of Ser-15, in response to DNA
damage (17). p53 is regarded as
a powerful suppressive factor that is capable of halting cell
proliferation and inhibiting migration (18,19). Previous studies have indicated
that p53 serves a key role in metformin-mediated growth inhibition
in numerous types of tumor cell (20–22). In addition, activated p53 can
activate 5′ adenosine monophosphate-activated protein kinase (AMPK)
(17). The suppressive effects of
metformin are closely associated with AMPK activation (14,23), which can induce cell cycle arrest
(23,24). However, the signaling pathways and
downstream effectors underlying the effects of metformin remain to
be elucidated.
As a member of the p200 protein family,
interferon-inducible protein 16 (IFI16), has been reported to have
a central role in regulating cell proliferation by interacting with
various cellular modulators (25). p53 can upregulate IFI16 in human
diploid fibroblasts (HDFs) (26),
whereas knockdown of IFI16 results in a reduction of AMPK in HDFs
(27). However, the molecular
mechanism by which IFI16 upregulation is induced by metformin
remains to be fully elucidated.
Based on these previous findings, the present study
demonstrated that metformin inhibits the proliferation and
migration of human aortic smooth muscle cells (HASMCs) via AMPK
activation. In addition, the roles of p53 and IFI16 in
metformin-mediated suppressive effects on HASMCs were identified.
Notably, the present study provided a novel insight into the
mechanisms underlying metformin-mediated HASMC proliferation arrest
and migratory inhibition in atherosclerosis development and
progression.
Materials and methods
Cell culture
Primary HASMCs were obtained from ScienCell Research
Laboratories, Inc. (San Diego, CA, USA). According to the
manufacturer's records, HASMCs were obtained from a donor, the
details of which are as follows: Age, 20 weeks; sex, female; race,
unknown. HASMCs were cultured in smooth muscle cell medium (SMCM;
ScienCell Research Laboratories, Inc.) supplemented with 5% fetal
bovine serum (ScienCell Research Laboratories, Inc.), 1% smooth
muscle cell growth supplement (ScienCell Research Laboratories,
Inc.), 100 U/ml penicillin and 100 U/ml streptomycin. Cells were
incubated at 37°C in a humidified atmosphere containing 5%
CO2.
RNA interference
p53 small interfering (si)RNA (sense,
5′-GCAUGAACCGGAGGCCCAU-3′ and antisense,
5′-AUGGGCCUCCGGUUCAUGC-3′), IFI16 siRNA (a pool of 3 different
siRNA duplexes: sc-35633A sense, 5′-CCACAAUCUACGAAAUUCA-3′ and
antisense, 5′-UGAAUUUCGUAGAUUGUGG-3′; sc-35633B sense,
5′-CCAUCCAGCAGUUUCUUCA-3′ and antisense, 5′-UGAAGAAACUGCUGGAUGG-3′;
sc-35633C sense, 5′-GGAAGGAGAUAAACUGAAA-3′ and antisense,
5′-UUUCAGUUUAUCUCCUUCC-3′), control siRNA, control siRNA
(fluorescein-conjugated), siRNA transfection reagent and siRNA
transfection medium were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Briefly, HASMCs (1×105 cells/ml)
were transiently transfected with siRNA (concentration, 0.06
mmol/l) according to the manufacturer's protocol. After adding the
siRNA, the cells were incubated for 5–7 h at 37°C in a
CO2 incubator. A total of 48 h post-transfection, cells
were processed for immunoblotting, cell cycle analysis and
immunofluorescence, or were treated with various concentrations of
metformin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany; dissolved
in 1X PBS) and incubated for an additional 24 h at 37°C.
RNA-silenced HASMCs were treated with various concentrations of
metformin (0, 10 mM) at 37°C for 24 h (RNA-silenced group;
RNA-silenced + metformin group).
Senescence-associated β-galactosidase
staining (SA-β-gal)
The SA-β-gal activities of young (passage number 3)
and old (passage number 7) HASMCs were detected using a SA-β-gal
staining kit (Beyotime Institute of Biotechnology, Shanghai, China)
according to the manufacturer's protocol. The number of blue cells
(SA-β-gal-positive cells) and the total number of cells in six
randomly chosen fields were counted to calculate the percentage of
senescent cells using an ordinary optics microscope (Olympus
Corporation, Tokyo, Japan). Percentage of senescent cells = (number
of SA-β-gal positive cells/number of total cells) x 100%.
Cell cycle analysis
Cells from each group (1×106 cells) were
washed with PBS, trypsinized, centrifuged at 167.7 × g at 4°C for 5
min, resuspended in 3 ml 75% ethanol and stored at 4°C overnight.
The cells were then centrifuged, resuspended in 200 μl PBS
supplemented with 10 μg/ml RNase A and were incubated at
37°C for 30 min. Subsequently, the cells were stained with
propidium iodide (PI) solution (0.1% sodium citrate, 0.1% Triton
X-100, and 50 μg/ml PI; BestBio, Shanghai, China) at 4°C for
30 min. Cell cycle progression was analyzed using a BD FACSCalibur
flow cytometer (BD Biosciences, San Jose, CA, USA), and data
analysis was conducted using FlowJo 7.6 software (FlowJo, LLC,
Ashland, OR, USA).
Cell viability assay
Cells were seeded at a density of 8,000 cells/well
in 96-well plates containing 0.1 ml complete medium in triplicate.
On the following day, cells were exposed to various concentrations
of metformin and were cultured for 24 h. Cell viability was
determined using a Cell Counting kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Shanghai, China) assay. After incubation with
CCK-8 solution for 2 h at 37°C, absorbance was determined at a
wavelength of 450 nm using a SpectraMax 190 Microplate Reader
(Molecular Devices, LLC, Sunnyvale, CA, USA).
Cell counting
Cell proliferation was evaluated by cell counting
after cells seeded into 6-well culture plates (1×105
cells/well) had been incubated at 37°C with different treatments.
The total cell number was then counted in 4 fields using a
hemocytometer and the mean values were calculated from 3
replicates. The cells were counted using a phase contrast
microscope (Olympus Corporation).
Transwell assay
HASMCs (0.5×105 cells) were resuspended
in 0.1 ml serum-free SMCM and were seeded into the upper Transwell
chamber (8 μm pore size, 24-well; Corning Life Sciences,
Tewksbury, MA, USA), whereas 0.8 ml complete SMCM was used as a
chemoattractant, which was added into the lower chamber. After 24 h
at 37°C, the cells that migrated through the membrane to the lower
surface were fixed with methanol for 20 min and were then stained
with 0.1% crystal violet solution for 20 min. Finally, the stained
cells were counted under an inverted microscope. Six random fields
were chosen for analysis of cell motility.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total cellular RNA was extracted from the untreated
controls, metformin-treated, control siRNA-transfected, p53
siRNA-transfected and IFI16 siRNA-transfected HASMCs using Total
RNA fast extraction kit (Fastagen, Shanghai, China) according to
the manufacturer's protocol. Total RNA (1 μg) underwent cDNA
synthesis using Takara PrimeScript™ RT reagent kit (Takara Bio,
Inc., Otsu, Japan) according to the manufacturer's protocol.
RT-qPCR was performed in a 10 μl volume, using SYBR-Green
PCR Master mix (Takara Bio, Inc.) on a Roche
LightCycler® 480 system (Roche Diagnostics GmbH,
Mannheim, Germany) according to the manufacturers' protocols.
Thermal cycling conditions were as follows: One cycle at 95°C for
10 min, followed by 45 cycles at 95°C for 10 sec and 60°C for 30
sec. A PCR reaction mixture was prepared. The reaction then started
(stage 1, 1 cycle at 95°C for 10 min; stage 2, 45 cycles at 95°C
for 10 sec, 60°C for 30 sec). PCR amplification was conducted using
the following primer sequences: p53 (239 bp), forward
5′-CTCCTCAGCATCTTATCCGAG-3′, reverse 5′-GCTGTTCCGTCCCAGTAGATTA-3′;
IFI16 (80 bp), forward 5′-GAAGTGCCAGCGTAACTCCTA-3′, reverse
5′-TACCTCAAACACCCCATTCAC-3′; AMPK (132 bp), forward
5′-TGTGACAAGTACATACTCCAA-3′, reverse 5′-GATCTCTGTGGAGTAGCAG-3′; and
GAPDH (697 bp), forward 5′-TCACCATCTTCCCAGGAGCGAG-3′ and reverse
5′-TGTCGCTGTTGAAGTCAGAG-3′. Relative quantification values were
calculated using the 2−ΔΔCq method (28).
Western blot analysis
Whole-cell extracts were homogenized in extraction
buffer (Beyotime Institure of Biotechnology). Equal amounts of
lysate were separated by 8–12% SDS-PAGE and were transferred onto
polyvinylidene fluoride membranes (Beyotime Institute of
Biotechnology). The protein was quantified using BCA Protein Assay
kit (Beyotime Institure of Biotechnology). A total of 40 μg
(4 μg/μl ×10 μl) protein was loaded onto the
gels. The membranes were blocked with 5% nonfat dry milk and were
then incubated with the recommended concentration of primary
antibody overnight at 4°C. The mem branes (needless primary
antibodies) were washed three times (15 min/wash) and were then
incubated with a 1:5,000–1:10,000 dilution of peroxidase-conjugated
goat anti-rabbit (1:10,000; cat. no. 9101) and goat anti-mouse
(1:10,000; cat. no. 9100) secondary antibodies (OriGene
Technologies, Inc., Beijing, China) for 1 h at room temperature.
Finally, the membranes (needless secondary antibodies) were washed
three times (15 min/wash). Blots were visualized using an enhanced
chemiluminescence buffer and an Amersham Imager 600 (GE Healthcare,
Chicago, IL, USA). The data were analyzed with ImageJ software
v2.1.4.7 (National Institutes of Health, Bethesda, MD USA). Each
experiment was repeated three times to ensure consistency of the
results. The primary antibodies used in the present study were as
follows: Rabbit anti-human AMPKα monoclonal antibody (1:1,000; cat.
no. 2603), rabbit anti-human phosphorylated (p)-AMPKα (Thr-172)
monoclonal antibody (1:1,000; cat. no. 2535) (both Cell Signaling
Technology, Inc., Danvers, MA, USA), mouse anti-human IFI16
monoclonal antibody (1:200; cat. no. sc-8023), rabbit anti-human
GAPDH polyclonal antibody (1:200; cat. no. sc-25778) (both Santa
Cruz Biotechnology, Inc.), rabbit anti-human p53 polyclonal
antibody (1:1,000; cat. no. 10442-1-AP; Proteintech Group, Inc.,
Chicago, IL, USA), rabbit anti-human p-p53 (Ser-15) antibody
(1:1,000; cat. no. 9284), rabbit anti-human ATM monoclonal antibody
(1:1,000; cat. no. 2873) and mouse anti-human p-ATM (Ser-1981)
monoclonal antibody (1:1,000; cat. no. 4526) (all Cell Signaling
Technology, Inc.).
Immunofluorescence
Following the appropriate treatment in 24-well
plates, the cells were washed three times with PBS and were fixed
with 4% paraformaldehyde for 15 min at room temperature. The cells
were washed a further three times with PBS and were permeabilized
with 0.5% Triton X-100 for 20 min at room temperature, prior to
three further washes with PBS and blocking with 30% goat serum
(Beyotime Institure of Biotechnology) for 30 min at room
temperature. The cells were incubated with the recommended
concentration of primary antibodies [rabbit polyclonal anti-human
P53 (1: 200; cat. no. 10442-1-AP; Proteintech Group, Inc.) and
mouse monoclonal anti-human IFI16 (1: 50; cat. no. sc-8023; Santa
Cruz Biotechnology, Inc.)] overnight at 4°C. Subsequently, the
cells were washed three times with PBS and were incubated with
fluorescein isothiocyanate (FITC) (green)-conjugated goat
anti-rabbit (1:50; cat. no. 0311), and rhodamine (red)-conjugated
goat anti-mouse (1:50; cat. no. 0313) secondary antibodies (both
from OriGene Technologies, Inc.) at 37°C for 1 h. After washing
three times, the nuclei (blue) were counterstained with DAPI for 5
min. The cells were observed under an inverted fluorescence
microscope (Olympus Corporation) with appropriate optical filters.
At least six randomly selected fields were examined in each of the
three separate experiments. The ratio = (number of stained
cells/number of total cells x 100%
Statistical analysis
Data are presented as the means ± standard deviation
from at least three independent experiments. Comparisons between
two or more groups were subjected to a two-tailed Student's t-test
or ANOVA when appropriate. All statistical analyses were performed
using SPSS 17.0 statistical software (SPSS, Inc., Chicago, IL, USA)
and GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla,
CA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Senescent primary HASMCs exhibit a
decrease in proliferation and migration
To determine the proliferative and migratory
capabilities of HASMCs, HASMCs were investigated at different
passage numbers. Pre-senescent HASMCs (passage number 3; young)
exhibited a small and spindle-like morphology, whereas
post-senescent HASMCs (passage number 7; old) appeared
morphologically flattened with enlarged shapes; these cells were
more positive for the recognized biomarker of senescence (SA-β-gal)
(Fig. 1A and B). After cell
counting, the present study indicated that the number of old cells
was significantly lower compared with the number of young cells
after the first 24-h culture (Fig.
1C). In addition, the viability of senescent HASMCs was
markedly decreased after a 24-h culture, according to the results
of the CCK-8 assay (Fig. 1D). As
shown in Fig. 1E and F, the
proportion of cells arrested at G1 phase of the cell
cycle was evidently increased, and the percentage of cells at S
phase was markedly decreased among the old cells compared with the
young cells. The results of a cell migration assay indicated that
the migratory capabilities of the old cells were markedly reduced
compared with the young cells (Fig.
1G and H).
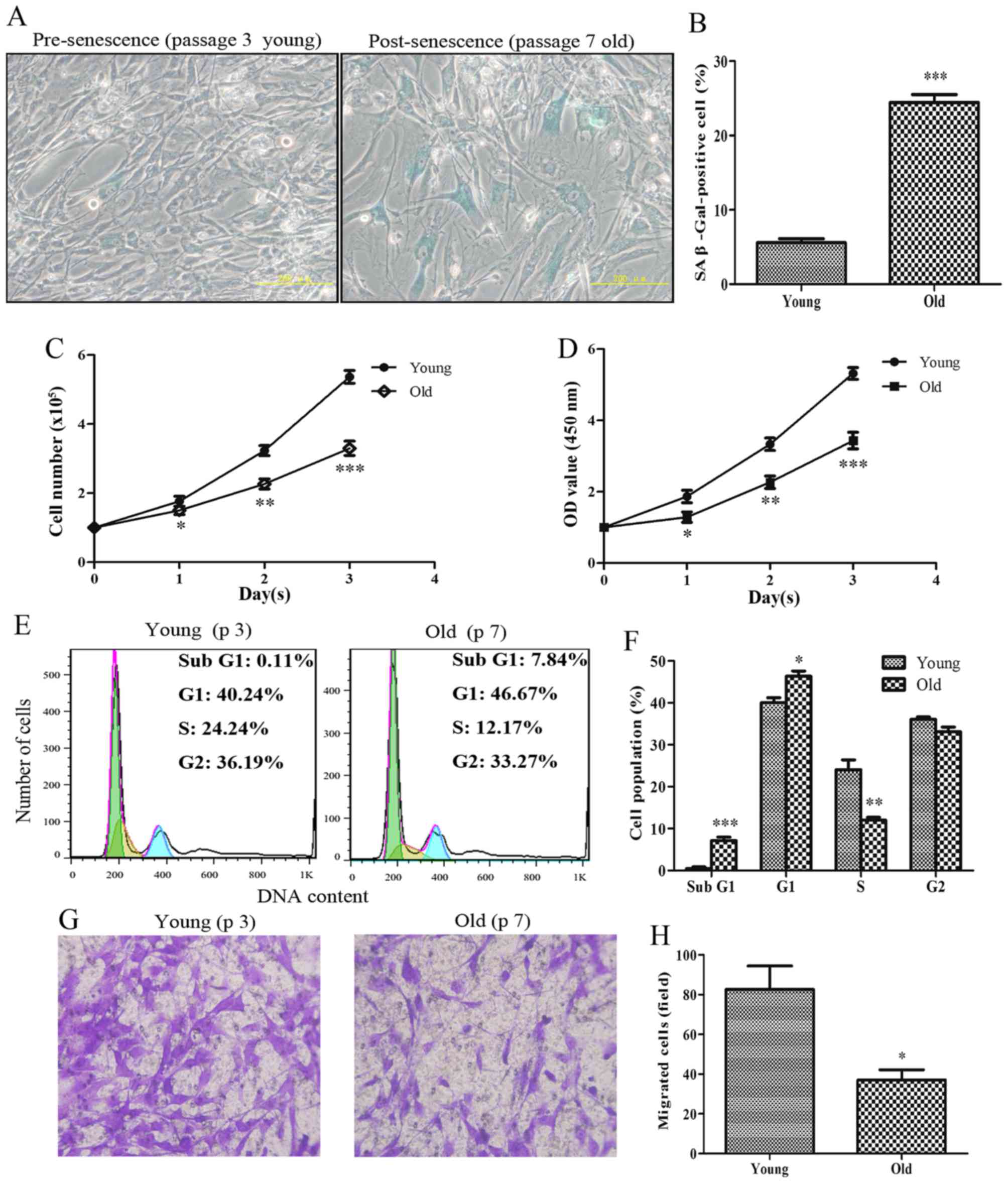 | Figure 1Senescent primary HASMCs exhibit a
decrease in proliferation and migration. Young (passage number 3)
and old (passage number 7) cells were seeded at a density of
1×105 cells/well in 6-well culture plates (SA-β-gal
staining assay, cell counting assay, CCK-8 assay and cell cycle
assay). (A) SA-β-gal activity was detected using a SA-β-gal
staining assay following culturing for 3 days. Blue cells were
considered SA-β-gal-positive cells. (B) Histogram represents the
percentage of SA-β-gal-positive cells. (C) Number of cells was
counted per well after 0, 1, 2 and 3 days. (D) Cell viability was
detected using the Cell Counting kit-8 assay after 0, 1, 2 and 3
days. (E) Cell cycle progression was detected using a cell cycle
assay following culturing for 3 days. (F) Histogram represents the
percentage of cells in each cell cycle phase. (G) After culturing
for 3 days, equal numbers of young and old cells
(0.5×105) were seeded into the upper Transwell chambers
and were cultured for 24 h. Representative images of stained and
migrated cells are presented; magnification, ×200. (H) Number of
migrated cells was quantified by counting the cells from six random
fields. All data are presented as the means ± standard deviation
from three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the young
group. HASMCs, human aortic smooth muscle cells; OD, optical
density; SA-β-gal, senescence-associated β-galactosidase. |
Protein expression levels of p53, IFI16,
AMPK and p-AMPK are increased in old HASMCs
The present study aimed to detect the relative
expression levels of proteins associated with growth arrest and
migratory inhibition in senescent HASMCs. As shown in Fig. 2A and B, the basal levels of p53,
IFI16, AMPK and p-AMPK were higher in the protein extracts derived
from old HASMCs compared with in those derived from young HASMCs.
Immunofluorescence analysis indicated that endogenous p53
expression (green) was increased in the old cells compared with in
the young cells (Fig. 2C and D).
In addition, the expression levels of endogenous IFI16 (red) were
increased in old HASMCs (Fig. 2E and
F).
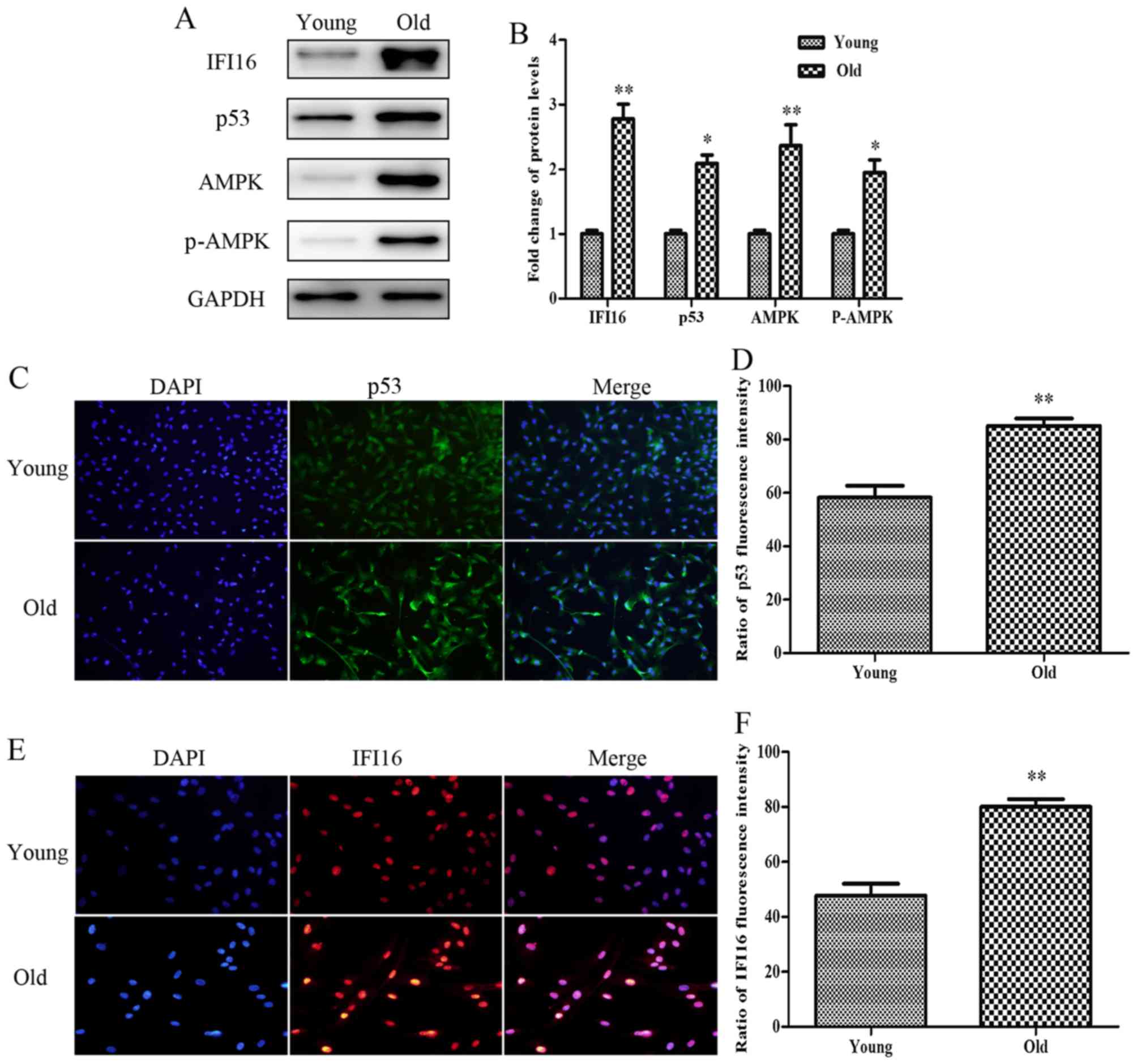 | Figure 2Protein expression levels of p53,
IFI16, AMPK and p-AMPK are increased in old (passage 7) HASMCs
compared with in young (passage 3) HASMCs. Young and old cells were
cultured for 3 days. (A) Western blot analysis was used to detect
IFI16, p53, AMPK and p-AMPK expression in young and old HASMCs.
GAPDH was used as a loading control. (B) Histogram represents the
fold change in IFI16, p53, AMPK and p-AMPK protein expression
normalized to GAPDH relative to the young group. (C) Young and old
cells were stained with a fluorescein isothiocyanate
(green)-conjugated secondary antibody to detect endogenous p53 and
nuclei were stained with DAPI (blue). Cells were observed under a
fluorescence microscope (magnification, ×100). (D) Histogram
represents the percentage of endogenous p53-positive cells. (E)
Young and old cells were stained with a rhodamine (red)-conjugated
secondary antibody to detect endogenous IFI16 and nuclei were
stained with DAPI (blue). Cells were observed under a fluorescence
microscope (magnification, ×200). (F) Histogram represents the
percentage of endogenous IFI16-positive cells. All data are
presented as the means ± standard deviation from three independent
experiments. *P<0.05 and **P<0.01 vs.
the young group. AMPK, 5′ adenosine monophosphate-activated protein
kinase; HASMCs, human aortic smooth muscle cells; IFI16,
interferon-inducible protein 16; p-AMPK, phosphorylated-AMPK. |
Metformin upregulates p53, IFI16 and
p-AMPK expression, and inhibits proliferation and migration of
HASMCs
To explore whether the suppressive effects of
metformin were associated with the extent of relative protein
activation in HASMCs, a dose-dependent immunoblot analysis was
conducted. HASMCs (passage 3) were treated with various
concentrations of metformin (0–10 mmol/l) for 24 h. As shown in
Fig. 3A, the mRNA expression
levels of p53 and IFI16 were increased in a dose-dependent manner
following treatment with metformin. The protein expression levels
of p53, p-p53 (Ser-15), IFI16 and p-AMPK (Thr-172) were also
steadily increased in response to increasing metformin
concentrations (Fig. 3B and
C).
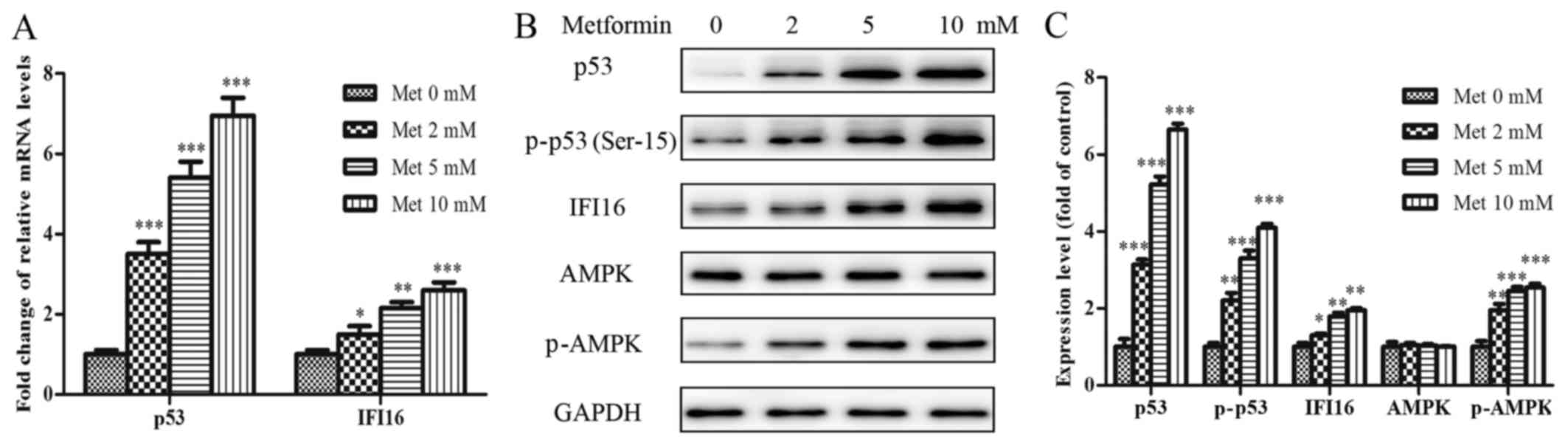 | Figure 3Met activates p53, p-p53, IFI16 and
p-AMPK in HASMCs (passage 3). HASMCs were treated with Met at
various concentrations (0, 2, 5 and 10 mmol/l) for 24 h. (A) mRNA
expression levels of p53 and IFI16 were measured by reverse
transcription-quantitative polymerase chain reaction. Histogram
represents the fold change in p53 and IFI16 mRNA expression
normalized to GAPDH relative to the control (met 0 mM). (B) Western
blotting was performed to detect p53, p-p53, IFI16, AMPK and p-AMPK
protein expression. GAPDH was used as a loading control. (C)
Histogram represents the fold change in p53, p-p53, IFI16, AMPK and
p-AMPK protein expression normalized to GAPDH relative to the
control (met 0 mM). All data are presented as the means ± standard
deviation of three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the control
group. AMPK, 5′ adenosine monophosphate-activated protein kinase;
HASMCs, human aortic smooth muscle cells; IFI16,
interferon-inducible protein 16; Met, metformin; p-,
phosphorylated. |
As shown in Fig. 4A
and B, incubation of the cells with increasing metformin
concentrations resulted in an inhibition in cell growth and reduced
cell viability. Similar to the effects of senescence on cell cycle
progression, metformin was able to arrest HASMCs in G1
phase and decreased the percentage of cells in S phase of the cell
cycle in a dose-dependent manner (Fig. 4C and E). Furthermore, increasing
metformin concentrations resulted in a gradual, significant
reduction in the migration of HASMCs (Fig. 4D and F).
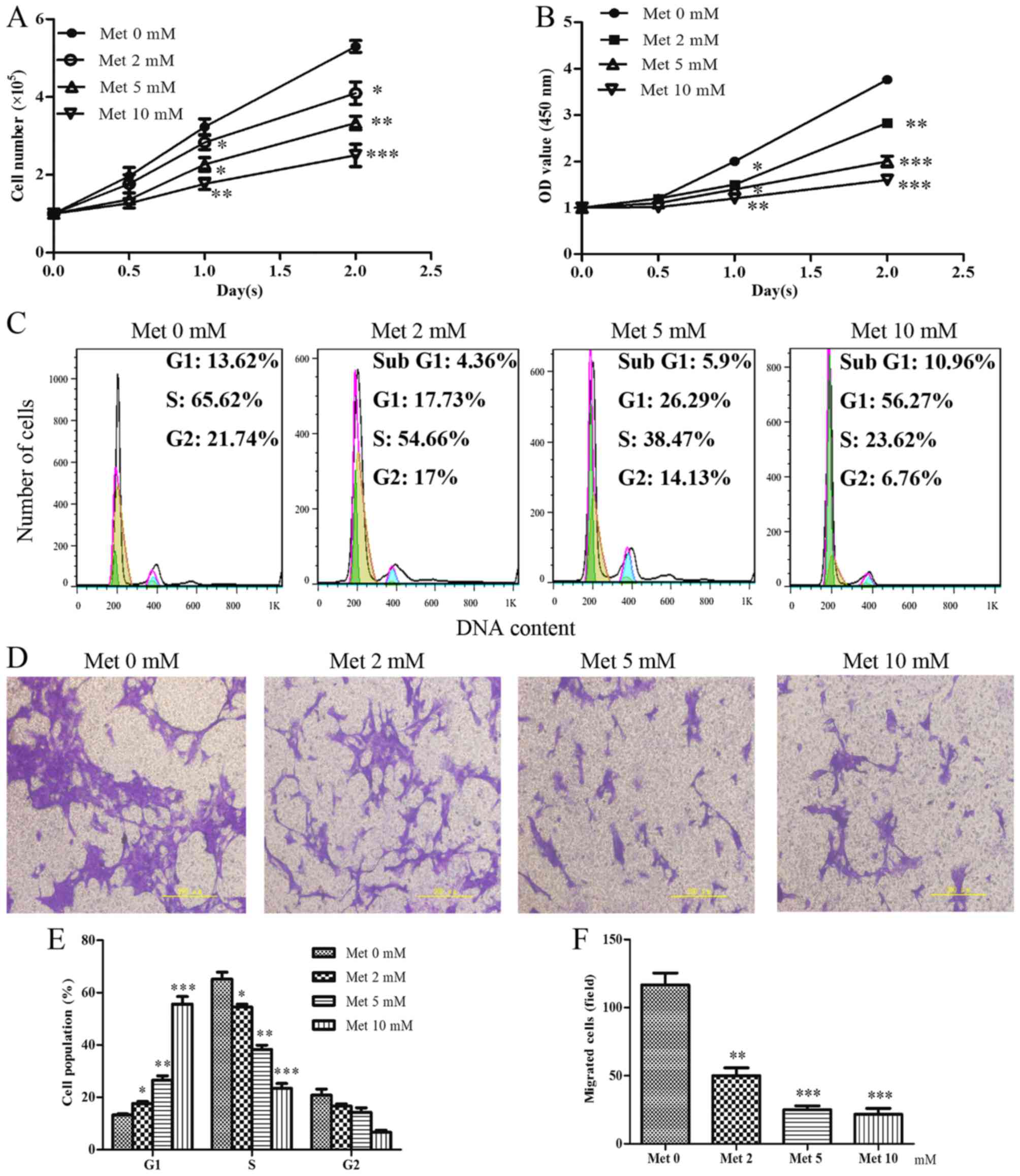 | Figure 4Met inhibits the proliferation and
migration of HASMCs (passage 3). HASMCs were seeded at a density of
1×105 cells/well in 6-well culture plates (cell counting
assay, CCK-8 assay and cell cycle assay) and were treated with Met
at various concentrations (0, 2, 5 and 10 mmol/l) for 0, 0.5, 1.0
and 2.0 day. (A) Cell number per well was counted following
treatment with various concentrations of Met for 0, 0.5, 1.0 and
2.0 days. (B) Cell viability was determined using the Cell Counting
kit-8 assay following treatment with various concentrations of Met
for 0, 0.5, 1.0 and 2.0 days. (C and E) Cell cycle progression was
determined using a cell cycle assay following treatment with
various concentrations of Met for 24 h. Histogram represents the
percentage of cells in each cell cycle phase. (D and F) Cell
migration was detected by Transwell assay following treatment with
various concentrations of Met for 24 h. Representative images of
stained and migrated cells are shown; magnification, ×200.
Histogram represents the number of migrated cells from six random
fields. All data are presented as the means ± standard deviation
from three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the control
group (Met 0 mM). HASMCs, human aortic smooth muscle cells; Met,
metformin; OD, optical density. |
p53-knockdown significantly suppresses
metformin-mediated growth arrest and migratory inhibition in
HASMCs
To explore the role of p53 in metformin-mediated
growth arrest and migratory inhibition, p53 was knocked down using
p53 siRNA, and the effects of metformin on p53-silenced HASMCs were
reassessed. The present study initially confirmed that p53 was
knocked down in p53 siRNA-transfected HASMCs. Cell
immunofluorescence analysis indicated that endogenous p53
expression was evidently attenuated in cells transfected with p53
siRNA (Fig. 5A and B).
Furthermore, p53-knockdown resulted in a significant reduction in
the expression levels of IFI16, AMPK and p-AMPK (Fig. 5C and D). With regards to mRNA
expression, p53, IFI16 and AMPK expression was significantly
reduced in p53 siRNA-transfected cells (Fig. 5E).
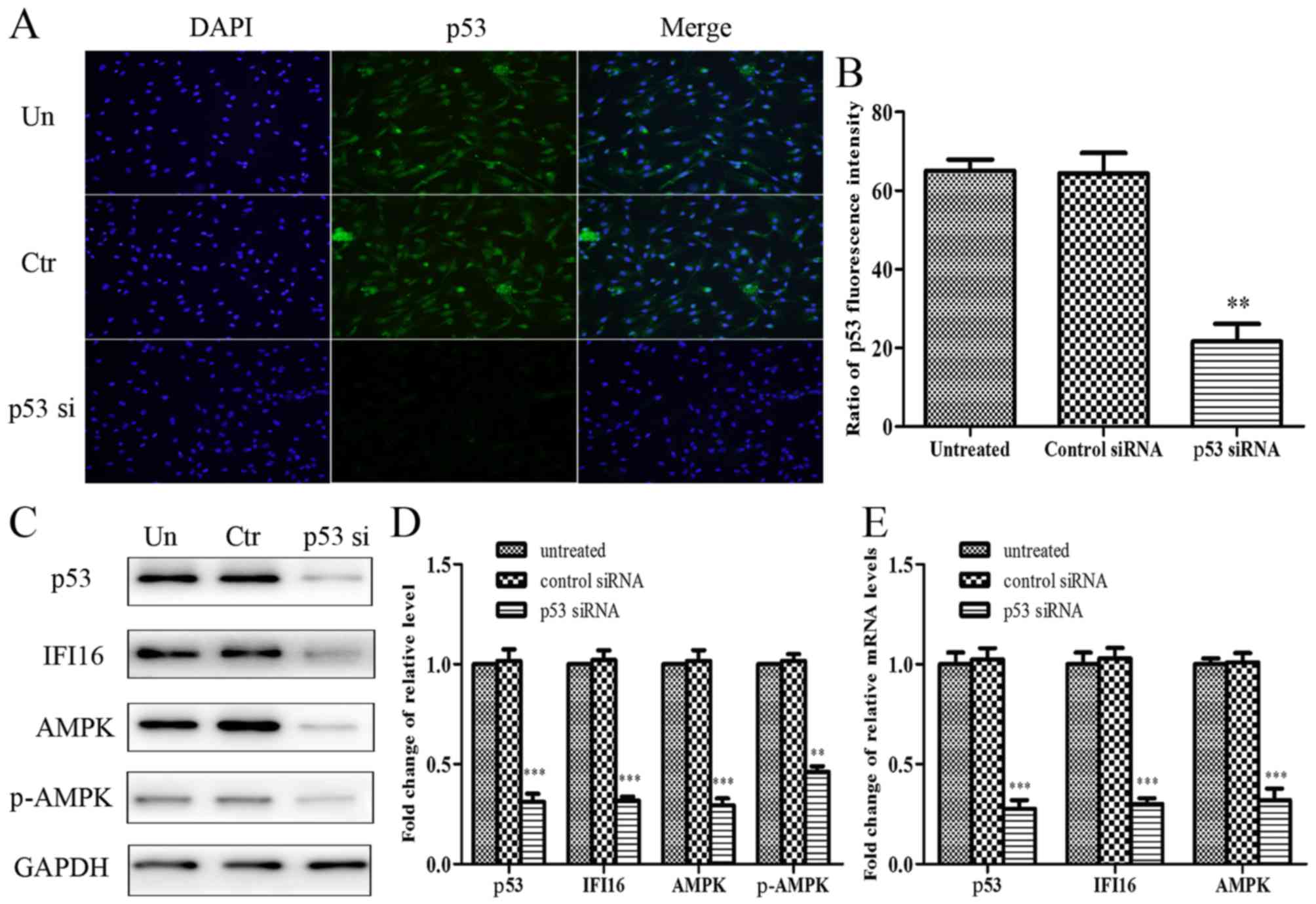 | Figure 5Effects of p53-knockdown on the
protein and mRNA expression levels of IFI16, AMPK and p-AMPK in
HAMSCs (passage 3). HASMCs were untreated, or were transfected with
control siRNA or p53 siRNA. (A) A total of 48 h post-transfection,
immunofluorescence analysis was performed to detect endogenous p53
expression. (B) Histogram represents the ratio of p53 fluorescence
intensity from six random fields. (C) A total of 48 h
post-transfection, cells were lysed and examined by western
blotting to detect p53, IFI16, AMPK and p-AMPK protein expression.
(D) Histogram represents the fold change in p53, IFI16, AMPK and
p-AMPK protein expression normalized to GAPDH relative to the
untreated group. (E) A total of 24 h post-transfection, the mRNA
expression levels of p53, IFI16 and AMPK were measured by reverse
transcription-quantitative polymerase chain reaction. Histogram
represents the fold change in p53, IFI16 and AMPK mRNA expression
normalized to GAPDH relative to the untreated group. All data are
presented as the means ± standard deviation from three independent
experiments. **P<0.01 and ***P<0.001
vs. the untreated group. AMPK, 5′ adenosine monophosphate-activated
protein kinase; Ctr, control; HASMCs, human aortic smooth muscle
cells; IFI16, interferon-inducible protein 16; p-, phosphorylated;
siRNA, small interfering RNA; Un, untreated. |
A total of 24 hours after culture, the results
indicated that p53-silenced cells exhibited increased
proliferation, which was resistant to metformin-induced growth
arrest (Fig. 6A). Analysis of
cell cycle progression indicated that the percentage of
G1 phase cells was significantly decreased and the
percentage of S phase cells was increased in p53-silenced HASMCs.
Notably, p53-knockdown evidently suppressed the metformin-mediated
increase in the accumulation of HASMCs in G1 phase of
the cell cycle (Fig. 6B and C).
Furthermore, cell viability was markedly increased in the
p53-silenced cells, whereas the metformin-mediated decrease in cell
viability was suppressed by knockdown of p53 (Fig. 6D). Cell migration analysis
indicated that knockdown of p53 promoted an increase in mobility.
As expected, metformin-induced migratory inhibition was markedly
suppressed in HASMCs when cells were transfected with p53 siRNA
prior to metformin-treatment (Fig. 6E
and F).
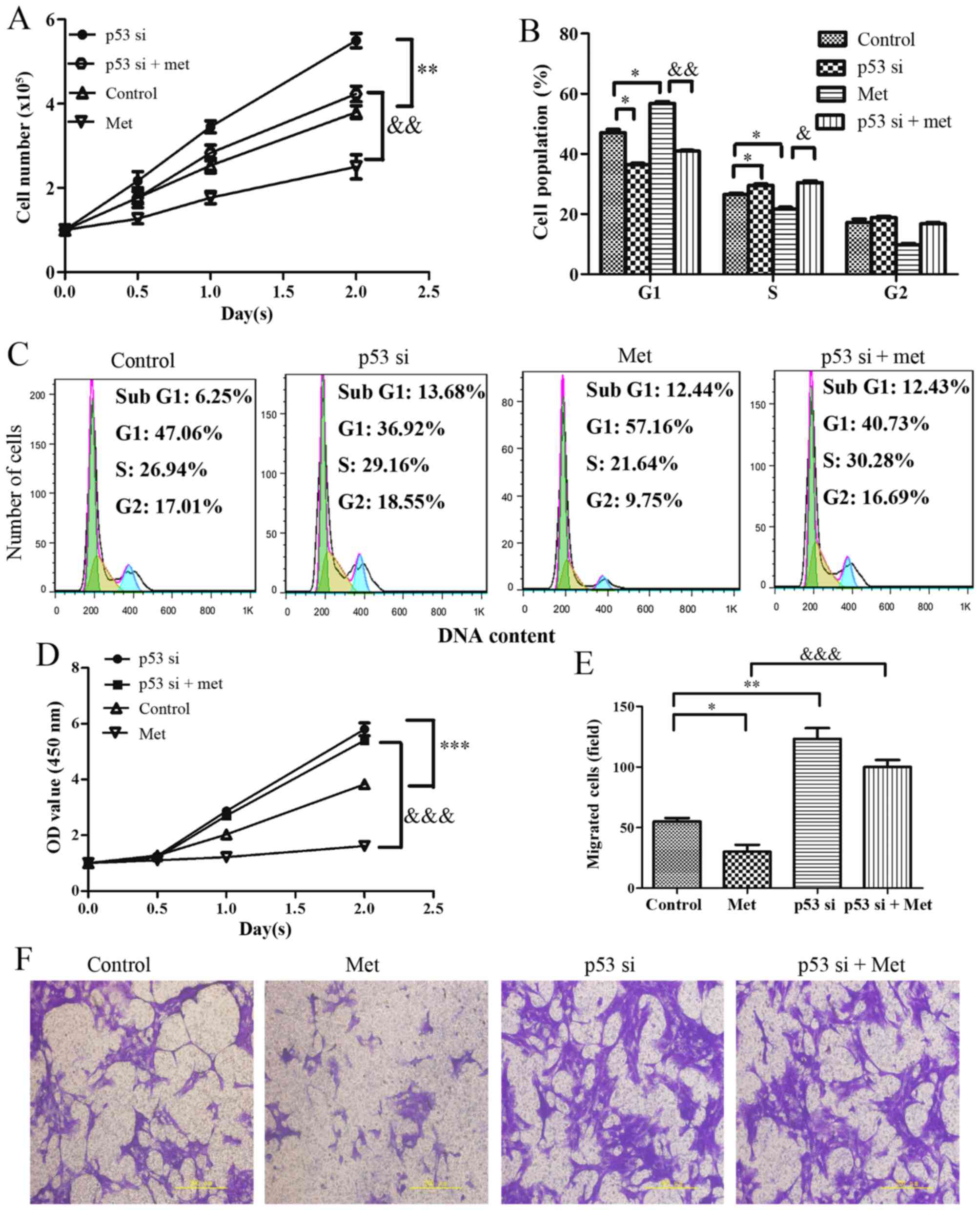 | Figure 6Effects of p53-knockdown on
Met-mediated growth arrest and migratory inhibition in HASMCs
(passage 3). Control HASMCs and p53-silenced HASMCs (48 h
post-transfection) were treated with or without 10 mM metformin for
24 h. (A) Control HASMCs (Control), p53-silenced HASMCs (p53 si),
control HASMCs treated with Met (Met) and p53-silenced HASMCs
treated with Met (p53 si + Met) were seeded at a density of
1×105 cells/well in 6-well culture plates (cell counting
assay, CCK-8 assay and cell cycle assay). Number of cells per well
was counted at 0, 0.5, 1.0 and 2.0 days. (B and C) Cell cycle
analysis was performed to evaluate the number of cells in each
phase of the cell cycle in each group. Histogram represents the
percentage of cells in each cell cycle phase. (D) Cell viability
was detected using the Cell Counting kit-8 assay at 0, 0.5, 1.0 and
2.0 days. (E and F) Cell migration was detected by Transwell assay
in each group. Representative images of stained and migrated cells
are shown; magnification, ×200. Histogram represents the number of
migratory cells per field from six random fields. All data are
presented as the means ± standard deviation from three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. the control group;
&P<0.05, &&P<0.01 and
&P<0.001 vs. the Met group. HASMCs, human aortic smooth
muscle cells; Met, metformin; OD, optical density; si, small
interfering RNA. |
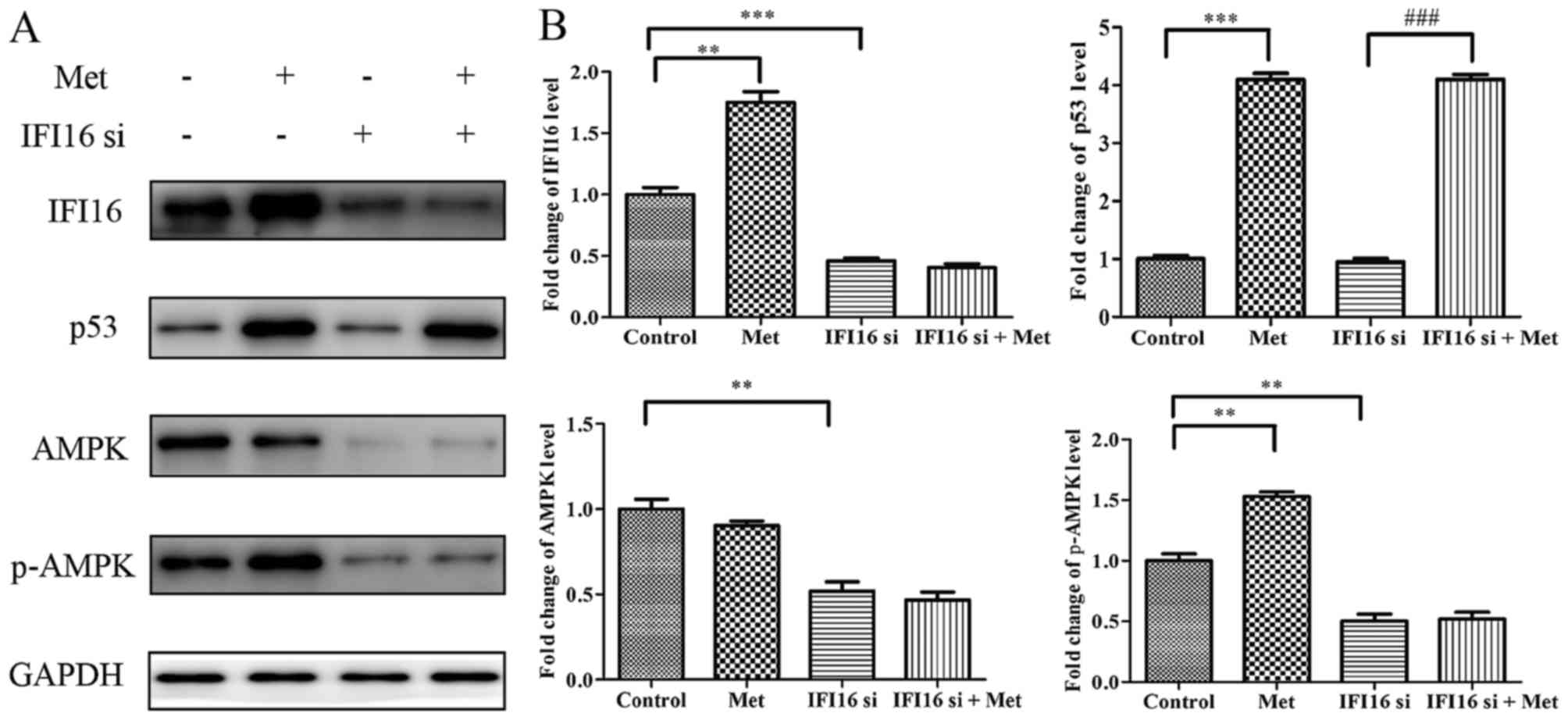
IFI16-knockdown significantly suppresses
metformin-mediated growth arrest and migratory inhibition in
HASMCs
Subsequently, the present study aimed to investigate
the role of IFI16 in metformin-mediated suppressive effects. IFI16
siRNA significantly attenuated endogenous IFI16 expression, thus
confirming that IFI16 was knocked down in HASMCs transfected with
IFI16 siRNA (Fig. 7A and B).
Furthermore, downregulation of IFI16 markedly decreased the
expression levels of AMPK and p-AMPK; however, the expression
levels of p53 were not significantly altered in response to IFI16
siRNA (Fig. 7C and D). Similarly,
the mRNA expression levels of IFI16 and AMPK were decreased in
IFI16 siRNA-transfected cells. IFI16 siRNA had no significant
effect on p53 mRNA expression (Fig.
7E).
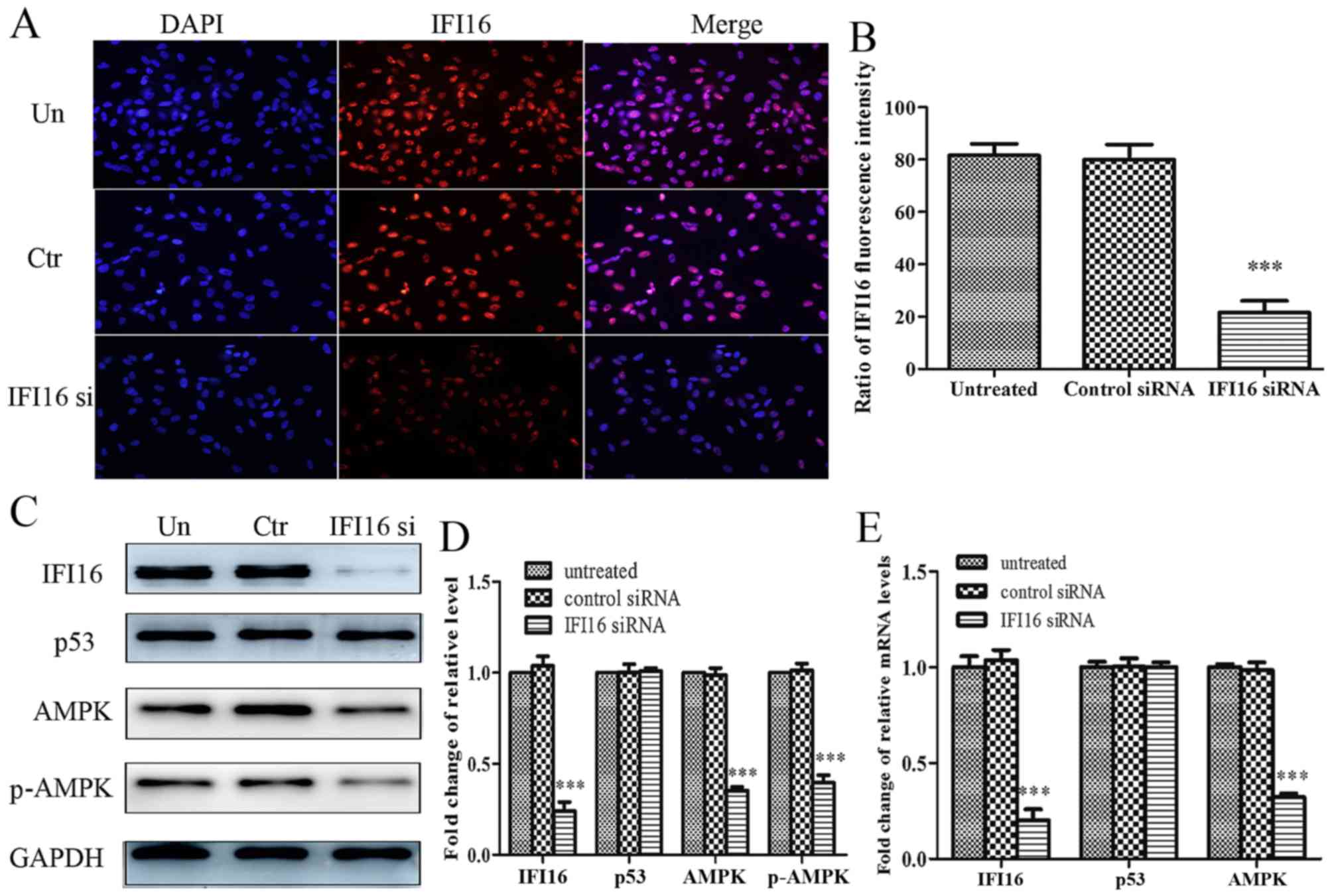 | Figure 7Effects of IFI16-knockdown on the
protein and mRNA expression levels of p53, AMPK and p-AMPK in
HAMSCs (passage 3). HASMCs were untreated, or were transfected with
control siRNA or IFI16 siRNA. (A) A total of 48 h
post-transfection, immunofluorescence analysis was performed to
detect endogenous IFI16 expression. (B) Histogram represents the
ratio of IFI16 fluorescence intensity from six random fields. (C) A
total of 48 h post-transfection, cells were lysed and examined by
western blotting to detect IFI16, p53, AMPK and p-AMPK protein
expression. (D) Histogram represents the fold change in IFI16, p53,
AMPK and p-AMPK protein expression normalized to GAPDH relative to
the untreated group. (E) A total of 24 h post-transfection, the
mRNA expression levels of IFI16, p53 and AMPK were measured by
reverse transcription-quantitative polymerase chain reaction.
Histogram represents the fold change in IFI16, p53 and AMPK mRNA
expression normalized to GAPDH relative to the untreated group. All
data are presented as the means ± standard deviation from three
independent experiments. ***P<0.001 vs. the untreated
group. AMPK, 5′ adenosine monophosphate-activated protein kinase;
Ctr, control; HASMCs, human aortic smooth muscle cells; IFI16,
interferon-inducible protein 16; p-, phosphorylated; siRNA, small
interfering RNA; Un, untreated. |
Through cell counting, cell cycle assay and CCK-8
assay, the present study demonstrated that IFI16-silenced cells
exhibited increased proliferation compared with the control cells.
Notably, IFI16-knockdown markedly suppressed metformin-induced cell
growth inhibition, G1 phase cell cycle arrest and cell
viability inhibition (Fig. 8A–D).
In addition, IFI16 siRNA transfection induced an increase in cell
migration. As expected, metformin hardly had an effect on the
migration of IFI16-silenced cells (Fig. 8E and F).
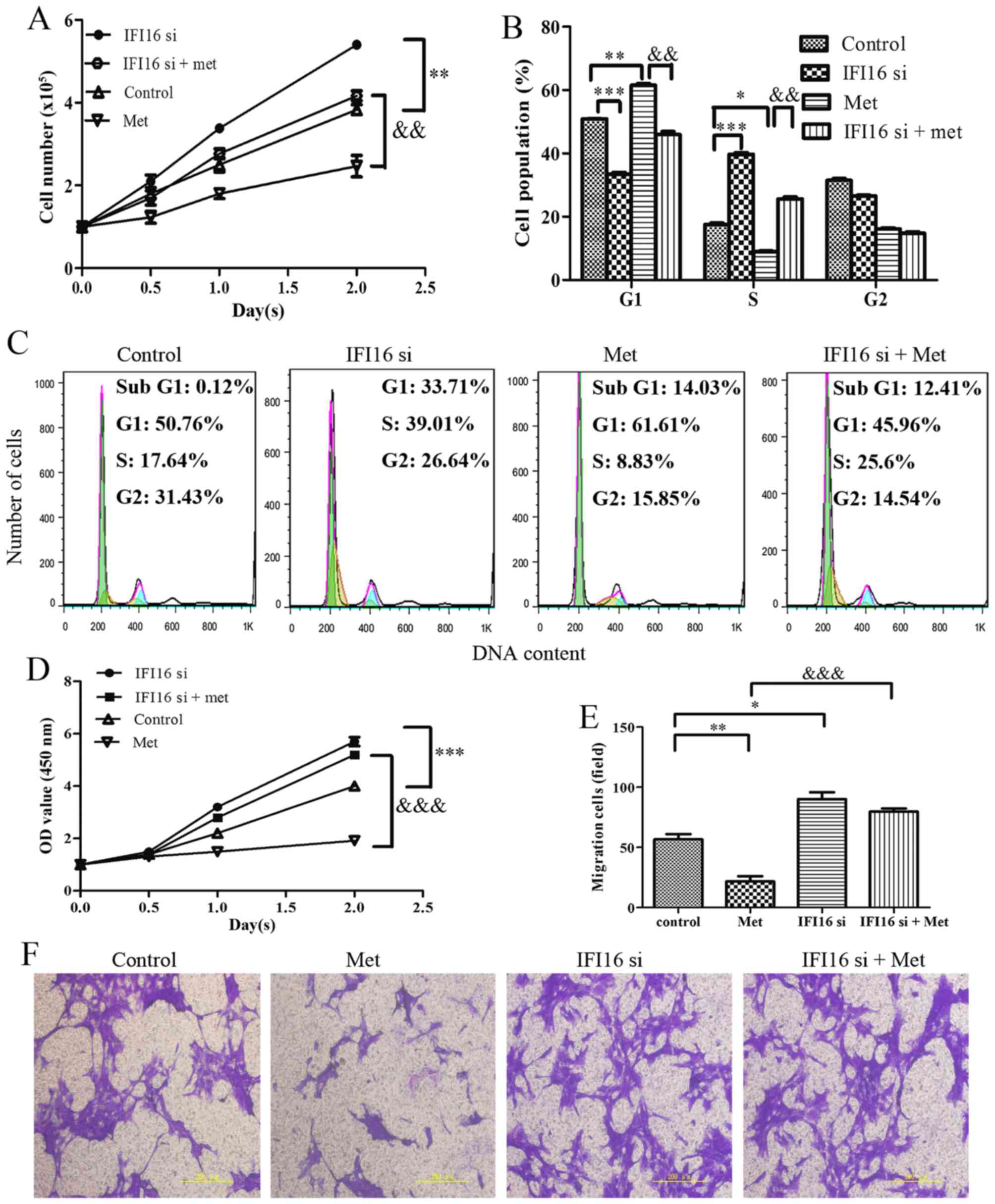 | Figure 8Effects of IFI16-knockdown on
Met-mediated growth arrest and migratory inhibition in HASMCs
(passage 3). Control HASMCs and IFI16-silenced HASMCs (48 h
post-transfection) were untreated or treated with 10 mM of Met for
24 h. (A) Control HASMCs (control), IFI16-silenced HASMCs (IFI16
si), control HASMCs treated with Met (Met) and IFI16-silenced
HASMCs treated with Met (IFI16 si + Met) were seeded at a density
of 1×105 cells/well in 6-well culture plates (cell
counting assay, CCK-8 assay and cell cycle assay). Number of cells
per well was counted at 0, 0.5, 1.0 and 2.0 days. (B and C) Cell
cycle analysis was performed to evaluate the number of cells in
each phase of the cell cycle in each group. Histogram represents
the percentage of cells in each cell cycle phase. (D) Cell
viability was detected using the Cell Counting kit-8 assay at 0,
0.5, 1.0 and 2.0 days. (E and F) Cell migration was detected using
a Transwell assay in each group. Representative images of stained
and migrated cells are shown; magnification, ×200. Histogram
represents the number of migratory cells per field from six random
fields. All data are presented as the means ± standard deviation
from three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the control
group; &&P<0.01 and
&&&P<0.001 vs. the Met group. HASMCs,
human aortic smooth muscle cells; IFI16, interferon-inducible
protein 16; Met, metformin; OD, optical density; si, small
interfering RNA. |
Metformin upregulates IFI16 and p-AMPK
expression through activating p53 in HASMCs
To investigate the molecular mechanism underlying
p53-knockdown-induced suppression of metformin-mediated growth
arrest and migratory inhibition, HASMCs were transfected with p53
siRNA followed by metformin stimulation for 24 h. Western blot
analysis indicated that metformin upregulated ATM, p-ATM
(Ser-1981), p53, p-p53 (Ser-15), IFI16 and p-AMPK expression in p53
siRNA-untreated cells; however, metformin had no significant effect
on IFI16 and p-AMPK expression in p53-silenced cells. Conversely,
metformin was able to potentiate ATM and p-ATM expression in
p53-silenced cells (Fig. 9).
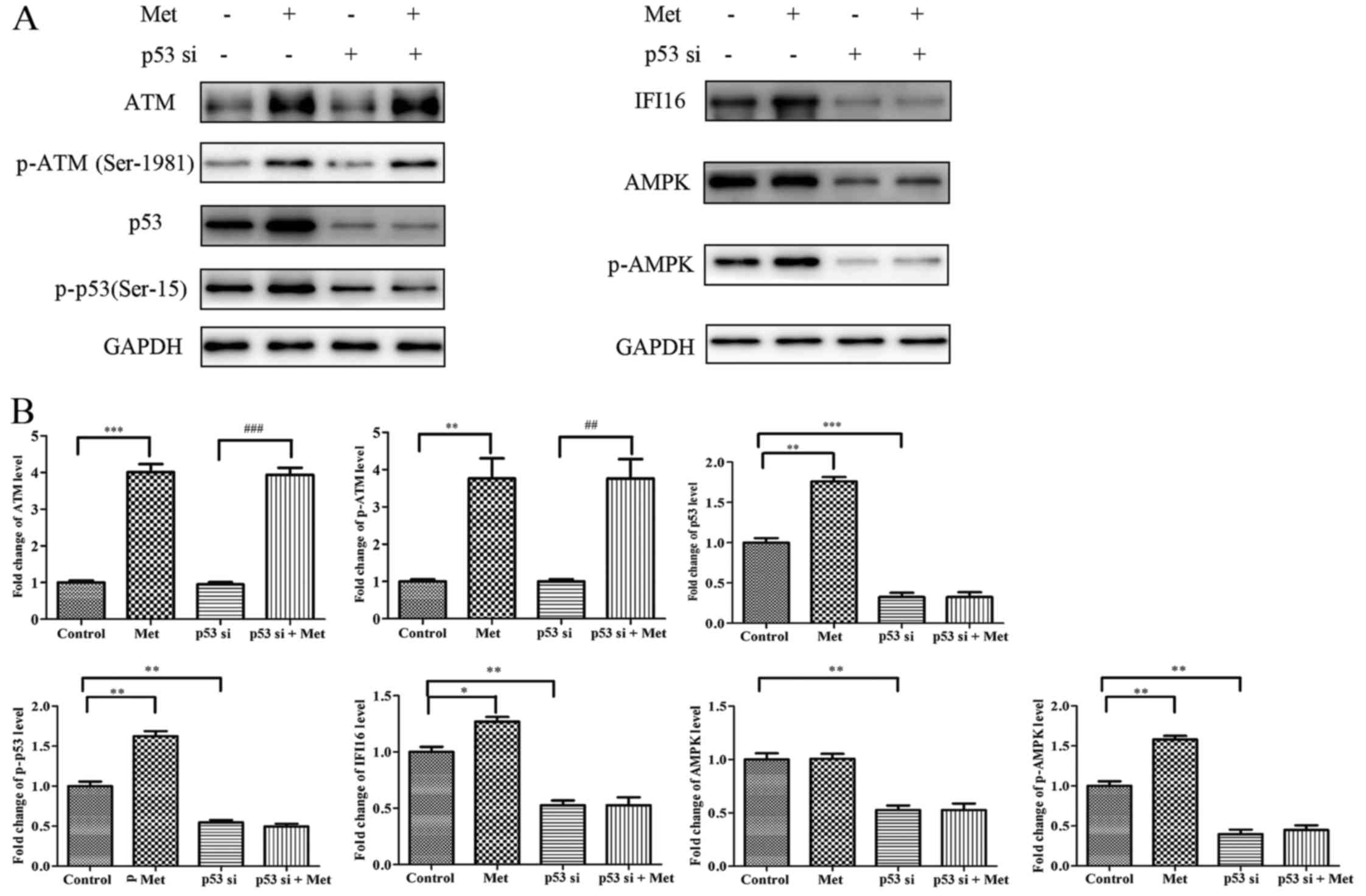 | Figure 9Met does not activate IFI16 and AMPK
in p53-silenced HASMCs. Control HASMCs and p53-silenced HASMCs (48
h post-transfection) were treated with or without 10 mM Met for 24
h. (A) Control HASMCs (control), control HASMCs treated with Met
(Met), p53-silenced HASMCs (p53 si) and p53-silenced HASMCs treated
with Met (p53 si + Met) were lysed and examined by western blot
analysis to detect ATM, p-ATM, p53, p-p53, IFI16, AMPK and p-AMPK
protein expression. (B) Histogram represents fold change in protein
expression normalized to GAPDH relative to the control. All data
are presented as the means ± standard deviation from three
independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the control
group; ##P<0.01 and ###P<0.001 vs. the
p53 si group. AMPK, 5′ adenosine monophosphate-activated protein
kinase; ATM, ataxia telangiectasia-mutated; HASMCs, human aortic
smooth muscle cells; IFI16, interferon-inducible protein 16; p-,
phosphorylated; si, small interfering RNA. |
IFI16 is required for metformin-induced
upregulation of p-AMPK
In order to investigate the molecular mechanism
underlying IFI16-knockdown-induced suppression of
metformin-mediated suppressive effects, HASMCs were transfected
with IFI16 siRNA followed by metformin stimulation for 24 h.
Western blot analysis indicated that, although p53 expression was
increased in IFI16-silenced cells in response to metformin,
IFI16-knockdown markedly suppressed the metformin-induced increase
in p-AMPK expression (Fig.
10).
Discussion
Previous studies have suggested that metformin
exerts significant cardioprotective effects alongside its
fundamental role as an antidiabetic agent (10,11). The mechanism of action of
metformin in atherosclerosis is associated with AMPK (29,30). It has previously been reported
that numerous cytokines, chemokines and growth factors regulate
smooth muscle cell proliferation and the progression of
atherosclerosis (31,32). Increased proliferation of smooth
muscle cells has been observed during early atherosclerosis
(1). Smooth muscle cell migration
into the intima is considered evidence that smooth muscle cell
migration from the media is important in atherogenesis (4).
Activation of AMPK by metformin antagonizes the
proliferation and migration of HASMCs (14). Consistent with the results of the
present study, treatment with metformin can significantly inhibit
the growth and migration of HASMCs via activation of AMPK.
Previous studies have reported that AMPK and p53
serve important roles in metformin-mediated cell growth arrest and
migratory inhibition (21,22,33).
The present study indicated that p53-knockdown resulted in
downregulation of IFI16, AMPK and p-AMPK. Furthermore, metformin
has been reported to activate p53 via ATM, in order to upregulate
IFI16 and p-AMPK; however, in the present study, metformin had no
significant effect on activation of IFI16 and p-AMPK in
p53-silenced HASMCs. These findings suggested that p53 is required
for metformin-mediated activation of IFI16 and AMPK, and is
essential for metformin-mediated suppressive effects in HASMCs.
The present study further examined whether
metformin-mediated growth arrest and migratory inhibition of HASMCs
in an IFI16-dependent manner. IFI16 siRNA had no significant
effects on p53 expression, whereas it decreased the levels of AMPK
and p-AMPK. Furthermore, metformin had no significant effects on
antagonizing cell proliferation and migration, and activating AMPK
in IFI16-silenced HASMC. These results indicated that the
metformin-induced upregulation of p53 may activate AMPK in an
IFI16-dependent manner, and IFI16 may be required for
metformin-mediated inhibition of HASMC proliferation and migration.
Notably, IFI16 has been reported to serve a central role in
suppressing cell proliferation (34). Therefore, in the present study, it
was also hypothesized that the antiproliferative and antimigratory
effects of metformin may be attributed to IFI16 activation. The
present study demonstrated that metformin inhibits cell
proliferation and migration of primary HASMCs via upregulation of
p53 and IFI16. To the best of our knowledge, the present study is
the first, to the best of our knowledge, to demonstrate that
activation of AMPK by metformin antagonizes proliferation and
migration, and these activities require the participation of p53
and IFI16 in primary HASMCs. Further in vivo studies are
required to clarify the molecular mechanisms underlying the
association between the suppressive effects of metformin and the
ATM-p53-IFI16-AMPK pathway in HASMCs.
In conclusion, the present study indicated that
metformin may inhibit HASMC proliferation and migration through
activating the ATM-p53-IFI16-AMPK pathway. In addition, p53 and
IFI16 may be required for metformin-mediated suppressive effects
and activation of AMPK in HASMCs. These findings suggested that
metformin could be considered a potential therapeutic agent used to
reduce the risk of atherosclerosis.
Acknowledgments
The authors of the present study would like to thank
the technological support of the Key Laboratory of Cardiovascular
Remolding and Function Research, Chinese Ministry of Education,
Chinese Ministry of Health in Qilu Hospital. The present study was
supported by the National Natural Science Foundation of China
(grant no. 81260030).
References
|
1
|
Lim S and Park S: Role of vascular smooth
muscle cell in the inflammation of atherosclerosis. BMB Rep.
47:1–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Virmani R, Kolodgie FD, Burke AP, Farb A
and Schwartz SM: Lessons from sudden coronary death: A
comprehensive morphological classification scheme for
atherosclerotic lesions. Arterioscler Thromb Vasc Biol.
20:1262–1275. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rafieian-Kopaei M, Setorki M, Doudi M,
Baradaran A and Nasri H: Atherosclerosis: Process, indicators, risk
factors and new hopes. Int J Prev Med. 5:927–946. 2014.PubMed/NCBI
|
|
4
|
Bennett MR, Sinha S and Owens GK: Vascular
smooth muscle cells in atherosclerosis. Circ Res. 118:692–702.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rudijanto A: The role of vascular smooth
muscle cells on the pathogenesis of atherosclerosis. Acta Med
Indones. 39:86–93. 2007.PubMed/NCBI
|
|
6
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ross R: The pathogenesis of
atherosclerosis: A perspective for the 1990s. Nature. 362:801–809.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Crandall JP, Knowler WC, Kahn SE, Marrero
D, Florez JC, Bray GA, Haffner SM, Hoskin M and Nathan DM; Diabetes
Prevention Program Research Group: The prevention of type 2
diabetes. Nat Clin Pract Endocrinol Metab. 4:382–393. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ajjan RA and Grant PJ: Cardiovascular
disease prevention in patients with type 2 diabetes: The role of
oral anti-diabetic agents. Diab Vasc Dis Res. 3:147–158. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wan X, Huo Y, Johns M, Piper E, Mason JC,
Carling D, Haskard DO and Boyle JJ: 5′-AMP-activated protein
kinase-activating transcription factor 1 cascade modulates human
monocyte-derived macrophages to atheroprotective functions in
response to heme or metformin. Arterioscler Thromb Vasc Biol.
33:2470–2480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Q, Zhang M, Torres G, Wu S, Ouyang C,
Xie Z and Zou MH: Metformin suppresses diabetes-accelerated
atherosclerosis via the inhibition of Drp1-mediated mitochondrial
fission. Diabetes. 66:193–205. 2017. View Article : Google Scholar :
|
|
13
|
Uitz E, Bahadori B, McCarty MF and
Moghadasian MH: Practical strategies for modulating foam cell
formation and behavior. World J Clin Cases. 2:497–506. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vigetti D, Clerici M, Deleonibus S,
Karousou E, Viola M, Moretto P, Heldin P, Hascall VC, De Luca G and
Passi A: Hyaluronan synthesis is inhibited by adenosine
mono-phosphate-activated protein kinase through the regulation of
HAS2 activity in human aortic smooth muscle cells. J Biol Chem.
286:7917–7924. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SA and Choi HC: Metformin inhibits
inflammatory response via AMPK-PTEN pathway in vascular smooth
muscle cells. Biochem Biophys Res Commun. 425:866–872. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Storozhuk Y, Hopmans SN, Sanli T, Barron
C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G and Tsakiridis T:
Metformin inhibits growth and enhances radiation response of
non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J
Cancer. 108:2021–2032. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rufini A, Tucci P, Celardo I and Melino G:
Senescence and aging: The critical roles of p53. Oncogene.
32:5129–5143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McCubrey JA and Demidenko ZN: Recent
discoveries in the cycling, growing and aging of the p53 field.
Aging (Albany NY). 4:887–893. 2012. View Article : Google Scholar
|
|
19
|
Wang J, Deng Y, Tan Y, Li K, Wen B and
Zhao Q: Cannabinoid WIN55, 212-2 inhibits proliferation, invasion
and migration of human SMMC-7721 hepatocellular carcinoma cells. Xi
Bao Yu Fen Zi Mian Yi Xue Za Zhi. 32:619–624. 2016.In Chinese.
PubMed/NCBI
|
|
20
|
Li P, Zhao M, Parris AB, Feng X and Yang
X: p53 is required for metformin-induced growth inhibition,
senescence and apoptosis in breast cancer cells. Biochem Biophys
Res Commun. 464:1267–1274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cerezo M, Tichet M, Abbe P, Ohanna M,
Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P,
Bertolotto C, et al: Metformin blocks melanoma invasion and
metastasis development in AMPK/p53-dependent manner. Mol Cancer
Ther. 12:1605–1615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cai X, Hu X, Tan X, Cheng W, Wang Q, Chen
X, Guan Y, Chen C and Jing X: Metformin induced AMPK activation,
G0/G1 phase cell cycle arrest and the inhibition of growth of
esophageal squamous cell carcinomas in vitro and in vivo. PLoS One.
10:e01333492015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen HP, Shieh JJ, Chang CC, Chen TT, Lin
JT, Wu MS, Lin JH and Wu CY: Metformin decreases hepatocellular
carcinoma risk in a dose-dependent manner: Population-based and in
vitro studies. Gut. 62:606–615. 2013. View Article : Google Scholar
|
|
24
|
Zhuang Y and Miskimins WK: Cell cycle
arrest in metformin treated breast cancer cells involves activation
of AMPK, downregulation of cyclin D1, and requires
p27Kip1 or p21Cip1. J Mol Signal. 3:182008.
View Article : Google Scholar
|
|
25
|
Kim EJ, Park JI and Nelkin BD: IFI16 is an
essential mediator of growth inhibition, but not differentiation,
induced by the leukemia inhibitory factor/JAK/STAT pathway in
medullary thyroid carcinoma cells. J Biol Chem. 280:4913–4920.
2005. View Article : Google Scholar
|
|
26
|
Song LL, Alimirah F, Panchanathan R, Xin H
and Choubey D: Expression of an IFN-inducible cellular senescence
gene, IFI16, is upregulated by p53. Mol Cancer Res. 6:1732–1741.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duan X, Ponomareva L, Veeranki S and
Choubey D: IFI16 induction by glucose restriction in human
fibroblasts contributes to autophagy through activation of the
ATM/AMPK/p53 pathway. PLoS One. 6:e195322011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Vasamsetti SB, Karnewar S, Kanugula AK,
Thatipalli AR, Kumar JM and Kotamraju S: Metformin inhibits
monocyte-to-macrophage differentiation via AMPK-mediated inhibition
of STAT3 activation: Potential role in atherosclerosis. Diabetes.
64:2028–2041. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding WX: Uncoupling AMPK from autophagy: A
foe that hinders the beneficial effects of metformin treatment on
metabolic syndrome-associated atherosclerosis? Focus on 'glucose
and palmitate uncouple AMPK from autophagy in human aortic
endothelial cells'. Am J Physiol Cell Physiol. 308:C246–C248. 2015.
View Article : Google Scholar
|
|
31
|
Rakesh K and Agrawal DK: Cytokines and
growth factors involved in apoptosis and proliferation of vascular
smooth muscle cells. Int Immunopharmacol. 5:1487–1506. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Selzman CH, Miller SA, Zimmerman MA,
Gamboni-Robertson F, Harken AH and Banerjee A: Monocyte chemotactic
protein-1 directly induces human vascular smooth muscle
proliferation. Am J Physiol Heart Circ Physiol. 283:H1455–H1461.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sarfstein R, Friedman Y, Attias-Geva Z,
Fishman A, Bruchim I and Werner H: Metformin downregulates the
insulin/IGF-I signaling pathway and inhibits different uterine
serous carcinoma (USC) cells proliferation and migration in
p53-dependent or -independent manners. PLoS One. 8:e615372013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Clarke CJ, Hii LL, Bolden JE and Johnstone
RW: Inducible activation of IFI 16 results in suppression of
telomerase activity, growth suppression and induction of cellular
senescence. J Cell Biochem. 109:103–112. 2010.
|