Parkinson's disease (PD) is the most common movement
disorder and is clinically characterized by motor symptoms,
including bradykinesia, resting tremors, rigidity and postural
instability, caused by the progressive degeneration of dopaminergic
neurons in the sustantia nigra (SN) (1). While the underlying mechanisms
contributing to the damage of dopaminergic neuron remains poorly
understood, oxidative stress has been considered to be strongly
linked to the loss of neurons in PD (2,3).
Studies in postmortem brains have shown the increased levels of
4-hydroxyl-2-nonenal (HNE), a by-product of lipid peroxidation,
carbonyl modifications of soluble proteins, and the DNA and RNA
oxidation products 8-hydroxy-deoxyguanosine and 8-hydroxyguanosine
in the SN of PD patients (4–8).
The link between oxidative stress and the pathogenesis of PD is
further supported by animal models induced by neurotoxins,
including 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP),
rote-none and 6-hydroxydopamine (6-OHDA), which cause the
production of ROS and the progressive loss of dopaminergic neurons
(9-11). Oxidative stress is an imbalance in
the rate of reactive oxygen species (ROS) production and the rate
of ROS scavenging, resulting in excessive accumulation of ROS
(12). These ROS attack all
macromolecules, including lipids, proteins and nucleic acids, and
trigger an inflammatory response, resulting in cellular damage,
mitochondrial dysfunction, oxidative DNA injury and
neuroinflammation, all of which have been considered as key
contributors in the neurodegenerative process of PD (13–16). Oxidative stress appears to be a
central event associated with the development of PD by activating
the cascade of events leading to the degeneration of these
dopaminergic neurons. The present review stresses the fundamental
pathological pathway of oxidative stress in the development of PD,
in order to gain a better understanding of the underlying
mechanisms, and to provide available evidence and future directions
for potential effective therapeutic targets with enhanced efficacy
in the prevention and treatment of PD.
Mitochondria are important organelles for the
maintenance of cellular homeostasis by generating and supplying
energy for the cells through oxidative phosphorylation. The process
of oxidative phosphorylation involves the interaction of unpaired
electrons with molecular O2, resulting in the generation
of a superoxide radical, an amphibolic radical that cannot easily
pass through biological membranes (17,18). Subsequently, the radical
O2− is converted by the mitochondrial
superoxide dismutase or manganese superoxide dismutase to form
hydrogen peroxide (H2O2) in the mitochondria
(19). H2O2
is a relatively inactive compound that is released from the
mitochondria into the cellular cytosol and nucleus where it
contributes to oxidative stress. In the presence of reduced ferrous
iron, H2O2 can be converted into the highly
reactive hydroxyl radical, leading to further oxidative damage
(20). It is widely accepted that
complex I inhibition and a subsequent increase in the production of
ROS is a leading cause responsible for the loss of dopaminergic
neurons in PD (3,14,21). The first evidence for the link
between complex I inhibition with subsequent oxidative stress and
the pathogenesis of PD was the recognition that complex I inhibitor
MPTP can cause acute and irreversible parkinsonian symptoms in
humans (22). Subsequently, the
molecular mechanism underling the neurotoxicity of MPTP was also
intensively studied. MPTP is a lipophilic molecule that can rapidly
cross the blood-brain barrier. In the brain it is oxidized to form
the toxic metabolite 1-methyl-4-phenylpyridinium (MPP+)
by type B monoamine oxidase (23). MPP+ is a substrate for
the dopamine transporter and can be taken up selectively into
dopaminergic neurons, accumulating in the mitochondria, where it
inhibits respiration complex I of the mitochondrial electron
transport chain (ETC), leading to the production of ROS (24). Postmortem studies in patients with
idiopathic PD have shown the disease-specific deficits in
mitochondrial complex I activity in the SN (25,26). This change is not limited to the
SN of the brain, and mitochondrial dysfunction and complex I
inhibition have also been reported in peripheral tissues, including
the striatum, cortical brain tissue, blood platelets, fibroblasts,
skeletal muscle and lymphocytes, in PD (27–33). Administration of rotenone, a
well-known complex I inhibitor, was previously shown to cause
selective nigral dopaminergic neuron loss and a significant
reduction in complex I activity, and this toxicity was
significantly attenuated by methylene blue, which functioned as an
alternative electron carrier to bypass complex I blockage, further
supporting the involvement of mitochondrial complex inhibition in
PD pathogenesis (34). Although
the downstream events of mitochondrial dysfunction that cause
neuronal cell death are not completely understood, oxidative stress
caused by ROS production is strongly suggested to be involved in
the neurodegenerative process (3,14,21). Mitochondria are the primary
intracellular source of ROS, and respiratory chain complexes,
especially complex I, are sites of ROS production (35–37). This production of ROS in turn
damages the components of the respiratory chain, particularly
complex I, leading to its further inhibition and greater ROS
production. Normally, the toxicity of ROS can be detoxified by
diverse defence mechanisms; for instance, as the primary ROS
superoxide radicals can be catalyzed into O2 and
H2O2 by the superoxide dismutase, which is
expressed in nearly all living organisms (14), H2O2 can be
catalized by glutathione peroxidase and catalase into
H2O and O2. Oxidative damage occurs when the
balance between the production of ROS and antioxidant defence is
perturbed, and excessive ROS accumulate (38). Excessive ROS can damage all
macromolecules, including lipids, proteins and nucleic acids,
leading to defects in their physiological functions. The central
nervous system (CNS), particularly dopaminergic neurons, is more
prone to oxidative damage, resulting in the degeneration of the
cell and PD pathogenesis (39).
The CNS contains a large number of mitochondria in
order to meet the demands of high levels of energy consumption.
Therefore, the iron content in CNS cells is particularly high,
since numerous mitochondrial enzymes require iron to function,
leading to the greater generation of ROS that contribute to
oxidative stress and subsequently the degeneration of neurons
(40). Iron promotes the
generation of highly reactive oxygen species, resulting in further
oxidative damage, particularly for nigral dopaminergic neurons that
appear to exhibit increased sensitivity to iron-induced oxidative
stress. Studies in postmortem brains of PD patients have shown
higher levels of iron in the SN compared with that in controls
(41,42). The link of oxidative iron
dysregulation with the neurodegenerative process is also supported
by PD animal models, where increased levels of iron and hyroxyl
radicals could be detected in the SN (43). Administration of desferrioxamine,
an iron chelator, significantly decreases the levels of iron in the
brain and protects against neurodegeneration induced by iron and
MPTP in PD mouse models (44),
further supporting the contribution of iron in the
neurodegenerative process of PD. Furthermore, the brain is enriched
in lipids that participate in membrane fluidity and permeability,
and mediate the inflammatory processes and apoptotic signals
(45). The lipids are susceptible
to ROS-mediated damage, particularly polyunsaturated fatty acids,
which are the most prone to lipid peroxidation, resulting in the
structural damage of membranes, consequent neuronal damage and
ultimately, mortality (14).
Oxidative stress-mediated death mechanism has been underlied in the
pathogenesis of PD (39). Higher
levels of malondialdehyde, a production of polyunsaturated fatty
acid peroxidation in oxidative conditions, have also been reported
in SN compared with that in other brain regions in PD (46). The lipid peroxidation marker,
cholesterol lipid hydroperoxide, is also detected as significantly
increased in PD brains compared with that in control subjects
(47). The reinforcement of
peroxidation of polyunsaturated fatty acids to oxidative damage of
dopaminergic neurons is also supported by the elevated levels of
HNE detected in the SN and the cerebrospinal fluid of PD patients
(5,48). HNE is a lipid peroxidation product
contributing to apoptotic cell death via the activation of the
caspase cascade and the subsequent induction of DNA fragmentation
(49). HNE can also reduce the
levels of glutathione (GSH) resulting in the vulnerability of the
neurons to oxidative attack, since GSH is a major non-enzymatic
antioxidant in the CNS (50).
Additionally, other causal factors that are associated with the
vulnerabilities of dopaminergic neurons to oxidative stress have
been well documented (39). Taken
together, these results indicate that the dopaminergic neurons are
more vulnerable to oxidative attack. Although the mechanisms of
oxidative damage in response to oxidative stress causing the
progressive degeneration of dopaminergic neurons in PD is unclear,
events such as mitochondrial dysfunction, the opening of the
mitochondrial permeability transition pore (mPTP),
neuroinflammation and oxidative DNA damage induced by oxidative
stress may serve crucial roles in the process of neurodegeneration.
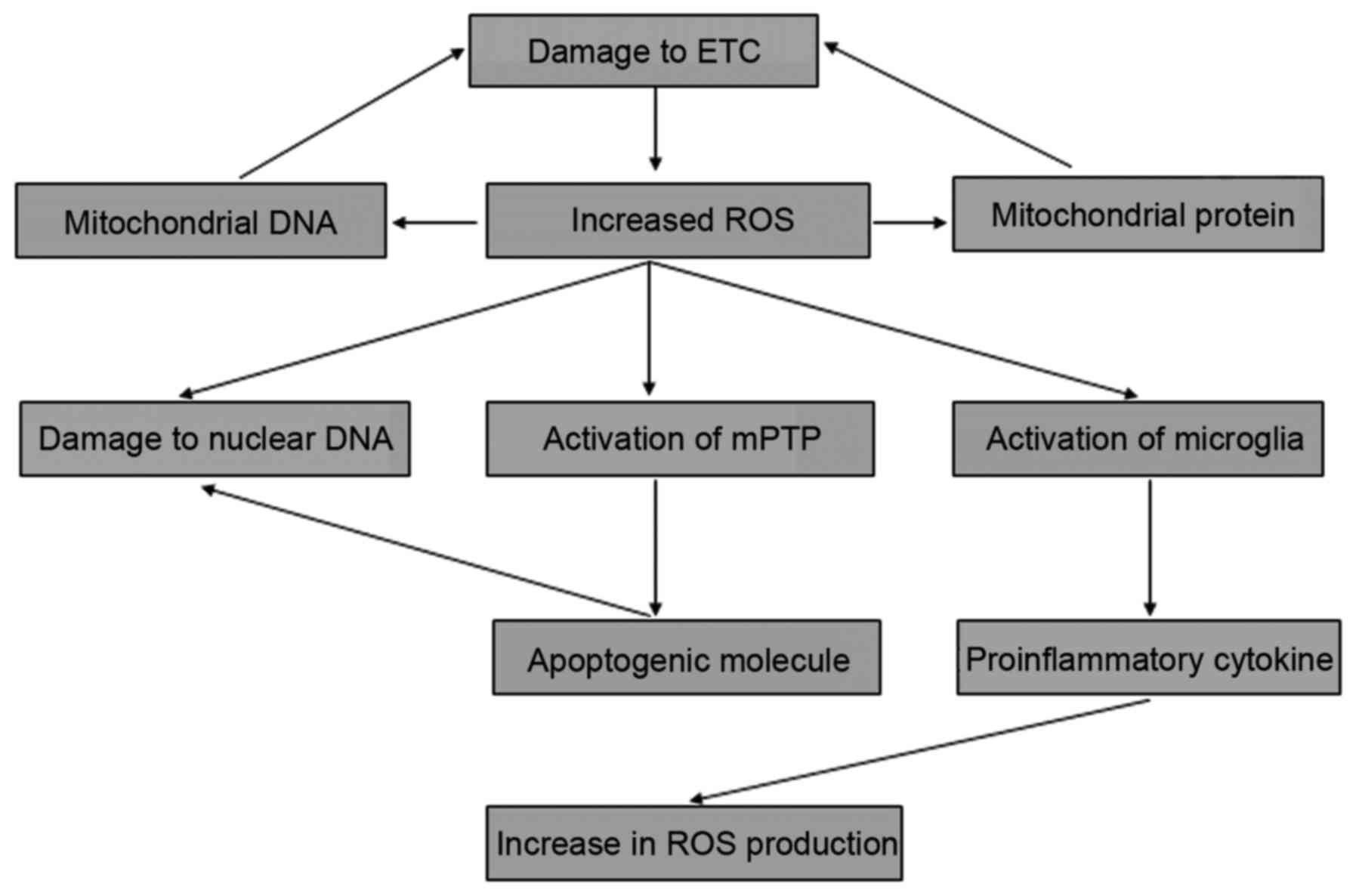
The interaction between these various mechanisms forms a positive
feedback loop that drives uncontrolled pathogenesis conditions,
resulting in the development of PD (Fig. 1).
Mitochondria are the primary intracellular source of
ROS, and for this reason the organelles are frequently exposed to
oxidative stress (51,52). The complex of the ETC is one of
the main cellular targets of ROS-induced oxidative stress, and
oxidative damage of the ETC leads to the inhibition of ATP
production and further generation of ROS (53). Consequently, the vicious cycle
between the defects in the ETC and the subsequent production of ROS
drive the uncontrolled oxidative stress that may play a central
role in the progressive degeneration of dopaminergic neurons and
have been underlied in PD pathogenesis (3). The proteins of the ETC complex are
encoded by mitochondrial and nuclear genomes. Mitochondrial DNA
(mtDNA) encodes 13 proteins that are all ETC complex subunits
involved in oxidative phosphorylation and ATP production (54). Due to the proximity to the ETC
complexes and the lack of histone protein protection, mtDNA is
vulnerable to ROS attack (55).
The damage to mtDNA and subsequent defects in the production of
these proteins could induce mitochondrial dysfunctions that are
implicated in a multitude of diseases or pathological conditions
(53). The accumulation of
defects in mtDNA has been detected in nigral dopaminergic neurons
of elderly individuals and sporadic PD subjects (56–58). Inhibition of mtDNA expression
leads to dysfunction in the respiratory chain in dopaminergic
neurons accompanied by progressive parkinsonism in mouse models
(59). High levels of mtDNA
deletions could be also detected in the midbrain brains of PD
models induced by rotenone, which inhibits ETC, resulting in the
production of ROS (60). These
studies suggest that oxidative ETC and mtDNA damage may be involved
in the degeneration of dopaminergic neurons in oxidative
conditions.
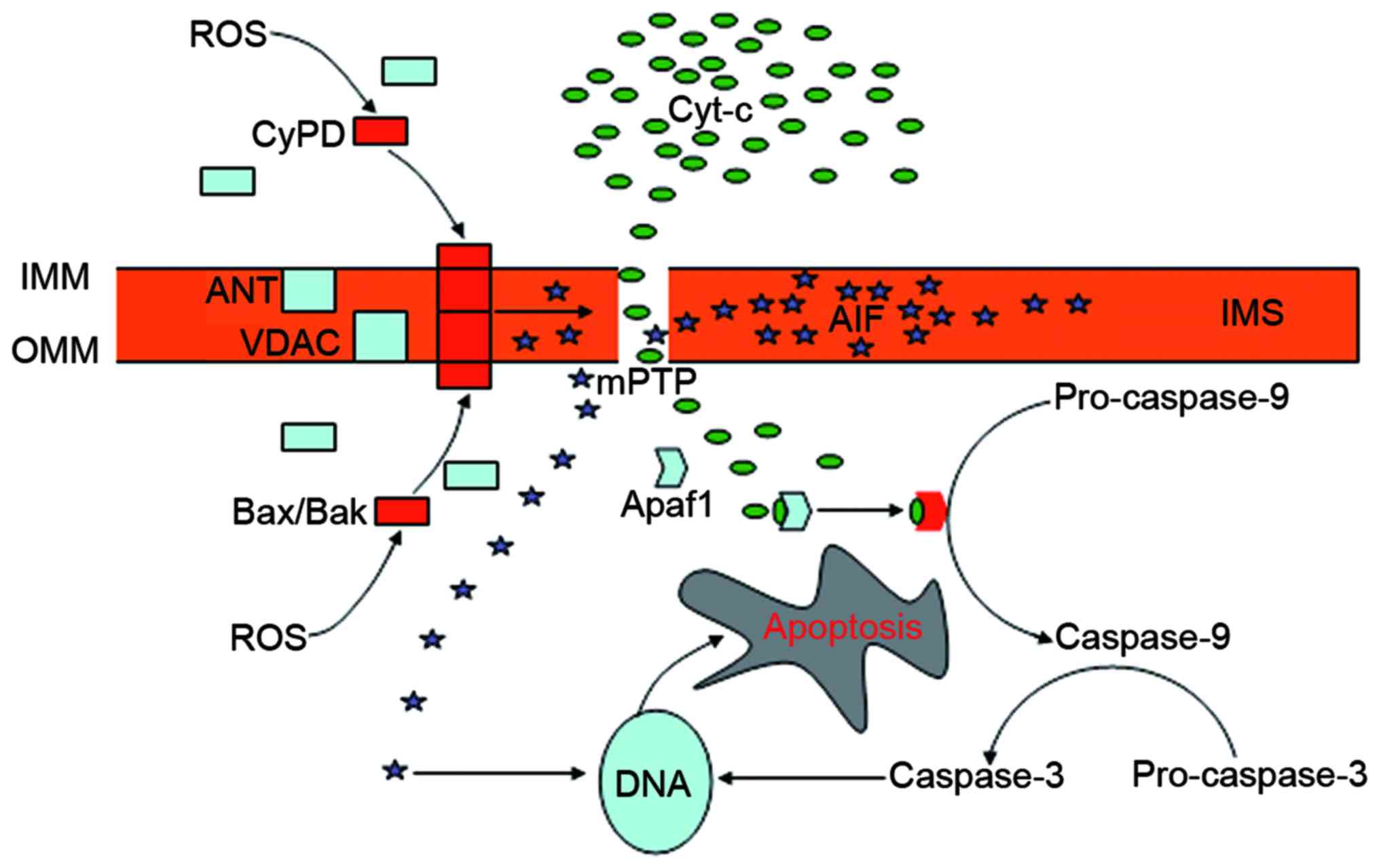
The mPTP is a poly-protein transmembrane channel
that is formed at contact sites between the outer mitochondrial
membrane (OMM) and the IMM. Despite controvery over the structural
constituents of the mPTP, the voltage-dependent anion channel
(VDAC) in the OMM, the adenine nucleotide translocator (ANT) in the
IMM, the B-cell lymphoma-2 (Bcl-2) family proteins in the cytosol
and cyclophilin D (CyPD) in the matrix appear to be essential
components (53). Normally, mPTP
is impermeable, and the VDAC and the ANT are separated by the IMS.
The opening of the mPTP occurs during the interaction of the ANT
with the VDAC, and CyPD serves a crucial role in this process
(75). Generally, CyPD is a
mitochondrial matrix protein. When activated under the condition of
oxidative stress, this protein can be translocated to the IMM where
it interacts with the ANT and changes its conformation, leading to
the binding of the ANT to the VDAC and the subsequent activation of
the mPTP (75). The permeation of
the OMM depends on the Bcl-2 family of proteins, including
Bcl-2-associated X protein (Bax) and Bcl-2 homologous killer (Bak)
(76). These proteins are located
in the cytosol, but translocate and oligomerize into the OMM in
response to oxidative stress. ROS promote the translocation of CyPD
to the IMM, and Bax and Bak to the OMM, thus serving a crucial role
in the opening of the mPTP (53).
The opening causes the collapse of the mitochondrial transmembrane
potential and the disturbance of the H+ gradient across
the IMM, which inhibits the production of ATP and causes further
generation of ROS, ultimately leading to cell death (77,78). The release of mitochondrial
apoptogenic proteins from the opening pore into the cytosol serves
a crucial role in mPTP-mediated cell death, of which cytochrome
c is the most potent apoptotic inducer (79,80). The released cytochrome c
triggers the activation of caspase-9 via the interaction with
apoptotic protease-activating factor 1 (Apaf1) (80). Apaf1 is a cytoplasmic protein that
contains several domains associated with its functional and
regulatory role (81). The
binding of cytochrome c with the special domain of Apaf1
results in the protein forming an oligomeric apoptosome that is
required for the activation of pro-caspase-9. Caspase-9 cleaves
pro-caspase-3 resulting in its activation and the subsequent
cleavage of DNA, the irreversible step toward apoptotic cell death
(79,80). Apoptosis-inducing factor (AIF) is
another apoptotic factor released from the mitochondria into the
cytosol triggering caspase-independent apoptosis (53). AIF is a mitochondrial protein
expressed in the IMS between the IMM and the OMM, and can be
released in response to apoptotic signaling (82). The cytosolic AIF then translocates
to the nucleus where it binds to DNA to instigate chromatin
condensation (83) (Fig. 2). The contribution of other
apoptotic mediators released from the opening of the mPTP to
apoptosis has been well documented (53,84). Several mechanisms have been
revealed to antagonize the opening of the mPTP. The translocation
and oligomerization of Bax and Bak into the OMM, for example, can
be antagonized by the antiapoptotic proteins Bcl-2 and Bcl-xL via
sequestration and inhibition of the activator proteins that are
required for the activation of these pro-apoptosis proteins
(84). Glycogen synthase
kinase-3β (GSK-3β) can be also implicated in the modulation of the
opening of mPTP (85,86). GSK-3β is a Ser/Thr protein kinase
expressed in the cytosol, nucleus and mitochondria of all
eukaryotic cells, and is involved in modulating a wide range of
biological functions (87,88).
GSK-3β activation promotes the upregulation of the levels of Bax
(89,90), and facilitates its mitochondrial
localization by directly phosphorylating Ser163 of this protein
(91). Mitochondrial GSK-3β
modulates the process of oxidative phosphorylation that is
implicated in the production of ROS, the key inducer of the opening
of the mPTP (92). Studies in
cell and animal models of PD have shown that GSK-3β inhibition can
protect dopaminergic neurons from the toxicity of
MPP+/MPTP (93–96),
and the blockage of the opening of the mPTP may be involved
(97). Overall, the oxidative
stress-mediated opening of the mPTP is one of the pathways
responsible for the apoptosis of dopaminergic neurons in PD, and
understanding the mechanisms involved is essential to the
development of effective therapies for neurodegenerative
diseases.
Neuroinflammation is a protective mechanism of the
CNS against infectious insults and injury by activation of the
innate immune system in the brain to destroy and remove the
detrimental agents and injured tissues (98). However, uncontrolled inflammation
can cause excessive cell and tissue damage, ultimately leading to
chronic inflammation and progressive destruction of normal tissue.
The elevated levels of ROS production serve an important role in
the activation of a strong proinflammatory response, and the link
between oxidative stress and inflammation and tissue injury has
been well documented (15). The
inflammatory damage has been underlied in the pathogenesis of
neurodegenerative diseases, including Alzheimer's disease,
Huntington's disease, multiple sclerosis and PD (99–102). The inflammatory response is a
complex process involved in a series of cellular and molecular
processes, including the activation of immune cells, the induction
of certain intracellular signaling pathways and the release of
inflammatory mediators in the brain (103). The activation of microglia is an
initiator in inflammation-mediated neuronal injury. Microglia are
the resident immune cells of the brain that become activated in
response to brain injury or immune challenge (104). Activated microgli are an
important source of superoxide and nitric oxide, triggers of
oxidative and nitrative stress in neurotoxicity; they can also
produce proinflammatory cytokines such as glutamate and tumor
necrosis factor-α (TNF-α), which are potentially toxic agents in
the brain microenvironment (104–106). Inflammation-derived oxidative
stress and cytokine-dependent toxicity have been suggested to be
involved in the loss of dopaminergic neurons in PD (107–109). Postmortem studies revealed the
presence of inducible NO synthase (iNOS) and proinflammatory
cytokines, including TNF-α, interleukin-1β (IL-1β), IL-2 and IL-6,
in the SN of PD patients (110–112). A series of proinflammatory
cytokines, including TNF-α, IL-1α, IL-1β and IL-6, and activated
microglia have also been identified in PD animal models (113–118). As one important cytokine, TNF
serves a crucial role in inflammation-mediated neurodegeneration,
since elavated levels of this cytokine can be persistently detected
in the affected areas of the SN in PD (119). In addition to the induction of
proinflammatory signaling pathways resulting in cell damage
(16), TNF can promote the
secretion of NO by increasing the expression of iNOS in microglia
(120). Furthermore, TNF can
activate NADPH oxidases, leading to the production of ROS, which
contribute to oxidative stress and in turn result in an
uncontrolled inflammatory response (16). The dopaminergic neurons are
particularly susceptible to microglia-mediated toxicity due to the
highest density of microglial cells being distributed in the SN of
the brain (121,122). Microglia activation promotes the
production of proinflammatory cytokines, which cause dopaminergic
nigrostriatal neuron degeneration in MPTP models of PD (123). Animal models of PD have shown
that suppression of the inflammatory response, results in the
protection of neurons from the damage induced by neurotoxin
(124,125). These data indicate a close
association between microglial activation and the degeneration of
dopaminergic neurons in PD pathogenesis. Dopaminergic neuronal
death releases noxious endogenous mediators, including oxidized
proteins, lipids and DNA, in the extracellular space, which can
also activate the microglia, resulting in the release of multiple
proinflammatory cytokines. Proinflammatory factor prodcution
subsequently exacerbates damage to the neurons via oxidative stress
and cytokine toxicity (19),
causing the injured neurons to release further noxious endogenous
mediators and resulting in a continuous inflammatory response
(104). This positive feedback
loop between activated microglia and damaged neurons forms a
neurotoxic vicious cycle and an uncontrolled, prolonged
inflammatory process, and is hypothesized to be partially
responsible for the gradual loss of dopaminergic neurons in PD
(126,127). Thereby, inhibiting the
inflammatory response generated by microglia activation may be show
benefits in neurodegenerative conditions.
DNA integrity is required for cell survival. Under
physiopathologycial conditions, DNA is often subjected to damage by
endogenous and environmental toxic agents, and unrepaired DNA
damage leads to genetic and protein instability, and subsequent
cell death. Nucleic acids, RNA and DNA, are particularly
susceptible to oxidative damage, with DNA damage being a key
contributor to a number of different diseases (128). Dopaminergic neurons are
frequently exposed to ROS attack, resulting in DNA oxidative damage
due to the high levels of ROS production. In PD, increased levels
of 8-hydroxyguanine, the marker of DNA oxidative damage, have been
detected selectively in the SN (7,129). The number of strand breaks in
nuclear DNA have also been reported to be elevated in the SN
compared with that in other areas of the brain, and evidence of
alterations to DNA conformation and stability in the SN has also
been documented (129). mDNA is
more susceptible to oxidative damage than nuclear DNA (130). Postmortem studies in the brains
of patients with PD have shown increased levels of mtDNA damage
marker abasic sites in the SN. Abasic sites were also shown in
brain tissue from PD mouse models treated with the neuronal toxin
rotenone, which causes oxidative stress by inhibiting the
mitochondrial complex (131).
Abasic sites are DNA segments that have lost a purine or pyrimidine
base, leading to the blockage of the polymerase during the
replication and transcription of DNA (132). These studies demonstrate that
dopaminergic neuron injury could be ascribed to the oxidative
damage of nuclear DNA and mtDNA, which alters its coding properties
or interferes with normal metabolic function, and subsequently
results in cell death (128).
ROS attack on DNA may be reversible or irreversible, dependent on
the efficiency of its repair. Effective repair of damaged DNA is
required to preserve its integrity and maintain the viability of
the cell, particularly in dopaminergic neurons. A number of
cellular mechanisms are devoted to the repair of DNA (133). A previous study determined an
association between variants in DNA repair and an increased risk of
PD (134). As a critical
regulatory protein for DNA repair, proliferating cell nuclear
antigen (PCNA) serves a central role in the repair of damaged DNA
in a variety of pathological conditions via the interaction with
numerous enzymes and regulatory proteins (135,136). PCNA-dependent repair of DNA has
been reported to contribute to the reserve in the DNA integrity of
the dopaminergic neurons under oxidative conditions (137,138). We previously studied in
vitro the mechanism of the degeneration of dopaminergic neurons
in PC12 cells induced by MPP+, which causes ROS
production by inhibiting complex I, leading to oxidative DNA damage
and subsequent neuronal cell death. The results showed that
MPP+ treatment significantly reduced PCNA expression in
the neuronal PC12 cells and increased the level of cell apoptosis.
The reversal of PCNA expression markedly promoted cell survival in
PC12 cells with MPP+-induced neurotoxicity, supporting
the hypothesis of the PCNA-dependent apoptotic pathway as a
potential molecular mechanism in PD pathogenesis associated with
DNA damage in oxidative conditions (139). These results may provide a
potential target for the reversal of oxidative DNA damage-mediated
neuronal death in PD pathogenesis.
The pathogenesis and progression of PD are complex
and involved in a series of diverse mechanisms that alone or
together contribute to the damage and gradual loss of dopaminergic
neurons. Oxidative stress appears to serve a central role in the
neurodegenerative process, since dopaminergic neurons are
frequently exposed to oxidative stress, which triggers a cascade of
events, including mitochondrial dysfunction, impairment of nuclear
DNA and mtDDA, and neuroinflammation, which in turn cause more ROS
production. The formation of this vicious cycle may serve a central
role in the progressive degeneration of dopaminergic neurons in PD,
therefore, the inhibition of the production of ROS and the blockage
of the interactions in the signaling pathway may alleviate the
severity and development of the disease. This require further
elucidation.
This study was supported by Technology Research of
the Education Department of Jilin Province, China (grant no.
JJ-KH20170047K) to Ji-Dong Guo and the Department of Health of
Jilin Province, China (grant no. 2016J078) to Ji-Dong Guo.
The authors declare that they have no competing
interests.
|
1
|
Sulzer D and Surmeier DJ: Neuronal
vulnerability, pathogenesis, and Parkinson's disease. Mov Disord.
28:715–724. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shukla V, Mishra SK and Pant HC: Oxidative
stress in neurodegeneration. Adv Pharmacol Sci.
2011:5726342011.PubMed/NCBI
|
|
3
|
Kim GH, Kim JE, Rhie SJ and Yoon S: The
role of oxidative stress in neurodegenerative diseases. Exp
Neurobiol. 24:325–340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jenner P: Oxidative stress in Parkinson's
disease. Ann Neurol. 53(Suppl 3): S26–S38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoritaka A, Hattori N, Uchida K, Tanaka M,
Stadtman ER and Mizuno Y: Immunohistochemical detection of
4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl
Acad Sci USA. 93:2696–2701. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Floor E and Wetzel MG: Increased protein
oxidation in human substantia nigra pars compacta in comparison
with basal ganglia and prefrontal cortex measured with an improved
dinitrophenyl-hydrazine assay. J Neurochem. 70:268–275. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alam ZI, Jenner A, Daniel SE, Lees AJ,
Cairns N, Marsden CD, Jenner P and Halliwell B: Oxidative DNA
damage in the parkinsonian brain: An apparent selective increase in
8-hydroxyguanine levels in substantia nigra. J Neurochem.
69:1196–1203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Isobe C, Abe T and Terayama Y: Levels of
reduced and oxidized coenzyme Q-10 and 8-hydroxy-2′-deoxyguanosine
in the cerebrospinal fluid of patients with living Parkinson's
disease demonstrate that mitochondrial oxidative damage and/or
oxidative DNA damage contributes to the neurodegenerative process.
Neurosci Lett. 469:159–163. 2010. View Article : Google Scholar
|
|
9
|
Callio J, Oury TD and Chu CT: Manganese
superoxide dismutase protects against 6-hydroxydopamine injury in
mouse brains. J Biol Chem. 280:18536–18542. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Perier C, Bové J, Vila M and Przedborski
S: The rotenone model of Parkinson's disease. Trends Neurosci.
26:345–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burns RS, Chiueh CC, Markey SP, Ebert MH,
Jacobowitz DM and Kopin IJ: A primate model of parkinsonism:
Selective destruction of dopaminergic neurons in the pars compacta
of the substantia nigra by
N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci
USA. 80:4546–4550. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhandary B, Marahatta A, Kim HR and Chae
HJ: An involvement of oxidative stress in endoplasmic reticulum
stress and its associated diseases. Int J Mol Sci. 14:434–456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Federico A, Cardaioli E, Da Pozzo P,
Formichi P, Gallus GN and Radi E: Mitochondria, oxidative stress
and neurodegeneration. J Neurol Sci. 322:254–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sanders LH and Greenamyre JT: Oxidative
damage to macromolecules in human Parkinson disease and the
rotenone model. Free Radic Biol Med. 62:111–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Peterson LJ and Flood PM: Oxidative stress
and microglial cells in Parkinson's disease. Mediators Inflamm.
2012:4012642012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fischer R and Maier O: Interrelation of
oxidative stress and inflammation in neurodegenerative disease:
Role of TNF. Oxid Med Cell Longev. 2015:6108132015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Andreyev AY, Kushnareva YE and Starkov AA:
Mitochondrial metabolism of reactive oxygen species. Biochemistry
(Mosc). 70:200–214. 2005. View Article : Google Scholar
|
|
18
|
Cadenas E and Davies KJ: Mitochondrial
free radical generation, oxidative stress, and aging. Free Radic
Biol Med. 29:222–230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rush JD and Koppenol WH: Oxidizing
intermediates in the reaction of ferrous EDTA with hydrogen
peroxide. Reactions with organic molecules and ferrocytochrome c. J
Biol Chem. 261:6730–6733. 1986.PubMed/NCBI
|
|
20
|
Gutteridge JM: Superoxide-dependent
formation of hydroxyl radicals from ferric-complexes and hydrogen
peroxide: An evaluation of fourteen iron chelators. Free Radic Res
Commun. 9:119–125. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Subramaniam SR and Chesselet MF:
Mitochondrial dysfunction and oxidative stress in Parkinson's
disease. Prog Neurobiol. 106–107:17–32. 2013. View Article : Google Scholar
|
|
22
|
Langston JW, Ballard P, Tetrud JW and
Irwin I: Chronic Parkinsonism in humans due to a product of
meperidine-analog synthesis. Science. 219:979–980. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiba K, Trevor A and Castagnoli N Jr:
Metabolism of the neurotoxic tertiary amine, MPTP, by brain
monoamine oxidase. Biochem Biophys Res Commun. 120:574–578. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Javitch JA, D'Amato RJ, Strittmatter SM
and Snyder SH: Parkinsonism-inducing neurotoxin,
N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: Uptake of the met
abolite N-methyl-4-phenylpyridine by dopamine neurons explains
selective toxicity. Proc Natl Acad Sci USA. 82:2173–2177. 1985.
View Article : Google Scholar
|
|
25
|
Schapira AH, Cooper JM, Dexter D, Clark
JB, Jenner P and Marsden CD: Mitochondrial complex I deficiency in
Parkinson's disease. J Neurochem. 54:823–827. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hattori N, Tanaka M, Ozawa T and Mizuno Y:
Immunohistochemical studies on complexes I, II, III, and IV of
mitochondria in Parkinson's disease. Ann Neurol. 30:563–571. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizuno Y, Ohta S, Tanaka M, Takamiya S,
Suzuki K, Sato T, Oya H, Ozawa T and Kagawa Y: Deficiencies in
complex I subunits of the respiratory chain in Parkinson's disease.
Biochem Biophys Res Commun. 163:1450–1455. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Parker WD Jr, Parks JK and Swerdlow RH:
Complex I deficiency in Parkinson's disease frontal cortex. Brain
Res. 1189:215–218. 2008. View Article : Google Scholar
|
|
29
|
Krige D, Carroll MT, Cooper JM, Marsden CD
and Schapira AH; The Royal Kings and Queens Parkinson Disease
Research Group: Platelet mitochondrial function in Parkinson's
disease. Ann Neurol. 32:782–788. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Haas RH, Nasirian F, Nakano K, Ward D, Pay
M, Hill R and Shults CW: Low platelet mitochondrial complex I and
complex II/III activity in early untreated Parkinson's disease. Ann
Neurol. 37:714–722. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mytilineou C, Werner P, Molinari S, Di
Rocco A, Cohen G and Yahr MD: Impaired oxidative decarboxylation of
pyruvate in fibroblasts from patients with Parkinson's disease. J
Neural Transm Park Dis Dement Sect. 8:223–228. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blin O, Desnuelle C, Rascol O, Borg M,
Peyro Saint Paul H, Azulay JP, Billé F, Figarella D, Coulom F,
Pellissier JF, et al: Mitochondrial respiratory failure in skeletal
muscle from patients with Parkinson's disease and multiple system
atrophy. J Neurol Sci. 125:95–101. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yoshino H, Nakagawa-Hattori Y, Kondo T and
Mizuno Y: Mitochondrial complex I and II activities of lymphocytes
and platelets in Parkinson's disease. J Neural Transm Park Dis
Dement Sect. 4:27–34. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan
LJ, Ju X, Liu R, Qian H, Marvin MA, et al: Alternative
mitochondrial electron transfer as a novel strategy for
neuroprotection. J Biol Chem. 286:16504–16515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kudin AP, Debska-Vielhaber G and Kunz WS:
Characterization of superoxide production sites in isolated rat
brain and skeletal muscle mitochondria. Biomed Pharmacother.
59:163–168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kudin AP, Bimpong-Buta NY, Vielhaber S,
Elger CE and Kunz WS: Characterization of superoxide-producing
sites in isolated brain mitochondria. J Biol Chem. 279:4127–4135.
2004. View Article : Google Scholar
|
|
37
|
Kussmaul L and Hirst J: The mechanism of
superoxide production by NADH:ubiquinone oxidoreductase (complex I)
from bovine heart mitochondria. Proc Natl Acad Sci USA.
103:7607–7612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Morán M, Moreno-Lastres D, Marín-Buera L,
Arenas J, Martín MA and Ugalde C: Mitochondrial respiratory chain
dysfunction: Implications in neurodegeneration. Free Radic Biol
Med. 53:595–609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson's disease. J Parkinsons Dis.
3:461–491. 2013.PubMed/NCBI
|
|
40
|
Morató L, Bertini E, Verrigni D, Ardissone
A, Ruiz M, Ferrer I, Uziel G and Pujol A: Mitochondrial dysfunction
in central nervous system white matter disorders. Glia.
62:1878–1894. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sian-Hülsmann J, Mandel S, Youdim MB and
Riederer P: The relevance of iron in the pathogenesis of
Parkinson's disease. J Neurochem. 118:939–957. 2011. View Article : Google Scholar
|
|
42
|
Kosta P, Argyropoulou MI, Markoula S and
Konitsiotis S: MRI evaluation of the basal ganglia size and iron
content in patients with Parkinson's disease. J Neurol. 253:26–32.
2006. View Article : Google Scholar
|
|
43
|
Sziráki I, Mohanakumar KP, Rauhala P, Kim
HG, Yeh KJ and Chiueh CC: Manganese: A transition metal protects
nigrostriatal neurons from oxidative stress in the iron-induced
animal model of parkinsonism. Neuroscience. 85:1101–1111. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lan J and Jiang DH: Desferrioxamine and
vitamin E protect against iron and MPTP-induced neurodegeneration
in mice. J Neural Transm Vienna. 104:469–481. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ruipérez V, Darios F and Davletov B:
Alpha-synuclein, lipids and Parkinson's disease. Prog Lipid Res.
49:420–428. 2010. View Article : Google Scholar
|
|
46
|
Dexter DT, Carter CJ, Wells FR, Javoy-Agid
F, Agid Y, Lees A, Jenner P and Marsden CD: Basal lipid
peroxidation in substantia nigra is increased in Parkinson's
disease. J Neurochem. 52:381–389. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dexter DT, Holley AE, Flitter WD, Slater
TF, Wells FR, Daniel SE, Lees AJ, Jenner P and Marsden CD:
Increased levels of lipid hydroperoxides in the parkinsonian
substantia nigra: An HPLC and ESR study. Mov Disord. 9:92–97. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Montine KS, Quinn JF, Zhang J, Fessel JP,
Roberts LJ II, Morrow JD and Montine TJ: Isoprostanes and related
products of lipid peroxidation in neurodegenerative diseases. Chem
Phys Lipids. 128:117–124. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu W, Kato M, Akhand AA, Hayakawa A,
Suzuki H, Miyata T, Kurokawa K, Hotta Y, Ishikawa N and Nakashima
I: 4-hydroxynonenal induces a cellular redox status-related
activation of the caspase cascade for apoptotic cell death. J Cell
Sci. 113:635–641. 2000.PubMed/NCBI
|
|
50
|
Schmidt H, Grune T, Müller R, Siems WG and
Wauer RR: Increased levels of lipid peroxidation products
malondialdehyde and 4-hydroxynonenal after perinatal hypoxia.
Pediatr Res. 40:15–20. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Starkov AA: The role of mitochondria in
reactive oxygen species metabolism and signaling. Ann NY Acad Sci.
1147:37–52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar
|
|
53
|
Camara AK, Lesnefsky EJ and Stowe DF:
Potential therapeutic benefits of strategies directed to
mitochondria. Antioxid Redox Signal. 13:279–347. 2010. View Article : Google Scholar :
|
|
54
|
Wallace DC: A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: A dawn for
evolutionary medicine. Annu Rev Genet. 39:359–407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Levy RJ and Deutschman CS: Deficient
mitochondrial biogenesis in critical illness: Cause, effect, or
epiphenomenon. Crit Care. 11:1582007. View
Article : Google Scholar
|
|
56
|
Kraytsberg Y, Kudryavtseva E, McKee AC,
Geula C, Kowall NW and Khrapko K: Mitochondrial DNA deletions are
abundant and cause functional impairment in aged human substantia
nigra neurons. Nat Genet. 38:518–520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bender A, Krishnan KJ, Morris CM, Taylor
GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock
T, et al: High levels of mitochondrial DNA deletions in substantia
nigra neurons in aging and Parkinson disease. Nat Genet.
38:515–517. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
58
|
Elstner M, Müller SK, Leidolt L, Laub C,
Krieg L, Schlaudraff F, Liss B, Morris C, Turnbull DM, Masliah E,
et al: Neuromelanin, neurotransmitter status and brainstem location
determine the differential vulnerability of catecholaminergic
neurons to mitochondrial DNA deletions. Mol Brain. 4:432011.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ekstrand MI, Terzioglu M, Galter D, Zhu S,
Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS,
Trifunovic A, et al: Progressive parkinsonism in mice with
respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci
USA. 104:1325–1330. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tanner CM, Kamel F, Ross GW, Hoppin JA,
Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR,
et al: Rotenone, paraquat, and Parkinson's disease. Environ Health
Perspect. 119:866–872. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Takeuchi A, Kim B and Matsuoka S: The
destiny of Ca(2+) released by mitochondria. J Physiol Sci.
65:11–24. 2015. View Article : Google Scholar
|
|
62
|
Jo H, Noma A and Matsuoka S:
Calcium-mediated coupling between mitochondrial substrate
dehydrogenation and cardiac workload in single guinea-pig
ventricular myocytes. J Mol Cell Cardiol. 40:394–404. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Satrústegui J, Pardo B and Del Arco A:
Mitochondrial transporters as novel targets for intracellular
calcium signaling. Physiol Rev. 87:29–67. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kirichok Y, Krapivinsky G and Clapham DE:
The mitochondrial calcium uniporter is a highly selective ion
channel. Nature. 427:360–364. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Alderton WK, Cooper CE and Knowles RG:
Nitric oxide synthases: Structure, function and inhibition. Biochem
J. 357:593–615. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jekabsone A, Ivanoviene L, Brown GC and
Borutaite V: Nitric oxide and calcium together inactivate
mitochondrial complex I and induce cytochrome c release. J Mol Cell
Cardiol. 35:803–809. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Gandhi S, Wood-Kaczmar A, Yao Z,
Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi
SJ, Wood NW, et al: INK1-associated Parkinson's disease is caused
by neuronal vulnerability to calcium-induced cell death. Mol Cell.
33:627–638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Surmeier DJ, Guzman JN, Sanchez-Padilla J
and Goldberg JA: The origins of oxidant stress in Parkinson's
disease and therapeutic strategies. Antioxid Redox Signal.
14:1289–1301. 2011. View Article : Google Scholar :
|
|
69
|
Muravchick S and Levy RJ: Clinical
implications of mitochondrial dysfunction. Anesthesiology.
105:819–837. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
O'Rourke B: Pathophysiological and
protective roles of mitochondrial ion channels. J Physiol.
529:23–36. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Di Lisa F and Bernardi P: A CaPful of
mechanisms regulating the mitochondrial permeability transition. J
Mol Cell Cardiol. 46:775–780. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jones SP, Teshima Y, Akao M and Marbán E:
Simvastatin attenuates oxidant-induced mitochondrial dysfunction in
cardiac myocytes. Circ Res. 93:697–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Vila M and Przedborski S: Targeting
programmed cell death in neurodegenerative diseases. Nat Rev
Neurosci. 4:365–375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Perier C, Tieu K, Guégan C, Caspersen C,
Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S
and Vila M: Complex I deficiency primes Bax-dependent neuronal
apoptosis through mitochondrial oxidative damage. Proc Natl Acad
Sci USA. 102:19126–19131. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Halestrap AP and Brenner C: The adenine
nucleotide trans-locase: A central component of the mitochondrial
permeability transition pore and key player in cell death. Curr Med
Chem. 10:1507–1525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Theruvath TP, Snoddy MC, Zhong Z and
Lemasters JJ: Mitochondrial permeability transition in liver
ischemia and reperfusion: Role of c-Jun N-terminal kinase 2.
Transplantation. 85:1500–1504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Theruvath TP, Zhong Z, Pediaditakis P,
Ramshesh VK, Currin RT, Tikunov A, Holmuhamedov E and Lemasters JJ:
Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate
storage/reperfusion injury after rat liver transplantation through
suppression of the mitochondrial permeability transition.
Hepatology. 47:236–246. 2008. View Article : Google Scholar
|
|
79
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex Initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Liu X, Kim CN, Yang J, Jemmerson R and
Wang X: Induction of apoptotic program in cell-free extracts:
Requirement for dATP and cytochrome c. Cell. 86:147–157. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kim GT, Chun YS, Park JW and Kim MS: Role
of apoptosis-inducing factor in myocardial cell death by
ischemia-reperfusion. Biochem Biophys Res Commun. 309:619–624.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Susin SA, Lorenzo HK, Zamzami N, Marzo I,
Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler
M, et al: Molecular characterization of mitochondrial
apoptosis-inducing factor. Nature. 397:441–446. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Candé C, Cecconi F, Dessen P and Kroemer
G: Apoptosis-inducing factor (AIF): Key to the conserved
caspase-independent pathways of cell death? J Cell Sci.
115:4727–4734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Martin LJ: Biology of mitochondria in
neurodegenerative diseases. Prog Mol Biol Transl Sci. 107:355–415.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Obame FN, Plin-Mercier C, Assaly R, Zini
R, Dubois-Randé JL, Berdeaux A and Morin D: Cardioprotective effect
of morphine and a blocker of glycogen synthase kinase 3 beta,
SB216763 [3-(2,4-dichlorophenyl)-4(1-methyl-1H-indol-3-yl)-1H-pyrro
le-2,5-dione], via inhibition of the mitochondrial permeability
transition pore. J Pharmacol Exp Ther. 326:252–258. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Nishihara M, Miura T, Miki T, Tanno M,
Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y and Shimamoto K:
Modulation of the mitochondrial permeability transition pore
complex In GSK-3beta-mediated myocardial protection. J Mol Cell
Cardiol. 43:564–570. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jope RS and Johnson GV: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kockeritz L, Doble B, Patel S and Woodgett
JR: Glycogen synthase kinase-3 - an overview of an over-achieving
protein kinase. Curr Drug Targets. 7:1377–1388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Tan J, Zhuang L, Leong HS, Iyer NG, Liu ET
and Yu Q: Pharmacologic modulation of glycogen synthase
kinase-3beta promotes p53-dependent apoptosis through a direct
Bax-mediated mitochondrial pathway in colorectal cancer cells.
Cancer Res. 65:9012–9020. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Watcharasit P, Bijur GN, Song L, Zhu J,
Chen X and Jope RS: Glycogen synthase kinase-3beta (GSK3beta) binds
to and promotes the actions of p53. J Biol Chem. 278:48872–48879.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Linseman DA, Butts BD, Precht TA, Phelps
RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML and
Heidenreich KA: Glycogen synthase kinase-3beta phosphorylates Bax
and promotes its mitochondrial localization during neuronal
apoptosis. J Neurosci. 24:9993–10002. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
King TD, Clodfelder-Miller B, Barksdale KA
and Bijur GN: Unregulated mitochondrial GSK3beta activity results
in NADH: Ubiquinone oxidoreductase deficiency. Neurotox Res.
14:367–382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wang W, Yang Y, Ying C, Li W, Ruan H, Zhu
X, You Y, Han Y, Chen R, Wang Y, et al: Inhibition of glycogen
synthase kinase-3beta protects dopaminergic neurons from MPTP
toxicity. Neuropharmacology. 52:1678–1684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
King TD, Bijur GN and Jope RS: Caspase-3
activation induced by inhibition of mitochondrial complex I is
facilitated by glycogen synthase kinase-3beta and attenuated by
lithium. Brain Res. 919:106–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Petit-Paitel A, Brau F, Cazareth J and
Chabry J: Involvement of cytosolic and mitochondrial GSK-3beta in
mitochondrial dysfunction and neuronal cell death of
MPTP/MPP-treated neurons. PLoS One. 4:e54912009. View Article : Google Scholar
|
|
96
|
Youdim MB and Ar raf Z: Prevention of MPTP
(N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic
neurotoxicity in mice by chronic lithium: Involvements of Bcl-2 and
Bax. Neuropharmacology. 46:1130–1140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Li DW, Liu ZQ, Chen W, Yao M and Li GR:
Association of glycogen synthase kinase-3β with Parkinson's disease
(Review). Mol Med Rep. 9:2043–2050. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Spencer JP, Vafeiadou K, Williams RJ and
Vauzour D: Neuroinflammation: Modulation by flavonoids and
mechanisms of action. Mol Aspects Med. 33:83–97. 2012. View Article : Google Scholar
|
|
99
|
Pimplikar SW: Neuroinflammation in
Alzheimer's disease: From pathogenesis to a therapeutic target. J
Clin Immunol. 34(Suppl 1): S64–S69. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Möller T: Neuroinflammation in
Huntington's disease. J Neural Transm Vienna. 117:1001–1008. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Frohman EM, Racke MK and Raine CS:
Multiple sclerosis - the plaque and its pathogenesis. N Engl J Med.
354:942–955. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Hirsch EC, Vyas S and Hunot S:
Neuroinflammation in Parkinson's disease. Parkinsonism Relat
Disord. 18(Suppl 1): S210–S212. 2012. View Article : Google Scholar
|
|
103
|
Zhang F and Jiang L: Neuroinflammation in
Alzheimer's disease. Neuropsychiatr Dis Treat. 11:243–256. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: Uncovering the molecular
mechanisms. Nat Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar
|
|
105
|
Ceulemans AG, Zgavc T, Kooijman R,
Hachimi-Idrissi S, Sarre S and Michotte Y: The dual role of the
neuroinflammatory response after ischemic stroke: Modulatory
effects of hypothermia. J Neuroinflammation. 7:742010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chéret C, Gervais A, Lelli A, Colin C,
Amar L, Ravassard P, Mallet J, Cumano A, Krause KH and Mallat M:
Neurotoxic activation of microglia is promoted by a nox1-dependent
NADPH oxidase. J Neurosci. 28:12039–12051. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
McGeer PL and McGeer EG: Glial reactions
in Parkinson's disease. Mov Disord. 23:474–483. 2008. View Article : Google Scholar
|
|
108
|
Frankola KA, Greig NH, Luo W and Tweedie
D: Targeting TNF-α to elucidate and ameliorate neuroinflammation in
neurodegenerative diseases. CNS Neurol Disord Drug Targets.
10:391–403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Qian L, Flood PM and Hong JS:
Neuroinflammation is a key player in Parkinson's disease and a
prime target for therapy. J Neural Transm Vienna. 117:971–979.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Hunot S, Dugas N, Faucheux B, Hartmann A,
Tardieu M, Debré P, Agid Y, Dugas B and Hirsch EC:
FcepsilonRII/CD23 is expressed in Parkinson's disease and induces,
in vitro, production of nitric oxide and tumor necrosis
factor-alpha in glial cells. J Neurosci. 19:3440–3447.
1999.PubMed/NCBI
|
|
111
|
Mogi M, Harada M, Narabayashi H, Inagaki
H, Minami M and Nagatsu T: Interleukin (IL)-1 beta, IL-2, IL-4,
IL-6 and transforming growth factor-alpha levels are elevated in
ventricular cerebrospinal fluid in juvenile parkinsonism and
Parkinson's disease. Neurosci Lett. 211:13–16. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hirsch EC, Breidert T, Rousselet E, Hunot
S, Hartmann A and Michel PP: The role of glial reaction and
inflammation in Parkinson's disease. Ann NY Acad Sci. 991:214–228.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Herrera AJ, Castaño A, Venero JL, Cano J
and Machado A: The single intranigral injection of LPS as a new
model for studying the selective effects of inflammatory reactions
on dopaminergic system. Neurobiol Dis. 7:429–447. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Iravani MM, Leung CC, Sadeghian M, Haddon
CO, Rose S and Jenner P: The acute and the long-term effects of
nigral lipopoly-saccharide administration on dopaminergic
dysfunction and glial cell activation. Eur J Neurosci. 22:317–330.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Iravani MM, Sadeghian M, Leung CC, Jenner
P and Rose S: Lipopolysaccharide-induced nigral inflammation leads
to increased IL-1β tissue content and expression of astrocytic
glial cell line-derived neurotrophic factor. Neurosci Lett.
510:138–142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Kohutnicka M, Lewandowska E,
Kurkowska-Jastrzebska I, Członkowski A and Członkowska A:
Microglial and astrocytic involvement in a murine model of
Parkinson's disease induced by
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).
Immunopharmacology. 39:167–180. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Członkowska A, Kohutnicka M,
Kurkowska-Jastrzebska I and Członkowski A: Microglial reaction in
MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced
Parkinson's disease mice model. Neurodegeneration. 5:137–143. 1996.
View Article : Google Scholar
|
|
118
|
Sriram K, Miller DB and O'Callaghan JP:
Minocycline attenuates microglial activation but fails to mitigate
striatal dopaminergic neurotoxicity: Role of tumor necrosis
factor-alpha. J Neurochem. 96:706–718. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Mogi M, Harada M, Riederer P, Narabayashi
H, Fujita K and Nagatsu T: Tumor necrosis factor-alpha (TNF-alpha)
increases both in the brain and in the cerebrospinal fluid from
parkinsonian patients. Neurosci Lett. 165:208–210. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Mir M, Tolosa L, Asensio VJ, Lladó J and
Olmos G: Complementary roles of tumor necrosis factor alpha and
interferon gamma in inducible microglial nitric oxide generation. J
Neuroimmunol. 204:101–109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Lawson LJ, Perry VH, Dri P and Gordon S:
Heterogeneity in the distribution and morphology of microglia in
the normal adult mouse brain. Neuroscience. 39:151–170. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu
B and Hong JS: Regional difference in susceptibility to
lipopolysac-charide-induced neurotoxicity in the rat brain: Role of
microglia. J Neurosci. 20:6309–6316. 2000.PubMed/NCBI
|
|
123
|
Liberatore GT, Jackson-Lewis V, Vukosavic
S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM and
Przedborski S: Inducible nitric oxide synthase stimulates
dopaminergic neurodegeneration in the MPTP model of Parkinson
disease. Nat Med. 5:1403–1409. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhang F, Qian L, Flood PM, Shi JS, Hong JS
and Gao HM: Inhibition of IkappaB kinase-beta protects dopamine
neurons against lipopolysaccharide-induced neurotoxicity. J
Pharmacol Exp Ther. 333:822–833. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Lofrumento DD, Nicolardi G, Cianciulli A,
De Nuccio F, La Pesa V, Carofiglio V, Dragone T, Calvello R and
Panaro MA: Neuroprotective effects of resveratrol in an MPTP mouse
model of Parkinson's-like disease: Possible role of SOCS-1 in
reducing pro-inflammatory responses. Innate Immun. 20:249–260.
2014. View Article : Google Scholar
|
|
126
|
Gao HM, Zhou H, Zhang F, Wilson BC, Kam W
and Hong JS: HMGB1 acts on microglia Mac1 to mediate chronic
neuroinflammation that drives progressive neurodegeneration. J
Neurosci. 31:1081–1092. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Gao HM and Hong JS: Why neurodegenerative
diseases are progressive: Uncontrolled inflammation drives disease
progression. Trends Immunol. 29:357–365. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Evans MD, Dizdaroglu M and Cooke MS:
Oxidative DNA damage and disease: Induction, repair and
significance. Mutat Res. 567:1–61. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Hegde ML, Gupta VB, Anitha M, Harikrishna
T, Shankar SK, Muthane U, Subba Rao K and Jagannatha Rao KS:
Studies on genomic DNA topology and stability in brain regions of
Parkinson's disease. Arch Biochem Biophys. 449:143–156. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Maynard S, de Souza-Pinto NC,
Scheibye-Knudsen M and Bohr VA: Mitochondrial base excision repair
assays. Methods. 51:416–425. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Sanders LH, McCoy J, Hu X, Mastroberardino
PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B and Greenamyre JT:
Mitochondrial DNA damage: Molecular marker of vulnerable nigral
neurons in Parkinson's disease. Neurobiol Dis. 70:214–223. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wilson DM III and Barsky D: The major
human abasic endo-nuclease: Formation, consequences and repair of
abasic lesions in DNA. Mutat Res. 485:283–307. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Larsen E, Reite K, Nesse G, Gran C,
Seeberg E and Klungland A: Repair and mutagenesis at oxidized DNA
lesions in the developing brain of wild-type and Ogg1−/−
mice. Oncogene. 25:2425–2432. 2006. View Article : Google Scholar
|
|
134
|
Gencer M, Dasdemir S, Cakmakoglu B,
Cetinkaya Y, Varlibas F, Tireli H, Kucukali CI, Ozkok E and Aydin
M: DNA repair genes in Parkinson's disease. Genet Test Mol
Biomarkers. 16:504–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Mailand N, Gibbs-Seymour I and
Bekker-Jensen S: Regulation of PCNA-protein interactions for genome
stability. Nat Rev Mol Cell Biol. 14:269–282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Moldovan GL, Pfander B and Jentsch S:
PCNA, the maestro of the replication fork. Cell. 129:665–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Burkovics P, Hajdú I, Szukacsov V, Unk I
and Haracska L: Role of PCNA-dependent stimulation of
3′-phosphodiesterase and 3′-5′ exonuclease activities of human Ape2
in repair of oxidative DNA damage. Nucleic Acids Res. 37:4247–4255.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Amoroso A, Concia L, Maggio C, Raynaud C,
Bergounioux C, Crespan E, Cella R and Maga G: Oxidative DNA damage
bypass in Arabidopsis thaliana requires DNA polymerase λ and
proliferating cell nuclear antigen 2. Plant Cell. 23:806–822. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Li DW, Li GR, Zhang BL, Feng JJ and Zhao
H: Damage to dopaminergic neurons is mediated by proliferating cell
nuclear antigen through the p53 pathway under conditions of
oxidative stress in a cell model of Parkinson's disease. Int J Mol
Med. 37:429–435. 2016. View Article : Google Scholar
|