Introduction
Dexmedetomidine is a potent, highly selective
α2-adrenoceptor agonist with an increased (8-fold) affinity for the
α2-adrenoceptor, compared with clonidine (1). Dexmedetomidine is noTable for its
ability to provide sedative, analgesic and anxiolytic effects
following intravenous administration to postsurgical patients under
intensive care (1–3) with no clinically apparent
respiratory depression (4,5).
In addition to these effects, dexmedetomidine is important in
various tissues via different mechanisms. For example,
intra-operative dexmedetomidine improves the quality of recovery
and postoperative pulmonary function (6), dexmedetomidine post-conditioning
reduces brain injury following brain hypoxia-ischemia in neonatal
rats (7,8), and it protects from intestinal
ischemia-reperfusion injury (9).
Although the cardiac protective effect of
dexmedetomidine has been reported to occur via various proximate
causes, including decreased heart rate, atrial arrhythmias and
lactate release from cardiomyocytes (10–12), the underlying mechanism remains to
be fully elucidated. Although a previous report demonstrated that
dexmedetomidine attenuates lung injury by inhibiting oxidative
stress, mitochondrial dysfunction and apoptosis in rats (13), whether these mechanisms mediate
cardiac protective effects remains to be fully elucidated. There is
previous evidence of the presence of α2-adrenoceptors in
cardiomyocytes, particularly the α2A/α2C subtype (14,15), which suggests potential
mitochondria-associated effects of dexmedetomidine directly on
cardiomyocytes.
Cardiomyocyte apoptosis has been considered to
contribute to end-stage cardiac remodeling and heart failure
(16,17). It is well known that mitochondrial
dynamics, mitochondrial respiratory complexes and mitochondrial
membrane potential (Δψm) are critical in cardiomyocyte
apoptosis (18,19). Reactive oxygen species (ROS), the
natural byproducts of the normal metabolism of oxygen, are
important in cellular homeostasis and cellular signal transduction.
However, excessive ROS caused by high metabolism has the adverse
effect of oxidizing DNA, proteins and lipids (20). As a consequence, ROS is considered
to be a key inducer of cellular apoptosis in normal and abnormal
cells (21). As ROS are
predominantly generated by mitochondria, which are also a target of
ROS, this indicates crosstalk between mitochondrial ROS and
apoptosis. However, the direct effects of dexmedetomidine on these
effects remain to be fully elucidated.
The present study used dexmedetomidine preincubated
neonatal rat cardiomyocytes, and the rate of apoptosis was detected
by using live cell observation, TUNEL staining and
fluorescence-activated cell sorting (FACS) following hydrogen
peroxide (H2O2) stimulation. To investigate
dexmedetomidine-associated mitochondrial function, western blotting
detected mitochondrial respiratory complexes, tetramethylrhodamine
ethyl ester (TMRE) assay detected Δψm and flux analyzer
detected cellular oxygen consumption rate (OCR). The present study
investigated the protective effect of dexmedetomidine pretreatment
against H2O2-induced cardiomyocyte apoptosis,
with H2O2 being a form of ROS often used as a
ROS simulator (22). The
protective effects occurred through decreased mitochondrial
respiratory complexes and reinforced Δψm, which
facilitated a reduction in the acute response sensitivity of
cardiomyocytes to H2O2 by suppressing the
H2O2-induced increase of ROS and the cellular
OCR. These results indicated a novel mitochondria-associated
protective mechanism against ROS-induced cardiomyocyte
apoptosis.
Materials and methods
Neonatal rat cardiomyocyte culture
Neonatal rat cardiomyocytes were cultured as
reported previously (23). The
whole hearts of 1–3-day-old neonatal Wistar rats were purchased
from Slac Laboratory Animals, Shanghai, China, and cut into ~1–2 mm
sections and digested in a digestion solution (0.025% collagen type
II, 0.06% trypsin, and 20 µg/ml DNase) at 37°C three times,
for 15 min each time. The homogenate was loaded onto a 45.5%
percoll gradient over another 58.5% percoll gradient, followed by
centrifugation at 15°C with 3,800 × g for 30 min. The dissociated
cardiomyocytes were collected and seeded at 1×105 cells
per cm2, followed by culture in Dulbecco's modified
Eagle medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
with 10% fetal bovine serum (Thermo Fisher Scientific, Inc.) at
37°C with 5% CO2 and 95% O2. In all
experiments, different concentrations of dexmedetomidine (10, 100
and 1,000 nM; Sigma; Merck Millipore, Darmstadt, Germany) were used
for cardiomyocyte treatment at 37°C for 24 h. All experiments were
approved by the Animal Care and Use Committee of Shanghai General
Hospital (Shanghai, China) and performed in accordance with the
relevant guidelines and regulations of the Bio-X Institutes of
Shanghai Jiao Tong University (Shanghai, China).
TUNEL assay
Cells were cultured in multi-glass slides. Following
dexmedetomidine treatment, cells were washed in sterilized PBS
three times (5 min each) and fixed in cold acetone for 20 min,
followed by washing with TBS three times. Following blocking with
BSA (Sigma; Merck Millipore) for 1 h at room temperature, the cells
were analyzed using a TdT-FragEL DNA Fragmentation Detection kit
(Sigma; Merck Millipore) to quantify apoptosis. Counterstaining
with fluorescence mounting medium containing DAPI (blue; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) was performed to
visualize normal nuclei. Sections were observed using a
fluorescence microscope (Olympus FluoView™ FV1000; Olympus
Corporation, Tokyo, Japan). Measurements of the apoptotic nuclei
percentage were obtained and analyzed using ImageJ (version 1.47;
National Institutes of Health) from five randomly selected visual
fields, and the average TUNEL-positive percentage (%) was
calculated.
Cell survival assay
Cardiomyocytes were cultured in a multi 6-well dish.
Cells were pretreated with dexmedetomidine at 37°C for 24 h, and
then incubated with H2O2 (200 µM) at
37°C for ~6 h. Cell were washed with PBS three times and then
images were captured using fluorescence microscopy as
aforementioned (Olympus FluoView™ FV1000; Olympus Corporation,
Tokyo, Japan). To determine cell survival, the area of live cells
from five randomly selected visual fields using ImageJ (version
1.47; National Institutes of Health, Bethesda, MD, USA) was
analyzed. The percentage of live cells area within a visual field
area was defined as the cell survival (% field).
FACS analysis
FACS analysis was performed to detect the apoptosis
of cardiomyocytes induced by H2O2, as
reported previously (24).
Briefly, the neonatal rat cardiomyocytes were cultured in 6-well
dishes. For cellular apoptosis, H2O2 (200
µM) was added to cells and incubated at 37°C for 4 h,
following exposure of the cells to dexmedetomidine pretreatment for
24 h. The cardiomyocytes (10×105 cells/500 µl)
were labeled fluorescently for the detection of apoptotic and
necrotic cells by adding 50 µl binding buffer and 5
µl Annexin V-FITC (BD Pharmingen, San Diego, CA, USA) and 2
µl of propidium iodide (Cedarlane Laboratories, Hornby, ON,
Canada). The samples were incubated at room temperature for 15 min
following gentle mixing. A minimum of 20,000 cells within a gated
region were analyzed using FACS (Coulter Epics Altra flow
cytometer; Beckman Coulter, Fullerton, CA, USA).
MitoSOX and TMRE assays
The primary neonatal rat cardiomyocytes were
cultured in collagen-coated 96-well dishes. MitoSOX (cat. no.
M36008; Invitrogen; Thermo Fisher Scientific, Inc.) or TMRE (cat.
no. ab113852; Abcam; Cambridge, UK) were used following
dexmedetomidine stimulation for 24 h. For the MitoSOX assay, on the
day of measurement, the cells were washed twice in sterilized PBS
followed by stimulation with H2O2 at 37°C for
15 min. The cells were then incubated with 5 µM
MitoSOX-containing HBSS medium at 37°C for 10 min and were measured
at Ex/Em: 510/580 nm. On the day of TMRE measurement, the cells
were incubated with 1 µM TMRE at 37°C for 15 min, and then
washed with PBS with 0.2% BSA three times to remove excess TMRE,
followed by measurement at Ex/Em: 510/580 nm. For the
carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP) inhibition
assay, prior to TMRE incubation, the cells were stimulated by 0.5
or 1 µM FCCP (Agilent Technologies, Inc., Santa Clara, CA,
USA) at 37°C for 30 min.
Cellular flux analyzer
Neonatal rat cardiomyocyte OCR was measured using a
Seahorse XF 24 extracellular analyzer (Agilent Technologies, Inc.).
The cells were seeded at a density of 0.5×105
cells/well. Cellular mitochondrial respiratory complexes were
inhibited by injecting 1 µm oligomycin (inhibitor of complex
V), 0.5 µm FCCP, and a combination of 0.5 µm rotenone
and 0.5 µm antimycin A (R/A; inhibitors of complex I and
complex III). Basic parameters were calculated as follows: Cellular
respiration, basic OCR prior to oligomycin injection; mitochondria
respiration, final rate measurement prior to oligomycin
injection-minimum rate measurement following R/A injection; ATP
production, final rate measurement prior to oligomycin
injection-minimum rate measurement following oligomycin injection;
maximal respiration, maximum rate measurement following FCCP
injection-minimum rate measurement following R/A injection; proton
leak, minimum rate measurement following oligomycin
injection-minimum rate measurement following R/A injection;
coupling efficiency, ATP production rate/cellular respiration rate.
For measurement of the cellular response to ROS,
H2O2 was injected three times following basic
measurements.
Western blot analysis
The cultured cardiomyocytes were scraped and
collected, and the centrifuged (1,500 × g at 4°C for 5 min)
cardiomyocytes were dissolved in lysis buffer as reported
previously (23). Protein
concentration was quantified using protein assay buffer (cat. no.
500–0006; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Briefly,
original protein solution and standard curve protein was diluted
(500-fold) with protein assay buffer in a 96 microplate and
analyzed using an iMark microplate reader (absorbance 595 nm;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). Total protein (10
µg) was loaded and separated using SDS-PAGE (8–20% gel) for
90 min, and then transferred onto PVDF membranes for another 90
min. Following blocking in 3% skim milk for 1 h, the membranes were
incubated in primary antibodies at 4°C overnight. The unbound
antibodies were then washed off in TBST 3–5 times the subsequent
day, followed by incubations with horseradish peroxidase-conjugated
sheep anti-rabbit immunoglobulin G (1:2,000; cat. no. HAF016;
Bio-Techne Ltd., Oxford, UK) at room temperature for 1 h. A
FluorChem E (Cell Biosciences, Inc., Shanghai, China) imaging
system was used to visualize the signals. The primary antibodies
used were as follows: α2a, α2b and α2c adrenergic receptor
(1:1,000; cat. nos. ab85570, ab151727 and ab151618, respectively;
Abcam, Cambridge, UK), B-cell lymphoma 2 (1:1,000; BCL2; cat. no.
ab59348; Abcam), Bcl-2-associated X protein (1:2,000; BAX; cat. no.
2772; Cell Signaling Technology, Inc., Danvers, MA, USA), total
OXPHOS rodent WB antibody cocktail (1:10,000; cat. no. ab110413;
Abcam) and GAPDH (1:2,000; cat. no. 2118; Cell Signaling
Technology, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from cultured cardiomyocytes
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.), and cDNA
(50 ng/µl) was synthesized using oligo (dT) primers with the
Transcriptor First Strand cDNA Synthesis kit (cat. no. 04896866001;
Roche Diagnostics, Shanghai, China). Total DNA was extracted from
the cultured cardiomyocytes using a QIAamp DNA Mini kit (Qiagen,
Inc., Shanghai, China). Following analysis using NanoDrop™
2000/2000c (Thermo Fisher Scientific, Inc.), DNA was diluted with
RNase-free distilled H2O (Takara Biotechnology Co.,
Ltd., Dalian, China) to a total volume of 20 ng/µl. The
mitochondrial DNA was then measured by detecting the cytochrome B
(CYTB) gene. The real-time PCR amplifications were quantified using
SYBR-Green (cat. no. 04887352001; Roche Diagnostics) and the
reaction system was composed of 10 µl SYBR premix Ex Taq II,
0.2 µl primers, 8.8 µl H2O, 1 µl
DNA. There thermocycling conditions were as follows: 5 sec at 95°C
followed by 30 sec at 60°C for 42 cycles using a thermal cycler
dice real-time system (Takara Biotechnology Co., Ltd.) (25). The results were normalized by the
gene expression of 18s rRNA. The primers used in the present study
are presented in Table I.
 | Table IRat primers used in reverse
transcription-polymerase chain reaction analysis. |
Table I
Rat primers used in reverse
transcription-polymerase chain reaction analysis.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| CYTB |
AACCACTCCTTTATCGACCTC |
CCTCATGGGAGTACATAGCCCAT |
| PGC1α |
ACCCACAGGATCAGAACAAACC |
GACAAATGCTGTTTGCTTTATTGC |
| PPARα |
TGGTGGACCTCCCCCA |
TCTTCTTGATGACCTGCACGA |
| NRF1 CC |
ACATTACAGGGCGGTGAA |
AGTGGCTCCCTGTTGCATCT |
| ERRα |
GTGGCCGACAGAAGTACAAG |
GGTTCAACCACCAGCAGATG |
Statistical analysis
All experiments were repeated two or three times.
All results are reported as the mean ± standard error of the mean.
The normality of distribution was analyzed using the
D'Agostino-Pearson omnibus normality test using GraphPad Prism
software (version 6.0; GraphPad Software, Inc., La Jolla, CA, USA).
Comparisons between two groups were analyzed using Student's
t-test. Multiple comparisons between groups were performed using
one-way analysis of variance with Tukey's multiple comparisons test
(GraphPad Prism version 6.0). P<0.05 was considered to indicate
a statistically significant difference.
Results
Dexmedetomidine prevents ROS-induced
cardiomyocyte apoptosis
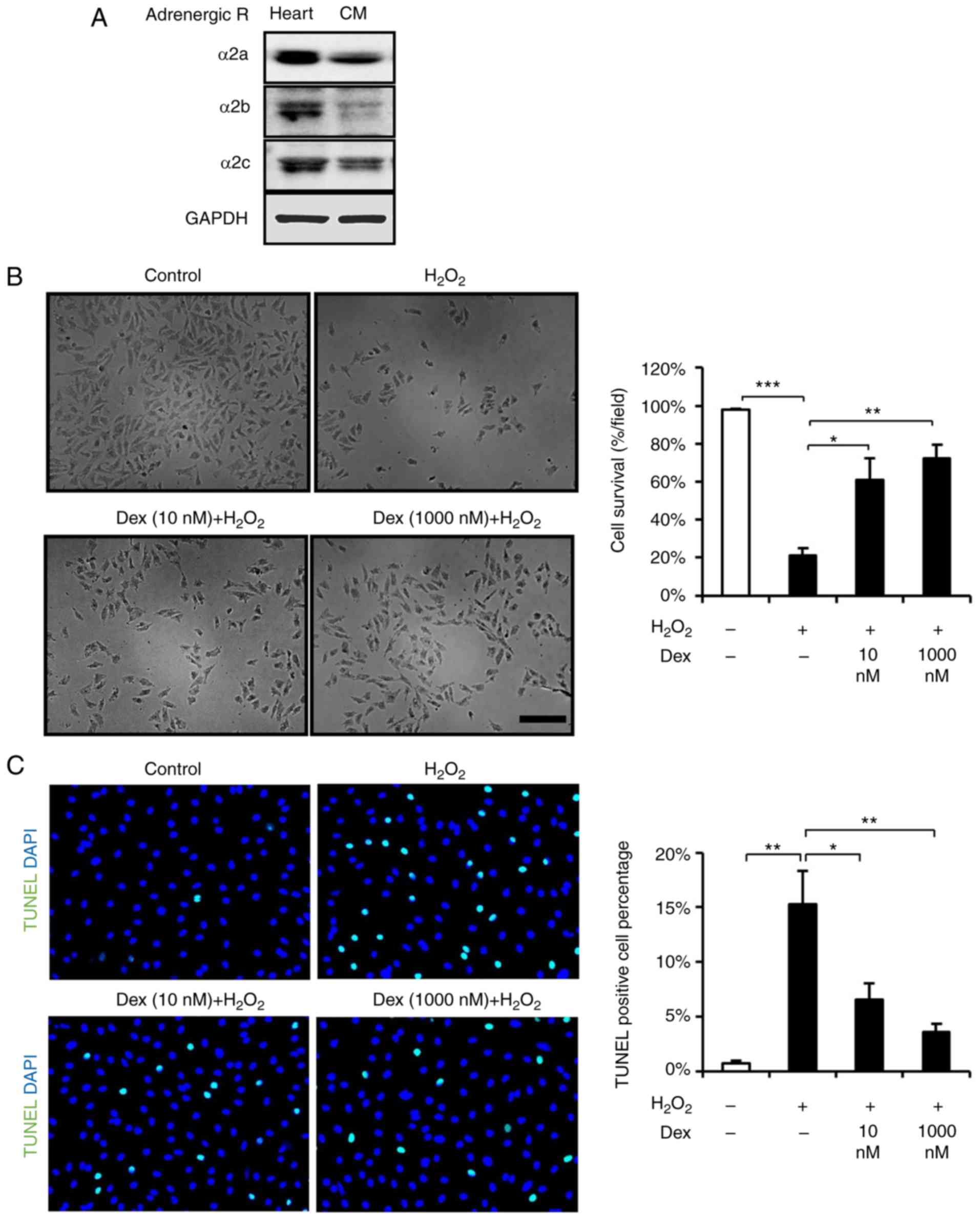
Prior to experiments, it was detected that α2
adrenergic receptors were expressed in cardiomyocytes using western
blot analysis (Fig. 1A), which is
consistent with a previous report (14). To determine whether
dexmedetomidine treatment is beneficial to cardiomyocytes, the
present study investigated the survival and apoptosis of
dexmedetomidine-pretreated cardiomyocytes following
H2O2 stimulation. First, the cardiomyocytes
were exposed to various doses of dexmedetomidine (26) and it was demonstrated that
dexmedetomidine pretreatment attenuated the loss of cardiomyocytes
induced by H2O2 (Fig. 1B). As the beneficial effect of
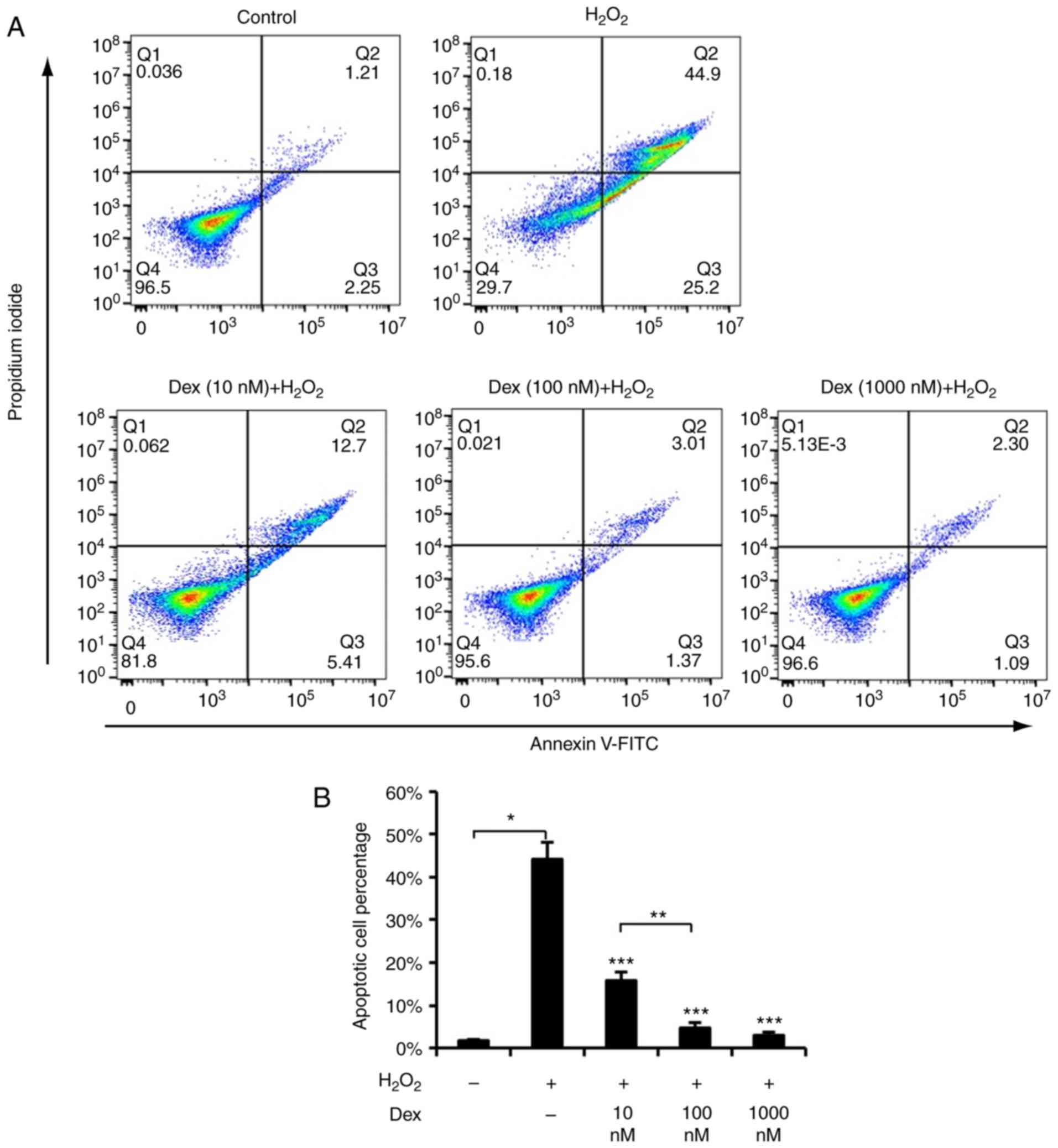
dexmedetomidine against ROS may suppressed apoptosis, apoptosis was
detected using TUNEL staining and FACS, and it was identified that
dexmedetomidine pretreatment suppressed
H2O2-induced cardiomyocyte apoptosis, as
presented in Figs. 1C, 2A and B. These results suggested that
dexmedetomidine pretreatment created protective cellular
conditions, which resisted ROS-induced apoptosis and increased
cellular survival.
Dexmedetomidine suppresses mitochondria
respiratory complexes
To verify the protective role of dexmedetomidine
pretreatment, the present study investigated several
mitochondria-associated parameters, as mitochondria are the primary
site for ROS generation and the main target of ROS (27,28). As mitochondrial DNA and biogenesis
are crucial in the maintenance of cellular and mitochondrial
function under oxidative stress (29), the results demonstrated that
dexmedetomidine did not affect mitochondrial DNA synthesis
(Fig. 3A), but significantly
decreased (P<0.05) the expression of genes involved in
mitochondrial biogenesis (Fig.
3B), in addition to respiratory complex I, II and IV-related
proteins, in a dose-dependent manner (Fig. 3C). However, marginal changes were
observed in integrated mitochondrial respiratory complexes
(Fig. 3D). These results led to
the investigation of whether suppressed mitochondria were
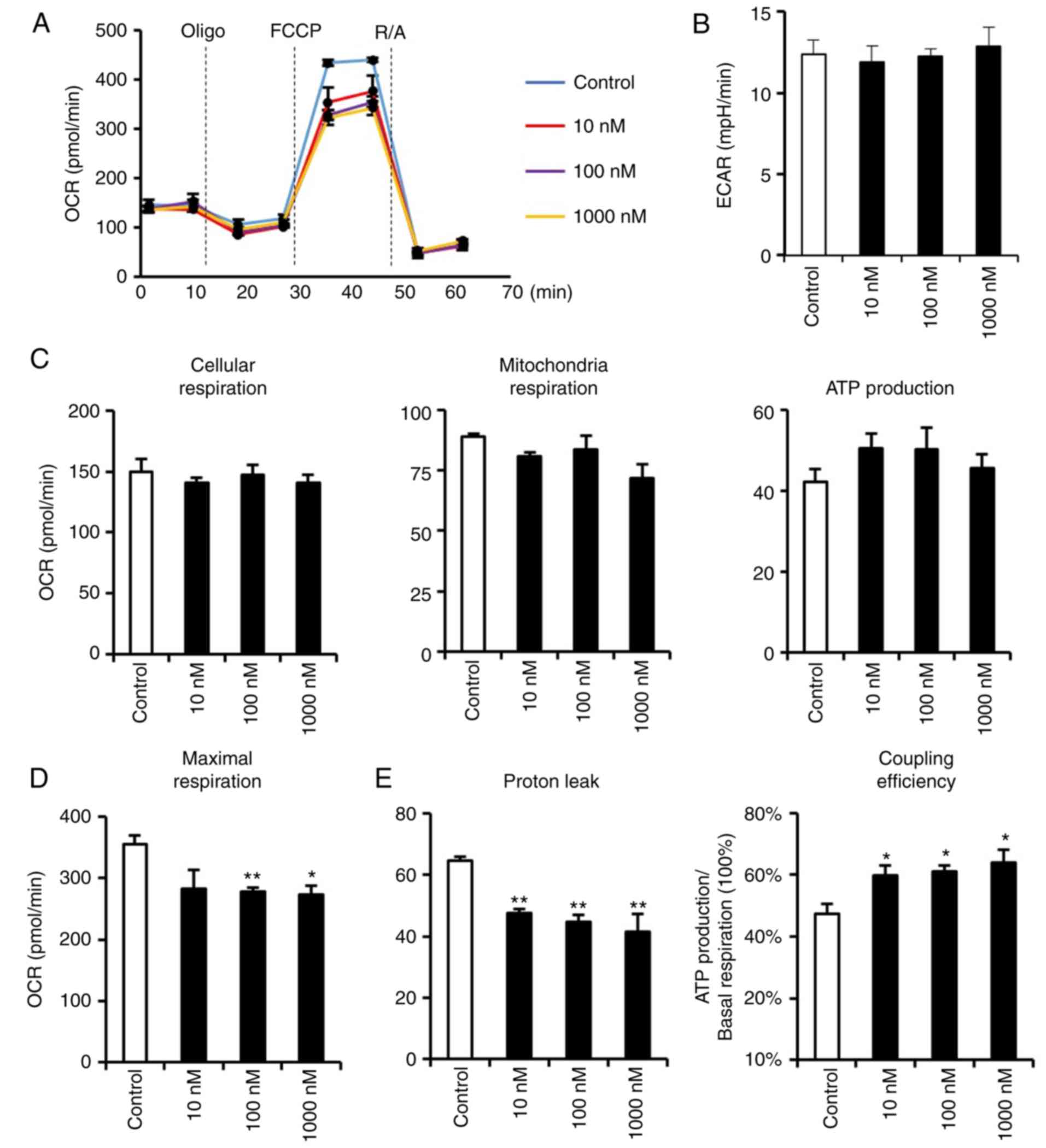
accompanied by any adverse functional phenotypes. Therefore, an
extracellular flux analyzer was used to detect the respiratory
functions of dexmedetomidine-treated cardiomyocytes (Fig. 4). Dexmedetomidine pretreatment did
not affect extracellular acidification rate, nor cellular,
mitochondria or ATP-linked respiration (Fig. 4B and C), indicating that the
decreased mitochondria biogenesis and respiratory complexes induced
by dexmedetomidine were not accompanied by any pathological
alterations. However, dexmedetomidine decreased FCCP-induced
cellular maximal respiration (Fig.
4D), indicating a potential role for dexmedetomidine in
resistance to FCCP. It was hypothesized that mitochondria
respiratory complexes declined but with no respiratory impairment
due to elevated respiratory efficiency. As expected,
dexmedetomidine decreased proton leakage, which reduced the
uncoupling proton influx and increased coupling efficiency
(Fig. 4E).
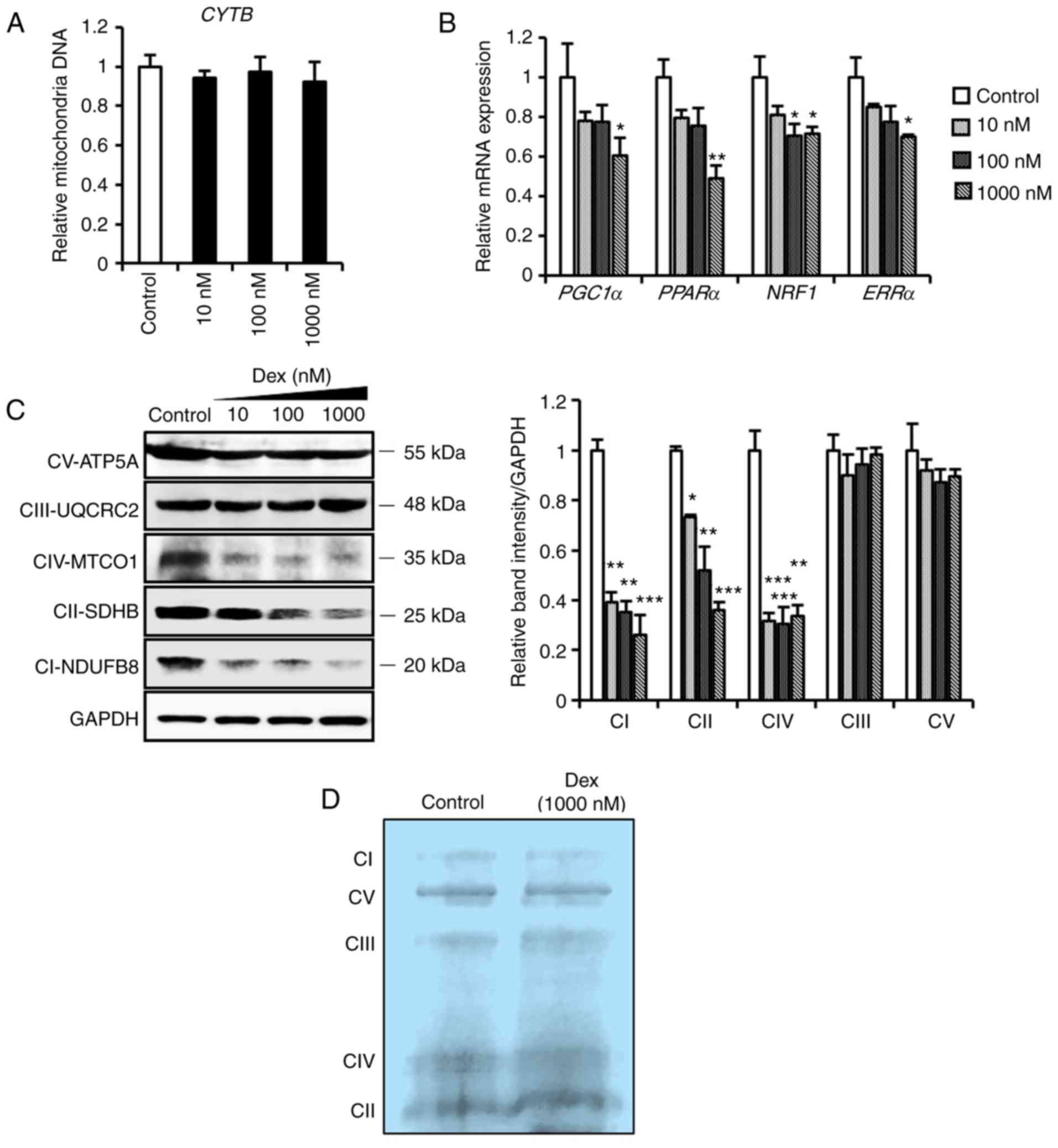 | Figure 3Mitochondrial profiles of
cardiomyocytes pretreated with dexmedetomidine. (A) Relative
mitochondrial DNA expression of CYTB in cardiomyocytes following
dexmedetomidine treatment (n=5 per group). (B) Relative mRNA
expression of mitochondrial biogenesis genes of cardiomyocytes
following dexmedetomidine treatment (n=5 per group). (C)
Representative mitochondrial complex protein expression and
quantification of band intensity following dexmedetomidine
treatment (n=4 per group). (D) Representative blue negative page of
mitochondria respiratory complexes following dexmedetomidine
treatment. Statistical significance was determined using one-way
analysis of variance (*P<0.05;
**P<0.01; ***P<0.001, each group vs.
control). Dex, dexmedetomidine; CYTB, cytochrome B; PPARα,
peroxisome proliferator-activated receptor α; PGC1α, PPARγ
coactivator 1α; NRF1, nuclear respiratory factor 1; ERR,
estrogen-related receptor α; ATP5A, ATP synthase subunit alpha;
UQCRC2, ubiquinol-cytochrome c reductase core protein I;
MTCO1, mitochondrially encoded cytochrome c oxidase I; SDHB,
succinate dehydrogenase complex iron sulfur subunit B; NDUFB8, NADH
dehydrogenase [ubiquinone] 1 β subcomplex subunit 8. |
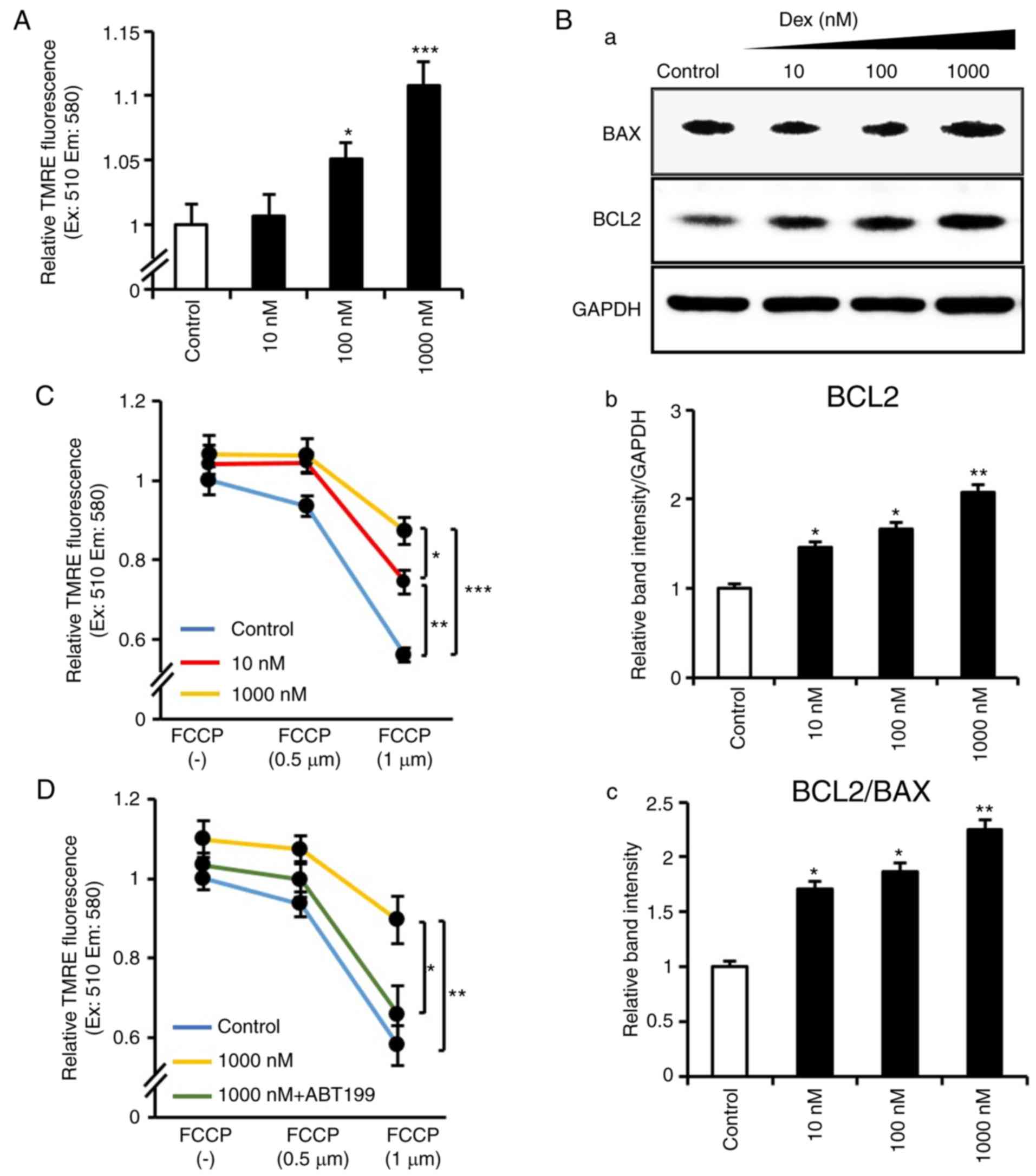
Dexmedetomidine attenuates Δψm
loss via the activation of Bcl2
The Δψm protects cells against ROS and
apoptosis (30), therefore, the
present study examined whether Δψm was affected by
dexmedetomidine. It was identified that dexmedetomidine upregulated
Δψm in a dose-dependent manner (Fig. 5A). As evidence indicates that BCL2
family members protect against Δψm depolarization and
apoptosis (31–33), the present study examined BCL2
family proteins and demonstrated that dexmedetomidine treatment
significantly induced the protein expression of BCL2 without any
change in BAX expression (Fig.
5B). Subsequently, whether the improvement in Δψm by
dexmedetomidine contributed to its anti-apoptotic role was
investigated. Cardiomyocytes were exposed to 0.5 and 1 µM
FCCP, a mitochondrial uncoupler reported to impair Δψm
(34). The results confirmed that
FCCP induced Δψm loss; however, dexmedetomidine
pretreatment inhibited FCCP-induced mitochondria Δψm
loss (Fig. 5C). These results
indicated that dexmedetomidine reinforced Δψm, which
suppressed its collapse induced by FCCP. To account for the crucial
role of BCL2 in regulating Δψm, the present study
examined tested the upregulated BCL2 induced by dexmedetomidine is
involved in resisting apoptosis. Cardiomyocytes were exposed to a
BCL2 inhibitor (ABT-199), and it was demonstrated that
dexmedeto-midine did not suppress FCCP-induced loss of
Δψm following BCL2 inhibition (Fig. 5D), indicating that BCL2 was
involved in the protective effect of dexmedetomidine against
Δψm loss.
Dexmedetomidine attenuates mitochondrial
response sensitivity to H2O2
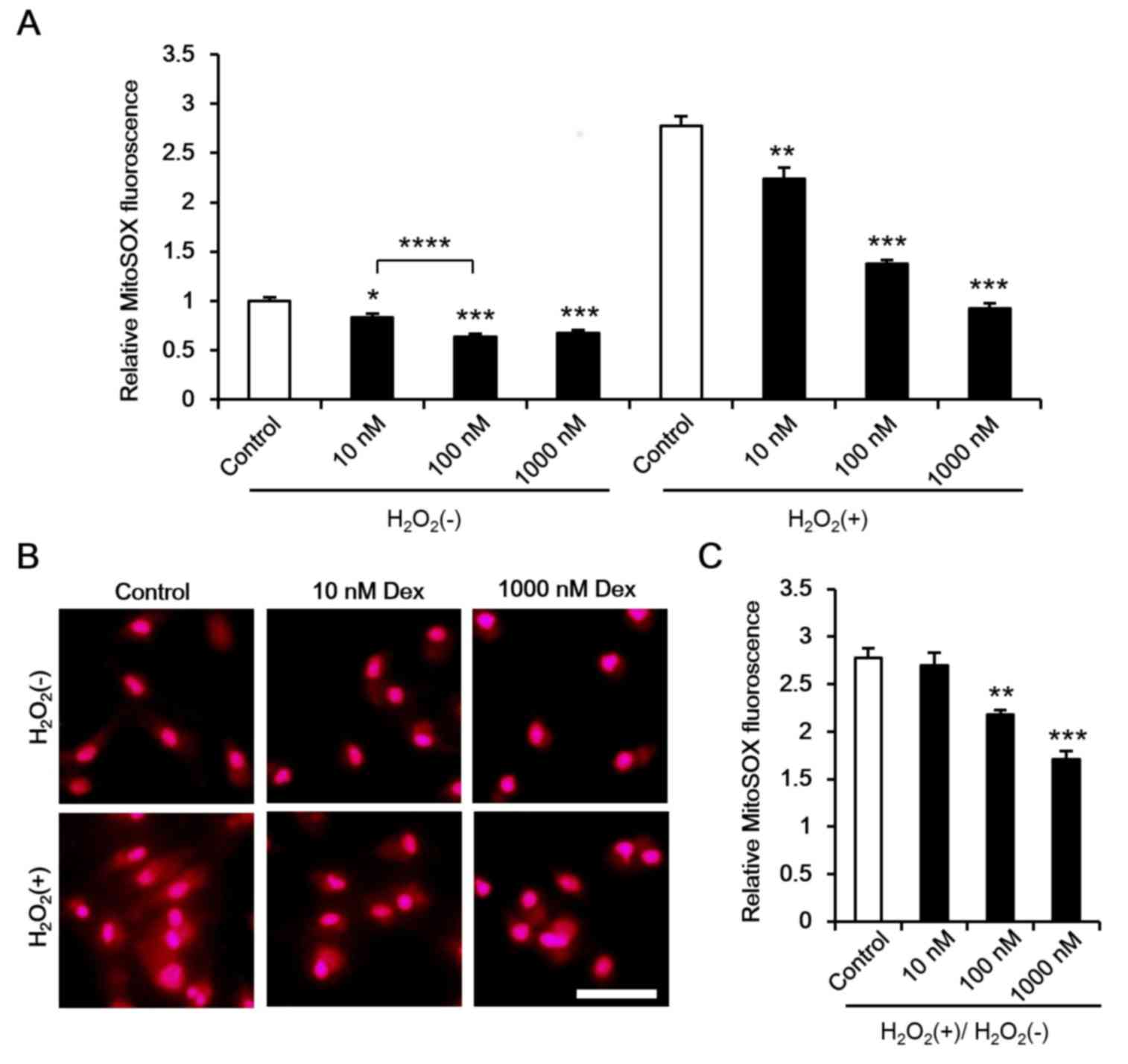
As mitochondria are a main target for ROS damage, it
was hypothesized that dexmedetomidine attenuates the mitochondrial
response to ROS, thus reducing ROS-induced damage. Additional ROS
has been reported to induce ROS release, which promotes
mitochondrial and cell damage (35). In the present study, ROS levels
were measured under normal conditions and following
H2O2 incubation. It was demonstrated that
dexmedetomidine marginally reduced cardiomyocyte ROS levels under
normal conditions and significantly reduced ROS levels following
H2O2 incubation (Fig. 6A and B). As different ROS levels
were inhibited by dexmedetomidine [H2O2(−) in
Fig. 6A] the ROS response rate to
ROS stimulation was calculated
[H2O2(+)/H2O2(−)], and
a reduced ROS response towards to H2O2 was
observed (Fig. 6B and C).
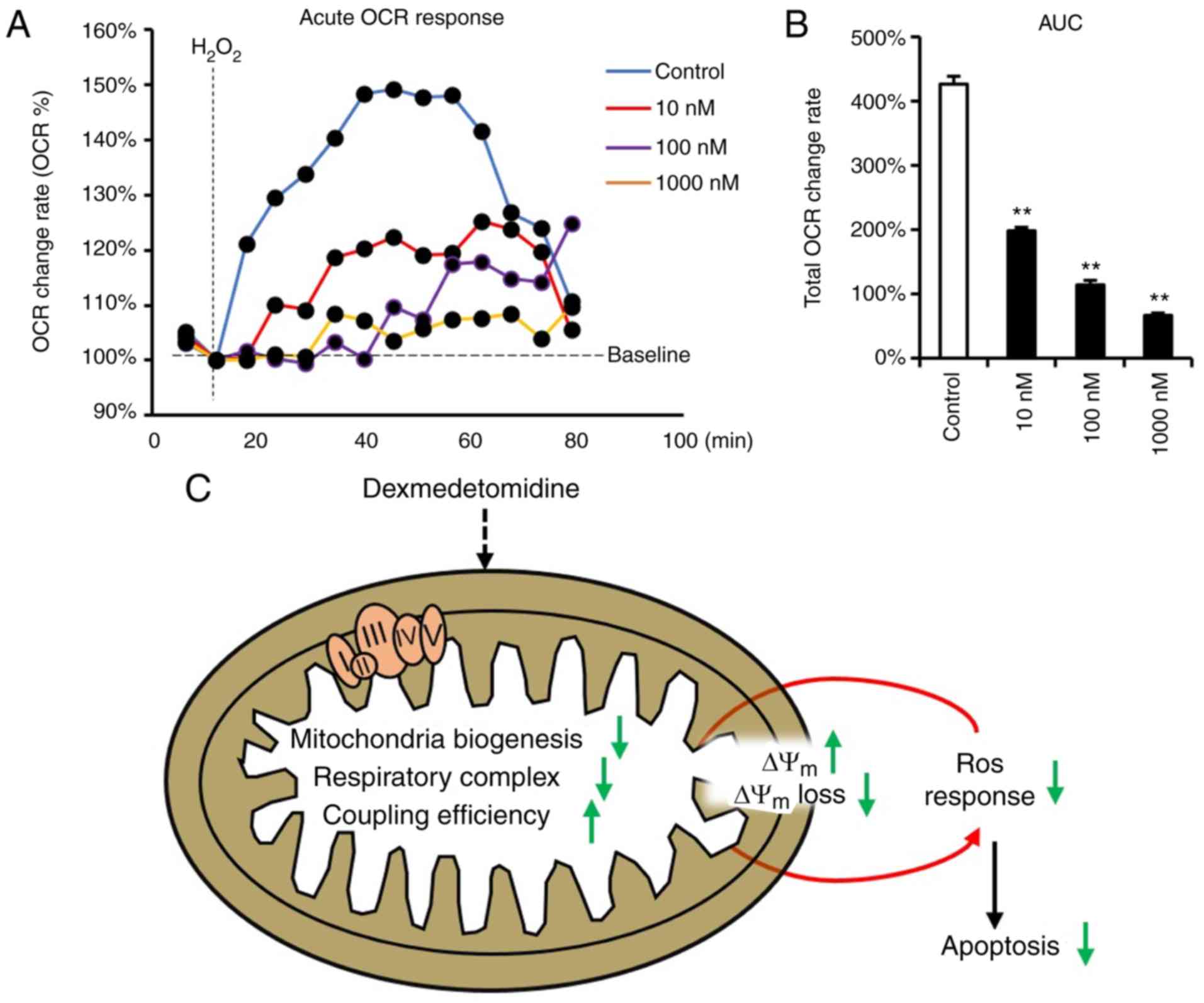
Mitochondrial ROS generation is accompanied by the improved
function of respiratory complexes (27,28), which leads to high cellular OCR,
and ROS has been reported to induce cellular OCR (36). Therefore, the present study
subsequently measured the OCR response toward
H2O2 in cardiomyocytes incubated with
dexmedetomidine. The dexmedetomidine-treated cardiomyocytes
significantly inhibited the cellular increase of OCR following
H2O2 incubation (Fig. 7A and B).
In summary, all results indicate that
dexmedetomidine preconditioning protects against ROS-induced
apoptosis of cardiomyocytes by reducing sensitivity of the
mitochondrial response towards ROS through decreasing mitochondrial
respiratory complexes, reinforcing Δψm, causing
resistance to Δψm loss (Fig. 7C).
Discussion
The present study is the first, to the best of our
knowledge, to demonstrate that dexmedetomidine preconditioning of
cardiomyocytes decreases mitochondrial respiratory complexes with
high coupling efficiency, and reinforces Δψm, causing
resistance to Δψm loss. These effects are beneficial and
may contribute to the reduced sensitivity of the mitochondria
response towards to ROS, thus protecting against ROS-induced
apoptosis (Fig. 7C).
Previously, it was reported that dexmedetomidine
preconditioning or post conditioning increases cardiomyocytes
viability and activity under hypoxia/reoxygenation conditions
(37), possibly by increasing the
levels of phosphorylated extracellular signal-regulated kinase
(Erk)1/2 and Akt, which are well known survival proteins that
inhibit cell death and promote survival (38), which significantly reduces
myocardial infarction size and improves functional recovery
(14). It has also been
previously reported that dexmedetomidine offers cardioprotection
against myocardial apoptotic injury via a decrease of caspase-12,
glucose-regulated protein 78 and C/EBP homologous protein (39). These reports indicate that
dexmedetomidine has a cardioprotective effect by inducing the
expression of survival genes, but also by reducing
apoptosis-associated genes. In the present study, it was also
demonstrated that dexmedetomidine promoted the phosphorylation of
Erk1/2 and Akt (data not shown). The Akt-induced protective effect
against cell death can be explained by suppression of the
mitochondrial translocation of BAX and release of cytochrome
c from mitochondria (40,41). In the present study, no change in
the expression of BAX was detected; however, a significant increase
in the expression of BCL2 was observed following dexmedetomidine
pretreatment. Upregulated BCL2 can inhibit the release of
cytochrome c from mitochondria and inhibit FCCP-induced
apoptosis (31,42). Existing evidence suggests there is
crosstalk between Akt-BCL2-mitochondria and dexmedetomidine.
Previously, it was reported that the acquisition of
chemoresistance is associated with increased mitochondrial coupling
and decreased ROS production (43), indicating a potential association
between tighter mitochondria coupling and reduced ROS production.
In the present study, it was identified that dexmedetomidine
decreased respiratory complexes without any mitochondrial or
cellular respiration impairment. Although dexmedetomidine did not
promote any mitochondria respiratory function, it decreased
mitochondria respiratory complexes while maintaining normal
cellular respiratory function, indicating that the respiratory
complexes had increased coupling efficiency, which may be
associated with lower mitochondria ROS generation. Of note,
mitochondrial coupling and decreased ROS production have also been
previously associated with lower lactate production (43), which is another reported mechanism
underlying dexmedetomidine-induced cardiac protection (12) and indicates that
dexmedetomidine-induced mitochondrial respiratory complex tight
coupling is an additional mechanism underlying reduced lactate
release.
The new concept of ROS-induced ROS-release (RIRR)
suggests that exposure to ROS results in an increase in ROS
reaching a threshold level, leading to the simultaneous breakdown
of Δψm and then to increased ROS generation from
respiratory complexes. Mitochondria release ROS to induce RIRR,
which impairs other mitochondria. This forms a positive feedback
resulting in progressive mitochondria damage and cellular apoptosis
(35). In the process of RIRR,
Δψm and respiratory complexes are involved; therefore, a
high sensitivity of the mitochondria response towards ROS is
necessary to further the RIRR cycle. In the present study, it was
identified that dexmedetomidine-pretreated cardiomyocytes had
attenuated ROS levels following H2O2
incubation, indicating decreased ROS response sensitivity. Although
it is possible that upregulated antioxidant enzymes contributed to
the decreased ROS, the majority, including superoxide dismutase 2,
glutathione peroxidase 4, glutaredoxin 1, were marginally decreased
(data not shown), suggesting that the decreased ROS in the
dexmedetomidine-pretreated cardiomyocytes was independent of
antioxidant enzymes. The results also demonstrated that
dexmedetomidine induced Δψm and inhibited FCCP-induced
Δψm loss. Considering the RIRR concept, it was
hypothesized that dexmedetomidine protected against ROS-induced
Δψm collapse and withstood the ROS leakage from
respiratory complexes of damaged mitochondria.
It has also been reported that cardiomyocytes
preconditioned with phenylephrine, an α1-adrenoceptor agonist, have
enhanced cellular OCR but with low OCR response sensitivity towards
to 4-HNE, an ROS associated product, and are protected against
4-HNE-induced cellular apoptosis (36). However, although different from
α1-adrenoceptor agonists, the present study identified that
dexmedetomidine preconditioning reduced the sensitivity of the
response of OCR towards H2O2. The similarity
in the results of the present study with those of a previous study
(36) suggest that the low
sensitivity of the cellular OCR response may be a common mechanism
in α1 and α2-adrenoceptor activation.
However, there were limitations in the present
study. Although it was demonstrated that the adrenergic receptors
for dexmedetomidine exist in cardiomyocytes, it is not possible to
exclude the possibility that dexmedetomidine affects mitochondria
directly. Additionally, animal experiments are required to discuss
the protective role of dexmedetomidine in vivo. These
limitations are to be addressed in future investigations.
In conclusion, the present demonstrated that
dexmedetomidine pretreatment suppressed cardiomyocyte apoptosis by
inhibiting mitochondrial respiratory complexes and elevating
Δψm, which attenuated the sensitivity of the
mitochondrial response towards ROS stimulation. This suggests a
novel mitochondria-associated mechanism for
dexmedetomidine-inhibited apoptosis.
Acknowledgments
This study was supported by a grant from the
National Natural Science Foundation of China through the project
'Why muscle relaxant promotes occurrence of critical myopathy and
prevention' (grant no. 81171845).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Bhana N, Goa KL and McClellan KJ:
Dexmedetomidine. Drugs. 59:263–270. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martin E, Ramsay G, Mantz J and Sum-Ping
ST: The role of the alpha2-adrenoceptor agonist dexmedetomidine in
postsurgical sedation in the intensive care unit. J Intensive Care
Med. 18:29–41. 2003. View Article : Google Scholar
|
|
3
|
Szumita PM, Baroletti SA, Anger KE and
Wechsler ME: Sedation and analgesia in the intensive care unit:
Evaluating the role of dexmedetomidine. Am J Health Syst Pharm.
64:37–44. 2007. View Article : Google Scholar
|
|
4
|
Venn RM, Hell J and Grounds RM:
Respiratory effects of dexmedetomidine in the surgical patient
requiring intensive care. Crit Care. 4:302–308. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hsu YW, Cortinez LI, Robertson KM, Keifer
JC, Sum-Ping ST, Moretti EW, Young CC, Wright DR, Macleod DB and
Somma J: Dexmedetomidine pharmacodynamics: Part I: Crossover
comparison of the respiratory effects of dexmedetomidine and
remifentanil in healthy volunteers. Anesthesiology. 101:1066–1076.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee SH, Lee CY, Lee JG, Kim N, Lee HM and
Oh YJ: Intraoperative dexmedetomidine improves the quality of
recovery and postoperative pulmonary function in patients
undergoing video-assisted thoracoscopic surgery: A
CONSORT-prospective, randomized, controlled trial. Medicine.
95:e28542016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ren X, Ma H and Zuo Z: Dexmedetomidine
postconditioning reduces brain injury after brain hypoxia-ischemia
in neonatal rats. J Neuroimmune Pharmacol. 11:238–247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sifringer M, von Haefen C, Krain M,
Paeschke N, Bendix I, Bührer C, Spies CD and Endesfelder S:
Neuroprotective effect of dexmedetomidine on hyperoxia-induced
toxicity in the neonatal rat brain. Oxid Med Cell Longev.
2015:5303712015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun Y, Gao Q, Wu N, Li SD, Yao JX and Fan
WJ: Protective effects of dexmedetomidine on intestinal
ischemia-reperfusion injury. Exp Ther Med. 10:647–652. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gu H, Liu J and Wu C: Impact of
dexmedetomidine versus propofol on cardiac function of children
undergoing laparoscopic surgery. Int J Clin Exp Med. 7:5882–5885.
2014.
|
|
11
|
Turan A, Bashour CA, You J, Kirkova Y,
Kurz A, Sessler DI and Saager L: Dexmedetomidine sedation after
cardiac surgery decreases atrial arrhythmias. J Clin Anesth.
26:634–642. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Willigers HM, Prinzen FW, Roekaerts PM, de
Lange S and Durieux ME: Dexmedetomidine decreases perioperative
myocardial lactate release in dogs. Anesth Analg. 96:657–664, Table
of contents. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fu C, Dai X, Yang Y, Lin M, Cai Y and Cai
S: Dexmedetomidine attenuates lipopolysaccharide-induced acute lung
injury by inhibiting oxidative stress, mitochondrial dysfunction
and apoptosis in rats. Mol Med Rep. 15:131–138. 2017. View Article : Google Scholar :
|
|
14
|
Ibacache M, Sanchez G, Pedrozo Z, Galvez
F, Humeres C, Echevarria G, Duaso J, Hassi M, Garcia L, Díaz-Araya
G and Lavandero S: Dexmedetomidine preconditioning activates
pro-survival kinases and attenuates regional ischemia/reperfusion
injury in rat heart. Biochim Biophys Acta. 1822:537–545. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maltsev AV, Kokoz YM, Evdokimovskii EV,
Pimenov OY, Reyes S and Alekseev AE: Alpha-2 adrenoceptors and
imidazoline receptors in cardiomyocytes mediate counterbalancing
effect of agmatine on NO synthesis and intracellular calcium
handling. J Mol Cell Cardiol. 68:66–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kang PM and Izumo S: Apoptosis and heart
failure: A critical review of the literature. Circ Res.
86:1107–1113. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Narula J, Haider N, Virmani R, DiSalvo TG,
Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW and Khaw BA:
Apoptosis in myocytes in end-stage heart failure. N Engl J Med.
335:1182–1189. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Parra V, Eisner V, Chiong M, Criollo A,
Moraga F, Garcia A, Härtel S, Jaimovich E, Zorzano A, Hidalgo C and
Lavandero S: Changes in mitochondrial dynamics during
ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res.
77:387–397. 2008. View Article : Google Scholar
|
|
19
|
Molkentin JD: Calcineurin, mitochondrial
membrane potential, and cardiomyocyte apoptosis. Circ Res.
88:1220–1222. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lyras L, Cairns NJ, Jenner A, Jenner P and
Halliwell B: An assessment of oxidative damage to proteins, lipids,
and DNA in brain from patients with Alzheimer's disease. J
Neurochem. 68:2061–2069. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar
|
|
22
|
Finkel T: Signal transduction by reactive
oxygen species in non-phagocytic cells. J Leukoc Biol. 65:337–340.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tian Z, Miyata K, Kadomatsu T, Horiguchi
H, Fukushima H, Tohyama S, Ujihara Y, Okumura T, Yamaguchi S, Zhao
J, et al: ANGPTL2 activity in cardiac pathologies accelerates heart
failure by perturbing cardiac function and energy metabolism. Nat
Commun. 7:130162016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eguchi M, Liu Y, Shin EJ and Sweeney G:
Leptin protects H9c2 rat cardiomyocytes from
H2O2-induced apoptosis. FEBS J.
275:3136–3144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tian Z, Miyata K, Tazume H, Sakaguchi H,
Kadomatsu T, Horio E, Takahashi O, Komohara Y, Araki K, Hirata Y,
et al: Perivascular adipose tissue-secreted angiopoietin-like
protein 2 (Angptl2) accelerates neointimal hyperplasia after
endovascular injury. J Mol Cell Cardiol. 57:1–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Housmans PR: Effects of dexmedetomidine on
contractility, relaxation, and intracellular calcium transients of
isolated ventricular myocardium. Anesthesiology. 73:919–922. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cadenas E, Boveris A, Ragan CI and
Stoppani AO: Production of superoxide radicals and hydrogen
peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c
reductase from beef-heart mitochondria. Arch Biochem Biophys.
180:248–257. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hinkle PC, Butow RA, Racker E and Chance
B: Partial resolution of the enzymes catalyzing oxidative
phosphorylation. XV Reverse electron transfer in the
flavin-cytochrome Beta region of the respiratory chain of beef
heart submitochondrial particles. J Biol Chem. 242:5169–5173.
1967.PubMed/NCBI
|
|
29
|
Lee HC and Wei YH: Mitochondrial
biogenesis and mitochondrial DNA maintenance of mammalian cells
under oxidative stress. Int J Biochem Cell Biol. 37:822–834. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ly JD, Grubb DR and Lawen A: The
mitochondrial membrane potential (deltapsi(m)) in apoptosis; an
update. Apoptosis. 8:115–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dispersyn G, Nuydens R, Connors R, Borgers
M and Geerts H: Bcl-2 protects against FCCP-induced apoptosis and
mitochondrial membrane potential depolarization in PC12 cells.
Biochim Biophys Acta. 1428:357–371. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng X, Gao F and May WS Jr: Bcl2 retards
G1/S cell cycle transition by regulating intracellular ROS. Blood.
102:3179–3185. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang XQ, Feng JQ, Chen J, Chen PX, Zhi JL,
Cui Y, Guo RX and Yu HM: Protection of oxidative preconditioning
against apoptosis induced by H2O2 in PC12
cells: Mechanisms via MMP, ROS, and Bcl-2. Brain Res. 1057:57–64.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Perry SW, Norman JP, Barbieri J, Brown EB
and Gelbard HA: Mitochondrial membrane potential probes and the
proton gradient: A practical usage guide. Biotechniques. 50:98–115.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sansbury BE, Riggs DW, Brainard RE,
Salabei JK, Jones SP and Hill BG: Responses of hypertrophied
myocytes to reactive species: Implications for glycolysis and
electrophile metabolism. Biochem J. 435:519–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Peng K, Qiu Y, Li J, Zhang ZC and Ji FH:
Dexmedetomidine attenuates hypoxia/reoxygenation injury in primary
neonatal rat cardiomyocytes. Exp Ther Med. 14:689–695. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Horbinski C and Chu CT: Kinase signaling
cascades in the mitochondrion: A matter of life or death. Free
Radic Biol Med. 38:2–11. 2005. View Article : Google Scholar
|
|
39
|
Wang H, Zhang S, Xu S and Zhang L: The
efficacy and mechanism of dexmedetomidine in myocardial apoptosis
via the renin-angiotensin-aldosterone system. J Renin Angiotensin
Aldosterone Syst. 16:1274–1280. 2015. View Article : Google Scholar
|
|
40
|
Tsuruta F, Masuyama N and Gotoh Y: The
phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax
translocation to mitochondria. J Biol Chem. 277:14040–14047. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kennedy SG, Kandel ES, Cross TK and Hay N:
Akt/Protein kinase B inhibits cell death by preventing the release
of cytochrome c from mitochondria. Mol Cell Biol. 19:5800–5810.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oliva CR, Moellering DR, Gillespie GY and
Griguer CE: Acquisition of chemoresistance in gliomas is associated
with increased mitochondrial coupling and decreased ROS production.
PLoS One. 6:e246652011. View Article : Google Scholar : PubMed/NCBI
|