Introduction
Renal transplantation has distinct advantages over
other medical interventions for end-stage kidney diseases in
improving survival and the quality of life (1). Owing to the development of effective
antimicrobial prophylactics for peri- and post-operative care, the
early diagnosis of post-operative complications, and potent
immunosuppressants for reducing acute rejection, short-term
transplant results are now excellent with a steady increase in
patient and graft survival (2).
However, due to patients with a functioning graft succumbing due to
a range of etiologies, including cardiovascular diseases and
malignancies, the long-term outcomes of renal transplantation
remain a critical concern (3,4).
Notably, hypertension is one of the most common and serious
complications following renal transplantation, and is an important
risk factor for graft loss and other disorders (5,6).
In particular, increased stiffness and altered contractility of
blood vessels are observed following renal transplantation, and may
contribute to the pathogenesis of abnormal blood pressure (BP)
(7,8). It is generally considered that the
renin-angiotensin system (RAS) serves systemic endocrine or local
paracrine/autocrine functions, eliciting either beneficial or
detrimental effects on cardiovascular homeostasis (9,10).
Angiotensin II (Ang II), as the essential bioactive peptide of the
RAS, exerts crucial roles in the regulation of the cardiovascular
system, particularly BP, and in the pathogenesis of cardiovascular
diseases such as hypertension (11). The majority of the physiological
and pathological roles of Ang II in the control of vascular
contractility are largely mediated via its interaction with two
G-protein coupled receptors known as the type 1 and type 2 Ang II
receptors (AT1R and AT2R, respectively)
(12). In physiological
conditions, Ang II mainly binds to AT1R to activate the
contraction of smooth muscle cells and maintain the vascular tone
(13). In the pathogenesis of
cardiovascular diseases such as hypertension, Ang II binds to
AT1R and AT2R in smooth muscle cells and
endothelial cells, which increases the contractility and
phenotype/morphology of the vasculature and eventually leads to BP
elevation (14). A previous study
revealed that the contractility of blood vessels in response to Ang
II was markedly increased in the recipient following renal
transplantation (15). Notably,
clinical research has demonstrated that therapies using Ang II
blockers for the prevention and treatment of hypertension are
closely associated with higher survival following renal
transplantation (16,17). However, whether and to what extent
the cellular and molecular mechanisms underlying Ang II-induced
vascular contractility are changed following renal transplantation
have not yet been fully elucidated.
Notably, emerging studies have revealed that
epigenetic mechanisms, which are biological processes that cause
heritable changes in gene expression without altering the DNA
sequence, exert essential effects on the clinical outcomes
following renal transplantation (18). These epigenetic changes include
non-coding RNA, DNA methylation and post-translational histone
modifications (19). Typical
epigenetic alterations in the pathogenesis of certain disorders
following renal transplantation have been investigated (20). It has been suggested that the
epigenome of the donor may influence kidney graft survival,
particularly when the epigenetic modifications are associated with
early stresses and donor aging (21). In clinical applications, certain
epigenetic modifications associated with the processes of
ischemia-reperfusion injury, host immune responses to graft and
graft responses to injury have been tentatively suggested to serve
as potential tools for evaluating graft function, and as new
therapeutic targets for improving outcomes following renal
transplantation (22–24). However, whether the altered Ang
II-induced vascular contractility following renal transplantation
is associated with epigenetic changes of the AT1R and
AT2R genes is unclear.
In humans, factors including uremia, pre-existing
vascular diseases, volume load, abnormal calcium phosphate
metabolism, and the limitations of examination methods interfere
with the study of the explicit effects of renal transplantation on
recipient cardiovascular functions (25). However, experimental animal models
may effectively exclude the aforementioned obstacles (26). Thus, in the present study, an
experimental Fisher-Lewis rat renal transplantation model was used.
The changes of BP in the conscious state and the vascular
contractility of large and small blood vessels in response to Ang
II of the recipient animal following renal transplantation were
investigated. Additionally, the mechanism by which Ang II modulates
vascular contractility via the activation of AT1R and
AT2R in the recipient was determined following renal
transplantation. Furthermore, alterations in the patterns of DNA
methylation were investigated, so as to elucidate the effects of
renal transplantation on the epigenetic modifications affecting
gene expression at the transcriptional level. Accordingly, the
cellular and molecular impacts of renal transplantation on Ang
II-induced vasoconstriction in the transplant recipient were
extensively explored, and the results may provide potential
candidates that may be targeted to prevent and ameliorate
hypertension following transplantation.
Materials and methods
Experimental animals and treatment
Male Fisher rats (n=12) and Lewis rats (n=12)
weighing 250–300 g were supplied by the Laboratory Animal Research
Center of Harbin Medical University (Harbin, China). The rats were
individually housed in plastic cages in an animal room under
temperature-controlled conditions of 22–25°C and humidity of 50–60%
with a 12-h light/dark cycle and access to commercial pellets and
water ad libitum. The rats were maintained under these
standard conditions for ≥1 week prior to experimentation. The rats
were randomly assigned into groups (n=12 for each group), and the
following experiments and drug treatments were performed in a
double-blind manner. The control group (Con) was subjected to a
sham surgery. The renal transplantation (RT) group received the
typical Fisher-Lewis rat renal transplantation surgery. Both
surgeries are described in detail below. In the first 10 days
following the renal transplantation surgery, all experimental rats
were treated with cyclosporine by intraperitoneal injection (1.5
mg/kg/body weight) once every day. All procedures were approved by
Harbin Medical University Animal Ethics Committee and conformed to
the Guide for the Care and Use of Laboratory Animals (5th Edition,
2015).
Renal transplantation procedure
Following anesthesia with an intraperitoneal
injection of 3% sodium pentobarbital (50 mg/kg) under sterile
conditions, the kidneys and vessels of the rats were exposed
through an abdominal midline incision. Microsurgery was performed
simultaneously in the donor (Fisher) and recipient (Lewis) rats by
two investigators. In the two rats, blood flow through the small
branches of the abdominal aorta and vena cava was arrested by
ligation using vessel clips. The left kidney of the donor rat,
including the ureter, the renal artery and a short section of the
aorta and the renal vein, was perfused with ice-cold electrolyte
solution and immediately transferred to the recipient rat. The
aorta and renal vein of the donor kidney, which was kept in gauze
and superfused with ice cold electrolyte solution, were sutured to
the aorta and vena cava end-to-side of the recipient rat. Following
ligation of the distal end of the graft aorta, the vessel clips in
the recipient rat were released. During this process, the graft
experienced ≤40 min ischemia in total. The ureter of the donor
kidney was inserted into the apex of the recipient bladder.
Finally, following flushing of the abdominal cavity with isotonic
saline, the abdomen was closed in layers. The right native kidney
of the recipient rat was removed 10 days after the left kidney
transplantation surgery. In the sham surgery, following an
abdominal midline incision, the kidneys and vessels were exposed by
pulling the mesentery out of the abdomen. All visible renal nerves
were cut for renal denervation, and the renal artery and vein were
dissected free from connective tissue and clamped for 40 min for
warm ischemia. The mesentery was then put back into the abdominal
cavity, and the abdomen was closed in layers.
Determination of blood biochemical
indices
At 48 weeks after the transplantation surgery, the
rats were anesthetized with 3% sodium pentobarbital (50 mg/kg)
intraperitoneally and 2-ml blood samples were collected via the
abdominal aorta into iced tubes containing lithium heparin
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) as an
anticoagulant. Arterial blood pH, plus the hematocrit (Hct),
glucose (Glu) and lactate (Lac) concentrations, the sodium
concentration ([Na+]) and potassium concentration
([K+]) of the blood were evaluated using a Nova
eleven-electrode analyzer (Nova Biomedical, Waltham, MA, USA). The
samples were centrifuged at 2,000 × g at 4°C for 10 min to remove
erythrocytes, leukocytes and platelets. The plasma was then
extracted and its osmolality measured using an advanced diagnostic
osmometer (Model MO; Advanced Instruments, Inc., Norwood, MA,
USA).
Measurement of Ang II levels in plasma
and blood vessels
Blood samples were collected from the rats as
described above, with the exception that the collection tubes were
iced tubes containing ethylenediaminetetraacetic acid (EDTA) as the
anticoagulant. The plasma was extracted by centrifugation at 2,000
× g at 4°C for 10 min, and then aliquoted and frozen at −80°C until
required for testing. In addition, the aorta and mesenteric
arteries were excised and placed in chilled plastic tubes
containing EDTA-Na2, 8-hydroxyquinoline and
dimercaptopropanol. The tissue samples were boiled in water at
100°C for 10 min to provide a blood vessel tissue homogenate, which
was then centrifuged at 2,200 × g at room temperature for 10 min.
The supernatant was aliquoted and frozen at −80°C until required.
The levels of Ang II in the plasma and blood vessels were detected
by routine radioimmunoassay, using commercially available kits
(cat. no. HY-10058; Beijing Sino-UK Institute of Biological
Technology, Beijing, China) with a sensitivity of 0.01 ng/ml, and
intra- and inter-assay coefficients of variation of 10.5–12.0%.
Vascular contractility of the aortas in the absence of endothelium
was examined by allowing air bubbles blew into the vascular lumen
at 10-sec intervals for a total duration of 5 min.
Arterial BP measurements in conscious
rats
At 48 weeks after the transplantation surgery, the
rats were intraperitoneally anesthetized with 3% sodium
pentobarbital (50 mg/kg), and polyvinyl catheters were introduced
into the femoral artery and femoral vein under sterile conditions
for arterial BP recording and drug administration, respectively.
Following the surgery, antibiotics were administered subcutaneously
for 2 days. On the third day, the arterial BP of the conscious rats
was measured continuously. Following the recording of baseline
measurements for 60 min, Ang II (100 ng/kg body weight;
Sigma-Aldrich; Merck KGaA) was intravenously administered via the
implanted catheter in the femoral vein. The basal arterial BP was
then continuously recorded for 60 min. A selective AT1R
antagonist (losartan; 1 mg/kg, in 0.2 ml saline) and
AT2R antagonist (PD-123319; 0.1 mg/kg, in 0.2 ml saline)
(both Sigma-Aldrich; Merck KGaA) were respectively administered
intravenously via the implanted catheter. The resting arterial BP
was then continuously recorded for 60 min. Continuous recording and
data acquisition of the arterial systolic BP (SBP), diastolic BP
(DBP) and mean BP (MAP) was conducted using a PowerLab system and
software (LabChart software version 6; ADInstruments, Bella Vista,
Australia).
Aortic ring contractility
At 48 weeks after the transplantation surgery, the
rats were intraperitoneally injected with 3% sodium pentobarbital
(50 mg/kg). The thoracic aorta was then isolated and cut into 4-mm
rings, which were mounted in tissue baths (10 ml) containing
modified Krebs solution equilibrated with 95% O2 and 5%
CO2 at 37°C. Following a 60-min equilibration period,
the rings were stretched to an optimal resting tension and then
stimulated with increasing concentrations of Ang II
(10−11–10−5 mol/l) in one-half log
increments. In addition, losartan (10 μmol/l) or PD-123319
(10 μmol/l) were used to pretreat the rings for 30 min prior
to the addition of Ang II. The contractile tension was continuously
recorded, with normalization by the maximum contraction elicited by
60 mmol/l KCl. As the repeated exposure of arterial segments to Ang
II is considered to lead to tachyphylaxis (27), each ring was only tested in one
experiment and was not reused.
Contractions and intracellular
Ca2+ concentration of mesenteric arteries
At 48 weeks after the transplantation surgery, the
rats were intraperitoneally injected with 3% sodium pentobarbital
(50 mg/kg). The fourth-order mesenteric arterial branches were
isolated under a microscope and cut into 3-mm segments. These
segments were mounted in a Multi Myograph system (Danish Myo
Technology A/S, Midtjylland, Denmark) containing modified Krebs
solution equilibrated with 95% O2 and 5% CO2
at 37°C. Following a 60-min equilibration period, the segments were
stretched to the optimal resting tension, and then stimulated with
increasing concentrations of Ang II
(10−11–10−5 mol/l) in one-half log
increments. Losartan (10 μmol/l) and PD-123319 (10
μmol/l) were each used to pretreat the segments for 30 min
prior to the addition of Ang II. The contractile tension was
measured and recorded using a Power-Lab system with Chart 5
software (both from ADInstruments), which was normalized by the
maximum contraction elicited by the stimulation of the 60 mmol/l
KCl. Each arterial segment was only tested once.
The fourth-order mesenteric arterial branches were
isolated under a microscope and cut into 3-mm segments. These
segments were mounted and pressurized in an organ chamber (Living
Systems Instrumentation, Burlington, VT, USA) in a darkened room.
The intracellular Ca2+ concentration
([Ca2+]i) of each segment was measured using
a Radiance 2100 confocal system with the acetoxymethyl ester of
Fura-2 (Fura-2 AM; Calbiochem, San Diego, CA, USA) as a
[Ca2+]i indicator. The vessel
[Ca2+]i was determined on the basis of the
fluorescence ratio of Fura-2 AM at wavelengths of 340 and 380 nm.
The arteries were superfused with modified Hanks' solution and
pressurized to 45 mmHg with a mixture of 95% O2 and 5%
CO2 at 37°C. Following 60 min incubation with Fura-2 AM
loading, the constriction and [Ca2+]i of the
mesenteric artery were simultaneously monitored in the same vessel
segment. The pressurized arteries were stimulated by a single Ang
II concentration (10−7 mol/l) until the maximal
reduction in arterial diameter was achieved. Additionally, losartan
(10 μmol/l) or PD-123319 (10 μmol/l) was used to
pretreat the arteries for 30 min prior to the addition of Ang II.
The mesenteric artery diameter and [Ca2+]i
signal were determined from the ratio of fluorescence at 340 and
380 nm and recorded with a SoftEdge Data Acquisition Subsystem
(IonOptix, Westwood, MA, USA). The results were normalized using
the maximum contraction induced by 60 mmol/l KCl.
Western blot analysis
At 48 weeks after the transplantation surgery,
protein samples were routinely extracted from the aortic and
mesenteric arteries, which were routinely homogenized in NP-40
lysis buffer (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
with freshly added protease inhibitors using sonication. Samples
with an equal protein content (80 μg/lane) were subjected to
electrophoresis using a 0.1% sodium dodecylsulfate (SDS) and 10%
polyacrylamide separating gel with 4% polyacrylamide stacking gel.
The proteins were then transferred to an Immobilon P
polyvinyldifluoride membrane (EMD Millipore, Billerica, MA, USA).
The membrane was blocked with Tris-buffered phosphate-buffered
saline (PBS) containing 5% bovine serum albumin (BSA;
Sigma-Aldrich; Merck KGaA) plus 0.1% v/v Tween-20 with gentle
shaking for 1 h at room temperature prior to incubation with
primary antibodies overnight at 4°C. The antibodies were rabbit
polyclonal antibodies directed against AT1R (sc-1173;
1:500), AT2R (sc-9040; 1:1,000), cyclic AMP-responsive
element-binding protein 1 (CREB-1; sc-58; 1:500), estrogen receptor
(ER)-α (sc-544; 1:500) and ER-β (sc-8974; 1:500) (all from Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), respectively. The
membranes were washed three times for 10 min in a solution of TBS
and Tween-20, and then incubated at room temperature for 10 min
with a horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:5,000; A0545; Sigma-Aldrich; Merck KGaA). The proteins
were visualized using an ECL reagent (Amersham; GE Healthcare Life
Sciences, Little Chalfont, UK) with exposure to an X-ray film. The
β-actin protein (sc-47778, 1:1,000) was blotted in the same
membrane as internal control for the normalization of band density.
Results were quantified and analyzed using a Kodak electrophoresis
documentation & analysis system, and 1D image analysis software
version 2.0 (Kodak, Rochester, NY, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
At 48 weeks after the transplantation surgery, total
tissue RNA was routinely isolated from the aortic and mesenteric
arteries, by homogenization with TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), followed by addition of chloroform. After
the homogenate was separated by centrifugation at 12,000 × g for 10
min at 4°C, RNA was precipitated from the upper aqueous layer with
isopropanol from the tissues. First strand cDNA synthesis was then
performed using MMLV-RT reverse transcriptase (Invitrogen; Thermo
Fisher Scientific, Inc.). cDNA synthesis reaction was performed
under 50–70°C for 30 min and then heating at 90°C for 5 min. qPCR
analysis was then used to evaluate the gene expression of
AT1aR, AT2R, CREB-1, ER-α and ER-β.
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an
internal reference and serial dilutions of the positive control
were performed on each plate to create a standard curve. The primer
sequences were as follows: AT1aR forward,
5′-GCAGCCTCTGACTAAATGGC-3′ and reverse, 5′-TCCATCCAGCTCCTGACTCT-3′;
AT2R forward, 5′ CGGTGGCTTGCCGTTTCTT-3′ and reverse,
5′-GACTCATTGGTGCCAGTTGC-3′; CREB-1-1 forward, 5′
ACTCAGCCGGGTACTACCAT-3′ and reverse, 5′-ACTCAGCCGGGTACTACCAT-3′;
ER-α forward, 5′ GCCACTCGATCATTCGAGCA-3′ and reverse,
5′-CACCCTGCTGGTTCAAAAGC-3′; ER-β forward, 5′
CTGGACAGGGATGAGGGGAA-3′ and reverse, 5′-CGAAGCGTGTGAGCATTCAG-3′;
GAPDH forward, 5′ GGTGGTCTCCACGGACTTTA-3′ and reverse,
5′-CAAGGAGGGGCCTTTATTTC-3′. qPCR was performed in a Bio-Rad iCycler
iQ Real-Time PCR system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) with a 25-μl reaction system in a 96-well plate
(One-Step SYBR® PrimeScript™ RT-PCR kit II; cat. no.
RR086A; Takara Bio, Inc., Shiga, Japan). The thermo-cycling
conditions were as follows: 42°C for 30 min, 95°C for 15 min,
followed by 40 cycles of 95°C for 20 sec, 56°C for 1 min and 72°C
for 20 sec. The target gene quantity was normalized to the
reference GAPDH to obtain the relative quantification cycle (ΔCq).
The 2−ΔΔCq method (28) was then used to determine the
relative target gene expression for each group.
Methylation-specific qPCR (MS-qPCR)
DNA methylation of CpG sites at the promoter region
of the rat vascular AT1aR gene was evaluated using
MS-qPCR. At 48 weeks after the transplantation surgery, genomic DNA
was isolated from the aortic or mesenteric arteries using an
GenElute Mammalian Genomic DNA Miniprep kit (Sigma-Aldrich; Merck
KGaA), and purified with a Wizard DNA clean up system (Promega
Corporation, Madison, WI, USA). This involves treatment with sodium
bisulfite, which converts cytosine residues to uracil residues but
leaves the methylated cytosines at CpG unchanged. Following
denaturation of the DNA with 2 M NaOH at 42°C for 15 min, the DNA
was treated with sodium bisulfite at 55°C for 16 h and then used as
the template for MS-qPCR. Specific primers were designed (CpG
site-809 forward, 5′-AGGGTTGGAATTTGTAGAGTAGC-3′ and reverse,
5′-GAATAAAACAAAACTCAAACCACG-3′; CpG site-725 forward,
5′-GAGGGTTGGAATTTGTAGAGTAGC-3′ and reverse,
5′-GAATAAAACAAAACTCAAACCACG-3′; CpG site-484 forward,
5′-GGTTGGAATTTGTAGAGTAGCGA-3′ and reverse,
5′-GAATAAAACAAAACTCAAACCACG-3′; CpG site-150 forward,
5′-GAGGGTTGGAATTTGTAGAGTAGC-3′ and reverse,
5′-CCCCGATAATCGATAATCGA-3′; and CpG site-96 forward,
5′-TGAGGGTTGGAATTTGTAGAGTAGT-3′ and reverse,
5′-CCACCCCAATAATCAATAATCAA-3′) to amplify the target regions with
un-methylated CpG via the detection of uracils and those with
methylated CpG via the detection of cytosines, using GADPH as an
internal reference. MS-qPCR was conducted using an iCycler
Real-Time PCR system. Data are presented as the percentage
methylation of the target region as follows: Methylation (%) =
methylated CpG/(methylated CpG + un-methylated CpG) ×100.
Chromatin immunoprecipitation assay
(ChIP)
At 48 weeks after the transplantation surgery, ChIP
of the aortic and mesenteric arteries was conducted using a
ChIP-IT® kit (Active Motif, Carlsbad, CA, USA) according
to the manufacturer's protocol. This involved setting up the
immunoprecipitation reaction with sonicated chromatin and
sequentially adding CHIP buffer and protease inhibitor cocktail to
the 1.5-ml microcentrifuge tube (≥200 ng chromatin per CHIP
reaction). Rabbit monoclonal anti-CREB-1-1 (ab-31387; 1:500),
anti-ER-α (ab-32063; 1:200) and anti-ER-β (ab-3577; 1:500) (all
from Abcam, Cambridge, MA, USA) were added and the reaction mixture
was incubated overnight at 4°C. The next day, following extensive
blocking in 0.5% BSA, protein G agarose beads were added to the
mixture and incubated for 3 h. Following washing and the reversal
of cross-links, DNA was recovered and purified. Finally, the
SYBR-Green quantitative PCR was performed. Data are presented as
the percentage of input.
Statistical analysis
All data are expressed as the mean ± standard
deviation. BP and vascular contractility data were analyzed using
repeated-measures analysis of variance (ANOVA) with Tukey's post
hoc tests. For analysis of the differences of mRNA levels between
groups, statistical significance was accepted (P<0.05) when the
ratio of 2−ΔΔCq was >1.7. One-way ANOVA was used for
other data to evaluate the differences among groups. P<0.05 was
considered to indicate a statistically significant result. SPSS
statistical software (version 18; SPSS, Inc., Chicago, IL, USA) was
used to conduct the statistical analyses.
Results
Blood biochemical indices
At 48 weeks after the transplantation surgery, no
significant differences in blood biochemical indices, including pH,
pCO2, pO2, sO2%, Hct, the
concentrations of Glu and Lac, [Na+], [K+]
and plasma osmolality were detected between the RT and control
groups using the Nova eleven-electrode analyzer (Nova Biomedical)
(Table I).
 | Table IBlood biochemical indices at 48 weeks
after surgery. |
Table I
Blood biochemical indices at 48 weeks
after surgery.
| Blood values | Con | RT |
|---|
| Osm (mOsm/kg) | 292.13±2.11 | 293.05±2.29 |
| [Na+]
(mmol/l) | 136.86±1.17 | 137.15±0.87 |
| [K+]
(mmol/l) | 4.11±0.51 | 4.16±0.57 |
| pH | 7.40±0.02 | 7.41±0.01 |
|
pCO2 | 37.31±1.21 | 37.41±1.26 |
| pO2 | 105.77±2.19 | 106.13±2.31 |
|
sO2% | 96.55±1.33 | 96.13±2.03 |
| Hct (%) | 35.51±1.01 | 35.13±1.27 |
| Glu (mmol/l) | 7.51±0.32 | 7.47±0.37 |
| Lac (mmol/l) | 1.53±0.11 | 1.49±0.23 |
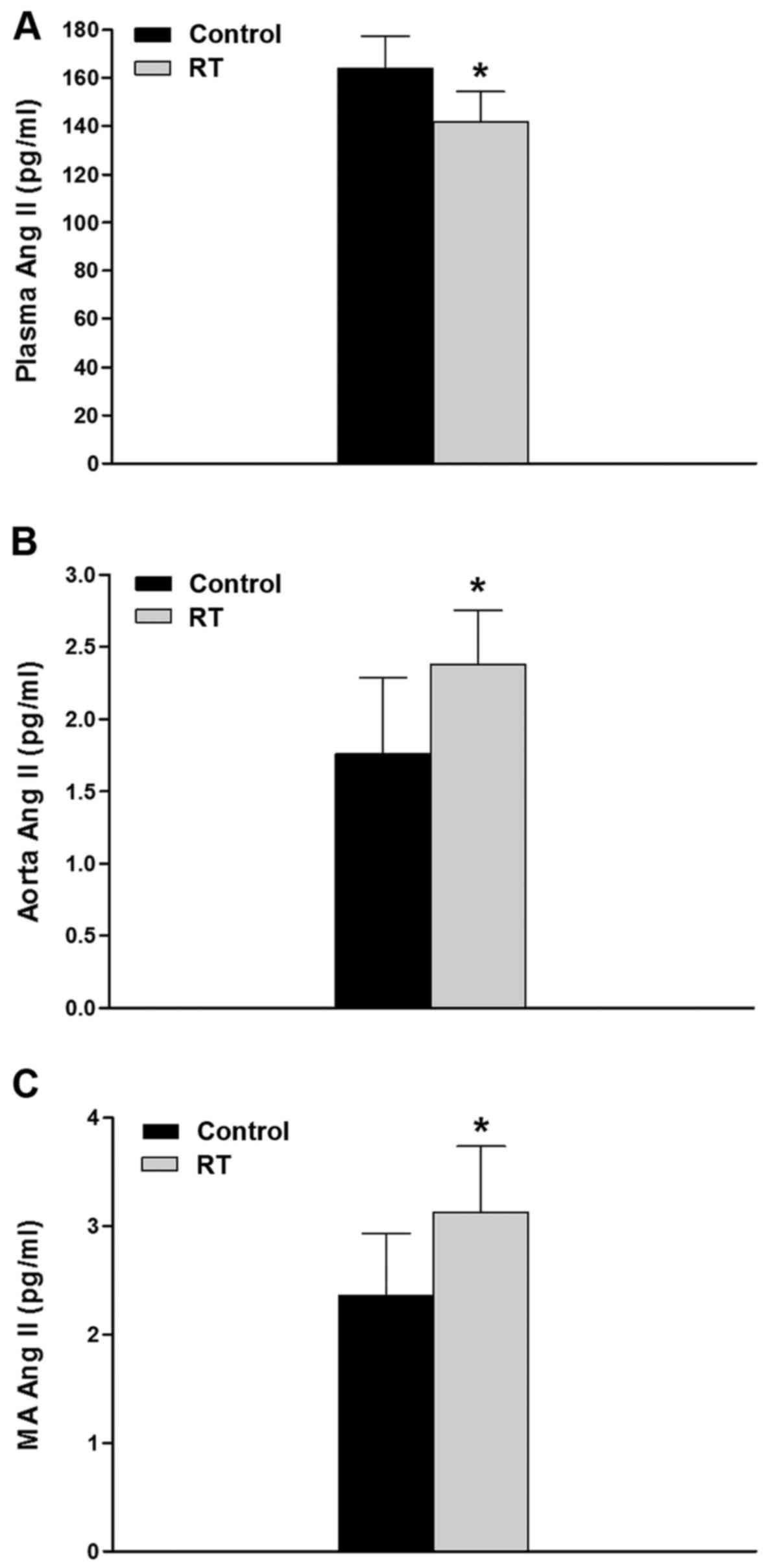
Concentrations of Ang II in plasma and
blood vessels
At 48 weeks after the transplantation surgery, the
plasma concentration of Ang II in the RT group was significantly
lower compared with that in the control group (P<0.05; Fig. 1A). However, in the aorta and
mesenteric artery tissues, the concentrations of Ang II were
significantly higher in RT group compared with the control group
(P<0.05; Fig. 1B and C).
Basal BP and Ang II-induced BP
response
At 48 weeks after the transplantation surgery, the
basal arterial SBP, DBP and MAP exhibited no significant difference
between the RT and control groups for rats in the conscious state
(Fig. 2A). Following the
intravenous administration of Ang II, the time-dependent arterial
BP response was in the range of several min. The Ang II-induced
arterial SBP, DBP and MAP values in the RT group were significantly
increased compared with those in the control group (P<0.05;
Fig. 2A).
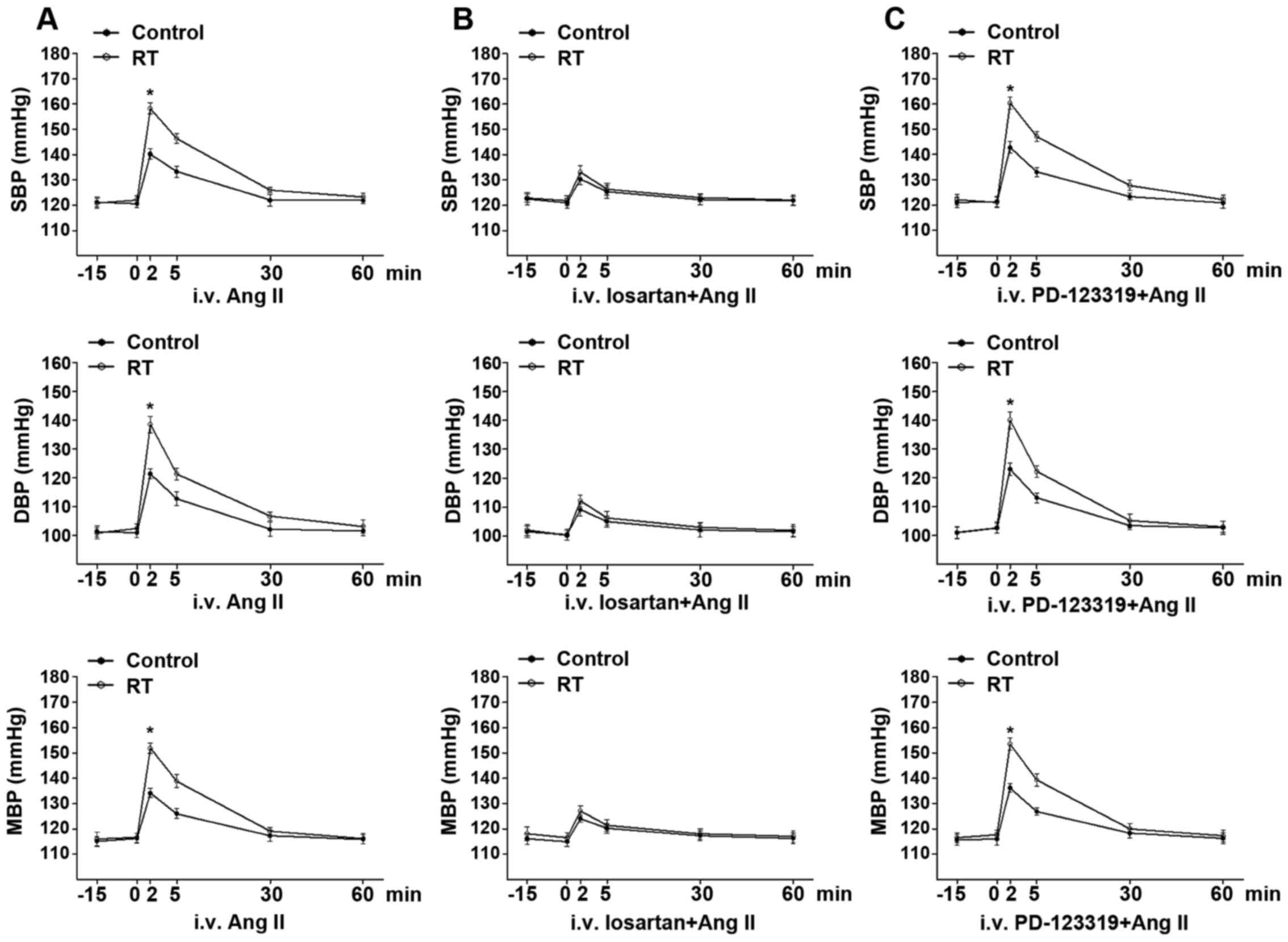 | Figure 2Basal BP and Ang II-induced BP
response at 48 weeks after transplantation surgery. (A) In the
conscious state, the basal arterial BP including SBP, DBP and MAP
exhibited no significant difference between the RT and control
groups. In addition, Ang II-induced SBP, DBP and MAP in the RT
group were significantly enhanced compared with those in the
control. (B) Following pretreatment with the selective
AT1R antagonist losartan, the time-dependent elevation
of SBP, DBP and MAP induced by Ang II was largely diminished and
exhibited no significant difference between the RT and control
groups. (C) Following pretreatment with the selective
AT2R antagonist PD-123319, the time-dependent elevation
of SBP, DBP and MAP induced by Ang II was not significantly changed
in the RT and control groups. Values are presented as the mean ±
standard deviation (n=6 per group). *P<0.05 vs.
control. BP, blood pressure; Ang II, angiotensin II; SBP, systolic
BP; DBP, diastolic BP; MAP, mean BP; RT, renal transplantation;
AT1R, angiotensin II receptor type 1; AT2R,
angiotensin II receptor type 2; i.v., intravenous. |
Following pretreatment with the selective
AT1R antagonist losartan, the time-dependent elevation
of arterial SBP, DBP and MAP induced by Ang II was largely
diminished and exhibited no significant difference between the RT
and control groups (Fig. 2B).
However, following pretreatment with the selective AT2R
antagonist PD-123319, the time-dependent elevation of arterial SBP,
DBP and MAP induced by Ang II was not significantly changed in the
RT and control groups (Fig.
2C).
Ang II-induced contraction of the
aorta
At 48 weeks after the transplantation surgery, the
aortal contractions induced by Ang II
(10−11–10−5 mol/l) were measured (Fig. 3). The contractions of the RT group
were significantly increased in tension compared with those of the
control (P<0.05; Fig. 3A). In
addition, following pretreatment with losartan, the Ang II-induced
aortal contractions in the RT and control groups were almost
completely eradicated (Fig. 3A).
However, pretreatment with PD-123319 did not significantly affect
the Ang II-induced aortal contractions in the RT and control groups
(Fig. 3C).
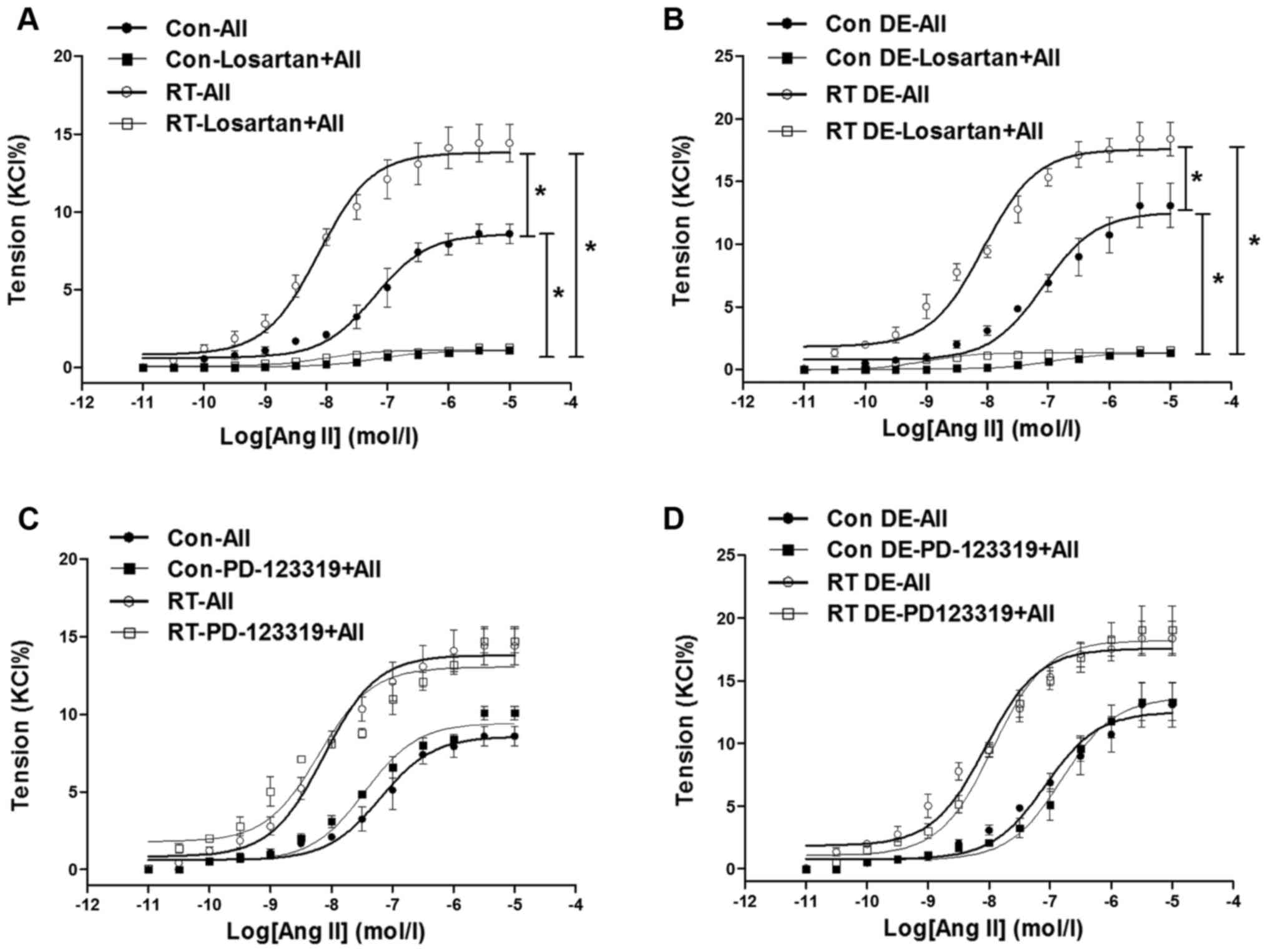 | Figure 3Ang II-induced contractions in rat
aortas at 48 weeks after transplantation surgery. (A) In the
presence of endothelium, Ang II-induced contractions of aortas of
the RT group were significantly increased compared with those of
the control group. In addition, following pretreatment with
losartan, Ang II-induced contractions of the aortas of the RT and
control groups were almost eradicated. (B) Without endothelium, Ang
II-induced vasoconstrictions in aortas of the RT and control groups
were significantly increased compared with those in
endothelium-intact aortas. Without endothelium, Ang II-induced
vasoconstrictions in aortas of the RT and control groups were
almost eradicated by losartan. (C) In the presence of endothelium,
following pretreatment with PD-123319, Ang II-induced contractions
in aortas of the RT and control groups were not significantly
changed. (D) Without endothelium, Ang II-induced vasoconstrictions
in aortas of the RT and control groups were not significantly
altered by PD-123319. Values are presented as the mean ± standard
deviation (n=6 per group). *P<0.05 as indicated. Ang
II (AII), angiotensin II; RT, renal transplantation; Con,
control. |
Vascular contractility of the aortas in the absence
of endothelium was examined to determine the effects of the
endothelium in vasodilation. Without endothelium, the Ang
II-induced aortal vasoconstrictions in the RT and control groups
were significantly increased compared with those in the
endothelium-intact aortas (P<0.05; Fig. 3B). However, Ang II-induced aortal
vasoconstrictions in the RT and control groups were almost
completely abolished by losartan, but hardly altered by PD-123319
in the absence of endothelium (Fig.
3B and D).
Ang II-induced contractions and
[Ca2+]i in small mesenteric arteries
At 48 weeks after transplantation surgery, the
contractions of the mesenteric arteries induced by Ang II
(10−11–10−5 mol/l) were measured (Fig. 4). The Ang II-induced contractions
of the mesenteric arteries of the RT group were significantly
increased in tension compared with those of the control (P<0.05;
Fig. 4A). In addition, following
pretreatment with losartan, the Ang II-induced contractions in the
mesenteric arteries of the RT and control groups were eliminated
(Fig. 4A). However, pretreatment
with PD-123319 did not have a significant effect on the Ang
II-induced contractions in the mesenteric arteries of the RT and
control groups (Fig. 4C).
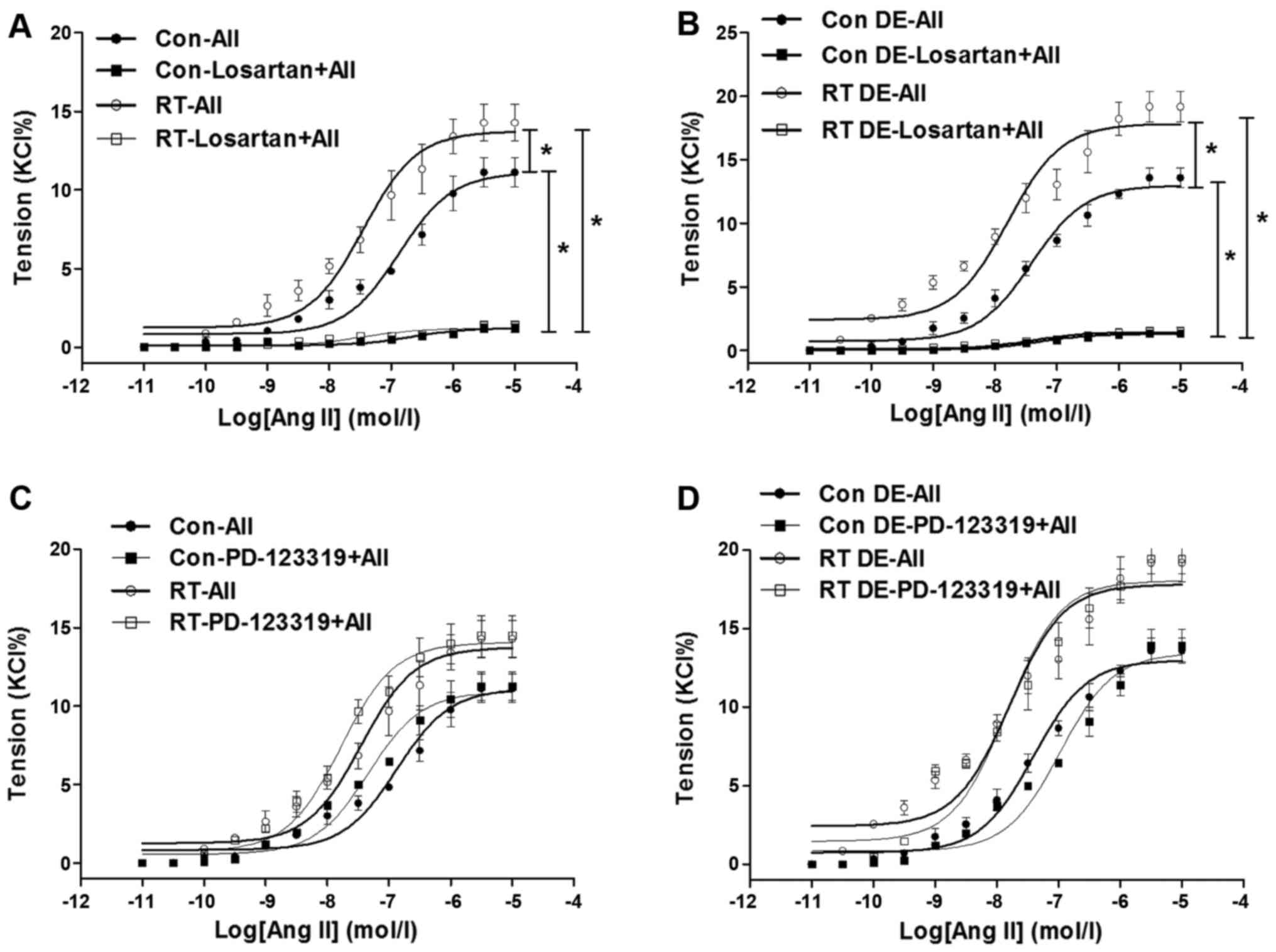 | Figure 4Ang II-induced contractions in
mesenteric arteries at 48 weeks after transplantation surgery. (A)
In the presence of endothelium, Ang II-induced contractions in
mesenteric arteries of the RT group were significantly increased
compared with those of the control. In addition, following
pretreatment with losartan, Ang II-induced contractions in
mesenteric arteries of the RT and control groups were almost
eradicated. (B) Without endothelium, Ang II-induced
vasoconstrictions in mesenteric arteries of the RT and control
groups were significantly increased compared with those in
endothelium-intact arteries. Without endothelium, Ang II-induced
vasoconstrictions in mesenteric arteries of the RT and control
groups were almost eradicated by losartan. (C) In the presence of
endothelium, following pretreatment with PD-123319, Ang II-induced
contractions in mesenteric arteries of the RT and control groups
were not significantly changed. (D) Without endothelium, Ang
II-induced vasoconstrictions in mesenteric arteries of the RT and
control groups were not significantly altered by PD-123319. Value
are the mean ± standard deviation (n=6 per group).
*P<0.05 as indicated. Ang II (AII), angiotensin II;
RT, renal transplantation; Con, control. |
The vasoconstriction of the mesenteric arteries in
the absence of endothelium was examined to further analyze the
effects of the endothelium in vasodilation. When the endothelium
was absent, the Ang II-induced vasoconstriction of the mesenteric
arteries was significantly increased in the RT and control groups
compared with the endothelium-intact arteries (P<0.05; Fig. 4B). However, as in the presence of
endothelium, in the absence of endothelium, the Ang II-induced
vascular contractility of the mesenteric arteries in the RT and
control groups was largely eradicated by losartan, but not markedly
changed by PD-123319 (Fig. 4B and
D).
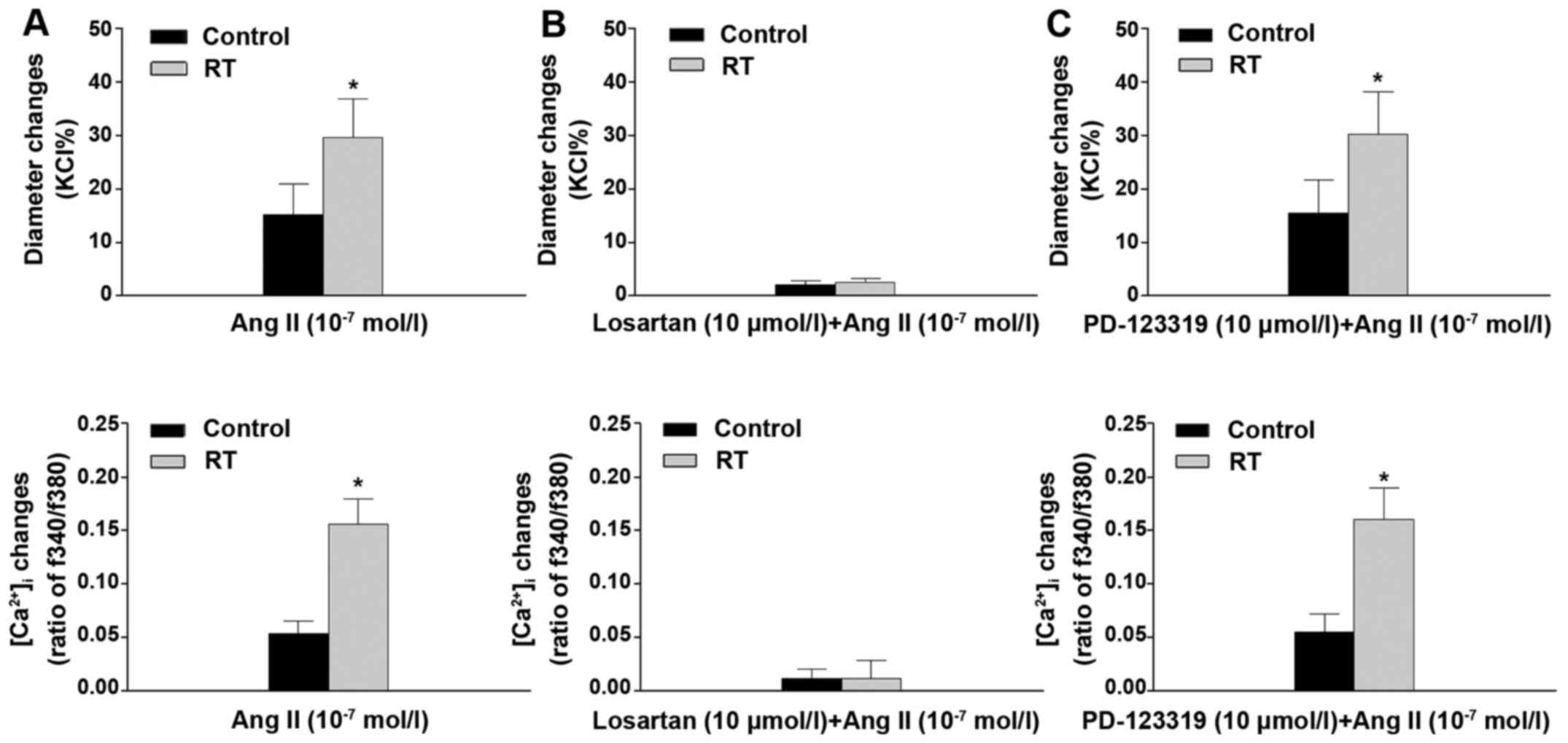
When the pressurized mesenteric arteries were
stimulated with Ang II (10−7 mol/l), the reductions in
diameter of the arteries were observed to be closely associated
with increases of [Ca2+]i. The results of the
organ chamber experiments demonstrated that the vascular
constriction and [Ca2+]i increases in these
arteries were significantly enhanced in the RT group compared with
the control group (P<0.05; Fig.
5A). In addition, following pretreatment with losartan, the Ang
II-induced vascular constriction and increase in
[Ca2+]i in the RT and control groups were
almost completely eradicated (Fig.
5B). However, pretreatment with PD-123319 did not significantly
alter the Ang II-induced vascular constriction and
[Ca2+]i increase in the RT and control groups
(Fig. 5C).
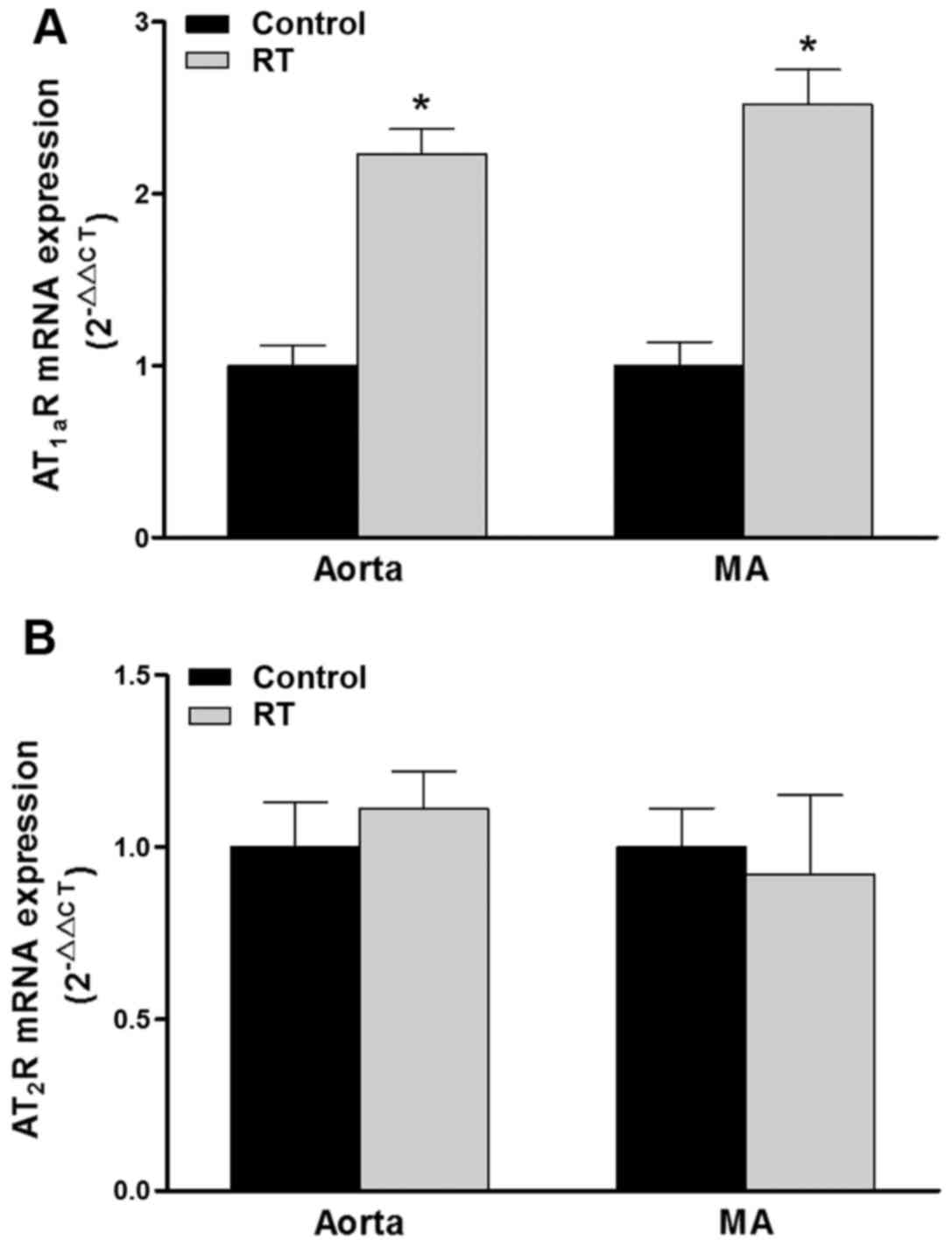
Expression of AT1aR and
AT2R mRNA in blood vessels
To investigation whether there was an association
between the Ang II-induced cardiovascular responses and changes in
AT1R and AT2R, the expression of these
receptors in the aorta and mesenteric arteries at the genetic level
was detected using RT-qPCR. In rodents, two subtypes of the
AT1R gene have been identified, namely AT1aR
and AT1bR, the former of which is the major subtype
localized in blood vessels (29).
At 48 weeks after the transplantation surgery, the expression
levels of AT1aR mRNA in the aorta and mesenteric
arteries of the RT group were significantly increased compared with
those in the control group (P<0.05; Fig. 6A). However, the mRNA expression
levels of AT2R in the aorta and mesenteric arteries
exhibited no significant difference between the RT and control
groups (Fig. 6B).
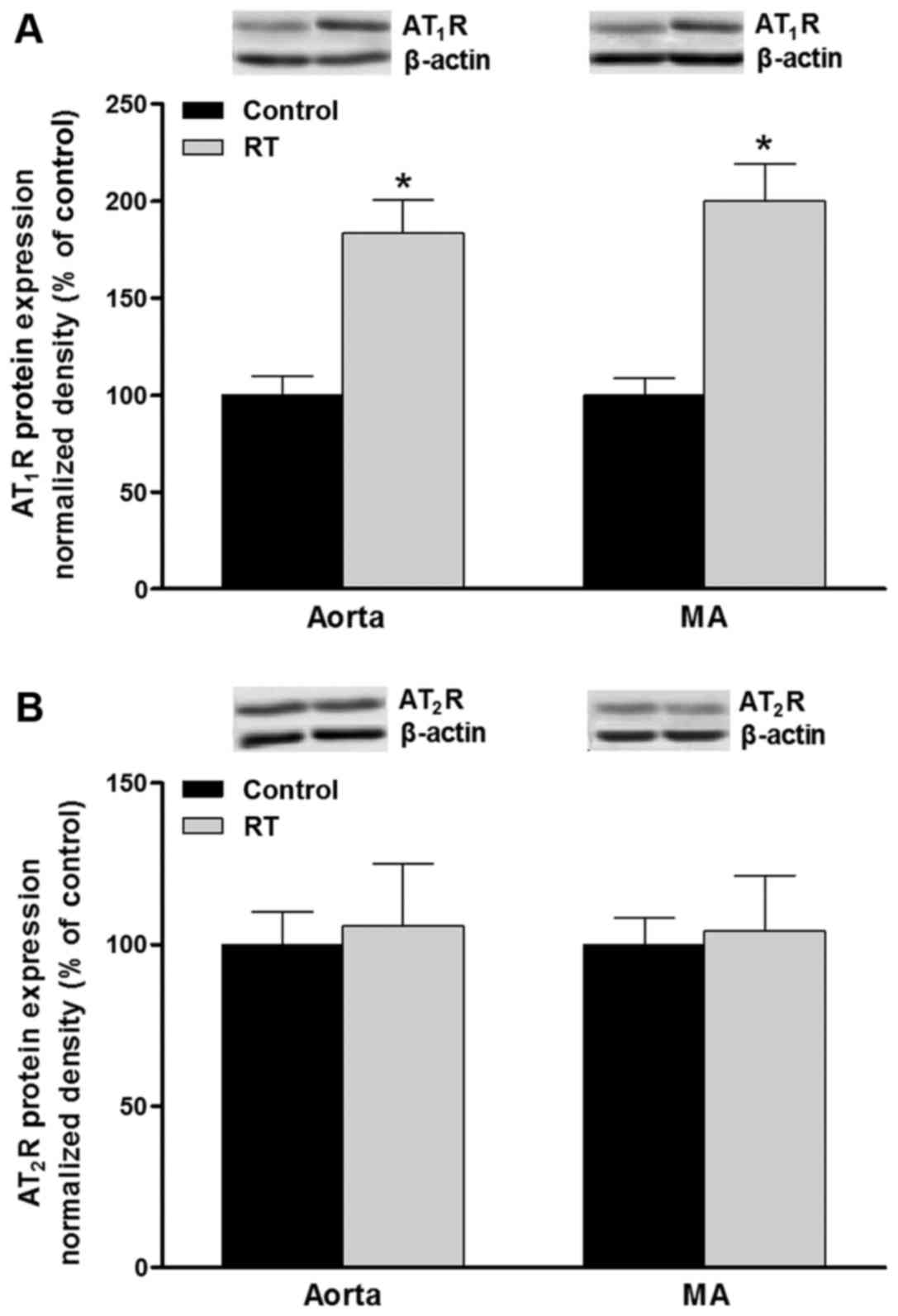
Protein levels of AT1R and
AT2R in blood vessels
The polypeptides expressed by the AT1aR
and AT1bR genes cannot be distinguished at the protein
level. Western blot analysis was used to evaluate the protein
expression of AT1R and AT2R in the blood
vessels. At 48 weeks after transplantation surgery, the protein
levels of AT1R in the aorta and mesenteric arteries of
the RT group were significantly increased compared with that in the
control group (P<0.05; Fig.
7A). However, no significant difference in the AT2R
protein levels of the aorta and mesenteric arteries were detected
between the RT and control groups (Fig. 7B).
DNA methylation of CpG loci in the
AT1aR gene promoter region in blood vessels
To evaluate the effects of DNA methylation patterns
on the transcriptional activity of the AT1aR gene, five
sequence-specific transcription factor binding sites containing CpG
loci were identified in the promoter region of the AT1aR
gene sequence from the rodent gene bank. The CpG sites at loci
−809, −725, −484, −150 and −96 upstream of the transcription start
site of the AT1aR gene correspond to the transcription
factor binding sites of erythroid transcription factor (GATA-1),
specificity protein 1 (Sp-1), ER-α/β, CREB-1 and Sp-1,
respectively. At 48 weeks after the transplantation surgery,
MS-qPCR analysis of the aorta and mesenteric arteries was
conducted. The results indicated that the DNA methylation levels of
CpG loci at the ER-α/β (−484) and CREB-1 (−150) transcription
factor binding sites in the AT1aR gene promoter region
were significantly decreased in the RT group compared with the
control group (P<0.05; Fig.
8). However, the DNA methylation status of the CpG loci at the
GATA-1 (−809) and Sp-1 (−725 and −96) transcription factor binding
sites of the AT1aR gene promoter region exhibited no
significant difference between the RT and control groups (Fig. 8).
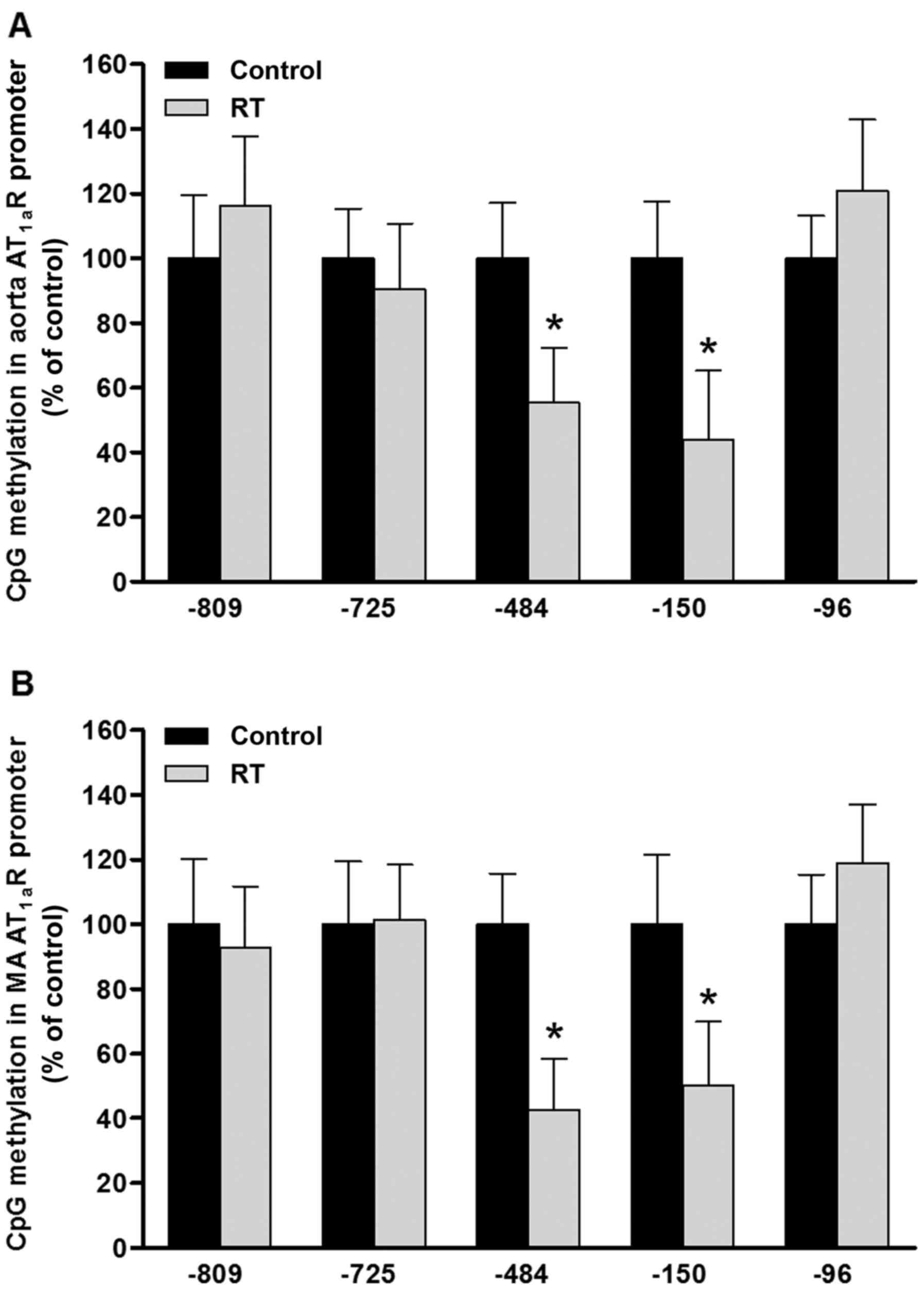 | Figure 8DNA methylation of CpG loci in the
AT1aR gene promoter in blood vessels at 48 weeks after
transplantation surgery. In the (A) aorta and (B) MA, the levels of
CpG methylation at ER-α/β (site −484) and CREB-1 (site −150)
transcription factor binding sites in the promoter region of the
AT1aR gene were significantly decreased in the RT group
compared with the control; however, the levels of CpG methylation
at the −809, −725 and −96 sites in the promoter region of the
AT1aR gene exhibited no significant difference between
the RT and control groups. Values are presented as the mean ±
standard deviation (n=6 per group), *P<0.05 vs.
control. AT1aR, angiotensin II receptor type 1a; MA,
mesenteric arteries; ER-α/β, estrogen receptor-α/β; CREB-1, cyclic
AMP-responsive element-binding protein 1; RT, renal
transplantation; Con, control. |
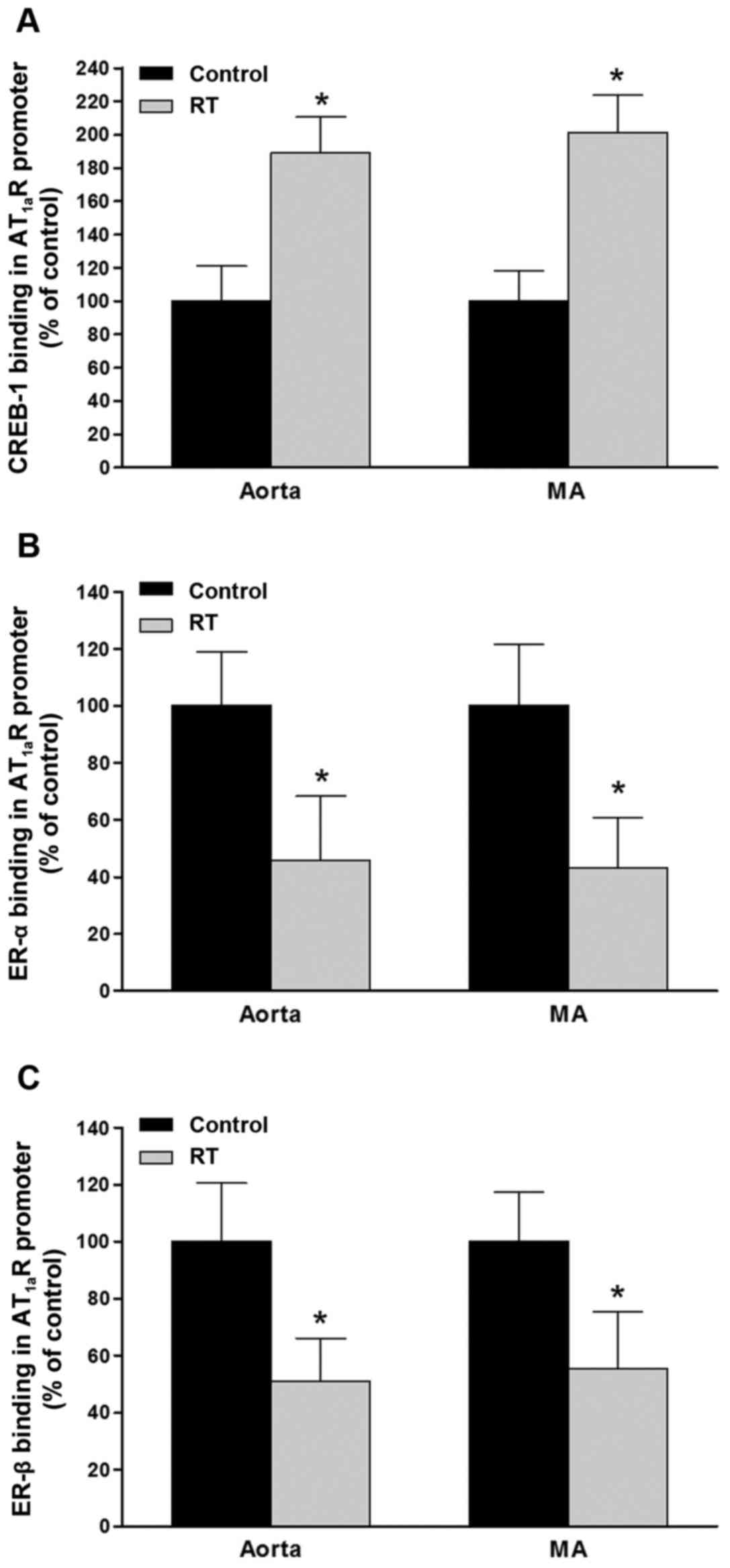
Binding capacity of CREB-1 and ER-α/β to
the AT1aR gene promoter region in blood vessels
To evaluate the functions of CREB-1 and ER-α/β in
the control of the transcriptional activity of the AT1aR
gene, the binding affinities of CREB-1 and ER-α/β for the specific
sequences in the promoter region of the AT1aR gene were
determined by ChIP assays of the blood vessels 48 weeks after
transplantation. The results demonstrated that in the aorta and
mesenteric arteries of the RT group, the binding affinities of
CREB-1 for the AT1aR gene promoter were significantly
increased compared with those in the control group (P<0.05;
Fig. 9A). However, the binding
affinities of ER-α and ER-β for the AT1aR gene promoter
in the aorta and mesenteric arteries were significantly decreased
in the RT group compared with the control (P<0.05; Fig. 9B and C).
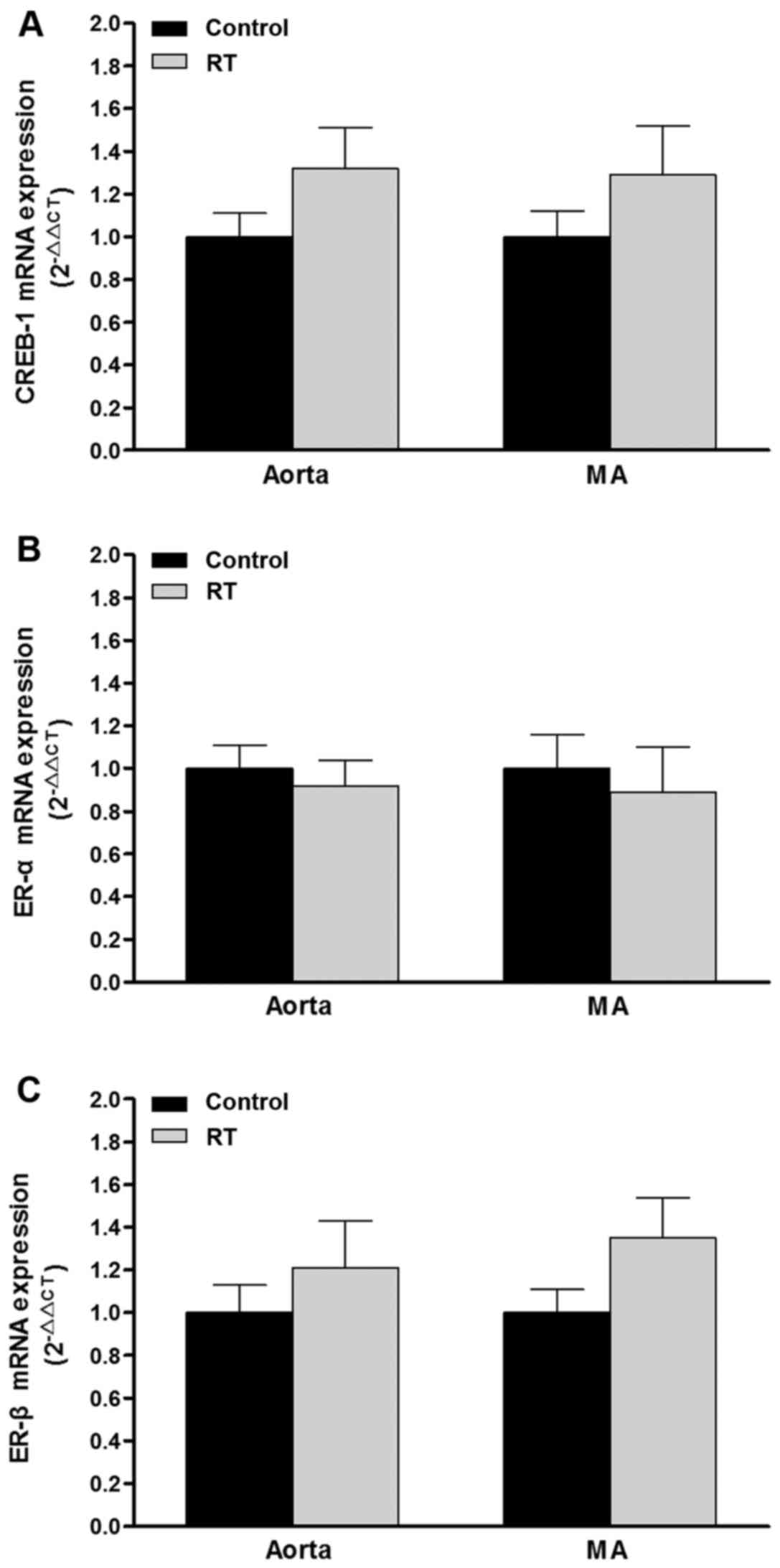
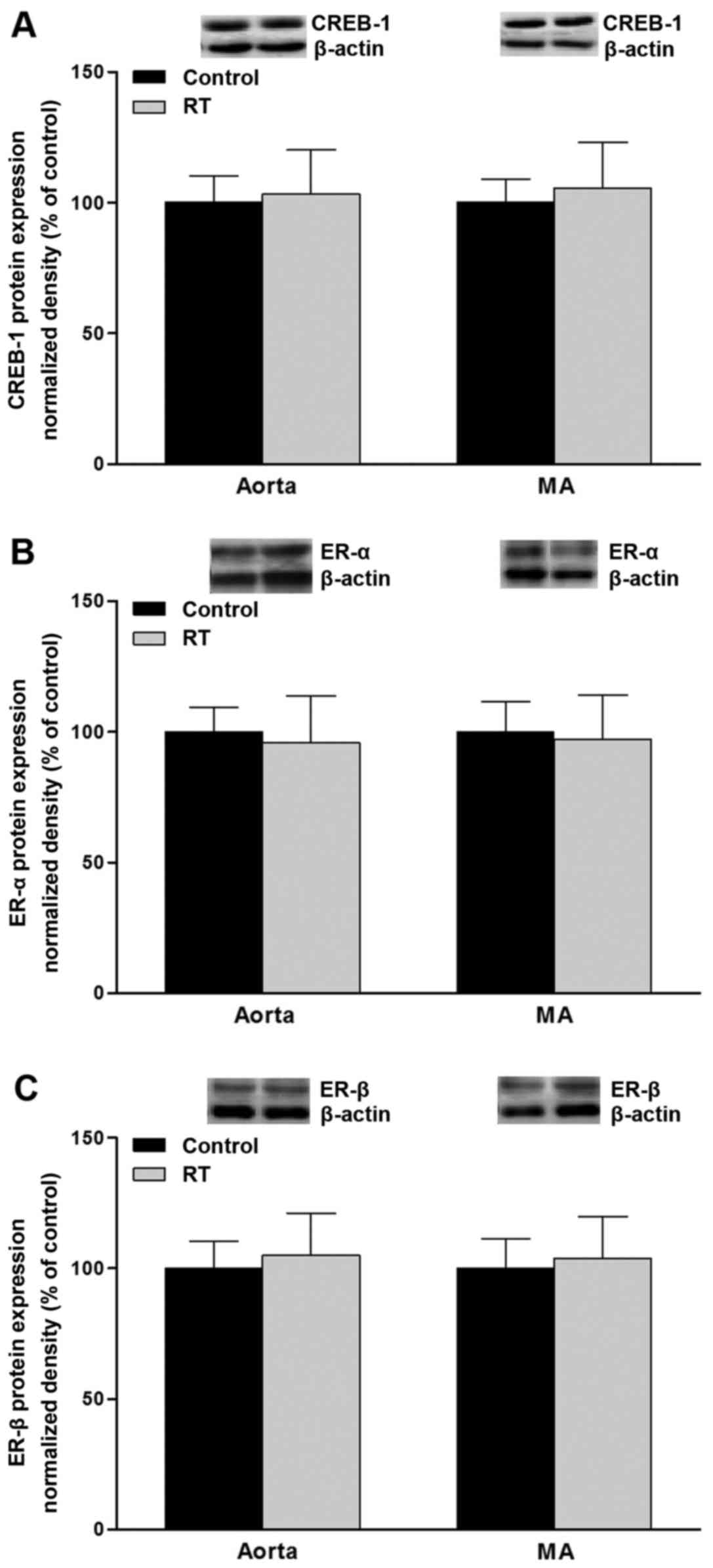
mRNA expression and protein levels of
CREB-1 and ER-α/β in blood vessels
To assess the association of transcription factor
levels with AT1aR gene expression, CREB-1, ER-α and ER-β
mRNA and protein levels were evaluated in the blood vessels of the
rats at 48 weeks after the transplantation surgery. The results of
RT-qPCR and western blotting demonstrated that the mRNA and protein
levels of CREB-1, ER-α and ER-β in the aorta and mesenteric
arteries did not differ significantly between the RT and control
groups (Figs. 10 and 11).
Discussion
Elevated BP is one of the most common and serious
complications following renal transplantation (30). In addition, epidemiological
evidence suggests that the development of hypertension is an
essential risk factor for morbidity and mortality following renal
transplantation, and is also a prominent predictor for impaired
graft survival independent of rejection episodes and transplant
functions (5). According to
current clinical recommendations, BP must be routinely measured
during every outpatient visit following surgery (31). Although it is widely considered
that the RAS serves definitive roles in the pathogenesis of primary
hypertension, the cellular and molecular mechanisms underlying the
progression of hypertension in response to the abnormal activation
of RAS following renal transplantation have not been fully
elucidated (32). Ang II-induced
vascular contractility is indicated to be altered following renal
transplantation (15).
Accordingly, it is hypothesized that the altered expression of
cardiovascular-responsive RAS-associated genes such as
AT1R and AT2R in blood vessels influences the
BP response to Ang II and increases the risk of hypertension
following renal transplantation. In the present study, using the
Fisher-Lewis rat renal transplantation model, a number of novel
findings were observed for the recipient rat following renal
transplantation: i) Ang II-induced BP response was prominently
increased; ii) AT1R-mediated vasoconstriction was
increased in association with increased
[Ca2+]i; and iii) the mRNA and protein
expression levels of vascular AT1R but not those of
AT2R, were significantly increased and were associated
with epigenetic mechanisms such as DNA methylation.
In the classic RAS system, Ang II is enzymatically
generated in the circulation from angiotensinogen, which is
produced and secreted by the liver (33). In general, Ang II mediates the
majority of its effects via binding to AT1R and
AT2R (12). These
receptors are widely distributed in almost all organs and tissues
(10). The regulation of
cardiovascular homeostasis by Ang II is extensively controlled by
the reactivity of AT1R and AT2R in the blood
vessels, heart, kidney and brain (11). In particular, the Ang II-induced
regulation of BP is largely mediated via AT1R and
AT2R activation in blood vessels (10). In physiological conditions, by
interacting with AT1R in the smooth muscle, Ang II is
able to elicit the contraction of large and small-resistance blood
vessels to maintain the vascular tone (12). However, in the pathogenesis of
primary hypertension, by binding to either AT1R or
AT2R, Ang II may exert detrimental effects on vascular
components, including smooth muscle cells, endothelial cells and
fibroblasts, which may increase vasoconstriction and induce
remodeling of the vascular structure (34). In the present study, the results
revealed that the time-dependent elevation of arterial BP (SBP and
DBP) induced by Ang II in the conscious state was significantly
increased in the RT group compared with the control group (Fig. 2). However, the basal arterial BP
(SBP and DBP) exhibited no significant difference between the RT
and control groups (Fig. 2). This
indicates that a recipient would be more susceptible to the
elevation of BP in response to Ang II following renal
transplantation. Furthermore, following pretreatment with the
selective AT1R antagonist losartan, the time-dependent
elevation of arterial BP (SBP and DBP) induced by Ang II was
largely attenuated, and exhibited no significant difference between
the RT and control groups (Fig.
2). However, following pretreatment with the selective
AT2R antagonist PD-123319, the time-dependent elevation
of arterial BP (SBP and DBP) induced by Ang II was not markedly
changed in the RT and control groups (Fig. 2). Thus, it is suggested that the
enhanced Ang II-induced BP elevation detected in the recipient
following renal transplantation is, at least in part, mediated by
the increased cardiovascular response activated by
AT1R.
The local vascular RAS is critical in the regulation
of vascular contractility and control of the arterial BP (10). From the morphological and
physiological perspectives, arteries may be roughly categorized
into large and small-resistance blood vessels, such as the aorta
and mesenteric arteries, respectively (35). Clinical study indicates that, in
the development and progression of hypertension, the increased
vasoconstriction of the aorta and other large blood vessels should
be tightly associated with abnormal increases in SBP; whereas, the
abnormal reactivity of small-resistance blood vessels such as the
mesenteric arteries should be largely correlated with an abnormal
increase in DBP (36). In the
present study, using aortic rings and mesenteric artery segments
in vitro as large and small-resistance blood vessels,
respectively, it was demonstrated that the Ang II-induced vascular
contractility of large and small-resistance blood vessels was
significantly increased in the RT group compared with the control
group (Figs. 3 and 4). This result may, at least in part,
explain why the arterial BP (SBP and DBP) elevation induced by Ang
II was increased in the recipient following renal transplantation
(Fig. 2). Furthermore, the
selective AT1R antagonist (losartan) almost completely
eradicated the Ang II-induced vasoconstriction of the aorta and
mesenteric arteries, whereas the selective AT2R
antagonist (PD-123319) had a minimal effect in the RT and control
groups (Figs. 3 and 4). This may explain why the Ang
II-induced arterial BP elevation in vivo was inhibited by
losartan but not PD-123319 in the recipient following renal
transplantation (Fig. 2).
Accordingly, these results suggest that the increased BP elevation
induced by Ang II in the recipient following renal transplantation
is partly due to increased AT1R-mediated
vasoconstriction in large and small-resistance blood vessels.
Contraction and dilation of the vasculature are
largely controlled by vascular smooth muscle cells and endothelial
cells (37). Studies have
revealed that the enhancement of endothelium-derived vasodilation
may serve an important compensatory/protective role in preventing
and ameliorating the development of hypertension (38). In the present study, to clearly
observe the contraction of vascular smooth muscle, the Ang
II-mediated vasoconstriction of the aorta and mesenteric arteries
in the absence of endothelium was evaluated. In comparison with
endothelium-intact vessels, Ang II-induced vasoconstriction in the
aorta and mesenteric arteries was significantly increased in the
absence of endothelium in the RT and control groups (Figs. 3 and 4). Additionally, in the absence of
endothelium, Ang II-induced vasoconstriction in the aorta and
mesenteric arteries was significantly increased in the RT group
compared with the control (Figs.
3 and 4). Furthermore,
without endothelium, as in the presence of endothelium, the Ang
II-induced vasoconstriction was almost completely eradicated by
losartan, but hardly altered by PD-123319 in the RT and control
groups (Figs. 3 and 4). These results indicate the following
transplantation, the AT1R-mediated contraction of
vascular smooth muscle is markedly increased in the large and
small-resistance blood vessels of the recipient. Furthermore, the
protective effects of endothelium-derived vasodilation may not be
impaired but could have lost the ability to compensate for the
over-activated vasoconstriction induced by Ang II following renal
transplantation.
At the cellular and subcellular levels, the
contraction of vascular smooth muscle mediated by AT1R
activation involves complex downstream signaling (11). The increase of
[Ca2+]i is generally considered to serve as
the fundamental determinant for vasoconstriction (39). In particular, following the
interaction between Ang II and AT1R, inositol
trisphosphate (IP3) is generated from
phosphatidylinositol-4,5-bisphosphate hydrolyzing when activated by
phospholipase C. IP3 then stimulates the sarcoplasmic reticulum to
mobilize intracellular Ca2+, leading to an increase in
[Ca2+]i (40). The production of diacylglycerol
(DAG) is increased and protein kinase C is activated, which further
promotes Ca2+ influx via certain ion channels (41). The increased
[Ca2+]i activates myosin light chain kinase
and the phosphorylation of 20-kDa myosin light chain
(MLC20), leading to the contraction of vascular smooth
muscle cells (40). In the
present study, due to the limitations of the IonOptix instrument
system, simultaneous measurement of the vascular contractility and
[Ca2+]i was possible in the mesenteric
arteries but not in the aorta. The results demonstrated that the
Ang II-induced vasoconstriction and [Ca2+]i
were simultaneously increased in the mesenteric arteries in the RT
group compared with the control (Fig.
5). Furthermore, the Ang II-induced increase in
[Ca2+]i was almost completely eliminated by
losartan, but hardly altered by PD-123319 in the RT and control
groups (Fig. 5). This suggests
that the increased AT1R-mediated vasoconstriction
observed in the mesenteric arteries was closely associated with
enhanced [Ca2+]i-dependent signaling in the
vascular smooth muscle of the recipient following renal
transplantation. However, it should be noted that, in addition to
the aforementioned Ca2+-dependent mechanism, the
inactivation of myosin light chain phosphatase and decreased
dephosphorylation of MLC20 may in turn increase the
sensitivity of Ca2+ at a fixed level of
[Ca2+]i to activate the contraction of
vascular smooth muscle cells induced by Ang II (42). This Ca2+-independent
mechanism is beyond the scope of the present study and requires
investigation in future studies.
The RAS may be anatomically and functionally
categorized into the systemic and local RAS, in the circulation and
tissues, respectively (10).
Furthermore, the abnormal activation of the local RAS may be
predominant in the pathogenesis of hypertension (11). In the present study, the levels of
Ang II in the RT group were significantly higher than those in the
control group in both the aorta and mesenteric arteries (Fig. 1). These results, together with the
aforementioned increased Ang II-induced vasoconstriction observed
in vitro, suggest that the local vascular RAS was
prominently over-activated in the recipient following renal
transplantation. However, with regard to the circulation, the
plasma concentration of Ang II in the RT group was significantly
lower than that in the control (Fig.
1), which indicates that the systemic RAS was inhibited in the
recipient following renal transplantation. Previous clinical and
experimental studies have found that, in the early phase of
hypertension, overactivation of the local RAS may provide negative
feedback signals causing inhibition of the systemic RAS (43,44). Thus, it is suggested in the
recipient after renal transplantation, the inhibited systemic RAS
serves a compensatory function that ameliorates the detrimental BP
elevation induced by the abnormal activation of the local vascular
RAS. This may explain why the basal BP was not observed to be
elevated in the recipient following renal transplantation (Fig. 2). Typically, the production Ang II
in the circulation and vascular tissue involves numerous enzymes,
including renin and angiotensin-converting enzymes (45). Whether and to what extent these
enzymes and functional products are affected by transplantation was
beyond the scope of the present study and requires elucidation in
the future.
Some uncertainty remains regarding the cellular and
molecular mechanisms underlying the increased Ang II-induced
vasoconstriction. There are at least two possible reasons for the
changes of Ang II-induced contraction in vascular smooth muscle: i)
AT1R and AT2R expression levels were changed;
ii) the sensitivity and/or effects of the downstream signaling of
AT1R and AT2R were altered in the vascular
smooth muscle cells. In the present study, only the analysis of
AT1R and AT2R expression levels in the blood
vessels was conducted. The mRNA and protein expression levels of
AT1R in the aorta and mesenteric arteries were
significantly increased in the RT group compared with the control
(Figs. 6 and 7), while those of AT2R
exhibited no significant difference between the RT and control
groups (Figs. 6 and 7), indicating that the effects of renal
transplantation on vascular RAS are gene-specific. In rodents,
there are two subtypes of the AT1R gene,
AT1aR and AT1bR, which are located on
different chromosomes (46).
AT1bR is mainly localized in the brain, for example, in
the pituitary and adrenal glands, whereas AT1aR is
widely dispersed in other organs and tissues, such as blood vessels
(47). Thus, it is proposed that
the elevated vascular levels of the AT1R gene in the
recipient following renal transplantation was partly attributable
to the increased transcriptional activity of the AT1aR
gene in large and small-resistance blood vessels. Notably, changes
of the transcriptional level of AT1R gene have been
indicated to be epigenetically affected in the pathogenesis of
hypertension, which may involve DNA methylation, post-translational
histone modifications and non-coding RNAs (47). In particular, DNA methylation,
occurring at the cytosine residues in CpG dinucleotide sequences,
may alter the chromatin structure and the long-term transcriptional
activity of the altered genes (48). Typically, the methylation
modification of just one CpG dinucleotide can change the expression
of the associated gene through affecting the binding affinity of
certain transcription factors (49). Several CpG sites have been
discovered in the sequence-specific transcription factor binding
sequences in the AT1aR gene promoter region of rodents,
including −809, −725, −484, −150 and −96 CpG loci (50). In the present study, MS-qPCR
demonstrated that the DNA methylation levels of CpG sites in the
−484 and −150 loci were significantly decreased in the aorta and
mesenteric arteries of the RT group compared with the control
(Fig. 8). Since the CpG loci of
−484 and −150 correspond to ER and CREB transcription factor
binding sequences, respectively, in the promoter region of the
AT1aR gene, it is speculated that renal transplantation
selectively decreased site-specific DNA methylation at the ER- and
CREB-binding sequences and thereby alters the transcriptional
efficacy of the vascular AT1aR gene in the
recipient.
To investigate the effects of ER and CREB in the
regulation of AT1aR gene transcriptional activity, ChIP
assays were conducted to detect the interactions of ER-α/β and
CREB-1 with the respective gene-specific transcription factor
binding sequences on promoter loci of the AT1aR gene in
the intact chromatin. The binding affinity of CREB-1 to the cAMP
response element in the promoter region of the vascular
AT1aR gene was observed to be significantly increased in
the aorta and mesenteric arteries of the RT group compared with the
control (Fig. 9). Additionally,
the expression of vascular CREB-1 (mRNA and protein) exhibited no
significant difference between the RT and control groups (Figs. 10 and 11). CREB-1 is known to be an essential
gene-specific factor for upregulating AT1aR gene
transcription (51). It may be
speculated that, due to the decreased DNA methylation level of the
−150 CpG site, the binding capacity of CREB-1 to the vascular
AT1aR gene promoter may be markedly increased, leading
to an increase in the transcriptional activity of the
AT1aR gene in the vasculature of the recipient following
renal transplantation. Furthermore, the binding affinity of ER-α/β
to the specific sequence in the promoter region of the
AT1aR gene was significantly decreased in the aorta and
mesenteric arteries of the RT group compared with the control
(Fig. 9). In addition, the
expression of vascular ER-α/β (mRNA and protein) exhibited no
significant difference between the RT and control groups (Figs. 10 and 11). Previous studies have demonstrated
that estrogen directly binds with ER to downregulate the
transcriptional activity of the AT1aR gene in the
vasculature, which prevents BP elevation in the pathogenesis of
hypertension (52). It may be
suggested that, due to the decreased DNA methylation level of the
−484 CpG site, the binding capacity of ER to the promoter region of
vascular AT1aR gene was markedly inhibited, so as to
further increase the transcriptional activity of the vascular
AT1aR gene in the recipient following renal
transplantation. Clinical evidence confirms that estrogen exerts
substantially protective roles that prevent the development of
hypertension (53–55). The beneficial effects of estrogen
in the reduction of vascular AT1aR gene expression may
be impaired by DNA demethylation in the recipient following renal
transplantation. However, why DNA demethylation at the −484 and
−150 sites differentially affected the binding capacity of ER and
CREB-1 to the promoter of the AT1aR gene is unknown and
requires investigation.
In conclusion, the present study demonstrated that
renal transplantation may cause Ang II-induced elevations of the BP
to increase and abnormally activate the local vascular RAS, leading
to the augmentation of AT1R-mediated vasoconstriction
associated with altered DNA methylation of the AT1aR
gene in large and small-resistance blood vessels in the recipient.
These results tentatively suggest that the susceptibility to
hypertension is increased in the recipient following renal
transplantation. Furthermore, epigenetic modifications such as DNA
methylation of vascular AT1aR gene may be potential
targets for preventing and treating hypertension.
Acknowledgments
The present study was supported by the Science and
Technology Research Project of Heilongjiang Province Department of
Education Project (grant no. 12531366).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Chandran S and Vincenti F: Clinical
aspects: Focusing on key unique organ-specific issues of renal
transplantation. Cold Spring Harb Perspect Med. 4:42014. View Article : Google Scholar
|
|
2
|
Lyerová L, Viklický O, Nemcová D and
Teplan V: The incidence of infectious diseases after renal
transplantation: A single-centre experience. Int J Antimicrob
Agents. 31(Suppl 1): S58–S62. 2008. View Article : Google Scholar
|
|
3
|
Saidi RF and Hejazii Kenari SK: Clinical
transplantation and tolerance: Are we there yet? Int J Organ
Transplant Med. 5:137–145. 2014.PubMed/NCBI
|
|
4
|
Holmberg C and Jalanko H: Long-term
effects of paediatric kidney transplantation. Nat Rev Nephrol.
12:301–311. 2016. View Article : Google Scholar
|
|
5
|
Mitsnefes MM, Khoury PR and McEnery PT:
Early post-transplantation hypertension and poor long-term renal
allograft survival in pediatric patients. J Pediatr. 143:98–103.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stoumpos S, Jardine AG and Mark PB:
Cardiovascular morbidity and mortality after kidney
transplantation. Transpl Int. 28:10–21. 2015. View Article : Google Scholar
|
|
7
|
Briese S, Claus M and Querfeld U: Arterial
stiffness in children after renal transplantation. Pediatr Nephrol.
23:2241–2245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cseprekál O, Kis E, Dégi AA, Kerti A,
Szabó AJ and Reusz GS: Bone metabolism and arterial stiffness after
renal transplantation. Kidney Blood Press Res. 39:507–515. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harrison-Bernard LM: The renal
renin-angiotensin system. Adv Physiol Educ. 33:270–274. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paul M, Poyan Mehr A and Kreutz R:
Physiology of local renin-angiotensin systems. Physiol Rev.
86:747–803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mehta PK and Griendling KK: Angiotensin II
cell signaling: Physiological and pathological effects in the
cardiovascular system. Am J Physiol Cell Physiol. 292:C82–C97.
2007. View Article : Google Scholar
|
|
12
|
Karnik SS, Unal H, Kemp JR, Tirupula KC,
Eguchi S, Vanderheyden PM and Thomas WG: International Union of
Basic and Clinical Pharmacology. XCIX Angiotensin receptors:
Interpreters of pathophysiological angiotensinergic stimuli
[corrected] Pharmacol Rev. 67:754–819. 2015.
|
|
13
|
de Gasparo M, Catt KJ, Inagami T, Wright
JW and Unger T: International union of pharmacology. XXIII The
angiotensin II receptors Pharmacol Rev. 52:415–472. 2000.
|
|
14
|
van Thiel BS, van der Pluijm I, te Riet L,
Essers J and Danser AH: The renin-angiotensin system and its
involvement in vascular disease. Eur J Pharmacol. 763:3–14. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gabriëls G, August C, Grisk O, Steinmetz
M, Kosch M, Rahn KH and Schlatter E: Impact of renal
transplantation on small vessel reactivity. Transplantation.
75:689–697. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Formica RN Jr, Friedman AL, Lorber MI,
Smith JD, Eisen T and Bia MJ: A randomized trial comparing losartan
with amlodipine as initial therapy for hypertension in the early
post-transplant period. Nephrol Dial Transplant. 21:1389–1394.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pilmore HL, Skeans MA, Snyder JJ, Israni
AK and Kasiske BL: Cardiovascular disease medications after renal
transplantation: Results from the patient outcomes in renal
transplantation study. Transplantation. 91:542–551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goldberg AD, Allis CD and Bernstein E:
Epigenetics: A landscape takes shape. Cell. 128:635–638. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zoghbi HY and Beaudet AL: Epigenetics and
human disease. Cold Spring Harb Perspect Biol. 8:a0194972016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mas VR, Le TH and Maluf DG: Epigenetics in
kidney transplantation: Current evidence, predictions, and future
research directions. Transplantation. 100:23–38. 2016. View Article : Google Scholar
|
|
21
|
Bontha SV, Maluf DG, Mueller TF and Mas
VR: Systems biology in kidney transplantation: The application of
multi-omics to a complex model. Am J Transplant. 17:11–21. 2017.
View Article : Google Scholar
|
|
22
|
Kim JI, Jung KJ, Jang HS and Park KM:
Gender-specific role of HDAC11 in kidney ischemia- and
reperfusion-induced PAI-1 expression and injury. Am J Physiol Renal
Physiol. 305:F61–F70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sui W, Lin H, Peng W, Huang Y, Chen J,
Zhang Y and Dai Y: Molecular dysfunctions in acute rejection after
renal transplantation revealed by integrated analysis of
transcription factor, microRNA and long noncoding RNA. Genomics.
102:310–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Betts G, Shankar S, Sherston S, Friend P
and Wood KJ: Examination of serum miRNA levels in kidney transplant
recipients with acute rejection. Transplantation. 97:e28–e30. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Becker-Cohen R, Nir A, Rinat C, Feinstein
S, Algur N, Farber B and Frishberg Y: Risk factors for
cardiovascular disease in children and young adults after renal
transplantation. Clin J Am Soc Nephrol. 1:1284–1292. 2006.
View Article : Google Scholar
|
|
26
|
Baumann M, Chang J, Thürmel K, Roos M, von
Eynatten M, Sollinger D, Lutz J and Heemann U: Fisher-Lewis kidney
transplantation model as a tool for investigation of
transplantation-induced cardiomyopathy. Transplant Proc.
41:2612–2615. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiao D, Xu Z, Huang X, Longo LD, Yang S
and Zhang L: Prenatal gender-related nicotine exposure increases
blood pressure response to angiotensin II in adult offspring.
Hypertension. 51:1239–1247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Gasc JM, Shanmugam S, Sibony M and Corvol
P: Tissue-specific expression of type 1 angiotensin II receptor
subtypes. An in situ hybridization study Hypertension. 24:531–537.
1994.
|
|
30
|
Bulum B, Ozcakar ZB, Kavaz A, Tutar E,
Ekim M and Yalcinkaya F: Hypertension in children after renal
transplantation. Pediatr Int. 57:1138–1142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weir MR, Burgess ED, Cooper JE, Fenves AZ,
Goldsmith D, McKay D, Mehrotra A, Mitsnefes MM, Sica DA and Taler
SJ: Assessment and management of hypertension in transplant
patients. J Am Soc Nephrol. 26:1248–1260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chatzikyrkou C, Eichler J, Karch A, Clajus
C, Scurt FG, Ramackers W, Lehner F, Menne J, Haller H, Mertens PR,
et al: Short- and long-term effects of the use of RAAS blockers
immediately after renal transplantation. Blood Press. 26:30–38.
2017. View Article : Google Scholar
|
|
33
|
Reudelhuber TL: The renin-angiotensin
system: Peptides and enzymes beyond angiotensin II. Curr Opin
Nephrol Hypertens. 14:155–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Steckelings UM, Rompe F, Kaschina E and
Unger T: The evolving story of the RAAS in hypertension, diabetes
and CV disease: Moving from macrovascular to microvascular targets.
Fundam Clin Pharmacol. 23:693–703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rizzoni D, De Ciuceis C, Salvetti M, Paini
A, Rossini C, Agabiti-Rosei C and Muiesan ML: Interactions between
macro- and micro-circulation: Are they relevant? High Blood Press
Cardiovasc Prev. 22:119–128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kaplan NM and Victor RG: Primary
hypertension: Pathogenesis (with a special section on renal
denervation and carotid barorecptor pacing). Kaplan's Clinical
Hypertension. 11th edition. Williams and Wilkins; Philadelphia, PA:
pp. 40–115. 2014
|
|
37
|
Triggle CR, Samuel SM, Ravishankar S,
Marei I, Arunachalam G and Ding H: The endothelium: Influencing
vascular smooth muscle in many ways. Can J Physiol Pharmacol.
90:713–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kang KT: Endothelium-derived relaxing
factors of small resistance arteries in hypertension. Toxicol Res.
30:141–148. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Touyz RM and Schiffrin EL: Signal
transduction mechanisms mediating the physiological and
pathophysiological actions of angiotensin II in vascular smooth
muscle cells. Pharmacol Rev. 52:639–672. 2000.PubMed/NCBI
|
|
40
|
Horowitz A, Menice CB, Laporte R and
Morgan KG: Mechanisms of smooth muscle contraction. Physiol Rev.
76:967–1003. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Perez-Vizcaino F, Cogolludo A and Moreno
L: Reactive oxygen species signaling in pulmonary vascular smooth
muscle. Respir Physiol Neurobiol. 174:212–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hansen PB: The complex field of interplay
between vasoactive agents. Kidney Int. 76:929–931. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Beevers DG, Morton JJ, Nelson CS, Padfield
PL, Titterington M and Tree M: Angiotensin II in essential
hypertension. Br Med J. 1:4151977. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Morton JJ, Garcia del Rio C and Hughes MJ:
Effect of acute vasopressin infusion on blood pressure and plasma
angiotensin II in normotensive and DOCA-salt hypertensive rats.
Clin Sci (Lond). 62:143–149. 1982. View Article : Google Scholar
|
|
45
|
Campbell DJ: Angiotensin II generation in
vivo: Does it involve enzymes other than renin and
angiotensin-converting enzyme? J Renin Angiotensin Aldosterone
Syst. 13:314–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kakar SS, Sellers JC, Devor DC, Musgrove
LC and Neill JD: Angiotensin II type-1 receptor subtype cDNAs:
Differential tissue expression and hormonal regulation. Biochem
Biophys Res Commun. 183:1090–1096. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Millis RM: Epigenetics and hypertension.
Curr Hypertens Rep. 13:21–28. 2011. View Article : Google Scholar
|
|
48
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 25:1010–1022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xiao D, Dasgupta C, Li Y, Huang X and
Zhang L: Perinatal nicotine exposure increases angiotensin II
receptor-mediated vascular contractility in adult offspring. PLoS
One. 9:e1081612014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Haack KK, Mitra AK and Zucker IH: NF-κB
and CREB are required for angiotensin II type 1 receptor
upregulation in neurons. PLoS One. 8:e786952013. View Article : Google Scholar
|
|
52
|
Nickenig G, Strehlow K, Wassmann S, Bäumer
AT, Albory K, Sauer H and Böhm M: Differential effects of estrogen
and progesterone on AT(1) receptor gene expression in vascular
smooth muscle cells. Circulation. 102:1828–1833. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wenger NK, Speroff L and Packard B:
Cardiovascular health and disease in women. N Engl J Med.
329:247–256. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Miller VM and Duckles SP: Vascular actions
of estrogens: Functional implications. Pharmacol Rev. 60:210–241.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
El-Mas MM and Abdel-Rahman AA:
Longitudinal assessment of the effects of oestrogen on blood
pressure and cardiovascular autonomic activity in female rats. Clin
Exp Pharmacol Physiol. 36:1002–1009. 2009. View Article : Google Scholar : PubMed/NCBI
|