Introduction
Oxygen is central to cellular respiration and energy
metabolism. However, hypoxia is common in the tissues of the
majority of individuals. Hypoxia-induced muscle wasting is a
condition frequently reported in several environmental and
pathological conditions, including exposure to high altitudes,
prolonged immobilization, chronic obstructive pulmonary disease,
exercise, and anemia (1–4). However, the mechanism underlying the
effects of hypoxia in skeletal muscle remains to be elucidated.
Cobalt chloride (CoCl2) is a well-known
hypoxia mimetic, which competes with the activity of bivalent ions
and suppresses the formation of oxygenated hemoglobin (5). In cell culture systems,
CoCl2 inhibits the catalysis of prolyl hydroxylases,
leading to an intracellular hypoxia-like state (6,7).
Therefore, CoCl2 was applied in the present study to
simulate a hypoxic condition.
In the mid-19th century, necrosis was the first
identified form of cell death. Necrosis is unregulated and is
considered to be a cause or consequence of disease; by contrast,
apoptosis is a highly regulated and programmed process, which is
manipulated by defined molecular pathways (8). However, in 1988, a pioneering
publication reported a type of necrotic cell death driven by
specific genes, which was defined as 'necroptosis' (9,10).
Necroptosis is initiated by tumor necrosis factor (TNF)/Fas ligand
under apoptosis-deficient conditions. Molecules that regulate
necroptosis include those involved in receptor-interacting protein
kinase (RIP)1, RIP3, and mixed lineage kinase domain-like protein
signaling. Necrostatin-1 (Nec-1) is a small molecule inhibitor,
which allosterically inhibits RIP1 by interacting with the T-loop,
which is essential for death domain receptor engagement (11). Accumulating evidence has indicated
that Nec-1 inhibits RIP1 but does not affect RIP3 (11,12).
Hypoxia inducible factor (HIF)-1α is a transcription
factor, which acts in response to hypoxia. The hypoxia- or
CoCl2-induced increase in HIF-1α is due to increased
protein stability, which is mediated by activation of the JNK,
extracellular signal-regulated kinase (ERK), p38 mitogen-activated
protein kinase (MAPK) pathways (13). The Raf/MAPK kinase (MEK)/ERK
cascade couples signals which regulate the activity of several
proteins involved in cell death (14). The activation of RIP1 results in
phosphorylation of the ERK signaling pathway through the activation
of MEK1/2. Nec-1 can inhibit the interaction between MEK and ERK1/2
(15). Another report showed that
the inhibitory effect on ERK1/2 was significantly augmented by
Nec-1 in K562 and HL60 cells.
HIF-1α also affects pro-death genes, including Bcl-2
adenovirus E1B 19-kDa interacting protein 3 (BNIP3) (16,17). Early reports suggested that BNIP3
is pivotal in the loss of skeletal muscle mass, and provides a
potential therapeutic target in muscle wasting disorders and other
diseases (18). BNIP3 readily
inserts into the mitochondrial membrane following a hypoxic
stimulus or reactive oxygen species (ROS) generation. Nec-1
effectively suppresses BNIP3-induced caspase-independent necrosis,
including cell death and lactate dehydrogenase leakage (19). However, the role of Nec-1 in the
response of HIF-1α to hypoxia remains to be fully elucidated. Based
on these findings, the present study hypothesized that HIF-1α/BNIP3
is involved in CoCl2-induced necroptosis.
Previous studies have shown that necroptosis can be
induced by CoCl2 and identified mitochondrion-generated
ROS as essential mediators of this process (5,20).
Several lines of evidence have shown that excessive ROS can
directly trigger mitochondrial dysfunction or activated tumor
necrosis factor (TNF)-α, which subsequently induces
caspase-dependent classical apoptosis or caspase-independent
necrosis (21,22). However, the mechanism underlying
the effects of CoCl2-induced necroptosis in skeletal
muscle remains to be elucidated.
Nec-1 has been used as evidence of the role of
necroptosis in different disorders (8,23,24). Nec-1 exhibits protective effects
against Parkinson's disease, cerebral ischemia injury,
cardiomyocyte hypertrophy, Alzheimer's disease, stroke, and
amyotrophic lateral sclerosis, in which oxidative stress or free
radical injury is considered a potent inducer (23–25). However, the effect of Nec-1 on
hypoxia-induced muscle wasting remains to be fully elucidated. In
the present study, it was shown that RIP1-dependent necroptosis
induced by CoCl2 was mediated through the ERK1/2 and
HIF-1α/BNIP3 pathways. Nec-1 may exert protective effects on C2C12
cell viability and differentiation under conditions of
CoCl2-induced hypoxia, which may provide a novel insight
into protecting against hypoxia-induced muscle wasting.
Materials and methods
C2C12 culture and drug
administration
The C2C12 mouse myoblast cell line (Stem Cell Bank,
Chinese Academy of Sciences, Shanghai, China) was cultured in
Dulbecco's modified Eagle's medium (DMEM) high glucose (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA), 100 U/ml of penicillin, and 100 µg/ml of
streptomycin in 5% CO2 at 37°C. When the cells reached
80–90% confluence, they were differentiated by incubation in DMEM
containing 2% horse serum (HyClone; GE Healthcare Life Sciences). A
stock solution of Nec-1 (Sigma-Aldrich; Merck Millipore, Munich,
Germany) was prepared in DMSO at a concentration of 150 mM and the
working solution was diluted into 0.1% with DMEM. CoCl2
(Sigma-Aldrich; Merck Millipore) was dissolved in DMEM as a 2,000 ×
concentrate and diluted to 200 µM for actual use.
Subsequently, 150 µM Nec-1 was added to cells for treatment
in the absence or presence of 200 µM CoCl2 for 48
h prior to cell collection.
Cell viability assays
Cell viability was assessed using CCK8 assays
(Beyotime Institute of Biotechnology, Jiangsu, China). Briefly, the
C2C12 cells were seeded onto 96-well plates at a density of
0.2×104 cells/well. Following drug treatment for 48 h,
the cells were treated with CCK8 solution. Quantification was
performed 1 h later by measuring the absorbance at 450 nm on a
microplate reader (BioTek Instruments, Inc., Midland, ON, Canada).
Data are presented as the percentage of the control.
Detection of necrosis and apoptosis
An Annexin V-fluorescein isothiocyanate apoptosis
detection kit (Sony Biotechnology Inc., San Jose, CA, USA) was used
in accordance with the manufacturer's protocol. The C2C12 myotubes
were incubated with CoCl2 or Nec-1 for 48 h. The cells
were then digested with trypsin and washed twice with cold
phosphate-buffered saline. The cells were subsequently resuspended
in 500 µl of binding buffer. Subsequently, 5 µl of
Annexin V and 5 µl of 7-aminoactinomycin D were added to the
cells and incubated in the dark for 15 min.
Transmission electron microscopy
(TEM)
The specimens were fixed in 2.5% glutaraldehyde and
post-fixed in 1% osmium tetroxide, dehydrated through a graded
ethanol series, and embedded in epoxy resin. Serial ultrathin
sections were cut to 70 µm on an LKB-III ultratome (Leica
Microsystems GmbH, Wetzlar, Germany), stained with uranyl acetate
(Ted Pella, Inc., Redding, CA, USA) and lead citrate (Ted Pella,
Inc.), and examined on an electron microscope (H7600; Hitachi,
Tokyo, Japan) at an acceleration voltage of 100 kV.
Western blot analysis
The C2C12 myotubes were lysed in RIPA buffer
containing protease inhibitor (Beyotime Institute of Biotechnology)
and phenylmethylsulfonyl fluoride to extract total proteins. An
equal quantity of protein (20 µg) was separated by 10–12%
sodium dodecyl sulfate polyacrylamide gel electrophoresis and
transferred onto polyvinylidene fluoride membranes. The membranes
were blocked with 5% non-fat milk and incubated with primary
antibodies targeting RIP1 (1:1,000; MAB3585, R&D Systems, Inc.,
Minneapolis, MN, USA), RIP3 (1:1,000; ab16090, Abcam, Cambridge,
UK), myogenin (1:500; MAB3876, EMD Millipore, Billerica, MA, USA),
myosin heavy chain (MyHC; 1:1,000; MAB4470, R&D Systems, Inc.),
atrogin-1 (1:1,000; ab168372, Abcam), phosphorylated ERK1/2
(p-ERK1/2, 1:1,000; 4370, Cell Signaling Technology, Inc., Danvers,
MA, USA), ERK1/2 (1:1,000; 4695, Cell Signaling Technology, Inc.),
HIF-1α (1:1,000; ab179483, Abcam), or BNIP3 (1:1,500; ab109362,
Abcam) overnight at 4°C. The membranes were then incubated with
goat anti-mouse (1:10,000; AS003, ABclonal Biotech, Co., Ltd.,
Woburn, MA USA) or anti-rabbit secondary antibodies (1:10,000;
AS014, ABclonal Biotech, Co., Ltd.) for 1 h at room temperature.
Band intensity was determined using a chemiluminescent imaging
system (Tanon Sciences and Technology Co., Ltd., Shanghai, China).
Tubulin (1:5,000; AC021, ABclonal Biotech, Co., Ltd.) was used as a
control for protein quantification. Band intensity was quantified
using ImageJ software (v1.4.3.67, National Institutes of Health,
Bethesda, MD, USA).
Determination of intracellular ROS
The level of intracellular ROS was measured by BD
Accuri C6 (BD Biosciences, Franklin Lakes, NJ, USA) using a
DCFDA-Cellular Reactive Oxygen Species Detection Assay kit (Abcam)
according to the manufacturer's protocol. The cells were incubated
with 2′,7′-dichlorofluorescin diacetate (DCFH-DA) at a final
concentration of 10 µM in 1X buffer for 30 min at 37°C, and
detected by fluorescence spectroscopy with maximum excitation and
emission spectra of 485 and 535 nm, respectively. For each
analysis, 10,000 events were recorded. Data were exported by Accuri
CFlow software (v1.0.227.4, BD Biosciences), and the intracellular
ROS levels were expressed as the average DCF fluorescence
intensity.
Measurement of mitochondrial membrane
potential (Δψm)
The Δψm of the C2C12 cells was determined by
CytoFLEX (Beckman Coulter, Brea, CA, USA) with the fluorescent dye
JC-1 (Nanjing KeyGEN Biotech Co., Ltd., Nanjing, China). The cells
were incubated with 1 µl JC-1 solution for 20 min at 37°C.
Fluorescence was measured on a flow cytometer with excitation and
emission spectra of 488 and 530 nm, respectively. Data were
exported by CyExpert (v1.2.11.0, Beckman Coulter).
Statistical analysis
Statistical analysis was performed using Prism 5
software (GraphPad Software, Inc., La Jolla, CA, USA). Data are
reported as the mean ± standard error of the mean. Statistical
significance was assessed by a one-way analysis of variance between
groups. When significant variations were found, Tukey's multiple
comparison test was performed. In all analyses, P<0.05 was
considered to indicate statistically significant difference.
Results
Nec-1 treatment increases cell viability
and reduces cell death in C2C12 cells under conditions of
CoCl2-induced hypoxia
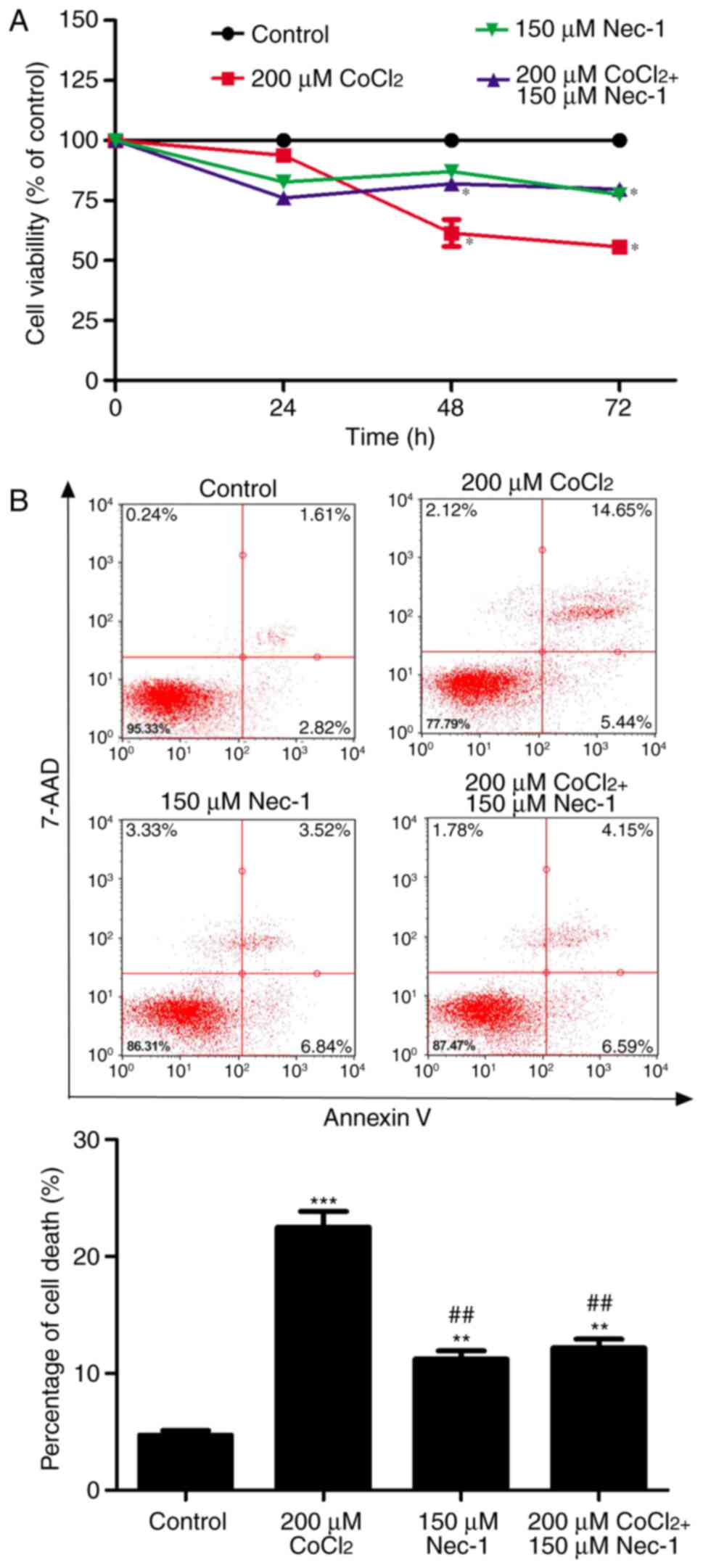
In the present study, the well-known hypoxia mimetic
CoCl2 was used to induce hypoxia, and C2C12 cell
viability was measured. As evidenced by the CCK8 assay,
CoCl2 produced toxic effects on the C2C12 cells in a
time-dependent manner. Cell viability was decreased by ~40%
following CoCl2 treatment for 48 h and had decreased to
~50% at 72 h. To determine the protective effects of Nec-1 on
hypoxia-induced cell viability, the C2C12 cells were cultured with
CoCl2 and Nec-1 for 72 h. The CoCl2-induced
cytotoxicity towards the C2C12 cells was significantly reversed by
Nec-1 treatment. This suggested that Nec-1 protected against
hypoxia-induced cell death in C2C12 myotubes (Fig. 1A).
The cytotoxic effects of CoCl2-induced
hypoxia were also analyzed by flow cytometry, which revealed that
the percentage of cells undergoing apoptosis and necrosis in
response to CoCl2 treatment was 22.52% (Fig. 1B). Nec-1 inhibited
CoCl2-induced cell death, suggesting that
CoCl2-induced cell death was RIP1-dependent.
Nec-1 treatment promotes C2C12
differentiation under conditions of CoCl2-induced
hypoxia
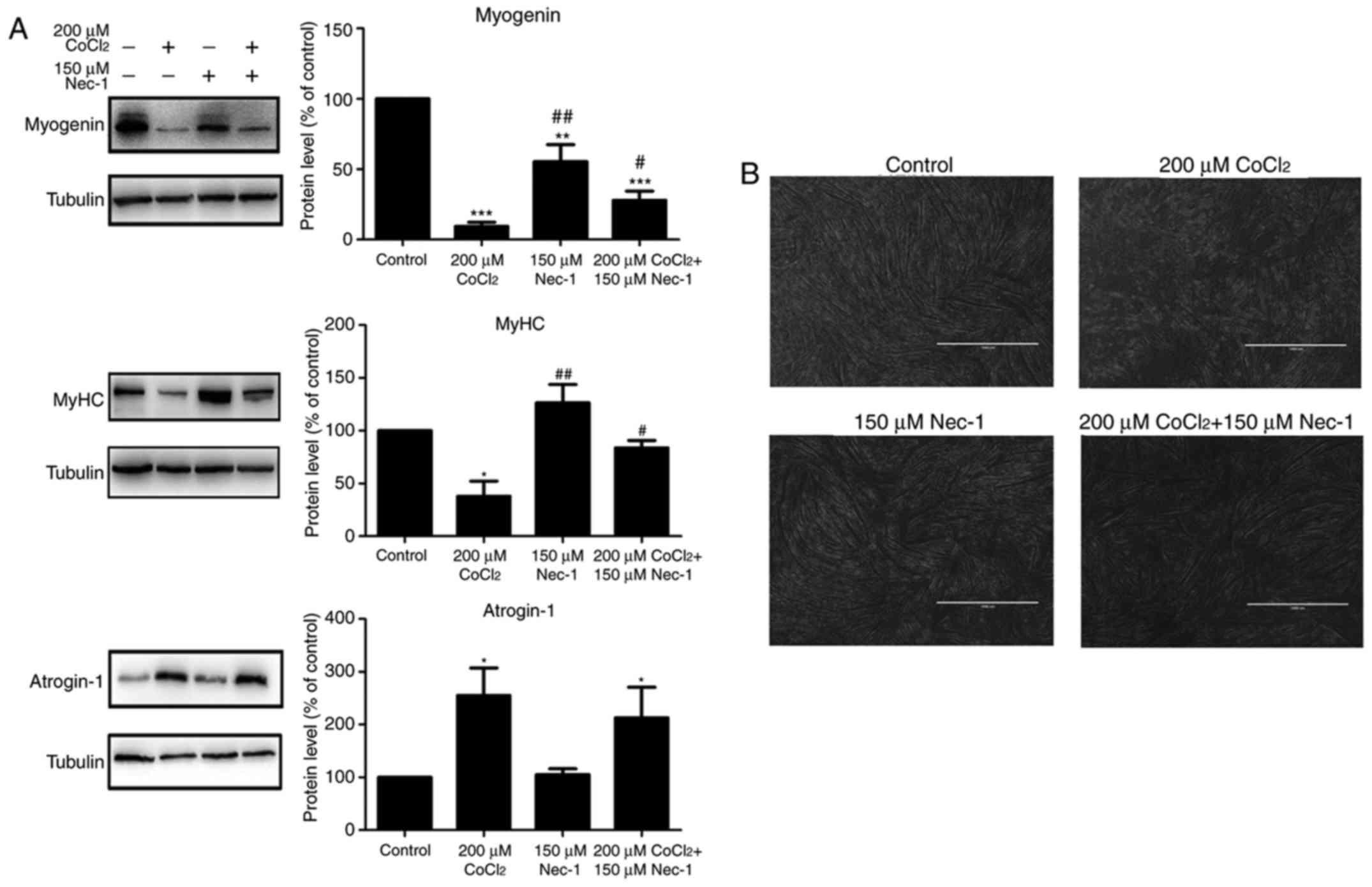
Myogenin and MyHC are specific differentiation
markers required for the fusion of myoblasts to form myotubes. As
shown in Fig. 2A, the expression
of myogenin and MyHC was inhibited by CoCl2 treatment
for 48 h. Nec-1 reversed the changes in the expression of myogenin
and MyHC under conditions of hypoxia. Spindled ring-shaped myotubes
were formed in all groups with the exception of the
CoCl2 group (Fig. 2B).
The level of atrogin-1, a muscle-specific protein that mediates
muscle degradation, was also detected. No significant decrease in
the protein level of atrogin-1 was found in the
Nec-1+CoCl2 group, compared with that in the
CoCl2 group, suggesting that Nec-1 did not reverse
CoCl2-induced muscular atrophy (Fig. 2A).
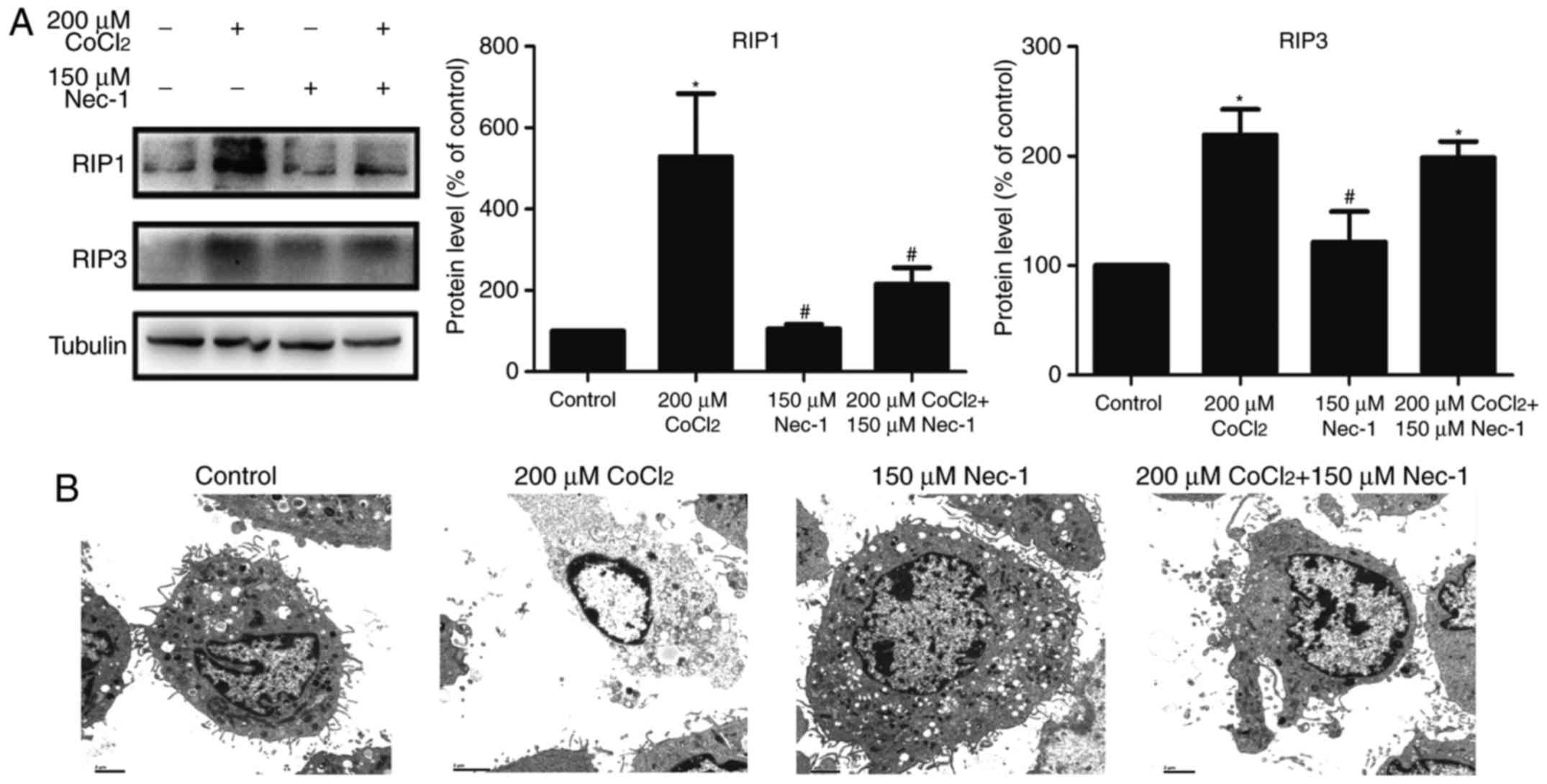
Nec-1 treatment inhibits necroptosis
induced by CoCl2 in C2C12 cells
RIP1 and RIP3 are key factors in triggering
necroptosis. The expression of RIP1 was upregulated following
treatment with CoCl2 and inhibited by Nec-1. However,
Nec-1 had minimal effect on RIP3 in the CoCl2-treated
cells (Fig. 3A). TEM was also
used to characterize the morphological characteristics of dead
cells at the ultrastructural level. CoCl2 induced
typical necroptotic morphological characteristics, including
mitochondrial swelling, membranolysis, organelle disappearance, and
mitochondrial damage (Fig. 3B).
Consistent with the results of the flow cytometric analysis, Nec-1
lessened the morphological damage to the C2C12 cells induced by
CoCl2. Collectively, these results indicated that
CoCl2 induced necroptosis in the C2C12 myotubes, and
that it was reversed by Nec-1 treatment.
Nec-1 treatment prevents the
phosphorylation of ERK1/2, and expression of HIF-1α and BNIP3 under
CoCl2-induced hypoxic conditions
To understand the protective mechanisms underlying
the effect of Nec-1 on C2C12 cells under hypoxic conditions, the
present study detected the levels of total and p-ERK1/2, which
regulate the activity of several proteins involved in cell death.
The increase in p-ERK1/2 induced by CoCl2 was inhibited
by Nec-1 (Fig. 4A). The
expression of HIF-1α and its downstream target, BNIP3, was also
evaluated. HIF-1α and BNIP3 were upregulated following
CoCl2 treatment, consistent with the results from our
previous study (26), whereas
culture with Nec-1 and CoCl2 reduced the expression of
HIF-1α and BNIP3, suggesting the involvement of the HIF-1α/BNIP3
signaling pathway in CoCl2-induced hypoxia in C2C12
cells (Fig. 4B).
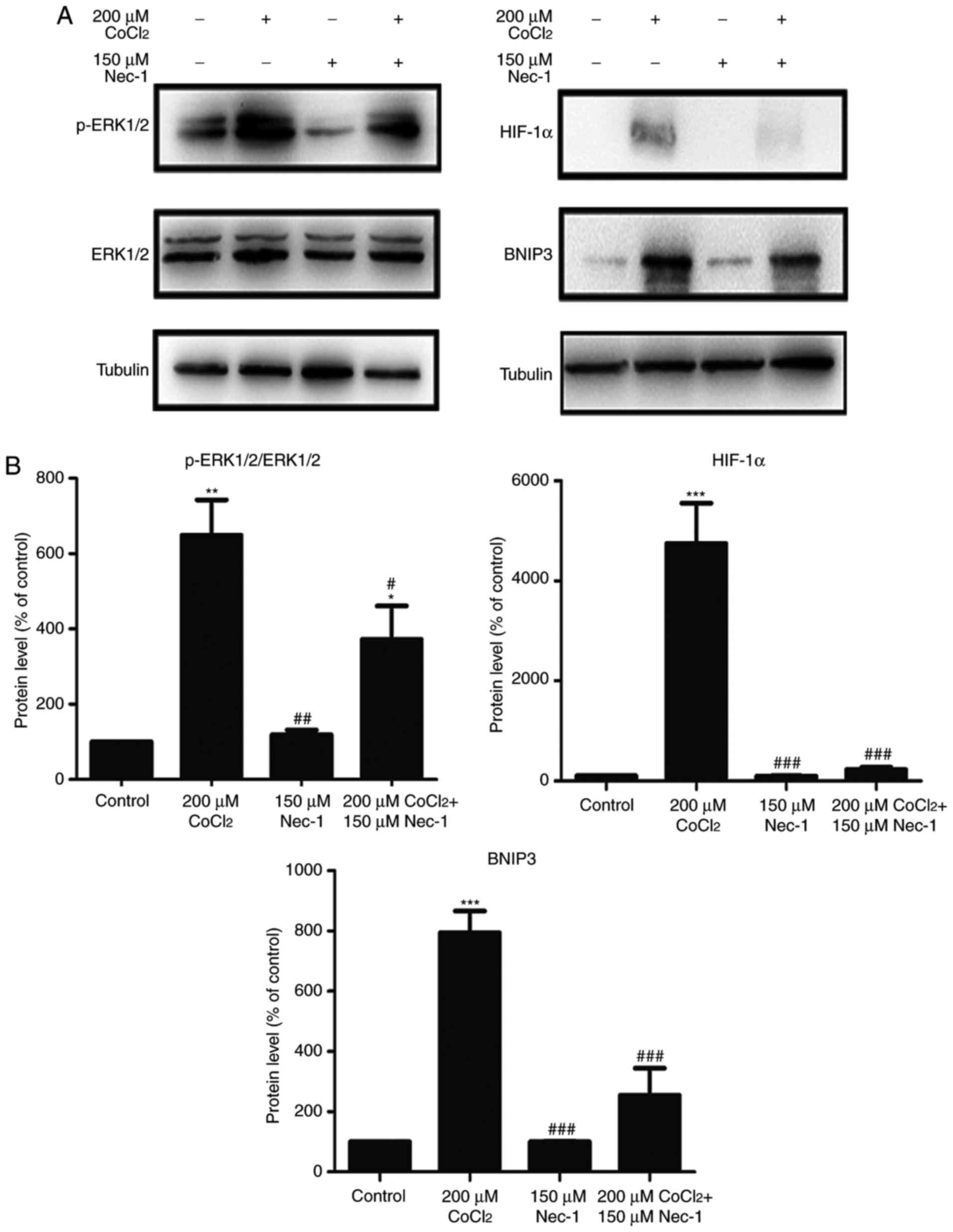 | Figure 4Nec-1 treatment prevents ERK1/2
phosphorylation and decreases the expression of HIF-1α and BNIP3 in
hypoxia induced by CoCl2. Western blot analysis was used
to examine the expression of (A) p-ERK1/2, ERK1/2, (B) HIF-1α and
its downstream target, BNIP, in C2C12 cells in the absence or
presence of CoCl2 and Nec-1. Band intensity was
quantified using ImageJ software. Tubulin was used as an internal
control. *P<0.05, **P<0.01 and
***P<0.001, compared with the control group;
#P<0.05, ##P<0.01 and
###P<0.01, compared with the CoCl2 group.
ERK, extracellular signal-regulated kinase; p-, phosphorylated;
HIF-1α, hypoxia-inducible factor-1α; BNIP3, Bcl-2 adenovirus E1B
19-kDa interacting protein 3; Nec-1, necrostatin-1;
CoCl2, cobalt chloride. |
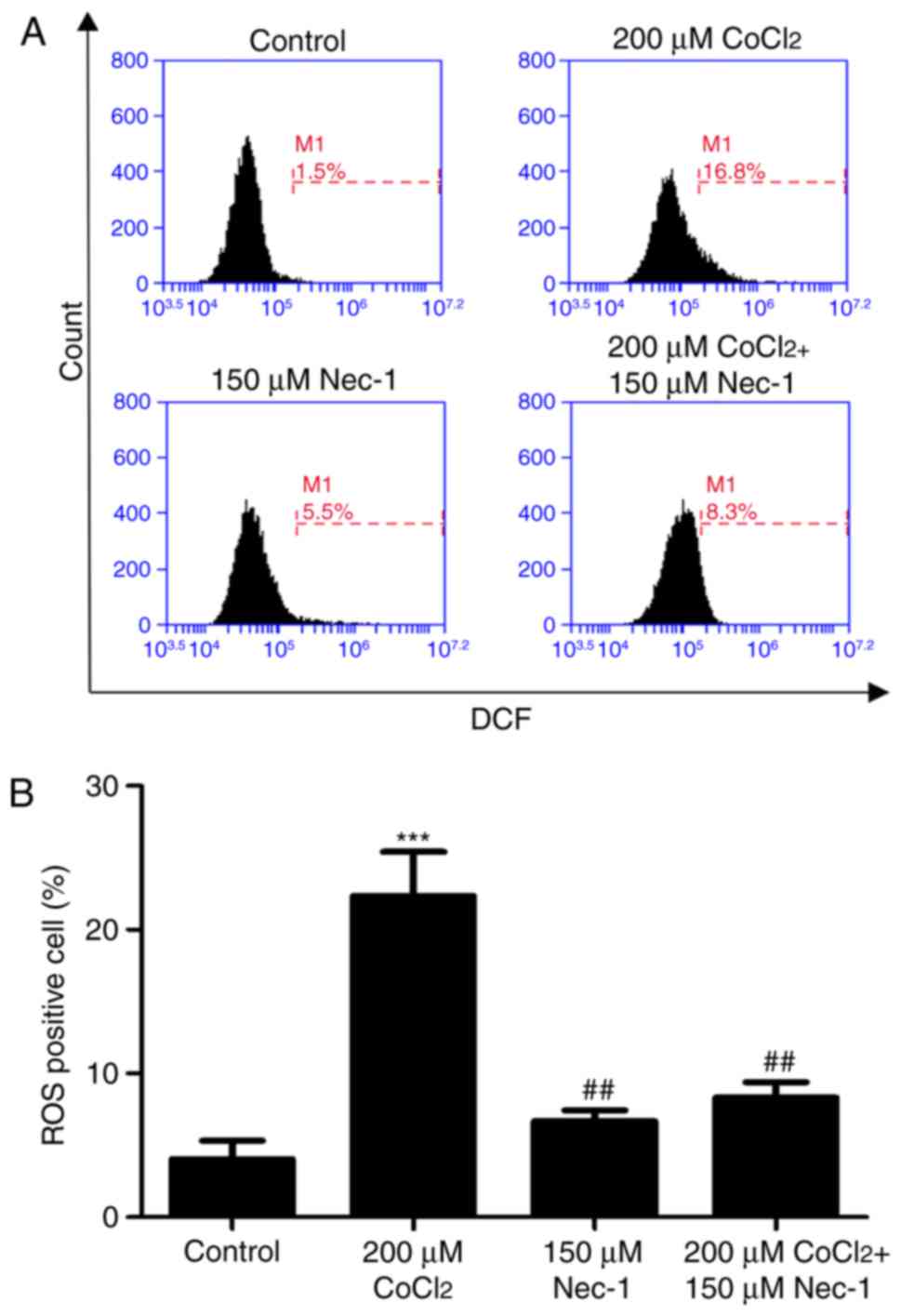
Nec-1 treatment decreases intracellular
ROS in C2C12 cells under CoCl2-induced hypoxic
conditions
Intracellular ROS production was measured by flow
cytometric analysis following DCFH-DA staining. DCFH-DA diffuses
into cells and is deacetylated by cellular esterase to
non-permeable and non-fluorescent DCFH, which is rapidly oxidized
to the fluorescent compound DCF and can be detected by fluorescence
spectroscopy. Compared with the control group, CoCl2
produced more ROS; however, this effect was inhibited by Nec-1
administration (Fig. 5A and
B).
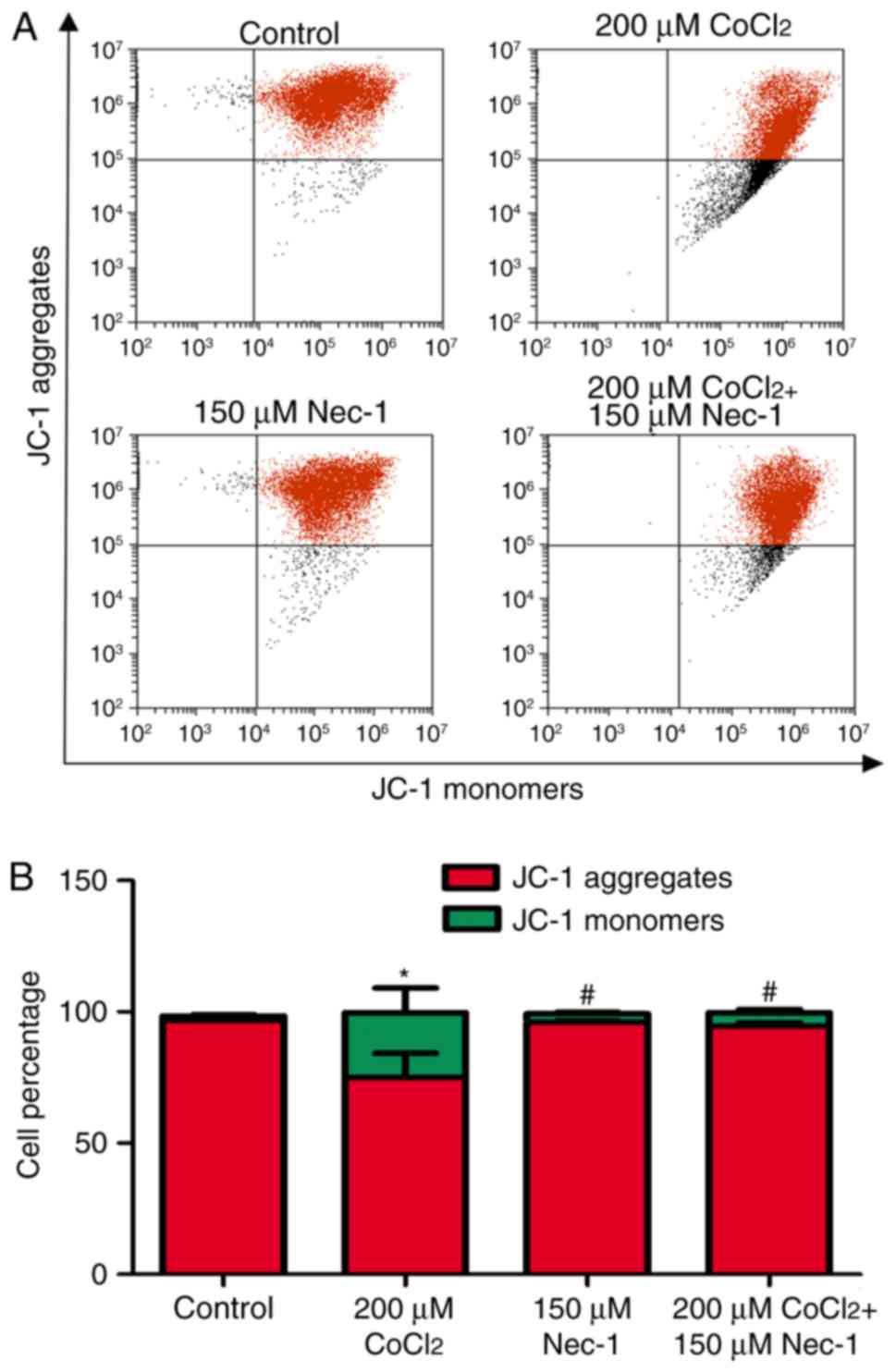
Nec-1 treatment decreases the Δψm of
C2C12 cells under CoCl2-induced hypoxic conditions
Δψm is an indicator of cell health, and the
dissipation of Δψm suggests a loss of mitochondrial membrane
integrity, reflecting the initiation of a pro-apoptotic signal. A
red fluorescent aggregate and monomer fluorescence were detected in
the control group. Following the administration of
CoCl2, the ratio of JC-1 aggregate/monomer fluorescence
intensity decreased, suggesting the earliest event in the process
of apoptosis and mitochondrial dysfunction. There was a significant
increase in aggregate fluorescence in the CoCl2+Nec-1
group, compared with that in the CoCl2 group (Fig. 6A and B).
Discussion
In the present study, it was shown that necroptosis
elicited by CoCl2 was mediated through the ERK1/2 and
HIF-1α/BNIP3 pathways, and that this was associated with an
increase in ROS. The production of ROS in the mitochondria of C2C12
cells resulted in a decreased Δψm, which led to cell death and
inhibited differentiation. Nec-1 decreased the expression of RIP1,
HIF-1α and BNIP3, and the phosphorylation of ERK1/2. The inhibition
of CoCl2-induced necroptosis by Nec-1 promoted myogenic
differentiation.
Necroptosis has been demonstrated in a variety of
disease models, particularly neurological disorders and cancer
(27–29). The present study aimed to
investigate the effects of CoCl2 on C2C12 myotubes,
which may be associated with necroptosis; additionally, it has been
reported that the viability of C2C12 myotubes was affected by
CoCl2, which may be attenuated by Nec-1 (5). In the present study, it was
demonstrated that CoCl2 treatment upregulated the
expression of RIP1 and RIP3, members of the RIP family, which are
key factors in triggering necroptosis. In addition, TEM revealed
necroptotic characteristics in dead cells, including mitochondrial
swelling, membranolysis and organelle disappearance. Nec-1 is an
allosteric inhibitor, which interacts with the RIP1-RIP3 complex.
The results also showed that Nec-1 markedly inhibited the
expression of RIP1 but only marginally decreased that of RIP3,
whereas Nec-1 increased cell viability and reversed morphological
damage in the cells treated with CoCl2. Together, these
results suggested that Nec-1 exerted a protective effect against
CoCl2-induced cytotoxicity. In our previous study, it
was demonstrated that CoCl2-induced hypoxia
downregulated the expression of myogenin and impaired myoblast
fusion (26). To further examine
the protective effect of Nec-1 on skeletal C2C12 cells, the present
study examined the specific differentiation markers, myogenin and
MyHC, which are required for the fusion of myoblasts to form
myotubes. The expression levels of myogenin and MyHC were increased
following Nec-1 treatment. However, no significant difference in
the muscle degradation-mediated protein, atrogin-1 was found,
indicating that Nec-1 facilitated muscle differentiation but did
not attenuate CoCl2-induced hypoxia-induced myofibrillar
degradation.
Several lines of evidence have shown that the
MEK/ERK pathway regulates cell survival and death (30-32). Xie et al (32) investigated the effects of dimethyl
fumarate on different gastrointestinal cancer cell lines and found
that it induced necroptosis in colon cancer cells; the mechanism
involved glutathione depletion, an increase in ROS, and activation
of MAPK-mediated signaling. Locatelli et al (31) reported that the PI3K/AKT and
RAF/MEK/ERK pathways were constitutively activated in patients with
Hodgkin's lymphoma. AEZS-136, a PI3K/ERK dual inhibitor, markedly
promoted the dephosphorylation of MAPK and PI3K/AKT pathway
components, leading to caspase-independent necroptosis with ROS
generation (31). Hypoxia or
CoCl2 increased HIF-1α due to increased protein
stability, which was mediated by activation of the ERK pathway; the
inhibition of RIP1 kinase activity by Nec-1 or the knockdown of
RIP1 using small interfering RNA significantly inhibited the
activation of ERK (11). The
results of the present study indicated that the phosphorylation of
ERK1/2 induced by CoCl2-induced hypoxia was inhibited by
Nec-1.
ROS are also required for TNF-induced necroptosis
(33). In the present study, a
lower Δψm and a simultaneous increase in ROS production was
observed in the CoCl2 group, suggesting dysfunction and
decreased activity of the respiratory chain. Nec-1 decreased ROS
accumulation and prevented the execution of programmed necrosis.
The present study also investigated the expression of BNIP3, which
integrates into the mitochondrial membrane under conditions
triggering ROS accumulation, resulting in necroptosis associated
with energy failure (19,28). Kim et al (19) reported that BNIP3 induced
caspase-independent necrosis-like cell death, which was
significantly inhibited by Nec-1, whereas the pan-caspase inhibitor
zVAD-FMK did not. BNIP3 contains a hypoxia response element and
appears to be a direct target of transcriptional activation by
HIF-1α. Of note, HIF-1α was downregulated following Nec-1
treatment, suggesting that the molecular mechanism of Nec-1 in
C2C12 cells under CoCl2-induced hypoxia involved the
HIF-1α/BNIP3 pathway.
In conclusion, the findings of the present study
revealed that necroptosis was initiated by CoCl2, which
was mainly associated with the phosphorylation of ERK1/2,
upregulation of HIF-1α/BNIP3, and mitochondrion-generated ROS.
Therefore, Nec-1 may protect C2C12 cells under conditions of
CoCl2-induced hypoxia.
Acknowledgments
Not applicable.
Notes
[1]
Funding
The study was supported by grants from the Medical
Scientific Research Foundation of Guangdong Province (grant no.
A2016612), the Administration of Traditional Chinese Medicine of
Guangdong Province (grant no. 20172004), the Science Foundation of
Guangdong Second Provincial General Hospital (grant nos. YQ2015-017
and YN2017-003), the National Natural Science Foundation of China
(grant no. 81101866) and the Sci-tech Development Program of
Guangdong Province (grant no. 2014A020212581).
[2] Availability
of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
RC, JX and YS conceived and designed the
experiments. RC, JX, YS, TJ, SZ and HS performed the experiments.
TJ and CL analyzed the data. RC and YS wrote the paper. All authors
read and approved the final manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Kawamura I, Takemura G, Kanamori H,
Takeyama T, Kawaguchi T, Tsujimoto A, Goto K, Maruyama R, Watanabe
T, Shiraki T, et al: Repeated phlebotomy augments angiogenesis to
improve blood flow in murine ischemic legs. Am J Physiol Heart Circ
Physiol. 299:H372–H378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Masschelein E, Van Thienen R, D'Hulst G,
Hespel P, Thomis M and Deldicque L: Acute environmental hypoxia
induces LC3 lipidation in a genotype-dependent manner. FASEB J.
28:1022–1034. 2014. View Article : Google Scholar
|
|
3
|
Samaras N, Samaras D, Chambellan A,
Pichard C and Thibault R: Pulmonary rehabilitation: The reference
therapy for undernourished patients with chronic obstructive
pulmonary disease. Biomed Res Int. 2014:2484202014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saxena S, Shukla D, Saxena S, Khan YA,
Singh M, Bansal A, Sairam M and Jain SK: Hypoxia preconditioning by
cobalt chloride enhances endurance performance and protects
skeletal muscles from exercise-induced oxidative damage in rats.
Acta Physiol (Oxf). 200:249–263. 2010. View Article : Google Scholar
|
|
5
|
Rovetta F, Stacchiotti A, Faggi F,
Catalani S, Apostoli P, Fanzani A and Aleo MF: Cobalt triggers
necrotic cell death and atrophy in skeletal C2C12 myotubes. Toxicol
Appl Pharm. 271:196–205. 2013. View Article : Google Scholar
|
|
6
|
Cervellati F, Cervellati C, Romani A,
Cremonini E, Sticozzi C, Belmonte G, Pessina F and Valacchi G:
Hypoxia induces cell damage via oxidative stress in retinal
epithelial cells. Free Radical Res. 48:303–312. 2014. View Article : Google Scholar
|
|
7
|
Yuan Y, Hilliard G, Ferguson T and
Millhorn DE: Cobalt inhibits the interaction between
hypoxia-inducible factor-alpha and von Hippel-Lindau protein by
direct binding to hypoxia-inducible factor-alpha. J Biol Chem.
278:15911–15916. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Linkermann A and Green DR: Necroptosis.
New Engl J Med. 370:455–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Laster SM, Wood JG and Gooding LR: Tumor
necrosis factor can induce both apoptic and necrotic forms of cell
lysis. J Immunol. 141:2629–2634. 1988.PubMed/NCBI
|
|
10
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
11
|
Han W, Xie J, Fang Y, Wang Z and Pan H:
Nec-1 enhances shikonin-induced apoptosis in leukemia cells by
inhibition of RIP-1 and ERK1/2. Int J Mol Sci. 13:7212–7225. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen JK, Zhan YJ, Yang CS and Tzeng SF:
Oxidative stress-induced attenuation of thrombospondin-1 expression
in primary rat astrocytes. J Cell Biochem. 112:59–70. 2011.
View Article : Google Scholar
|
|
14
|
Locatelli SL, Cleris L, Stirparo GG,
Tartari S, Saba E, Pierdominici M, Malorni W, Carbone A, Anichini A
and Carlo-Stella C: BIM upregulation and ROS-dependent necroptosis
mediate the antitumor effects of the HDACi Givinostat and Sorafenib
in Hodgkin lymphoma cell line xenografts. Leukemia. 28:1861–1871.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Obitsu S, Sakata K, Teshima R and Kondo K:
Eleostearic acid induces RIP1-mediated atypical apoptosis in a
kinase-independent manner via ERK phosphorylation, ROS generation
and mitochondrial dysfunction. Cell Death Dis. 4:e6742013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi H, Merceron C, Mangiavini L, Seifert
EL, Schipani E, Shapiro IM and Risbud MV: Hypoxia promotes
noncanonical autophagy in nucleus pulposus cells independent of
MTOR and HIF1A signaling. Autophagy. 12:1631–1646. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Feng CC, Lin CC, Lai YP, Chen TS,
Marthandam Asokan S, Lin JY, Lin KH, Viswanadha VP, Kuo WW and
Huang CY: Hypoxia suppresses myocardial survival pathway through
HIF-1α-IGFBP-3-dependent signaling and enhances cardiomyocyte
autophagic and apoptotic effects mainly via FoxO3a-induced BNIP3
expression. Growth Factors. 34:73–86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mammucari C, Milan G, Romanello V, Masiero
E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J,
et al: FoxO3 controls autophagy in skeletal muscle in vivo. Cell
Metab. 6:458–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim JY, Kim YJ, Lee S and Park JH: BNip3
is a mediator of TNF-induced necrotic cell death. Apoptosis.
16:114–126. 2011. View Article : Google Scholar
|
|
20
|
Wang H and Zhang B: Cobalt chloride
induces necroptosis in human colon cancer HT-29 cells. Asian Pac J
Cancer Prev. 16:2569–2574. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang YC, Tsai MS, Hsieh PC, Shih JH, Wang
TS, Wang YC, Lin TH and Wang SH: Galangin ameliorates
cisplatin-induced nephrotoxicity by attenuating oxidative stress,
inflammation and cell death in mice through inhibition of ERK and
NF-kappaB signaling. Toxicol Appl Pharmacol. 329:128–139. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu S, Ai Q, Feng K, Li Y and Liu X: The
cardioprotective effect of dihydromyricetin prevents
ischemia–reperfusion-induced apoptosis in vivo and in vitro via the
PI3K/Akt and HIF-1α signaling pathways. Apoptosis. 21:1366–1385.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang M, Li J, Geng R, Ge W, Zhou Y, Zhang
C, Cheng Y and Geng D: The inhibition of ERK activation mediates
the protection of necrostatin-1 on glutamate toxicity in HT-22
cells. Neurotox Res. 24:64–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao S, Andreeva K and Cooper NG:
Ischemia-reperfusion injury of the retina is linked to necroptosis
via the ERK1/2-RIP3 pathway. Mol Vis. 20:1374–1387. 2014.PubMed/NCBI
|
|
25
|
Zhao M, Lu L, Lei S, Chai H, Wu S, Tang X,
Bao Q, Chen L, Wu W and Liu X: Inhibition of receptor interacting
protein kinases attenuates cardiomyocyte hypertrophy induced by
palmitic acid. Oxid Med Cell Longev. 2016:14516762016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen R, Jiang T, She Y, Xu J, Li C, Zhou
S, Shen H, Shi H and Liu S: Effects of cobalt chloride, a
hypoxia-mimetic agent, on autophagy and atrophy in skeletal C2C12
myotubes. Biomed Res Int. 2017:70975802017.PubMed/NCBI
|
|
27
|
Chen G, Cheng X, Zhao M, Lin S, Lu J, Kang
J and Yu X: RIP1-dependent Bid cleavage mediates TNFα-induced but
Caspase-3-independent cell death in L929 fibroblastoma cells.
Apoptosis. 20:92–109. 2015. View Article : Google Scholar
|
|
28
|
Das A, McDonald DG, Dixon-Mah YN, Jacqmin
DJ, Samant VN, Vandergrift WA III, Lindhorst SM, Cachia D, Varma
AK, Vanek KN, et al: RIP1 and RIP3 complex regulates
radiation-induced programmed necrosis in glioblastoma. Tumor Biol.
37:7525–7534. 2016. View Article : Google Scholar
|
|
29
|
Chavez-Valdez R, Martin LJ, Flock DL and
Northington FJ: Necrostatin-1 attenuates mitochondrial dysfunction
in neurons and astrocytes following neonatal hypoxia-ischemia.
Neuroscience. 219:192–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Festjens N, Vanden Berghe T, Cornelis S
and Vandenabeele P: RIP1, a kinase on the crossroads of a cell's
decision to live or die. Cell Death Differ. 14:400–410. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Locatelli SL, Careddu G, Stirparo GG,
Castagna L, Santoro A and Carlo-Stella C: Dual PI3K/ERK inhibition
induces necroptotic cell death of Hodgkin Lymphoma cells through
IER3 downregulation. Sci Rep. 6:357452016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie X, Zhao Y, Ma CY, Xu XM, Zhang YQ,
Wang CG, Jin J, Shen X, Gao JL, Li N, et al: Dimethyl fumarate
induces necroptosis in colon cancer cells through GSH depletion/ROS
increase/MAPKs activation pathway. Brit J Pharmacol. 172:3929–3943.
2015. View Article : Google Scholar
|
|
33
|
Zhao W, Feng H, Sun W, Liu K, Lu JJ and
Chen X: Tert-butyl hydroperoxide (t-BHP) induced apoptosis and
necroptosis in endothelial cells: Roles of NOX4 and mitochondrion.
Redox Biol. 11:524–534. 2017. View Article : Google Scholar : PubMed/NCBI
|