Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive
and irreversible lung disease of unknown etiology, characterized by
sequential episodes of acute lung injury with subsequent scarring.
Pulmonary fibrosis may lead to respiratory failure due to damage to
the lung structure and reduced gas exchange (1). No drugs are currently available that
are able to control the accumulation of collagens in the lung once
fibrosis has been established. Although several drugs have been
applied to treat IPF, their clinical efficacy remains poor and the
associated serious adverse effects pose problems during long-term
treatment. Therefore, the development of novel therapeutic agents
for this unmet medical need is required.
The NALP3 inflammasome represents the most
extensively investigated inflammasome, and comprises a cytoplasmic
multiprotein complex that consists of NACHT, LRR and PYD
domains-containing protein 3 (NALP3), the adaptor protein
apoptosis-associated Speck-like protein (ASC) containing a caspase
recruitment domain (CARD), and pro-caspase-1, and interacts with
various immune and cell metabolic pathways (2). Upon oligomerization, the NALP3
inflammasome activates the caspase-1 cascade, which produces the
active pro-inflammatory cytokines interleukin (IL)-1β and IL-18
when triggered by a range of molecules, ranging from
pathogen-associated molecular pattern molecules to
damage-associated molecular pattern molecules, which are involved
in infection, tissue injury and metabolic dysregulation. Evidence
suggests that the inflammasome is involved in the development of
fibrosis. Specifically, inorganic particulates associated with the
development of pulmonary fibrosis, such as asbestos (3), silica (4) and nanoparticles (5,6),
permeate lysosomal membranes, activating the NALP3 inflammasome,
with subsequent IL-1β and IL-18 production. Furthermore, recent
studies indicated that NF-κB mediates the reactive oxygen species
(ROS)-induced NALP3 inflammasome by promoting the transcription of
NALP3 and pro-IL-1β (7).
Bleomycin (BLM) is the chemotherapeutic agent most
widely used to induce lung fibrosis in animal models and to
identify the pathogenic mechanisms thereof, as the pathogenesis
characteristics are very similar to those of IPF (8). Recently, NALP3 inflammasome
activation in BLM-induced pulmonary fibrosis was reported by a
series of studies (9–11). Specifically, BLM treatment has
been shown to increase the production of ROS and uric acid
(11), thereby inducing the
development of lung fibrosis. In addition, these products have been
shown in several cases to trigger NALP3 inflammasome activation.
Furthermore, the purinergic receptor P2X7R is activated by ATP,
which is released by injured lung cells following BLM treatment,
leading to activation of the NALP3 inflammasome with subsequent
cleavage and secretion of IL-1β and IL-18 (11,12). Notably, previous studies have
demonstrated that a number of proinflammatory cytokines and
profibrotic growth factors, including IL-1β, IL-6, IL-18, tumor
necrosis factor-α and transforming growth factor (TGF)-β, are
involved in pulmonary inflammation and fibrosis (13,14). Recent studies, primarily focusing
on inflammasome/IL-1β secretion axis-mediated inflammatory actions,
further suggested that the NALP3 inflammasome mediates the
development of fibrosis in systemic sclerosis (15,16). NALP3 also appears to play a key
role in promoting TGF-β signaling and Smad2/3 activation in renal
epithelial cells (17). However,
although TGF-β represents one of the most extensively investigated
fibrogenic cytokines involved in the induction and development of
pulmonary fibrosis, the mechanism underlying the role of NALP3
inflammasome in pulmonary fibrosis remains unclear.
Notably, an increasing volume of evidence indicates
that NALP3 plays a key role in myofibroblast differentiation and
collagen production in an IL-1β/Toll-like receptor 4
(TLR4)/MyD88/NF-κB-dependent manner, or an inflammasome-independent
manner (18), as well as in liver
fibrosis (19). Thus, in the
present study, we hypothesized that the NALP3 inflammasome may
participate in collagen production, and that NF-κB may serve as a
link between NALP3 inflammasome activation and collagen
synthesis.
Materials and methods
Cell culture, transfection and
grouping
NIH-3T3 mouse embryonic fibroblasts were provided by
the Laboratory of Stem Cell Biology, State Key Laboratory of
Biotherapy, Sichuan University (Chengdu, China). The cells were
cultured in Dulbecco's modified Eagle's medium (HyClone; GE
Healthcare, Logan, UT, USA) supplemented with 10% fetal bovine
serum (Sijiqing, Zhejiang, China) in a humidified atmosphere
containing 5% CO2 at 37°C.
NALP3-siRNA (sense strand:
5′-CAGCCAGAGUGGAAUGACAdTdT-3′; antisense strand:
5′-UGUCAUUCCACUCUGGCUGdTdT-3′) and negative control (NControl)
siRNA were synthesized by RiboBio, Guangzhou, China. Transient
transfections were performed using the HiPerfect Transfection
Reagent (Qiagen GmbH, Hilden, Germany) together with either an
NControl siRNA (10 nM) or NALP3-siRNA (10 nM), according to the
HiPerFect Transfection Reagent instruction manual. At 12 h after
transfection, the cells were treated with
H2O2 (200 μM). The silencing
efficiency of the NALP3-siRNA was determined using reverse
transcription-polymerase chain reaction (RT-PCR) after 12 h of
treatment. Approximately 24 h after H2O2
stimulation, total RNA was extracted for RT-PCR and, 48 h later,
total protein was extracted for western blot analysis, and the cell
supernatants were frozen at -80°C for enzyme-linked immunosorbent
assay (ELISA).
Additional groups of cells were treated with or
without pyrrolidine dithiocarbamate (PDTC) (Sigma-Aldrich; Merck
KGaA, St. Louis, MO, USA) (50 μM) 1 h prior to
H2O2 stimulation. The cells and cell
supernatants were collected in the same manner as described
above.
Animals
A total of 42 male C57BL/6 mice (age, 8 weeks;
weight, 20–24 g) were purchased from the Laboratory Animal Center
of Sichuan University (Chengdu, China). The mice were housed (n=5
per cage) in an air-conditioned animal facility under constant
temperature and humidity, with a 12-h day-night cycle and free
access to food and water. The mice were allowed to acclimatize for
2 weeks prior to the initiation of the experimental procedures.
Animal experiments were performed according to protocols approved
by the Laboratory Animal Welfare and Ethics Committee of the
Sichuan University. Mice were randomly divided into three treatment
groups as follows: i) BLM (Nippon Kayaku, Takasaki, Japan) + PDTC:
On day 0, a single intratracheal injection of BLM (5 mg/kg in a
final volume of 50 μl) was performed and PDTC (100 mg/kg)
was intraperitoneally injected 2 h prior to the intratracheal
injection. From day 1 onwards, PDTC (100 mg/kg) was
intra-peritoneally injected once daily. ii) BLM +
phosphate-buffered saline (PBS): On day 0, a single intratracheal
injection of BLM (5 mg/kg in a final volume of 50 μl) was
performed, and an equal volume of PBS was intraperitoneally
injected 2 h prior to the surgery. From day 1 onwards, the same
volume of PBS was intraperitoneally injected once daily. iii)
Control group: A single intratracheal injection of PBS (50
μl) plus intraperi-toneal injections of PBS once daily. We
utilized 10% chloral hydrate (3.5 ml/kg intraperitoneally) to
anesthetize the mice. The mice (n=7/group) were sacrificed at 7 and
28 days after BLM intratracheal injection.
Western blotting
Whole protein from cells or lung tissue was lysed
with RIPA lysis buffer (Beyotime, Shanghai, China) in the presence
of protease inhibitor cocktail (Roche, Mannheim, Germany) for 30
min and centrifuged at 12,000 × g for 20 min at 4°C. Protein
concentrations were determined using the bicinchoninic acid protein
assay (Beyotime, Shanghai, China) according to the manufacturer's
instructions. Equal amounts of protein (60 μg) were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred to polyvinylidene difluoride
membranes (Millipore, Billerica, MA, USA). The membranes were
blocked in 5% non-fat dry milk in 0.1% Tween-20, 1X Tris-buffered
saline (TBST; pH 7.4) for 1 h at room temperature, and then
incubated with goat anti-NALP3 antibody (ab4207; Abcam, Cambridge,
MA, USA), rabbit anti-IκBα antibody (cat. no. 4812, CST, Danvers,
MA, USA), rabbit anti-pNF-κB antibody (cat. no. 3033, CST), rabbit
anti-collagen I antibody (ab34710; Abcam), rabbit anti-α-SMA
antibody (ab5694, Abcam), or rabbit anti-β-actin antibody
(bs-0061R; Bioss, Beijing, China) in blocking solution overnight at
4°C, and washed three times with TBST at 10-min intervals. All
above mentioned antibodies were diluted by 1:1,000. The membranes
were then incubated with horseradish peroxidase-conjugated rabbit
anti-goat (1:5,000; ZB-2306; ZSGB-Bio, Beijing, China) or mouse
anti-rabbit IgG antibody (1:5,000; ZDR-5306; ZSGB-Bio) for 1 h at
room temperature. After washing with TBST, antibody binding was
detected by electro-chemiluminescence using fluorescence detection
equipment (ChemiDoc MP; Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The membranes were stripped using a buffer (10% sodium
dodecyl sulfate, 25 mM glycine, pH 2.0) at room temperature for 30
min, followed by washing in TBST for 30 min. The membranes were
blocked and reprobed for β-actin as a loading control.
Relative gene expression analysis
Total RNA was extracted from the lung tissue and
from cells, using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Carlsbad, CA, USA) according to the manufacturer's
instructions. RNA was reverse-transcribed into cDNA, using the
ReverTra Ace qPCR RT kit (Toyobo Co., Ltd., Osaka, Japan). PCR was
performed in a final volume of 10 μl using a Thunderbird
SYBR qPCR Mix (Toyobo Co., Ltd.). The cycling program involved
initial denaturation at 95°C for 60 sec, followed by 40 cycles at
95°C for 15 sec and 60°C for 60 sec. The β-actin gene was used as
an internal control. NALP3, ASC, caspase-1, type I collagen and
α-smooth muscle actin (SMA) were detected. The sequences of the
primers and products are listed in Table I. The relative expression of the
genes was calculated by the 2−ΔΔq method.
 | Table IReverse transcription-polymerase
chain reaction primers and products. |
Table I
Reverse transcription-polymerase
chain reaction primers and products.
| Gene name | S/AS | Primer sequence
(5′–3′) | Product size
(bp) |
|---|
| NALP3 | S |
ATTACCCGCCCGAGAAAGG | 83 |
| AS |
TCGCAGCAAAGATCCACACAG | |
|
Caspase-1 | S |
AATACAACCACTCGTACACGTC | 78 |
| AS |
AGCTCCAACCCTCGGAGAAA | |
| ASC | S |
GACAGTGCAACTGCGAGAAG | 106 |
| AS |
CGACTCCAGATAGTAGCTGACAA | |
|
Collagen-1 | S |
GCTCCTCTTAGGGGCCACT | 91 |
| AS |
ATTGGGGACCCTTAGGCCAT | |
| α-SMA | S |
CCCAGACATCAGGGAGTAATGG | 104 |
| AS |
TCTATCGGATACTTCAGCGTCA | |
| β-actin | S |
GTGACGTTGACATCCGTAAAGA | 245 |
| AS |
GCCGGACTCATCGTACTCC | |
ELISA
The content of IL-1β in the lung tissue on day 28
after BLM instillation and in the cell supernatants was determined
using an ELISA kit (ExCell Bio, Shanghai, China) according to the
manufacturer's instructions.
Histopathological analysis
The tissue from the left lung was excised and
immediately fixed with 4% paraformaldehyde for 48 h, and then
embedded in paraffin. Serial 4-μm paraffin sections were
prepared using a rotator microtome. Sirius Red staining is
considered as the optimal method for identifying tissue collagen,
as it can differentiate between collagen types I and III, whereas
Masson's trichrome staining is the classical method for staining
collagen used by numerous studies. Masson's trichrome and
hematoxylineosin staining were selected in the present study to
estimate the degree of fibrosis, rather than differentiate between
collagen types. The tissues were visualized under a Zeiss AX10
imager A2 microscope and captured using a Zeiss AX10 cam HRC
(Zeiss, Oberkochen, Germany). The criteria for grading lung
fibrosis were based on the modified Ashcroft score (20). The grade of fibrotic changes in
each lung section was assessed as a mean score of severity from 10
randomly selected high-power fields.
Hydroxyproline assay
The levels of lung collagen were determined by
analysis of the hydroxyproline content on day 28 after BLM infusion
using a hydroxyproline assay kit (Jiancheng Institute of
Biotechnology, Nanjing, China) according to the manufacturer's
instructions.
Statistical analysis
All the results are shown as mean ± standard error
of the mean. Comparisons for multiple groups were performed by
one-way analysis of variance followed by Tukey's multiple
comparison test. For all analyses, a P-value of <0.05 was
considered to indicate statistically significant differences.
Results
H2O2-induced
collagen synthesis in mouse fibroblasts is mediated by the NF-κB
signaling pathway
The IκBs is the most important inhibitor of NF-κB
activation and the degradation of IκBs triggers NF-κB activation.
Phosphorylation of NF-κB p65 at Ser536 is an indirect indicator of
NF-κB activation, which results in translocation of the p65 subunit
of NF-κB to the nucleus (21). In
the present study, detection of the IκBα protein, which is the best
studied IκB protein, and pNF-κB, were used to estimate the degree
of NF-κB activation. Western blot analysis of cell lysates revealed
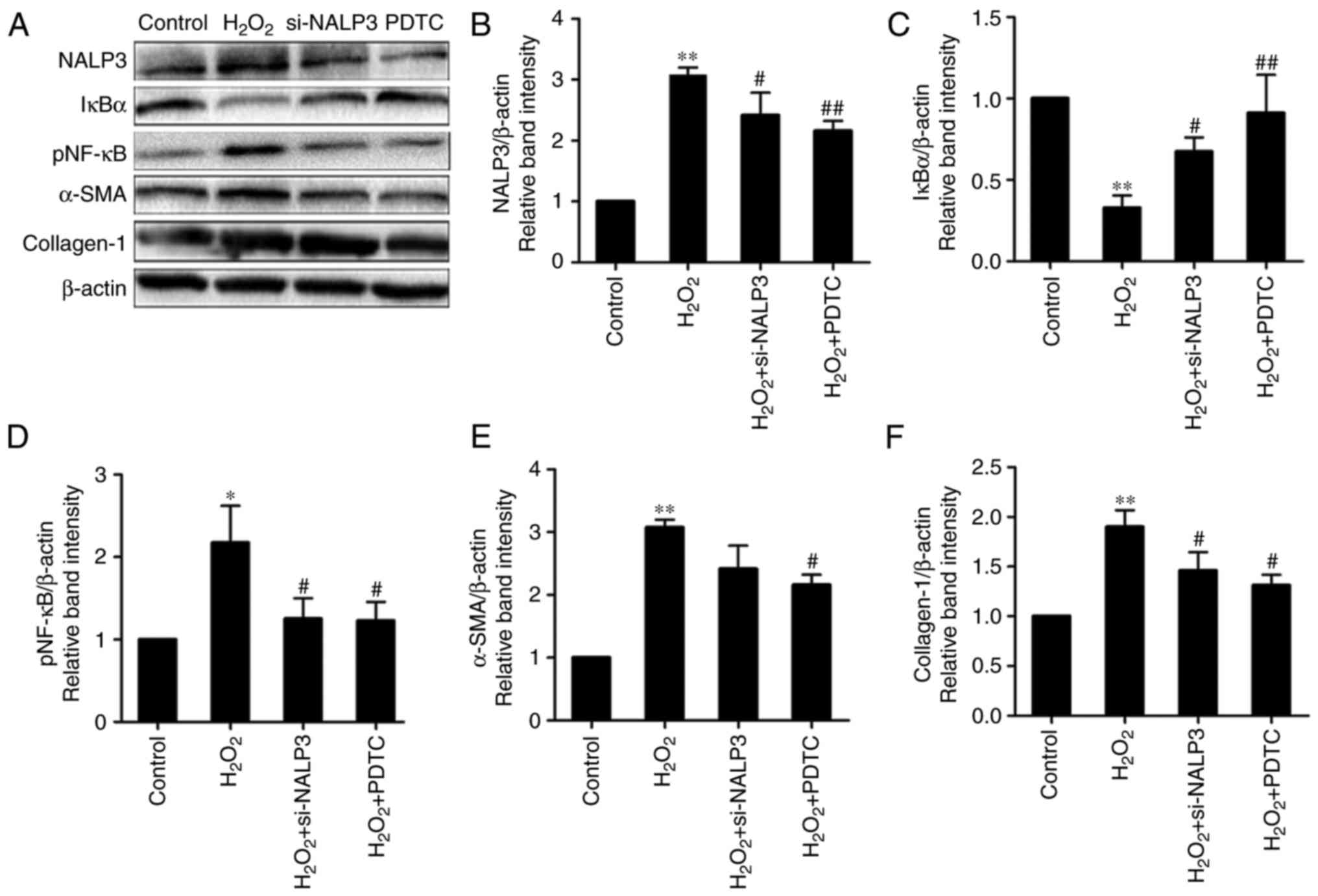
degradation of the IκBα protein and increase in pNF-κB in the
H2O2-treated group compared with controls. By
contrast, the PDTC+H2O2 group exhibited
preserved IκBα levels and decreased NF-κB p65 phosphorylation
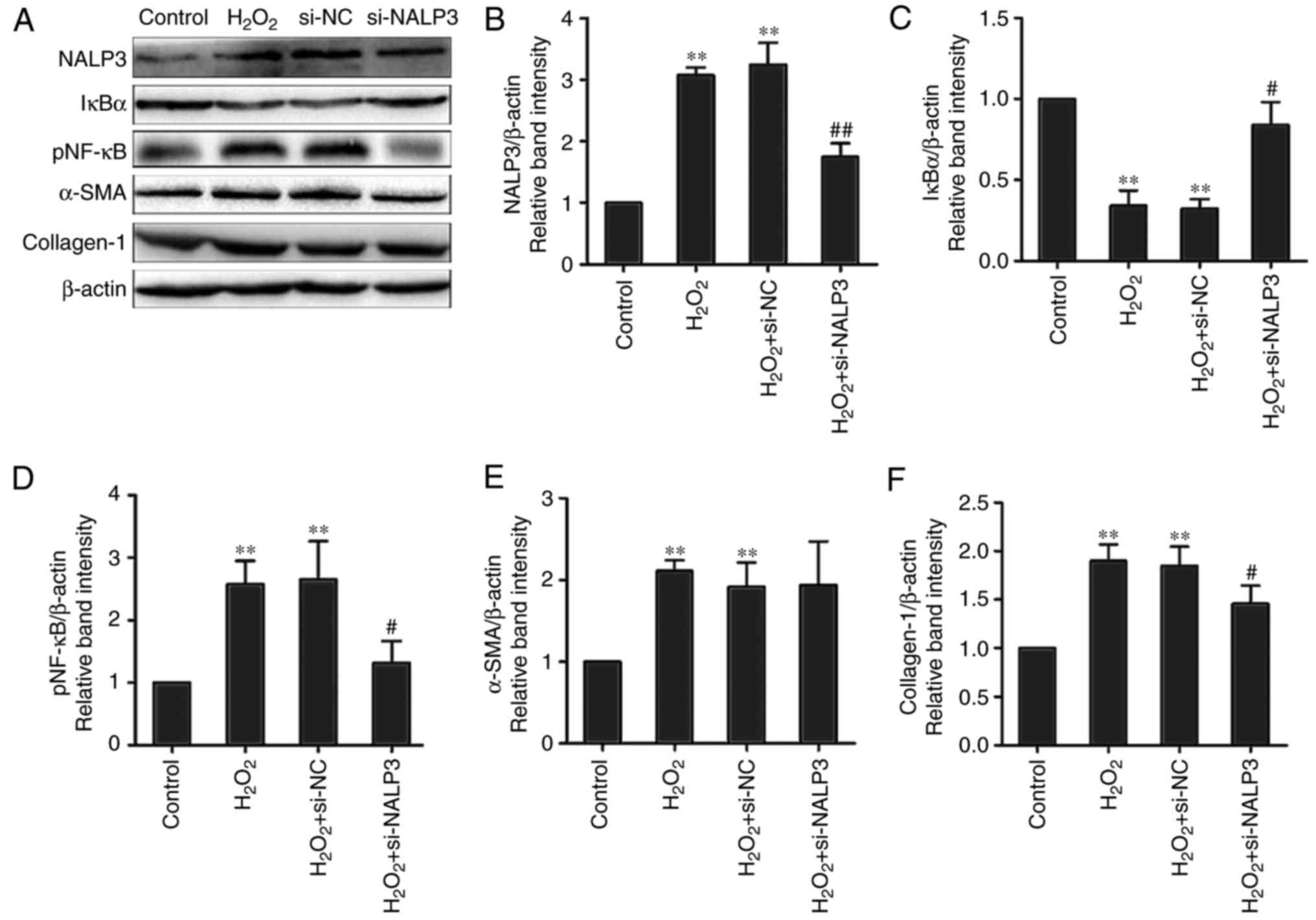
(Fig. 1A, C and D).
H2O2 is known to be an important regulator of
oxidative stress. The results confirmed that NF-κB was activated
(Fig. 1A) via IκBα degradation
during H2O2-mediated oxidative stress in
fibroblasts. Stimulation with H2O2
significantly increased the expression of type I collagen at the
mRNA and protein levels in NIH-3T3 cells (Figs. 1A and F, and 2A). Additionally, similar results for
α-SMA at the mRNA and protein levels were obtained at 24 h after
treatment in the H2O2 group (Figs. 1A and E, and 2A). These findings demonstrated that
H2O2 serves as an important regulator of
extracellular matrix (ECM) deposition for fibroblasts by increasing
type I collagen expression, and may be involved in the transition
from fibroblasts to myofibroblasts by promoting α-SMA expression.
Furthermore, the upregulation of α-SMA and type I collagen was
inhibited by the antioxidant and NF-κB inhibitor PDTC (Figs. 2A and 3A, E and F). Taken together, these data
suggest that H2O2-induced oxidative stress
stimulates type I collagen and α-SMA production in mouse
fibroblasts, and that NF-κB signaling is required in this
process.
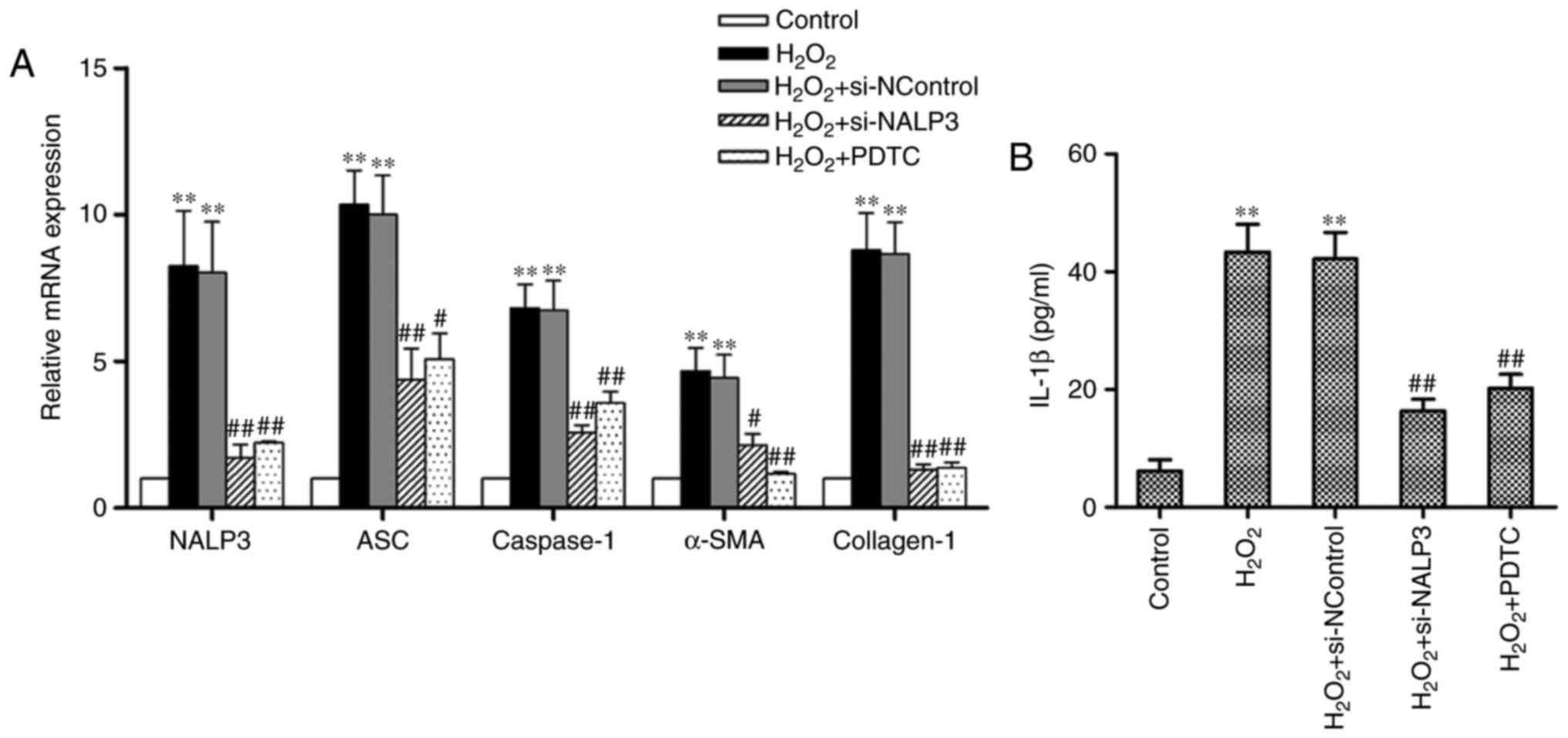 | Figure 2NALP3 gene silencing reduces NALP3
inflammasome activation during H2O2-mediated
oxidative stress and decreases gene expression of α-SMA and type I
collagen. (A) RT-PCR analysis was used to assess the relative mRNA
expression of NALP3, ASC, caspase-1, α-SMA, and collagen-1. (B) The
IL-1β content in cell supernatants was determined using ELISA. Data
are presented as the means ± standard error of the mean from three
independent experiments. The significance among groups was analyzed
by one way analysis of variance. **P<0.01 vs. the
control; #P<0.05, ##P<0.01 vs. the
H2O2 group. α-SMA, α-smooth muscle actin;
RT-PCR, reverse transcription-polymerase chain reaction; ASC,
apoptosis-associated Speck-like protein; IL, interleukin; NControl,
negative control; PDTC, pyrrolidine dithiocarbamate. |
The NALP3 inflammasome plays an important
role in H2O2-induced collagen synthesis in
mouse fibroblasts via the NF-κB signaling pathway
Cells treated with H2O2
exhibited overexpression of NALP3, ASC and caspase-1 at the mRNA
level (Fig. 2A). In addition,
H2O2 induced an increase in IL-1β content in
cell supernatants, whereas cells treated with PDTC exhibited
reduced expression of NALP3, ASC and caspase-1 mRNA and IL-1β
content. The effect of PDTC on NALP3 protein levels was confirmed
by western blot analysis. The results suggested that activation of
the NF-κB signaling pathway promoted NALP3 inflammasome activation
by increasing NALP3, ASC and caspase-1 expression during
H2O2-mediated oxidative stress (Figs. 2B and 3A–D). Furthermore, we demonstrated that
the expression of NALP3 could be effectively knocked down using
siRNA-NALP3, and that the mRNA levels of ASC and caspase-1 were
downregulated when exposed to H2O2 under
conditions of NALP3 knockdown. In addition, the secretion of IL-1β
was also decreased. Inhibition of NALP3 abolished
H2O2-mediated type I collagen synthesis and
increased the mRNA levels of α-SMA in fibroblasts. However, no
effect on α-SMA protein was observed following administration of
siRNA-NALP3 (Figs. 1E and
3E). The IκBα protein remained at
a relatively high level and pNF-κB level was reduced compared with
the siRNA-negative and H2O2 groups (Fig. 1A, C and D). Collectively, these
findings suggest that activation of the NALP3 inflammasome is
involved in H2O2-induced type I collagen
production in fibroblasts via the NF-κB signaling pathway.
The NF-κB pathway is required for NALP3
inflammasome activation in BLM-induced pulmonary fibrosis
A single intratracheal injection of BLM constitutes
a well-established animal model that results in airway epithelial
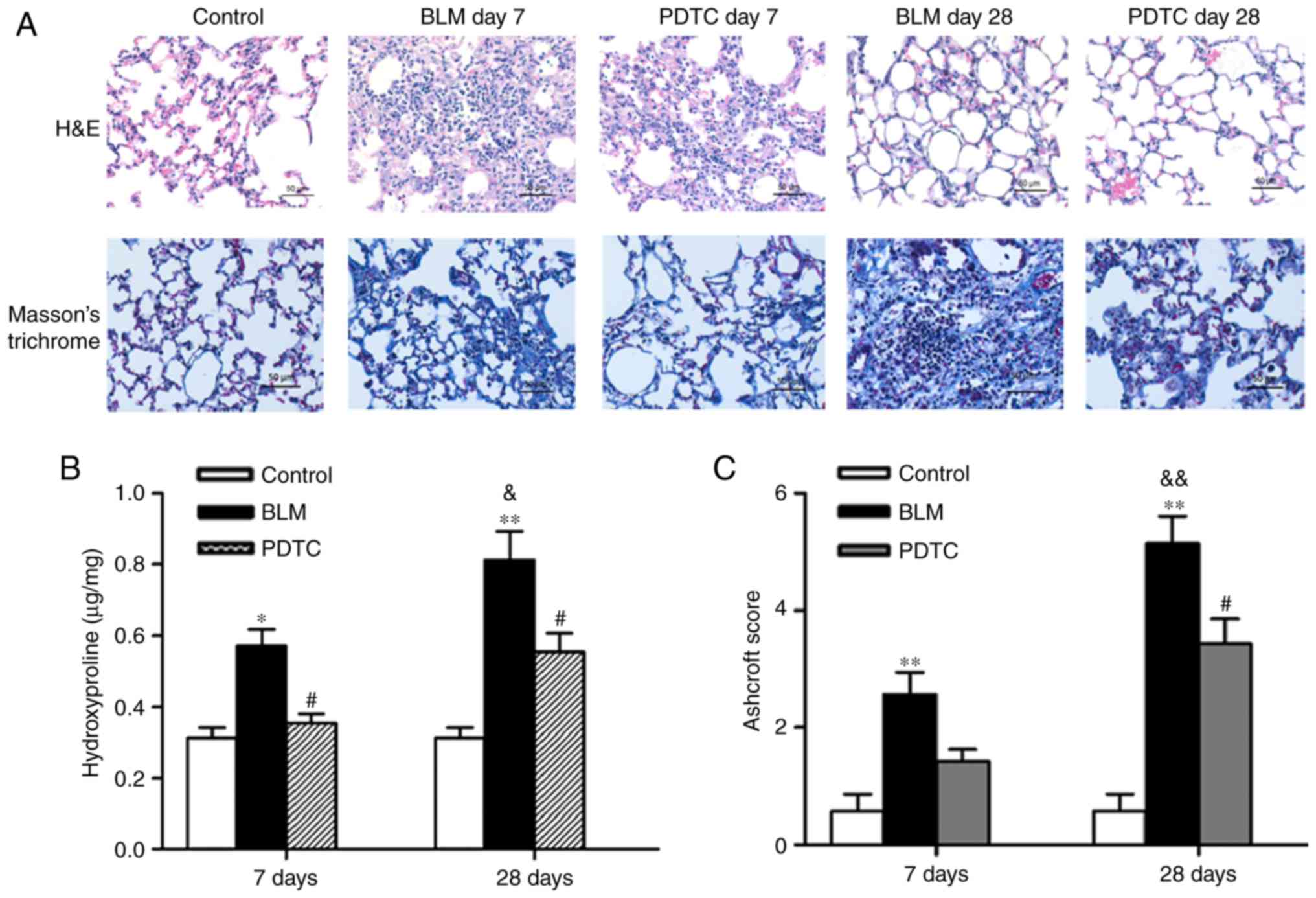
cell damage, inflammation and formation of fibrotic lesions. In the
present study, a well-alveolized normal histology was observed in
the control group. By contrast, obvious alveolar wall thickening,
massive infiltration of leukocytes and excessive deposition of
mature collagen in the interstitium was observed in BLM-treated
mice. Lung inflammation was contained on day 28, although the
fibrotic changes became more severe. These histopathological
changes were improved by PDTC pretreatment (Fig. 4A).
BLM administration induced a significant increase in
Ashcroft scores compared with the control on day 7, and these
scores were further increased by day 28 (Fig. 4C). Conversely, the scores of the
mice administered PDTC were significantly lower. In addition, a
similar trend was observed in the measurements of lung
hydroxyproline content (Fig. 4B).
Thus, these results indicate that the BLM-induced pulmonary
fibrosis model was successfully established, and that the NF-κB
signaling pathway played a key role in the process of fibrosis.
To examine the activation of NALP3 inflammasomes in
BLM-induced pulmonary fibrosis, the mRNA levels of NALP3, ASC,
caspase-1, α-SMA and type I collagen were measured and NALP3
protein levels were determined in the lungs of mice. BLM-treated
mice exhibited significantly elevated levels of NALP3, ASC,
caspase-1, α-SMA and type I collagen mRNA compared with the control
group when analyzed on day 28 (Fig.
5E). The level of the NALP3 protein was markedly higher
compared with that of the control on both days 7 and 28, whereas
NALP3 protein expression was reduced on day 28 compared with day 7
after BLM injection (Fig. 5A and
B). PDTC-pretreated mice exhibited a relatively lower
expression of NALP3, ASC, caspase-1, α-SMA and type I collagen mRNA
compared with the BLM group. Furthermore, PDTC reduced NALP3
protein levels to a statistically significant extent in the lungs
of BLM-treated mice on days 7 and 28. In addition, ELISA analysis
demonstrated that BLM administration resulted in a large increase
in IL-1β production, and that PDTC pretreatment was able to
attenuate the BLM-induced production of IL-1β in the lung tissues
on day 28 (Fig. 5D). Western blot
analysis revealed a decrease in IκBα levels in the BLM group as
opposed to their preservation in the BLM+PDTC group (Fig. 5A and C). However, a more
significant change in the level of IκBα was not observed over time.
These results suggest that the NALP3 inflammasome is activated
during the stages of early inflammation and fibrosis and,
therefore, may play a role in fibrogenesis.
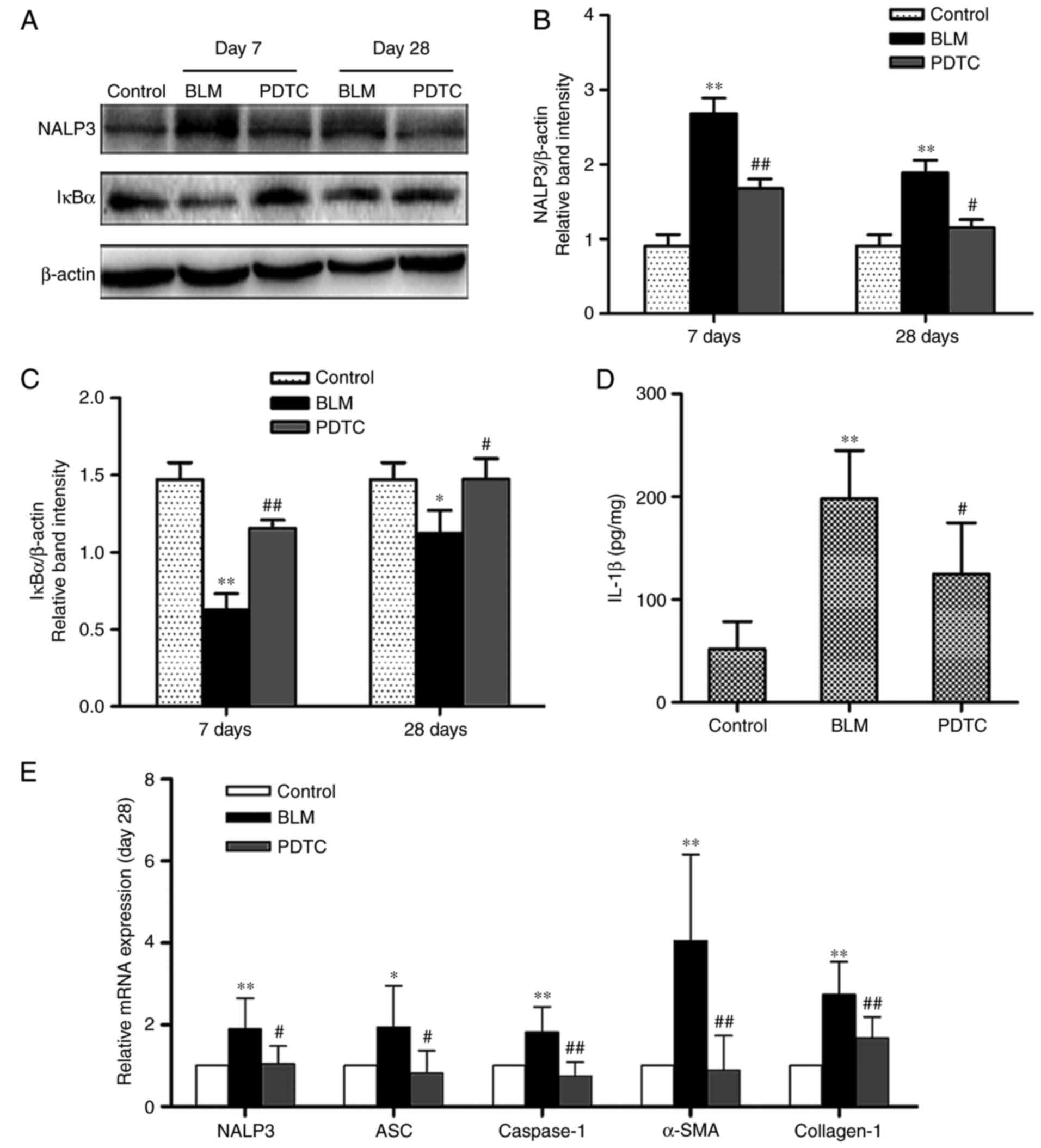 | Figure 5NALP3 inflammasome is activated in
BLM-induced pulmonary fibrosis after day 28 in mice. (A–C) Protein
levels of NALP3 and IκBα in lung tissues were determined on day 7
and day 28 by western blotting. Content of IL-1β in lung tissues
was examined on day 28 by ELISA (D). Relative mRNA expression of
NALP3, ASC, caspase-1, α-SMA, and type I collagen (E). Data are
presented as the means ± standard error of the mean, n=7.
*P<0.05, **P<0.01 vs. the control group
at day 28; #P<0.05, ##P<0.01 vs. the
BLM group at day 28. BLM, bleomycin; IκBα, nuclear factor of kappa
light polypeptide gene enhancer in B-cells inhibitor α; α-SMA,
α-smooth muscle actin; IL, interleukin; PDTC, pyrrolidine
dithiocarbamate; ASC, apoptosis-associated Speck-like protein. |
Discussion
NALP3 has been widely investigated with respect to
immune response over the past several decades, and it is considered
to act as a general sensor for cellular stress. In recent years,
NALP3 has been found to play important roles in various
pathological processes, including diabetes mellitus (22,23), non-alcoholic steatohepatitis
(24,25), chronic kidney diseases (26) and IPF (27). Furthermore, an increasing volume
of evidence indicates that NALP3 serves as an important factor in
organ fibrosis. Pulmanary fibrosis develops as a consequence of
abnormalities occurring in multiple biological pathways that affect
inflammation and wound repair, which involve a series of cells and
cytokines. Studies on the process of fibrogenesis have focused
primarily on cell injury, macrophage activation, inflammation and
ECM deposition. Tian et al (28) reported that siNALP3 may rescue
A549 from BLM-induced pulmonary fibrosis. Accordingly, the
biological characteristics of the NALP3 inflammasome during
collagen metabolism in pulmonary fibrosis remain unclear. To
further elucidate the role of the NALP3 inflammasome during type I
collagen synthesis in fibroblasts, the effects of NALP3-siRNA on
H2O2-treated mouse embryonic fibroblasts were
examined.
In the present study, it was suggested that NALP3
plays an important role in H2O2-mediated type
I collagen synthesis via the NF-κB signaling pathway. NF-κB
activation and type I collagen production were shown to be
significantly decreased by NALP3-siRNA. Additionally, the
inhibition of NF-κB resulted in downregulation of type I collagen
and the NALP3 inflammasome. In addition, consistent with the
results from previous studies, we demonstrated that the NALP3
inflammasome was activated in BLM-induced pulmonary fibrosis and
that this activation was attenuated by PDTC.
There are two essential criteria for triggering
NALP3 inflammasome activation. First, NALP3 expression per
se must be transcriptionally induced, which requires NF-κB. A
second, post-transcriptional step then leads to the activation of
NALP3, allowing for NALP3 inflammasome assembly. In the first step,
NALP3 expression is considered as the limiting factor for
inflammasome priming (29). The
corresponding NF-κB binding sites (nt −1,303 to −1,292 and −1,238
to −1,228) are located in the NALP3 promoter in macrophages
(30) and TLR2/MyD88/NF-κB and
TLR4/MyD88/NF-κB signaling is required for pro-IL-1β and NALP3 gene
expression (7,31). Our data further demonstrated that
PDTC inhibits the expression of NALP3, ASC and caspase-1 to varying
degrees. Taken together, this evidence suggests that NF-κB serves
as a critical upstream mediator for NALP3 inflammasome priming.
Accordingly, our study provides a new viewpoint regarding the
NF-κB/inflammasome pathway. The partial protection of NF-κB
activation by NALP3 silencing suggests that the inflammasome may
function upstream of NF-κB. In previous studies, inflammasome
activation leading to IL-1β maturation and release, IL-1β will also
activate the IL-1R1/MyD88/NF-κB pathway (10). This may contribute to an
autocrine/paracrine amplification loop of IL-1β and NF-κB during
the process of collagen metabolism (19).
The second criterion for triggering NALP3
inflammasome activation involves intracellular ROS and potassium
(K+) efflux due to the stimulation of ATP-sensitive ion
channels, which promote inflammasome assembly leading to caspase-1
activation and subsequent IL-1β release. Oxidative stress is a
strong NF-κB activator. We thus hypothesized that oxidative stress
acts both up- and downstream of the NALP3 inflammasome, and it also
plays a contributory role in the pathogenesis of NALP3 inflammasome
activation and pulmonary fibrosis by inducing genetic
overexpression of fibrogenic cytokines (32). Artlett et al indicated that
the NALP3 inflammasome plays important roles in collagen synthesis
via type IA and 3A1 collagen, and connective tissue growth factor
production and myofibroblast differentiation (15). Inhibition of caspase-1 abrogated
the expression of collagens, IL-1β and α-SMA in systemic sclerosis
dermal and lung fibroblasts. As an NALP3 inflammasome effector,
cytokine IL-1β exerts comprehensive biological effects associated
with inflammation and fibrosis. The association between IL-1β and
fibrosis has been widely investigated. IL-1 receptor antagonists or
IL-1R deficiency may reduce liver or lung fibrosis (10,24). IL-1β mediates collagen expression
mainly via the induction of TGF-β, the downstream cytokine of
IL-1β, which is also known as the most essential cytokine in the
biochemical processes of fibrosis. TGF-β directly increases the
transcriptional activation of collagen genes and also stimulates
the expression of a number of proinflamatory and fibrogenic
cytokines, such as TNF-α, PDGF, IL-1β, or IL-13, thereby further
enhancing the fibrotic response in macrophages, fibroblasts and
myofibroblasts. TGF-β is also a direct mediator of
epithelial-to-mesenchymal transition (EMT), which is one of the
most important sources of myofibrosis. Another important TGF-β
feature in increasing ECM deposition is the creation of a
microenvironment that favors ECM deposition. Redox balance
modulation may affect the NALP3 inflammasome/Smad pathway in terms
of collagen synthesis. Smads are the most crucial intracellular
proteins that transduce extracellular signals from TGF-β ligands to
activate downstream gene transcription. Thus, the mechanism
underlying collagen synthesis may be the NALP3/IL-1β/TGF-β pathway.
Of note, when NALP3−/− mouse primary cardiac fibroblasts
(CFs) were treated with TGF-β, which plays a direct role in
fibroblast differentiation into myofibroblasts and EMC deposition,
they displayed impaired and delayed myofibroblast differentiation
with reduced α-SMA expression (18). Furthermore, the expression of
α-SMA and MMP-9 was significantly decreased in mouse
NALP3−/− renal tubular epithelial cells that had been
stimulated by TGF-β. Taken together, these findings may suggest an
upstream role for TGF-β to NALP3 in myofibroblast differentiation.
Cai et al demonstrated that angiotensin 2 (Ang-2) increased
NALP3 and pro-IL-1β levels by activating the TLR4/MyD88/NF-κB
pathway, and first demonstrated that Ang-2-induced collagen
synthesis in hepatic stellate cells could be inhibited by NALP3
depletion. NF-κB may therefore affect inflammasome activation and
downstream IL-1β-mediated collagen metabolism.
Consistent with prior findings, our data suggest
that NALP3 may act as a new mediator in the pathomechanism of
fibrosis via regulating type I collagen and α-SMA expression. NALP3
also affects the collagen synthesis rate-limiting enzyme (P4Hα1)
and collagen breakdown enzymes (MMP-2 and MMP-9) (28). However, our data demonstrated
decreased mRNA expression with inhibition at the protein level of
α-SMA after transfection. A possible explanation for this apparent
discrepancy is that H2O2-mediated
myofibroblast differentiation may involve multiple pathways in
which NALP3 acts upon only a subset. Another possible explanation
may be associated with limitations of cell culture studies
including the short duration of transfection and the transfection
efficiency. To resolve this issue, we must design a more durable
transfection system in our future experiment.
H2O2 generates excessive
amounts of ROS, and both factors represent the primary mediators of
the effects of TGF-β in various cells (33–35). In particular,
H2O2-mediated collagen synthesis is
associated with TGF-β (36). The
association between NALP3 inflammasome and TGF-β is mentioned
above. Notably, the TGF-β effects involving NALP3 have been
identified as occurring in another inflammasome-independent manner.
The involvement of NALP3 in TGF-β-induced R-Smad phosphorylation,
nuclear accumulation and myofibroblast differentiation was
demonstrated; however, TGF-β did not induce the upregulation or
secretion of activated IL-1, IL-18, or caspase-1 in CFs. By
contrast, the protective effect of NALP3 deficiency was
consistently reported; simultaneously, IL-1β, IL-18, ASC, and
caspase-1 were shown to be less important under certain conditions
(37,38). It was suggested that ASC can
induce MAPK phosphorylation independently of cytokine production in
macrophages (39) to regulate
mRNA stability; this, in turn, affected DOCK-2 protein expression
and phagocytosis in leukocytes (40). These findings demonstrated that
the physiological roles of NALP3 and ASC are not limited to the
caspase-1/IL-1β axis. As TGF-β represents one of the most critical
cytokines in the EMT and in ECM deposition, whether the associated
regulatory mechanisms involving both NALP3 and TGF-β act via an
inflammasome-dependent or -independent manner remains unclear. The
independent biological effects of these components were mainly
reported in non-monocytes/macrophages, such as renal tubular
epithelial cells or CFs; thus, we may infer that the
inflammasome-independent pathway may be a mechanism distinguishing
epithelial cell lines from monocytes/macrophages, but this
hypothesis requires further investigation.
As TGF-β represents one of the most critical
cytokines in EMT and in ECM deposition, whether the associated
regulatory mechanisms involving both NALP3 and TGF-β act in an
inflammasome-dependent or -independent manner remains unclear.
In the present study, it was first demonstrated that
NALP3 deficiency alleviates H2O2-induced type
I collagen synthesis via the NF-κB signaling pathway. However,
further research is required to identify the exact mechanism
through which the NALP3 inflammasome plays a role in collagen
production. As fibroblast activation represents the main checkpoint
for ECM deposition, these results suggest that modulation of the
effects of the NALP3 inflammasome on fibroblasts may have important
therapeutic implications in pulmonary fibrosis.
Abbreviations:
|
NF-κB
|
nuclear factor κB
|
|
IL-1β
|
interleukin 1β
|
|
IL-18
|
interleukin 18
|
|
TGF-β
|
transforming growth factor-β
|
|
TNF-α
|
tumor necrosis factor-α
|
|
PDTC
|
pyrrolidine dithiocarbamate
|
|
ASC
|
apoptosis-associated Speck-like
protein
|
|
IκBα
|
nuclear factor of kappa light
polypeptide gene enhancer in B-cells inhibitor α
|
|
pNF-κB
|
phosphorylated NF-κB
|
|
α-SMA
|
α-smooth muscle actin
|
|
BLM
|
bleomycin
|
|
IPF
|
idiopathic pulmonary fibrosis
|
|
ROS
|
reactive oxygen species
|
|
ECM
|
extracellular matrix
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
TLR
|
toll-like receptor 4
|
|
Ang-2
|
angiotensin 2
|
|
MMP-9
|
matrix metalloproteinase 9
|
|
CFs
|
cardiac fibroblasts
|
|
TECs
|
renal tubular epithelial cells
|
Acknowledgments
The present was supported by a grant from the
Sichuan Provincial Department of Science and Technology (no.
0040205301A53).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Harari S and Caminati A: IPF: New insight
on pathogenesis and treatment. Allergy. 65:537–553. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Latz E, Xiao TS and Stutz A: Activation
and regulation of the inflammasomes. Nat Rev Immunol. 13:397–411.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hillegass JM, Miller JM, MacPherson MB,
Westbom CM, Sayan M, Thompson JK, Macura SL, Perkins TN, Beuschel
SL, Alexeeva V, et al: Asbestos and erionite prime and activate the
NLRP3 inflammasome that stimulates autocrine cytokine release in
human mesothelial cells. Part Fibre Toxicol. 10:392013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Peeters PM, Eurlings IM, Perkins TN,
Wouters EF, Schins RP, Borm PJ, Drommer W, Reynaert L and Albrecht
C: Silica-induced NLRP3 inflammasome activation in vitro and in rat
lungs. Part Fibre Toxicol. 11:582014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Naji A, Muzembo BA, Yagyu K, Baba N,
Deschaseaux F, Sensebé L and Suganuma N: Endocytosis of
indiumtin-oxide nanoparticles by macrophages provokes pyroptosis
requiring NLRP3-ASC-Caspase1 axis that can be prevented by
mesenchymal stem cells. Sci Rep. 6:261622016. View Article : Google Scholar
|
|
6
|
Sun B, Wang X, Ji Z, Wang M, Liao YP,
Chang CH, Li R, Zhang H, Nel AE and Xia T: NADPH oxidase-dependent
NLRP3 inflammasome activation and its important role in lung
fibrosis by multiwalled carbon nanotubes. Small. 11:2087–2097.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Segovia J, Sabbah A, Mgbemena V, Tsai SY,
Chang TH, Berton MT, Morris IR, Allen IC, Ting JP and Bose S:
TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux
activates NLRP3/ASC inflammasome during respiratory syncytial virus
infection. PLoS One. 7:e296952012. View Article : Google Scholar
|
|
8
|
Mouratis MA and Aidinis V: Modeling
pulmonary fibrosis with bleomycin. Curr Opin Pulm Med. 17:355–361.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
dos Santos G, Rogel MR, Baker MA, Troken
JR, Urich D, Morales-Nebreda L, Sennello JA, Kutuzov MA, Sitikov A,
Davis JM, et al: Vimentin regulates activation of the NLRP3
inflammasome. Nat Commun. 6:65742015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gasse P, Mary C, Guenon I, Noulin N,
Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF,
Lagente V, et al: IL-1R1/MyD88 signaling and the inflammasome are
essential in pulmonary inflammation and fibrosis in mice. J Clin
Invest. 117:3786–3799. 2007.PubMed/NCBI
|
|
11
|
Gasse P, Riteau N, Charron S, Girre S,
Fick L, Pétrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B
and Couillin I: Uric acid is a danger signal activating NALP3
inflammasome in lung injury inflammation and fibrosis. Am J Respir
Crit Care Med. 179:903–913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gicquel T, Victoni T, Fautrel A, Robert S,
Gleonnec F, Guezingar M, Couillin I, Catros V, Boichot E and
Lagente V: Involvement of purinergic receptors and NOD-like
receptor-family protein 3-inflammasome pathway in the adenosine
triphosphate-induced cytokine release from macrophages. Clin Exp
Pharmacol Physiol. 41:279–286. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hoshino T, Okamoto M, Sakazaki Y, Kato S,
Young HA and Aizawa H: Role of proinflammatory cytokines IL-18 and
IL-1beta in bleomycin-induced lung injury in humans and mice. Am J
Respir Cell Mol Biol. 41:661–670. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Phan SH and Kunkel SL: Lung cytokine
production in bleomycin-induced pulmonary fibrosis. Exp Lung Res.
18:29–43. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Artlett CM, Sassi-Gaha S, Rieger JL,
Boesteanu AC, Feghali-Bostwick CA and Katsikis PD: The inflammasome
activating caspase 1 mediates fibrosis and myofibroblast
differentiation in systemic sclerosis. Arthritis Rheum.
63:3563–3574. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martínez-Godínez MA, Cruz-Domínguez MP,
Jara LJ, Domínguez-López A, Jarillo-Luna RA, Vera-Lastra O,
Montes-Cortes DH, Campos-Rodríguez R, López-Sánchez DM,
Mejía-Barradas CM, et al: Expression of NLRP3 inflammasome,
cytokines, and cascular mediators in the skin of systemic sclerosis
patients. Isr Med Assoc J. 17:5–10. 2015.
|
|
17
|
Wang W, Wang X, Chun J, Vilaysane A, Clark
S, French G, Bracey NA, Trpkov K, Bonni S, Duff HJ, et al:
Inflammasome-independent NLRP3 augments TGF-β signaling in kidney
epithelium. J Immunol. 190:1239–1249. 2013. View Article : Google Scholar
|
|
18
|
Bracey NA, Gershkovich B, Chun J,
Vilaysane A, Meijndert HC, Wright JR Jr, Fedak PW, Beck PL, Muruve
DA and Duff HJ: Mitochondrial NLRP3 protein induces reactive oxygen
species to promote smad protein signaling and fibrosis independent
from the inflammasome. J Biol Chem. 289:19571–19584. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai S, Yang R, Li Y, Ning ZW, Zhang LL,
Zhou GS, Luo W, Li DH, Chen Y, Pan MX and Li X: Angiotensin-(1–7)
improves liver fibrosis by regulating the NLRP3 inflammasome via
redox balance modulation. Antioxid Redox Sign. 24:795–812. 2016.
View Article : Google Scholar
|
|
20
|
Hübner RH, Gitter W, El Mokhtari NE,
Mathiak M, Both M, Bolte H, Freitag-Wolf S and Bewig B:
Standardized quantification of pulmonary fibrosis in histological
samples. Biotechniques. 44:507–511. 514–517. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Q, Lenardo MJ and Baltimore D: 30
Years of NF-κB: A blossoming of relevance to human pathobiology.
Cell. 168:37–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Henriksbo BD, Lau TC, Cavallari JF, Denou
E, Chi W, Lally JS, Crane JD, Duggan BM, Foley KP, Fullerton MD, et
al: Fluvastatin causes NLRP3 inflammasome-mediated adipose insulin
resistance. Diabetes. 63:3742–3747. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo B, Li B, Wang W, Liu X, Xia Y, Zhang
C, Zhang M, Zhang Y and An F: NLRP3 gene silencing ameliorates
diabetic cardio-myopathy in a type 2 diabetes rat model. PLoS One.
9:e1047712014. View Article : Google Scholar
|
|
24
|
Wree A, Eguchi A, McGeough MD, Pena CA,
Johnson CD, Canbay A, Hoffman HM and Feldstein AE: NLRP3
inflammasome activation results in hepatocyte pyroptosis, liver
inflammation, and fibrosis in mice. Hepatology. 59:898–910. 2014.
View Article : Google Scholar :
|
|
25
|
Wree A, McGeough MD, Peña CA, Schlattjan
M, Li H, Inzaugarat ME, Messer K, Canbay A, Hoffman HM and
Feldstein AE: NLRP3 inflammasome activation is required for
fibrosis development in NAFLD. J Mol Med. 92:1069–1082. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vasileiou E, Montero RM, Turner CM and
Vergoulas G: P2X7 receptor at the heart of disease.
Hippokratia. 14:155–163. 2010.PubMed/NCBI
|
|
27
|
Lasithiotaki I, Giannarakis I, Tsitoura E,
Samara KD, Margaritopoulos GA, Choulaki C, Vasarmidi E, Tzanakis N,
Voloudaki A, Sidiropoulos P, et al: NLRP3 inflammasome expression
in idiopathic pulmonary fibrosis and rheumatoid lung. Eur Res J.
47:910–918. 2016. View Article : Google Scholar
|
|
28
|
Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie
H and Wang R: NLRP3 participates in the regulation of EMT in
bleomycin-induced pulmonary fibrosis. Exp Cell Res. 357:328–334.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Simard J, Cesaro A, Chapeton-Montes J,
Tardif M, Antoine F, Girard D and Tessier PA: S100A8 and S100A9
induce cytokine expression and regulate the NLRP3 inflammasome via
ROS-dependent activation of NF-κB1. PLoS One.
8:e721382013. View Article : Google Scholar
|
|
30
|
Qiao Y, Wang P, Qi J, Zhang L and Gao C:
TLR-induced NF-κB activation regulates NLRP3 expression in murine
macrophages. FEBS Lett. 586:1022–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bauernfeind FG, Horvath G, Stutz A,
Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks
BG, Fitzgerald KA, et al: Cutting edge: NF-kappaB activating
pattern recognition and cytokine receptors license NLRP3
inflammasome activation by regulating NLRP3 expression. J Immunol.
183:787–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Poli G and Parola M: Oxidative damage and
fibrogenesis. Free Radic Biol Med. 22:287–305. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hong YH, Peng HB, La Fata V and Liao JK:
Hydrogen peroxide-mediated transcriptional induction of macrophage
colony-stimulating factor by TGF-beta1. J Immunol. 159:2418–2423.
1997.PubMed/NCBI
|
|
34
|
Junn E, Lee KN, Ju HR, Han SH, Im JY, Kang
HS, Lee TH, Bae YS, Ha KS, Lee ZW, et al: Requirement of hydrogen
peroxide generation in TGF-beta 1 signal transduction in human lung
fibroblast cells: Involvement of hydrogen peroxide and
Ca2+ in TGF-beta 1-induced IL-6 expression. J Immunol.
165:2190–2197. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koo HY, Shin I, Lee ZW, Lee SH, Kim SH,
Lee CH, Kang HS and Ha KS: Roles of RhoA and phospholipase
A2 in the elevation of intracellular
H2O2 by transforming growth factor-beta in
Swiss 3T3 fibroblasts. Cell Signal. 11:677–683. 1999. View Article : Google Scholar
|
|
36
|
Park SK, Kim J, Seomun Y, Choi J, Kim DH,
Han IO, Lee EH, Chung SK and Joo CK: Hydrogen peroxide is a novel
inducer of connective tissue growth factor. Biochem Biophys Res
Commun. 284:966–971. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mizushina Y, Shirasuna K, Usui F, Karasawa
T, Kawashima A, Kimura H, Kobayashi M, Komada T, Inoue Y, Mato N,
et al: NLRP3 protein deficiency exacerbates hyperoxia-induced
lethality through Stat3 protein signaling independent of
interleukin-1β. J Biol Chem. 290:5065–5077. 2015. View Article : Google Scholar
|
|
38
|
Shigeoka AA, Mueller JL, Kambo A, Mathison
JC, King AJ, Hall WF, Correia Jda S, Ulevitch RJ, Hoffman HM and
McKay DB: An inflammasome-independent role for epithelial-expressed
Nlrp3 in renal ischemia-reperfusion injury. J Immunol.
185:6277–6285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Taxman DJ, Holley-Guthrie EA, Huang MT,
Moore CB, Bergstralh DT, Allen IC, Lei Y, Gris D and Ting JP: The
NLR adaptor ASC/PYCARD regulates DUSP10, mitogen-activated protein
kinase (MAPK), and chemokine induction independent of the
inflammasome. J Biol Chem. 286:19605–19616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ippagunta SK, Malireddi RK, Shaw PJ, Neale
GA, Vande Walle L, Green DR, Fukui Y, Lamkanfi M and Kanneganti TD:
The inflammasome adaptor ASC regulates the function of adaptive
immune cells by controlling Dock2-mediated Rac activation and actin
polymerization. Nat Immunol. 12:1010–1016. 2011. View Article : Google Scholar : PubMed/NCBI
|