Introduction
The incidence of diabetes mellitus (DM) is estimated
to rise to 7.7% by 2030 globally (1) and is predicted to affect 591,900,000
individuals by 2035 (2). DM is a
major and increasing health problem worldwide due to its increasing
incidence, which can lead to a variety of complications, including
diabetic retinopathy, diabetic nephropathy, diabetic neuropathy,
diabetic foot and cardiovascular complications. Of note,
cardiovascular complications are the major cause of
diabetes-associated mortality (3). The vascular endothelium is central
to the pathogenesis of diabetic complications, with vascular
endothelial cells mainly involved in maintaining endothelial
dysfunction; cardiovascular homeostasis is considered an important
factor in the pathogenesis of diabetes-associated vascular
complications (4,5). Studies have revealed that
hyperglycemia induces numerous pathological changes in vascular
endothelial cell injury, including oxidative stress (6), inflammation (7), increased endothelial cell apoptosis
(6,8) and mitochondrial membrane
permeabilization (9). However,
the associated molecular mechanism by which hyperglycemia results
in vascular endothelial cell injury in DM remains to be
elucidated.
As one of four protein-tyrosine kinases, Janus
kinase (JAK)1, JAK2, JAK3 and Tyk2, JAK2 is an essential factor in
cellular proliferation, differentiation, survival and senescence.
JAK2 also regulates other signaling molecules, including the RAS,
signal transducer and activator of transcription (STAT)5, STAT3 and
phosphoinositide 3-kinase/Protein kinase B (PI3K/AKT) pathways
(10). STAT3 is a member of the
STAT family (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6),
and its phosphorylation is induced by the activation of JAK2;
phosphorylated (p-)STAT3 forms a dimer and translocates into the
nucleus from the cytoplasm, where it binds to related sequences and
alters the expression of various target genes. Therefore, the
JAK2/STAT3 pathway is gradually being recognized as a
membrane-to-nucleus pathway for a variety of stimulating responses
in vitro and in vivo (11-13). Accumulating evidence has shown
that the JAK2/STAT3 pathway is a cell survival signal, which
contributes to cell proliferation, differentiation, growth and
apoptosis (14-16). The JAK2/STAT3 pathway is also
important in the progress of cardiovascular diseases. Manea et
al (17) demonstrated that
the JAK2/STAT3 pathway is a crucial regulator of the response of
EAhy926 endothelial cells to diabetes-associated cardiovascular
dysfunction. Hyperglycemia increases angiotensin (Ang)-II-induced
vascular smooth muscle cell proliferation by increasing JAK2/STAT3
pathway transduction (18). These
findings indicate that the JAK2/STAT3 pathway is important in
cardiovascular diseases and endothelial dysfunction. However, the
effects of the JAK2/STAT3 pathway on high glucose (HG)-induced
endothelial dysfunction in vivo, and the associated
mechanisms, remain to be elucidated.
There has been increasing focus on the protective
effects of Ang-(1-7) against hyperglycemia-induced cardiovascular
complication. The secretory level of plasma Ang-(1-7) was
demonstrated to be low in patients with diabetic-induced cardiac
dysfunction (19). The expression
level of angiotensin-converting enzyme 2 (ACE2), which is
responsible for the tissue degradation of Ang-II into Ang-(1-7),
was also observed to be decreased in patients with diabetes
(20). An increase in the
expression levels of ACE2 markedly improved control of blood
glucose levels and alleviated glomerular injury in streptozotocin
(STZ)-induced diabetic mice (21), and Ang-(1-7) was reported to exert
protective effects against diabetes-induced cardiovascular events
(22,23). In addition, the Ang-(1-7) and
ACE2/Ang-(1-7)/Mas receptor axis was revealed to have additional
beneficial effects in preventing diabetes-induced cardiac
dysfunction (24). These findings
indicate that Ang-(1-7) is closely associated with
hyperglycemia-induced cardiovascular dysfunction. However, the
underlying mechanisms between Ang-(1-7) and hyperglycemia-induced
cardiovascular dysfunction remain to be fully elucidated.
A previous study reported that Ang-(1-7) produced an
inhibitory effect on the activation of JAK2/STAT3 (25). In diabetic nephropathy, Ang-(1-7)
exerts renoprotective effects on diabetic nephropathy via
inhibiting the STAT3 pathway, apparent as a reduction in
inflammation, fibrosis, oxidative stress and lipotoxicity (26). The present study tested the
hypothesis that exogenous Ang-(1-7) protects human umbilical vein
endothelial cells (HUVECs) against HG-induced injury and
inflammation by inhibiting the JAK2/STAT3 pathway.
Materials and methods
Materials
Ang-(1-7) was purchased from Sigma; Merck KGaA
(Darmstadt, Germany) and stored at −20°C. The following
2′,7′-dichlorofluorescein diacetate (DCFH-DH), Hoechst 33258,
5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imida-carbocyanine iodide
(JC-1) and AG490 (an inhibitor of the STAT3/JAK2 pathway) were
obtained from Sigma-Aldrich; Merck KGaA). The Cell Counting Kit-8
(CCK-8) was supplied by Dojindo Molecular Technologies, Inc.
(Kumamoto, Japan). Fetal bovine serum (FBS) and Dulbecco's modified
Eagle's medium (DMEM) were purchased from Gibco; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Anti-p-STAT3 antibody (cat.
no. SAB4300033), anti-total (t-)STAT3 antibody (cat. no.
SAB4300708), anti-p-JAK2 antibody (cat. no. SAB4300124),
anti-t-JAK2 antibody (cat. no. SAB4501599), anti-caspase-3 antibody
(cat. no. C5737) anti-NADPH oxidase 4 (Nox4) antibody (cat. no.
SAB4503153) and anti-endothelial nitric oxide synthase (eNOS)
antibody (cat. no. N2643) were supplied by Cell Signaling
Technology, Inc. (Boston, MA, USA), horseradish peroxidase
(HRP)-conjugated secondary antibody (cat. no. KC-5A08) and a BCA
protein assay kit were obtained from KangChen Biotech, Inc.
(Shanghai, China). Enhanced chemiluminescence (ECL) solution was
purchased from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China).
The reagents for reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) analysis was obtained from Invitrogen;
Thermo Fisher Scientific, Inc.).
Cell culture and treatments
The HUVECs were supplied by Sun Yat-sen University
Experimental Animal Center (Guangzhou, China). The HUVECs were
cultured in DMEM supplemented with 10% FBS under an atmosphere of
5% CO2 and 37°C with 95% air.
To establish a model of HG-induced HUVEC injury, the
cells were cultured in DMEM (5.5 mM glucose) for 12 h prior to the
administration of 40 mM glucose (final concentration) for 24 h. The
glucose concentration of the control group was 5.5 mM. To
investigate the protective effect of exogenous Ang-(1-7) against HG
(40 mM glucose)-induced injury, the cells were seeded at a density
of 1×104/ml and treated with HG in the presence or
absence of Ang-(1-7) for 24 h at 37°C. In order to examine whether
the STAT3/JAK2 pathway contributed to the protective effects of
Ang-(1-7), and to further determine the mechanisms underlying the
protective effects of Ang-(1-7), the HUVECs at a density of
1×104/ml were co-treated with HG, 2 µmol/l
Ang-(1-7) and 20 µmol/l AG490 (an inhibitor of the
STAT3/JAK2 pathway), and incubated at 37°C.
RNA interference
HUVECs were transfected at 70% confluency using
Lipofectamine transfection reagent (Life Technologies; Thermo
Fisher Scientific, Inc.), with siRNA against NLRP3 (Ribo
Biotechnology, Shanghai, China) or a physiologically irrelevant
negative control siRNA. The siRNA sequences used in the present
study were as follows: Sense, 5′-GCU UCA GCC ACA UGA CUU UTT-3′;
and antisense, 5′-AAA GUC AUG UGG CUG AAG CTT-3′. Each siRNA was
dissolved in nuclease-free water to achieve a final concentration
of 20 µM. A total of 5 µl siRNA (20 µM) and 5
µl Lipofectamine were added to a 500 µl buffer
system. The mixtures were kept at room temperature for 30 min to
form complexes, and equal amounts were added into wells of a 6-well
plate. The cultures were incubated at 37°C in a 5% CO2
incubator. The medium was replaced after 12 h with DMEM without the
presence of siRNA or transfection reagent. Cells were collected at
12 h for analyses.
Western blot analysis
Following the indicated treatments, the HUVECs were
harvested using a cell scraper and lysed with cell lysis solution
(Beyotime Institute of Biotechnology, Haimen, China), at 4°C for 30
min. The total proteins were quantified using the BCA protein assay
kit. Loading buffer was added to the cytosolic extracts and boiled
for 5 min. Equal quantities of supernatant from each sample (20
µg were fractionated by 10% sodium dodecyl
sulphate-polyacrylamide gel electrophoresis, following which the
total proteins were transferred onto polyvinylidene difluoride
membranes. The membranes were blocked with 5% fat-free milk for 60
min in fresh blocking buffer containing 0.1% Tween20 in
Tris-buffered saline (TBS-T) at room temperature, and incubated
with either anti-p-STAT3 (1:1,000 dilution), anti-t-STAT3 (1:1,000
dilution), anti-Nox4 (1:1,000 dilution), anti-p-JAK2 (1:1,000
dilution), anti-t-JAK2 (1:1,000 dilution), anti-caspase-3 (1:1,000
dilution) and anti-eNOS (1:1,000 dilution) in freshly prepared
TBS-T with 3% fat-free milk, overnight with gentle agitation at
4°C. The membranes were then washed for 15 min with TBS-T and
incubated with horseradish peroxidase (HRP)-conjugated goat
anti-rabbit secondary antibody (Kangchen Biotech, Inc.), at a
1:3,000 dilution, in TBS-T with 3% fat-free milk for 90 min at room
temperature. The membranes were then washed three times with TBS-T
for 15 min and the immunoreactive signals were visualized using an
ECL detection. In order to quantify protein expression, the X-ray
films were scanned and analyzed with ImageJ 1.47i software
(National Institutes of Health, Bethesda, MA, USA). The experiment
was repeated three times.
Measurement of cell viability
The HUVECs were seeded in 96-well plates at a
concentration of 1×104/ml, and incubated at 37°C,
following which a CCK-8 assay was used to assess the cell viability
of HUVECs. Following the indicated treatments, 10 µl CCK-8
solution at a 1/10 dilution was added to each well and the plate
was incubated for 1.5 h in the incubator. The absorbance at 450 nm
was determined using a microplate reader (Molecular Devices LLC,
Sunnyvale, CA, USA). The mean optical density (OD) of three wells
in the indicated groups was used to calculate the percentage of
cell viability according to the following formula: Cell viability
(%) = (OD treatment group/OD control group) x 100%. The experiment
was performed five times.
Hoechst 33258 nuclear staining for the
analysis of apoptosis
Apoptotic cell death was analyzed using Hoechst
33258 staining followed by photofluorography. Firstly, the HUVECs
were plated in 35 mm dishes at a density of 1×106
cells/well. Following the indicated treatments, the cells were
fixed with 4% paraformaldehyde in 0.1 mol/l phosphate-buffered
saline (PBS; pH 7.4) for 10 min at 4°C. The slides were washed 5
times with PBS. Following staining by incubation with 5 mg/ml
Hoechst 33258 for 30 min, the cells were washed 5 times with PBS.
Finally, the cells were visualized under a fluorescence microscope
(Bx50-FLA; Olympus Corporation, Tokyo, Japan). Viable HUVECs
exhibited a uniform blue fluorescence throughout the nucleus and
normal nuclear size, whereas apoptotic HUVECs demonstrated
condensed, distorted or fractured nuclei. The experiment was
repeated 5 times.
Examination of intracellular reactive
oxygen species (ROS) generation
Intracellular ROS generation was determined by the
oxidative conversion of cell-permeable oxidation of DCF-DH to
fluorescent DCF. The HUVECs were cultured on a slide with DMEM.
Following the above treatments, the slides were washed twice with
PBS. Subsequently, 10 µmol/l DCFH-DA solution in serum-free
medium was added to the slides, and the cells were incubated at
37°C for a further 30 min. The slides were washed 5 times with PBS,
and DCF fluorescence was measured over the entire field of vision
using a fluorescence microscope connected to an imaging system
(BX50-FLA; Olympus Corporation). The mean fluorescence intensity
(MFI) from five randomly selected fields was measured using ImageJ
1.47i software and the MFI was used as an index of the level of
ROS. The experiment was repeated 5 times.
Measurement of the mitochondrial membrane
potential (MMP)
The MMP was determined using the fluorescent dye,
JC-1, a cell-permeable cationic dye, which preferentially enters
mitochondria based on the highly negative MMP. Depolarization of
MMP results in a loss of MMP from the mitochondria and a decrease
in green fluorescence. The HUVECs were cultured on a slide with
DMEM at a density of 1×106 cells/well. Following the
indicated treatments, the slides were washed 3 times with PBS; and
the cells were incubated with 1 mg/l JC-1 at 37°C for 30 min in the
incubator, washed briefly 3 times with PBS, and air-dried. The
fluorescence was measured over the entire field of vision using a
fluorescent microscope connected to an imaging system (BX50-FLA).
The MFI of JC-1 from three randomly selected fields was analyzed
using ImageJ 1.47i software, and the MFI was measured as an index
of the levels of MMP. The experiment was repeated 3 times.
Measurement of superoxide dismutase (SOD)
activity
SOD activity was analyzed using an SOD assay kit.
Following the indicated treatments, the HUVECs were washed using
PBS and lysed in ice-cold 0.1 M Tris/HCl (pH 7.4) containing 0.5%
Triton, 5 mmol/lβ-mercaptoethanol and 0.1 mg/ml
phenylmethylsulfonyl fluoride. The lysates were clarified by
centrifugation at 14, 000 × g at 4°C for 5 min and cell debris was
discarded. SOD activity was detected using a commercial SOD Assay
kit according to the manufacturer's protocol (Sigma-Aldrich; Merck
KGaA). The absorbance values at 450 nm were measured using a
microplate reader. The experiment was repeated 3 times.
Enzyme-linked immunosorbent assay (ELISA)
for the detection of interleukin (IL)-1β, IL-10, IL-12 and tumor
necrosis factor (TNF)-α in culture supernatant
The HUVECs were seeded at 1×104
cells/well and cultured in 96-well plates. Following the indicated
treatments, the levels of IL-1β, IL-10, IL-12 and TNF-α in culture
media were analyzed using ELISA according to the manufacturer's
protocol. The experiments were repeated 5 times.
RT-qPCR analysis
Total cellular RNA was extracted from cell cultures
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The extracted RNAs were
DNase-treated with RQ1 RNase-Free DNase (Promega Corporation,
Madison, WI, USA). First strand cDNA was prepared using Moloney
Murine Leukemia Virus Reverse Transcriptase (M-MLV RT; cat. no.
1701; Promega Corporation). The RT-qPCR analysis was performed
using the Rotor-Gene™ SYBR Green PCR kit (Qiagen GmbH, Dusseldorf,
Germany) and the QuantiNova SYBR Green PCR kit (Qiagen GmbH) on a
Rotor-Gene 6000 Rotary Analyzer (Qiagen GmbH), and determined using
Rotor-Gene 6000 software version 2.3.3 (Qiagen GmbH). For each
assay, a total of 8 ng cDNA was added to a final reaction volume of
25 µl containing 1X Rotor-Gene SYBR Green PCR master mix or
QuantiNova SYBR Green PCR master mix and 1 µM of each
forward and reverse primer. Quantitative gene amplifications were
performed using the following thermocycling conditions: Initial
denaturation for 5 min at 95°C, 40 cycles of denaturation at 95°C
for 5 sec, and annealing and extension at 60°C for 20 sec. The
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as
endogenous control (or reference gene). A non-template reaction was
used as a negative control. Three replicates were performed for all
analysis.
The selection of the threshold intensity was set at
a fixed intensity on the log-linear phase of the amplification
curve for all the samples tested. Validation experiments, which
included the generation of standard curves using a series of
diluted cDNA samples, were performed to ensure primer efficiency,
and target and reference gene amplification compatibility. Melt
curve analysis and conventional agarose gel analysis were used
alongside to verify the presence of a single amplicon. An
interassay calibration scheme was used to minimize loading
variation and to detect possible contamination with the inclusion
of duplicate reactions and 'no-template' control, respectively, in
each qPCR assay. Relative expression levels of the gene of interest
were calculated using 2−ΔΔCq method (27). All samples were normalized to
GAPDH as the endogenous control (28).
The primers used were as follows: STAT3, forward
5′-CTT TGA GAC CGA GGT GTA TCA CC-3′; and reverse 5′-GGT CAG CAT
GTT GTA CCA CAG G-3′; JAK2, forward 5′-CCG GAA TTC GCT TTG AGT CGG
TTT CTC CGG TTC C-3′; and reverse 5′-TGC TCT AGA CCT CAT GCA GTC
GCT GAA TAA GTC C-3′. GAPDH, forward, 5′CCA CCC ATG GCA AAT TCC ATG
GCA3′; and reverse 5′TCT AGA CGG CAG GTC AGG TCC ACC3′.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Differences between groups were analyzed using one-way
analysis of variance with SPSS 13.0 (SPSS, Inc., Chicago, IL, USA)
software, and followed by an LSD post hoc comparison test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Ang-(1-7) and AG490 attenuate HG-induced
decreased cell viability in HUVECs
The HUVECs were treated with different
concentrations of glucose (10, 20, 30, 40, 50 and 60 mmol/l
glucose) for 24 h, and it was found that glucose induced cell
cytotoxicity (Fig. 1A). A glucose
concentration of 40 mmol/l was considered a suitable concentration
for use in the following experiments. To examine the cytoprotective
effect of Ang-(1-7) against HG-induced cytotoxicity in HUVECs, a
dose-response study with varying doses of Ang-(1-7) (0.5, 1, 2, 4,
6, 8 and 10 µM) was performed to calculate the
cardioprotection dose of Ang-(1-7). As shown in Fig. 1B, exposure of the HUVECs to 40 mM
glucose (HG) for 24 h induced cytotoxicity, as indicated by the
decrease in cell viability. However, the cytotoxic effect of HG on
HUVECs was markedly inhibited by pre-treatment of the cells with
Ang-(1-7) for 30 min. The maximum inhibitory effect was observed
with 2 µM Ang-(1-7). Alone, 10 µM Ang-(1-7) did not
significantly alter the viability of the HUVECs. For this reason,
the HUVECs were pre-treated with 2 µM Ang-(1-7) for 30 min
prior to exposure to HG in all subsequent experiments.
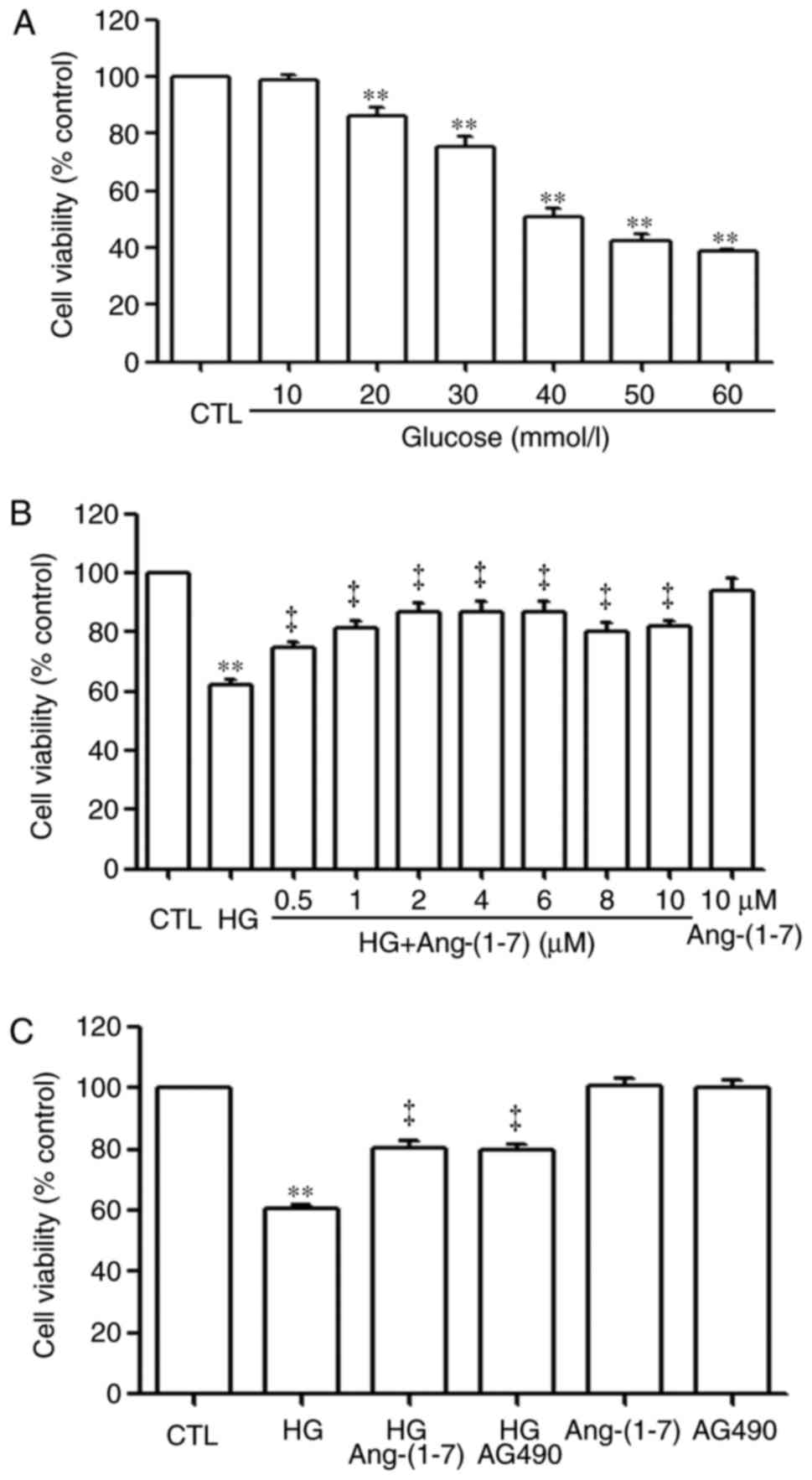 | Figure 1Ang-(1-7) and AG490 alleviate
HG-induced cardiomyocyte cyto-toxicity in HUVECs. Cell viability
was detected using the Cell Counting Kit-8 assay. (A) HUVECs were
treated with different concentrations of glucose (10, 20, 30, 40,
50 and 60 mmol/l glucose) for 24 h. (B) HUVECs were treated with HG
for 24 h in the absence or presence of pre-treatment with the
indicated concentrations (0.5, 1, 2, 4, 6, 8 and 10 µmol/l)
of Ang-(1-7) for 30 min prior to exposure of cells to HG for 24 h.
(C) Cells were pre-treated with or without 2 µmol/l
Ang-(1-7) or 20 µmol/l AG490 (inhibitor of Janus kinase
2/signal transducer and activator of transcription 3 pathway) for
30 min prior to exposure of cells to 40 mM glucose for 24 h.
**P<0.01, vs. CTL group; ‡P<0.01, vs.
HG group. HG, high glucose (40 mM glucose); Ang-(1-7),
angiotensin-(1-7); HUVECs, human umbilical vein
endothelial cells. |
As shown in Fig.
1C, exposure of the HUVECs to HG for 24 h induced cytotoxicity,
which led to a decrease in cell viability. However, this decreased
cell viability was markedly repressed by pre-treatment with 2
µM Ang-(1-7) or 20 µM AG490. Alone, neither 2
µM Ang-(1-7) or 20 µM AG490 affected the viability of
HUVECs.
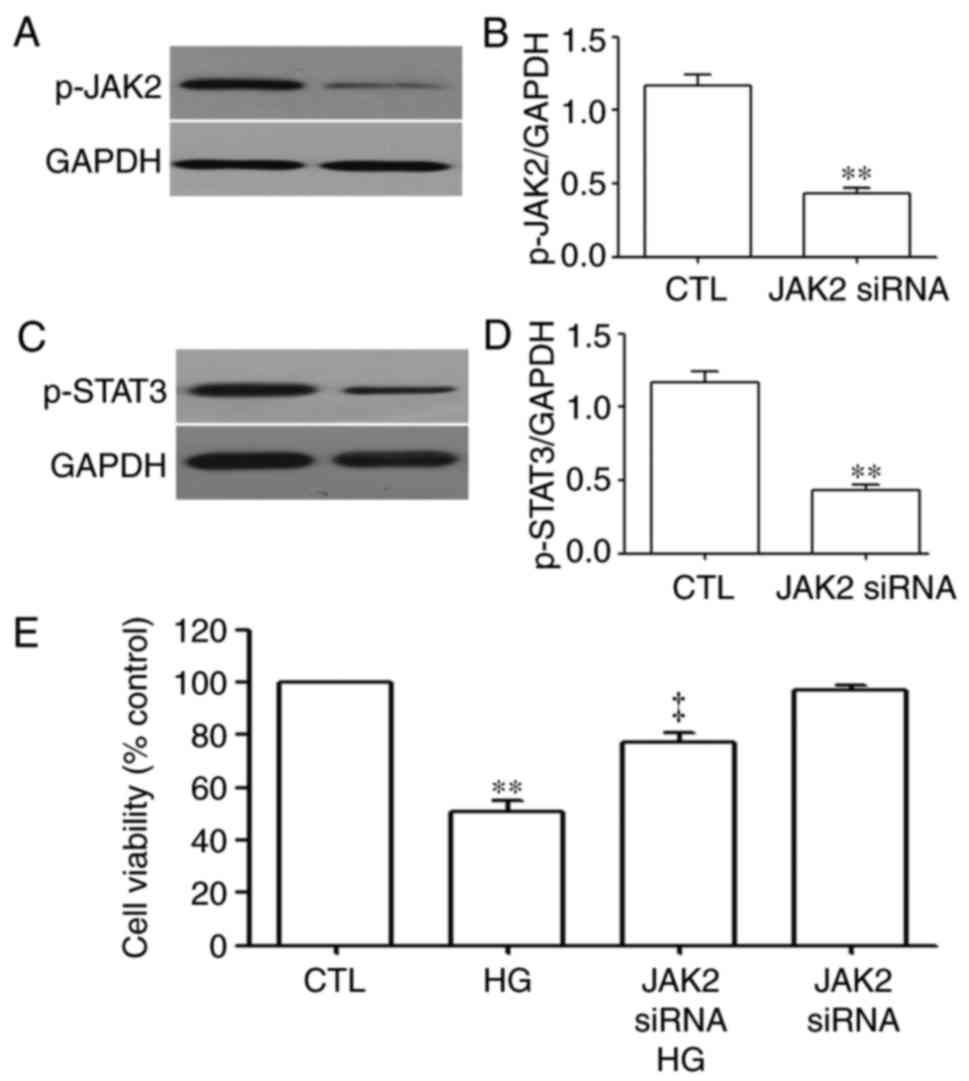
JAK2 siRNA inhibits expression of the
JAK2/STAT3 pathway and attenuates the HG-induced decrease in cell
viability of HUVECs
To observe the effects of JAK2 siRNA on the
expression of the JAK2/STAT3 pathway, the HUVECs were treated with
JAK2 siRNA. As shown in Fig. 2,
JAK2 siRNA significantly inhibited the expression levels of p-JAK2
(Fig. 2A and B) and p-STAT3
(Fig. 2C and D). In addition,
JAK2 siRNA attenuated the HG-induced decrease in cell viability in
the HUVECs, as shown in Fig.
2E.
HG activates the JAK2/STAT3 pathway in
HUVECs
The present study also examined the effects of HG on
the STAT3/JAK2 pathway, including the phosphorylation of STAT3 and
JAK2. As shown in Fig. 3A–D, the
effect of glucose on the HUVECs were examined. The cells were
exposed to the indicated concentrations (20, 30, 40, 50 and 60 mM)
of glucose for 24 h, and exposure to glucose significantly
upregulated the expression levels of (Fig. 3A and C) pSTAT3, peaking at 60 mM
glucose. However, the expression of t-STAT3 was not affected by the
indicated concentrations of glucose. Based on these results, the
effect of time on the expression of p-STAT3 was examined. As shown
in Fig. 3B and D, following
exposure of HUVECs to 40 mM glucose for the indicated times (3, 6,
12, 18, 24, 36 and 48 h), the expression levels of p-STAT3 were
significantly upregulated, reaching a peak at 36 h, whereas the
expression of t-STAT3 remained unchanged. Similarly, exposure of
the cells to 40 mM glucose increased the expression levels of
p-JAK2, as shown in the dose-response experiment (Fig. 3E) and time-response experiment
(Fig. 3F), respectively. The mRNA
expression levels of STAT3 (Fig.
3G) and JAK2 (Fig. 3H) were
also markedly increased.
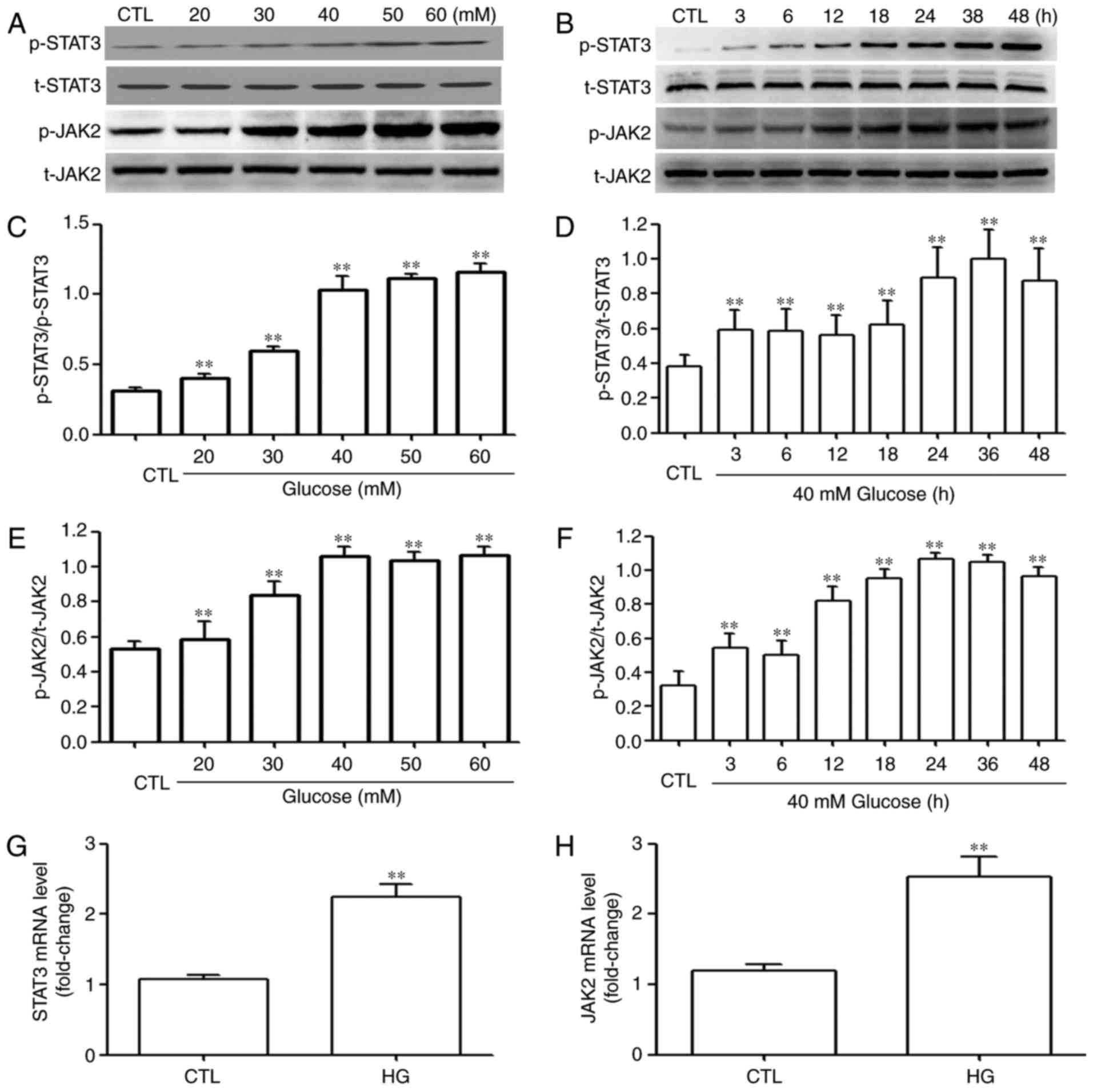 | Figure 3HG induces activation of the
STAT3/JAK2 pathway in HUVECs. (A) HUVECs were exposed to the
indicated concentrations of glucose (20, 30, 40, 50 and 60 mmol/l,
respectively) for 24 h or (B) were exposed to HG for the indicated
durations (3, 6, 12, 18, 24, 36 and 48 h, respectively), followed
by western blot analysis for the expression of STAT3 and JAK2.
Densitometric analysis was performed to determine levels of (C and
D) STAT3 and (E and F) JAK2 under different concentrations of
glucose and different durations of HG, respectively. mRNA levels of
(G) STAT3 and (H) JAK2 were assessed. **P<0.01 vs.
CTL group. JAK2, Janus kinase 2; STAT3, signal transducer and
activator of transcription 3; HUVECs, human umbilical vein
endothelial cells; HG, high glucose; siRNA, small interfering RNA;
CTL, control; p, phosphorylated; t, total. |
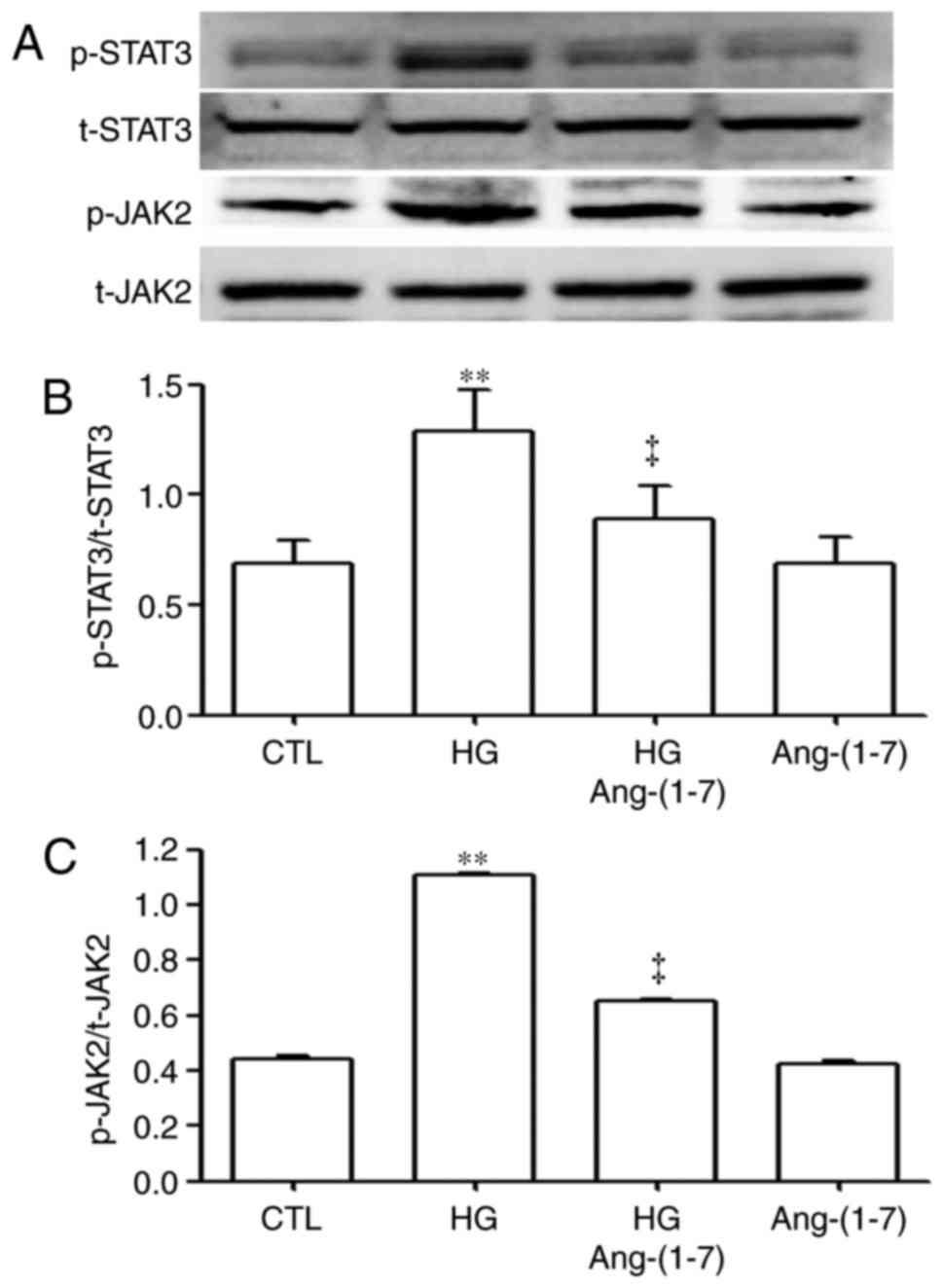
Ang-(1-7) downregulates HG-induced
activation of the JAK2/STAT3 pathway in HUVECs
To observe effects of Ang-(1-7) on the activation of
the JAK2/STAT3 pathway induced by HG, the HUVECs were pre-treated
with 2 µM Ang-(1-7) for 30 min, prior to exposure to HG for
24 h. As shown in Fig. 4,
exposure of the cells to 40 mM glucose significantly increased the
expression levels of p-STAT3 (Fig. 4A
and B) and p-JAK2 (Fig. 4A and
C). However, the increased phosphorylation of the JAK2/STAT3
pathway was reduced by pre-treatment with 2 µM
Ang-(1-7).
HG upregulates the expression level of
caspase-3 and downregulates the expression level of eNOS in
HUVECs
The present study also examined the effects of HG on
the expression levels of caspase-3 and eNOS in HUVECs. As shown in
Fig. 5A and E, The HUVECs were
exposed to (A) the indicated concentrations (20, 30, 40, 50 and 60
mM) of glucose for 24 h, or (B) with 40 mM glucose for the
indicated durations (3, 6, 12, 18, 24, 36 and 48 h). (C) Exposure
to different glucose concentrations markedly increased the
expression levels of caspase-3, peaking at 60 mM glucose. Based on
these results, the effect of time on the expression was examined.
As shown in Fig. 5D, when the
HUVECs were exposed to 40 mM glucose for the indicated durations
(3, 6, 12, 18, 24, 36 and 48 h), the expression levels of caspase-3
were significantly upregulated, reaching a peak at 12 and 18 h. By
contrast, exposure of the cells to HG decreased the expression
levels of eNOS, as shown in the dose-response experiment (Fig. 5E) and time-response experiment
(Fig. 5F), respectively.
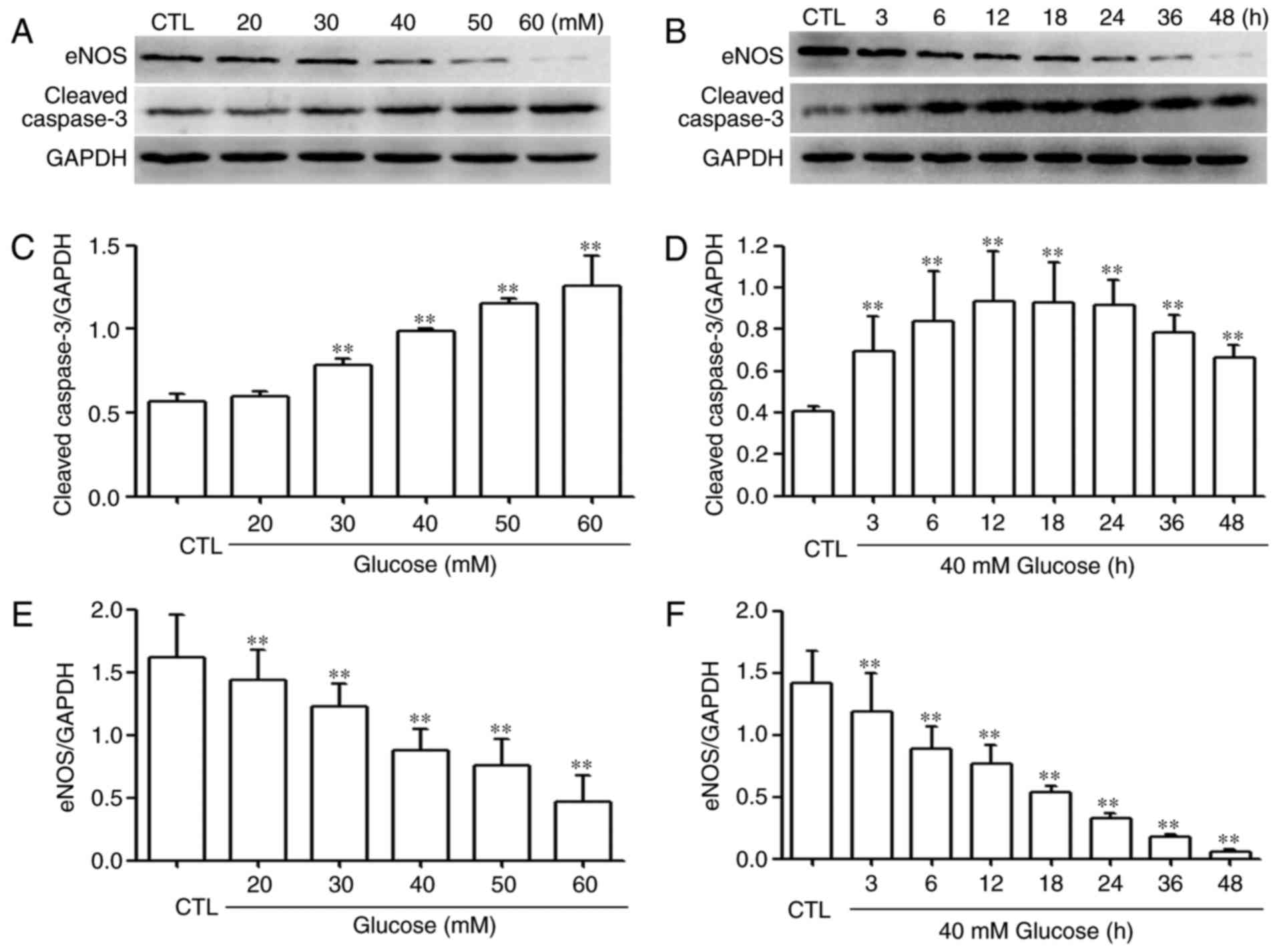 | Figure 5HG upregulates the expression levels
of caspase-3 and downregulates the expression levels of eNOS in
HUVECs. HUVECs were exposed to (A) the indicated concentrations of
glucose (20, 30, 40, 50 and 60 mmol/l, respectively) for 24 h, or
(B) HG for the indicated durations (3, 6, 12, 18, 24, 36 and 48 h,
respectively). Expression levels of caspase-3 and eNOS were
measured using western blot assays. Densitometric analysis of the
expression of caspase-3 under (C) different glucose concentrations
and (D) with HG for different durations. Densitometric analysis of
the expression of eNOS under (E) different glucose concentrations
and (F) with HG for different durations, respectively.
**P<0.01 vs. CTL group. HUVECs, human umbilical vein
endothelial cells; eNOS, endothelial nitric oxide synthase;
Ang-(1-7), angiotensin-(1-7);
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HG, high glucose
(40 mmol/l glucose); CTL, control. |
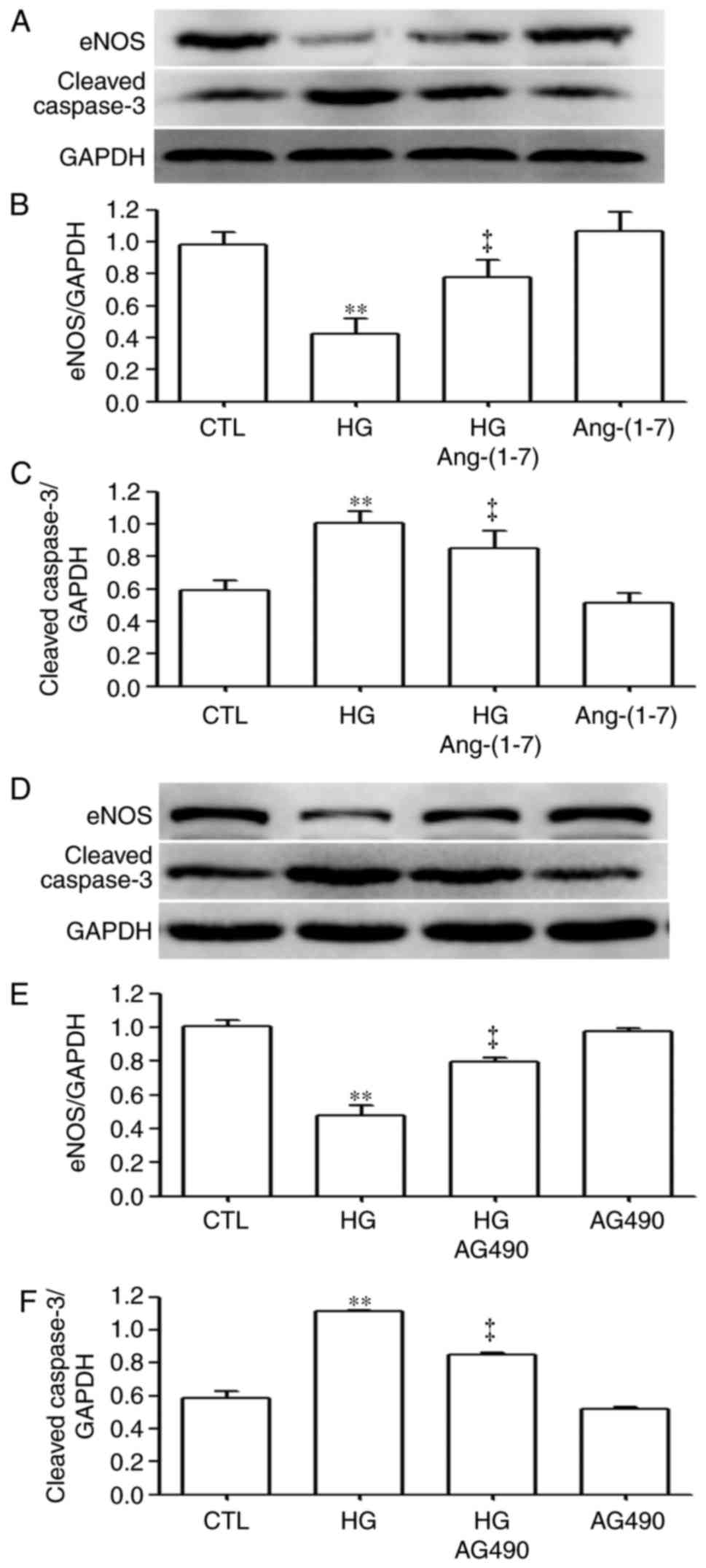
Ang-(1-7) and AG490 downregulate the increased
expression of caspase-3 and upregulate the decreased expression of
eNOS induced by HG in HUVECs. To observe the effects of Ang-(1-7)
and AG490 on the increased expression level of caspase-3 and
upregulating the decreased expression level of eNOS induced by HG,
the HUVECs were pre-treated with 2 µM Ang-(1-7), as shown in
Fig. 6A–C, or 20 µM AG490
for 30 min (Fig. 6D–F), prior to
exposure to HG for 24 h. As shown in Fig. 6, exposure of the cells to 40 mM
glucose significantly increased the expression levels of caspase-3
(Fig. 6A, C, D and F). However,
the increased expression level of caspase-3 was reduced by
pre-treatment with 2 µM Ang-(1-7) (Fig. 6A and B) or 20 µM AG490
(Fig. 6D and F) for 30 min prior
to exposure to HG for 24 h. Secondly, the exposure of cells to 40
mM glucose significantly decreased the expression levels of eNOS
(Fig. 6A, B, D and E). However,
the decreased expression levels of eNOS were upregulated following
pre-treatment with 2 µM Ang-(1-7) (Fig. 6A and B) or 20 µM AG490
(Fig. 6D and E) for 30 min prior
to exposure to HG for 24 h.
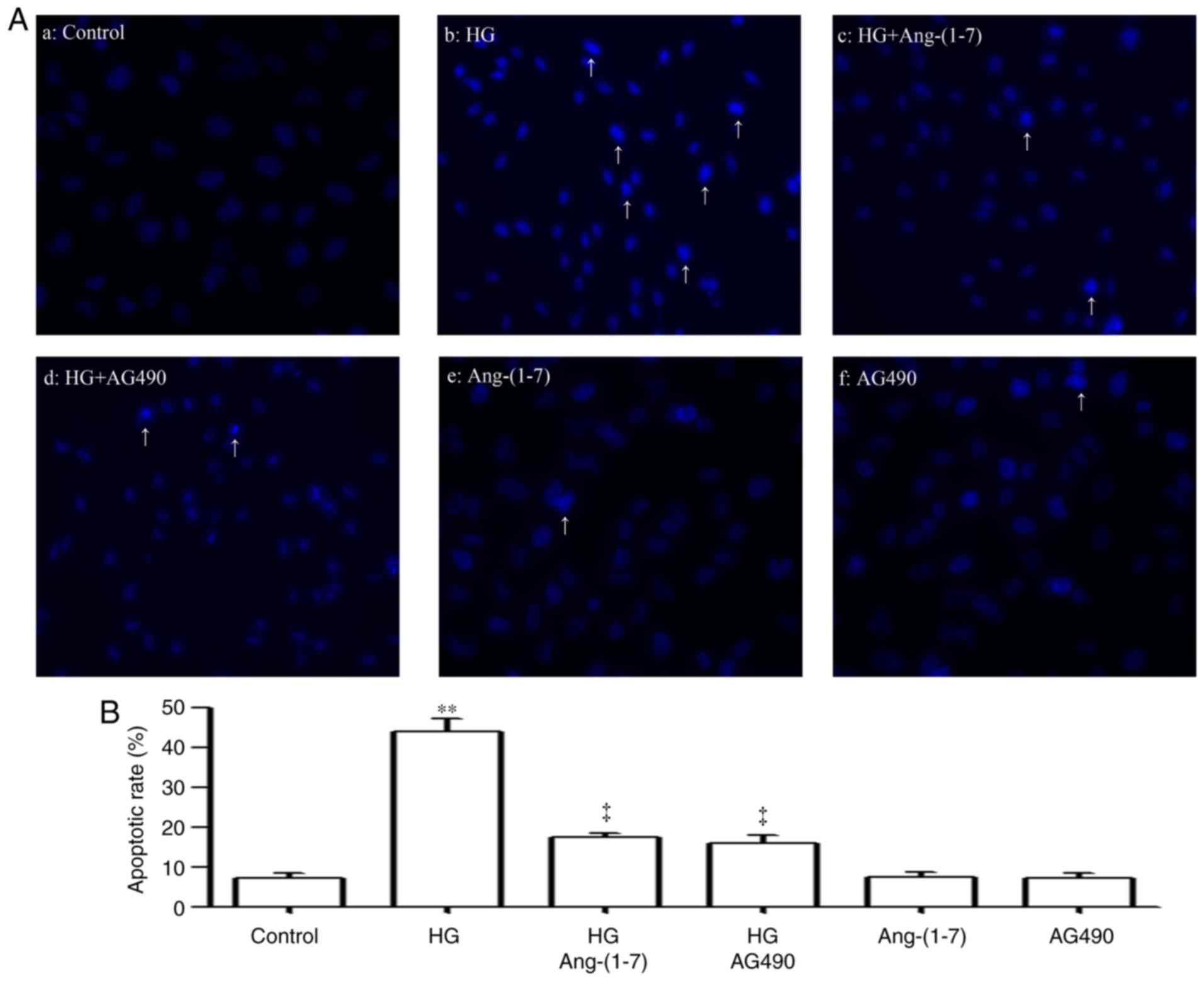
Ang-(1-7) and AG490 suppress HG-induced
apoptosis in HUVECs
As shown in Fig. 7Aa
and b, exposure of HUVECs to 40 mM glucose for 24 h induced
typical apoptosis, which was manifested as the nuclear condensation
and fragmentation condensation of chromatin, and the shrinkage of
nuclei and apoptotic bodies. However, pre-treatment of the cells
with 2 µM Ang-(1-7) for 30 min prior to exposure to HG for
24 h mitigated the HG-induced increase in the number of cells
undergoing apoptosis (Fig. 7Ac).
In addition, pre-conditioning of the cells with 20 µM AG490
for 30 min prior to exposure to HG for 24 h also ameliorated the
HG-induced apoptosis of cardiac cells (Fig. 7Ad). Alone, 2 µM Ang-(1-7)
(Fig. 7Ae) or 20 µM AG490
(Fig. 7Af) did not significantly
alter the number of apoptotic cells. Quantification of results is
shown in Fig. 7B.
Ang-(1-7) and AG490 reduce the oxidative
stress induced by HG in HUVECs
The results demonstrated that oxidative stress
contributed to HG-induced HUVEC injury. As shown in Figs. 8 and 9, exposure of the HUVECs to 40 mM
glucose for 24 h resulted in oxidative stress, as evidenced by an
increase in the generation of ROS (Fig. 8Ab and B), a decrease in SOD
activity (Fig. 9A) and increases
in the expression level of Nox4 in the dose-response experiment
(20, 30, 40, 50 and 60 mM glucose; Fig. 9B and C) and time-response
experiment (3, 6, 12, 18, 24, 36 and 48 h; Fig. 9D and E). However, pre-treatment of
the cells with 2 µM Ang-(1-7) for 30 min prior to exposure
to HG for 24 h mitigated the HG-induced increase in ROS generation
(Fig. 8Ac and B), increased the
HG-induced decrease in SOD activity (Fig. 9A) and decreased the expression
level of Nox4 (Fig. 9F and G). To
determine whether the JAK2/STAT3 pathway was involved in HG-induced
oxidative stress, the HUVECs were pre-treated cells with 20
µM AG490 for 30 min prior to exposure to HG for 24 h. The
resulting data showed that pre-treatment of the cells with 20
µM AG490 for 30 min prior to exposure to HG for 24 h
decreased the generation of ROS (Fig.
8Ad and B), upregulated SOD activity (Fig. 9A) and reduced the expression
levels of Nox4 (Fig. 9F and G) in
the HUVECs. Treatment with Ang-(1-7) or AG490 alone did not alter
the basal level of ROS, activity of SOD or expression level of Nox4
in the HUVECs.
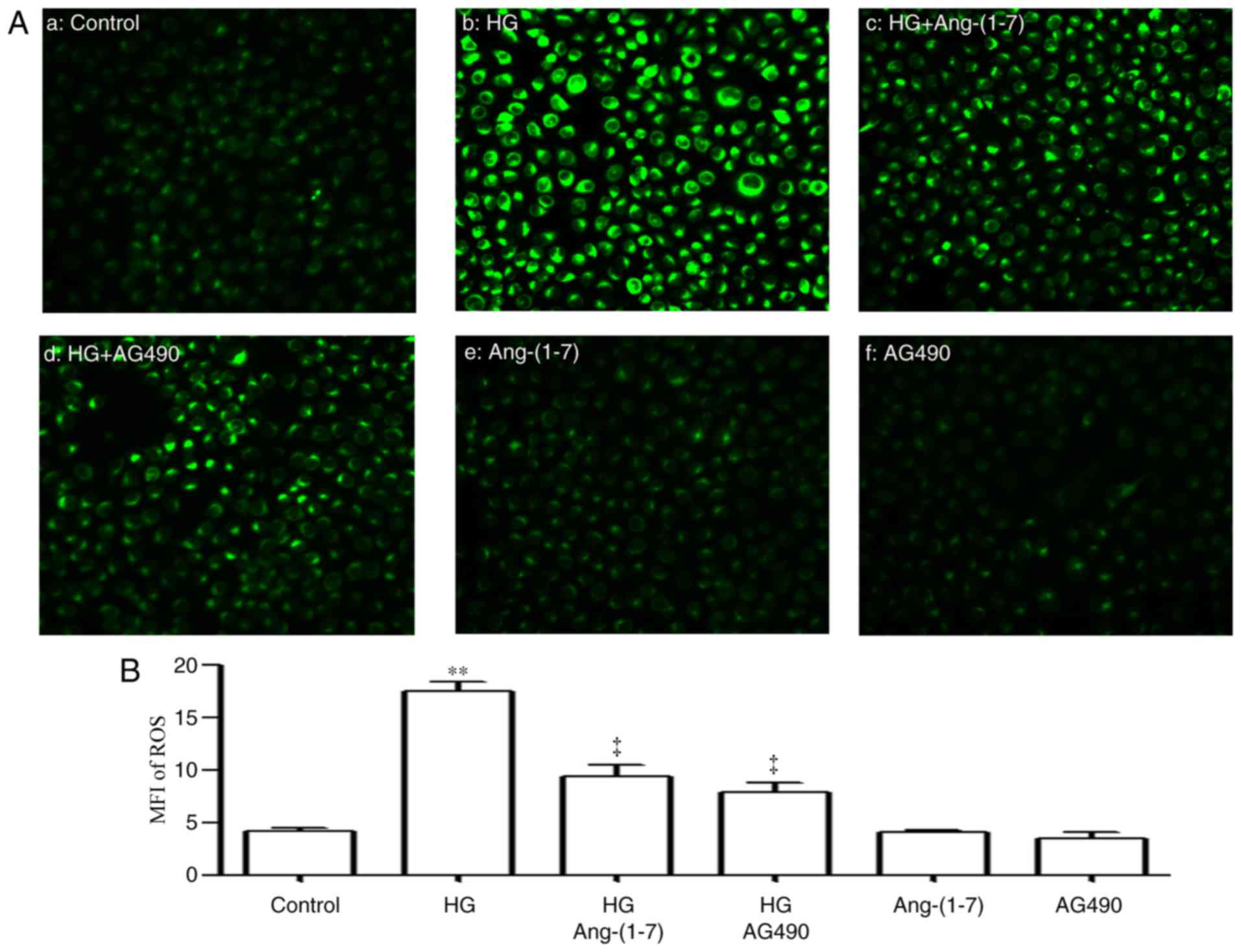 | Figure 8Ang-(1-7) and AG490 alleviate
HG-induced increased ROS generation in HUVECs. (A) DCFH-DA staining
followed by photofluorography was used to measure intracellular ROS
levels. (a) Control group; (b) HUVECs exposed to HG for 24 h; (c)
HUVECs co-treated with HG and 2 µmol/l Ang-(1-7) for 24 h;
(D) HUVECs co-treated with HG and 20 µmol/l AG490 (inhibitor
of the Janus kinase 2/signal transducer and activator of
transcription 3 pathway) for 24 h. (E) HUVECs treated with 2
µmol/l Ang-(1-7) for 24 h; (F) HUVECs treated with 2
µmol/l Ang-(1-7) for 24 h. (B) Quantitative analysis of the
MFI of DCFH-DA using ImageJ 1.47i software. Images are captured at
×100 magnification. **P<0.01, compared with the CTL
group; ‡P<0.01 vs. HG-treated group. HUVECs, human
umbilical vein endothelial cells; Ang-(1-7), angiotensin-(1-7);
HG, high glucose, 40 mmol/l glucose; DCFH-DA,
2′,7′-dichlorofluorescein diacetate; MFI, mean fluorescence
intensity; ROS, reactive oxygen species; CTL, control. |
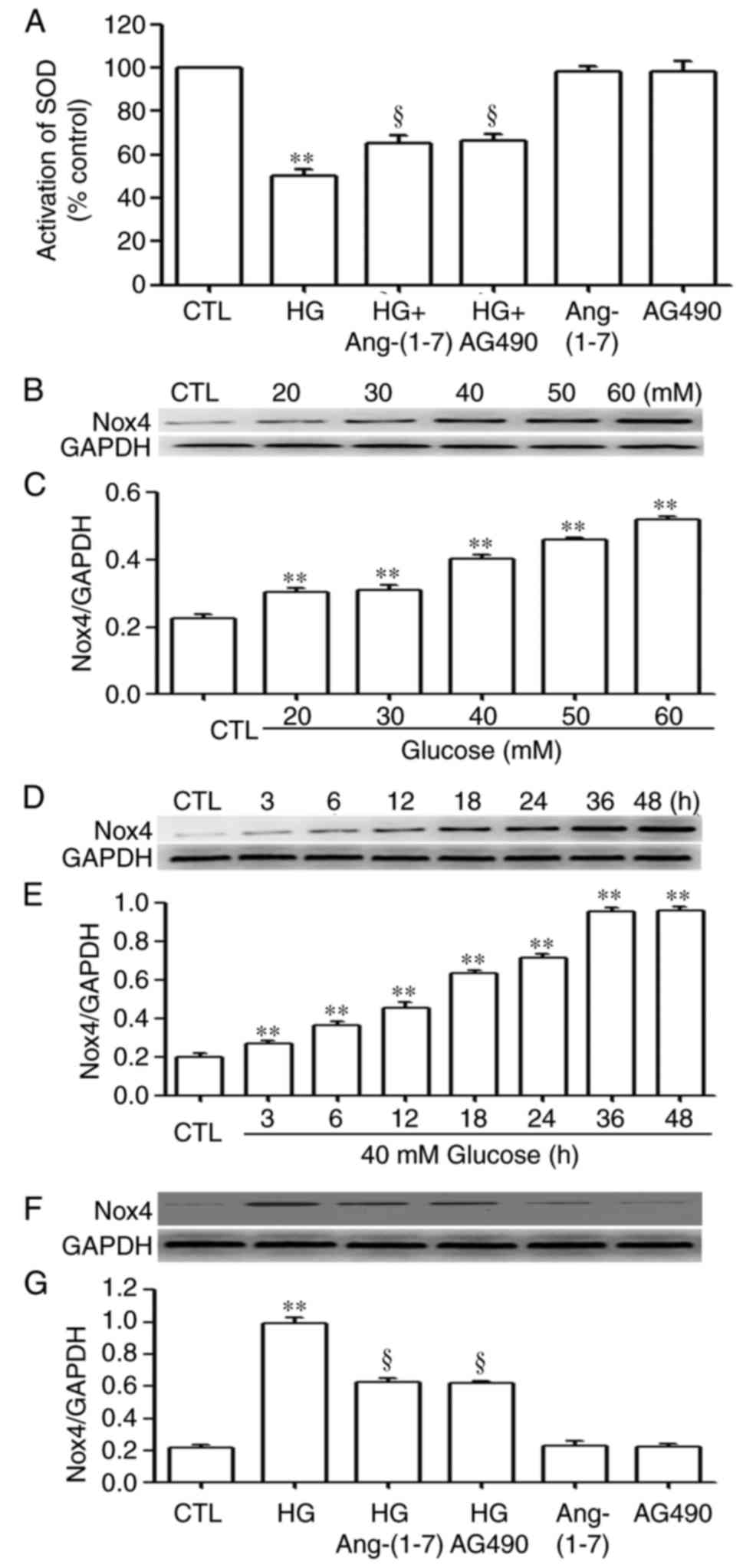 | Figure 9Ang-(1-7) and AG490 downregulate the
increased expression level of Nox4 and increase SOD activity
induced by HG in HUVECs. (A) SOD activity was examined using an SOD
assay kit. (B) HUVECs were exposed to the indicated concentrations
of glucose (20, 30, 40, 50 and 60 mmol/l, respectively) for 24 h
and (C) levels of Nox4 were determined using densitometric
analysis. (D) HUVECs were exposed to 40 mM glucose for the
indicated times (3, 6, 12, 18, 24, 36 and 48 h, respectively) and
(E) levels of Nox4 were determined using densitometric analysis.
(F) HUVECs were co-conditioned with HG and 2 µmol/l
Ang-(1-7) or 20 µmol/l AG490 (inhibitor of the Janus kinase
2/signal transducer and activator of transcription 3 pathway) for
24 h, and (G) expression levels of Nox4 were measured using western
blot analysis, followed by (F) densitometric analysis. Data are
presented as the mean ± standard error of the mean (n=3).
**P<0.01 vs. CTL group; ‡P<0.01,
compared with the HG-treated group. HUVECs, human umbilical vein
endothelial cells; Ang-(1-7), angiotensin-(1-7);
HG, high glucose (40 mmol/l glucose); Nox4, NADPH oxidase 4; SOD,
superoxide dismutase; CTL, control. |
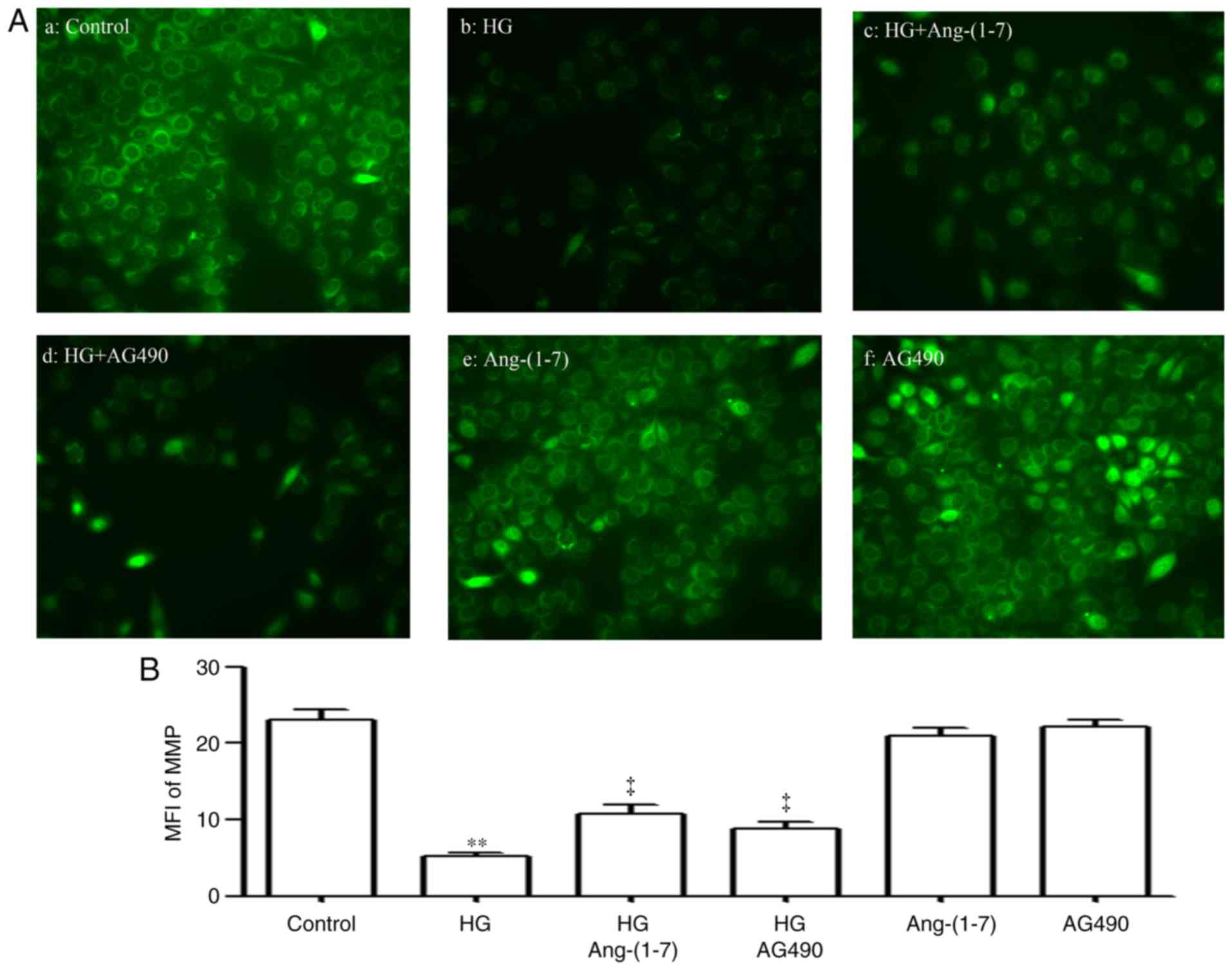
Ang-(1-7) and AG490 inhibit the
HG-induced dissipation of MMP in HUVECs
It was shown that the exposure of HUVECs to 40 mM
glucose for 24 h elicited mitochondrial damage, as manifested by
the dissipation of MMP (Fig. 10Aa
and b). The dissipation of MMP was reduced by pre-treatment of
the cells with 2 µM Ang-(1-7) for 30 min prior to exposure
to HG for 24 h (Fig. 10Ac),
which demonstrated that Ang-(1-7) protected the HUVECs against
HG-induced mitochondrial damage. Similarly, pre-treatment of HUVECs
with 20 µM AG490 for 30 min prior to exposure to HG for 24 h
attenuated the HG-induced dissipation of MMP (Fig. 10Ad). The quantitative results are
shown in Fig. 10B.
Ang-(1-7) and AG490 suppress the
HG-induced increased production of pro-inflammatory cytokines in
HUVECs
As shown in Fig.
11, the levels of IL-1β (Fig.
11A), IL-10 (Fig. 11B),
IL-12 (Fig. 11C) and TNF-α
(Fig. 11D) were markedly
increased in the HG-induced HUVECs, compared with those in the
control group (P<0.01). However, these increased levels of
IL-1β, IL-10, IL-12 and TNF-α were significantly suppressed by
pre-treatment of the HUVECs with 2 µM Ang-(1-7) for 30 min
prior to exposure to HG for 24 h. This suggested an inhibitory
effect of Ang-(1-7) on the production of pro-inflammatory
cytokines, including IL-1β, IL-10, IL-12 and TNF-α, induced by HG.
Similarly, pre-treatment of the HUVECs with 20 µM AG490 for
30 min prior to exposure to HG for 24 h decreased the enhanced
production of IL-1β, IL-10, IL-12 and TNF-α.
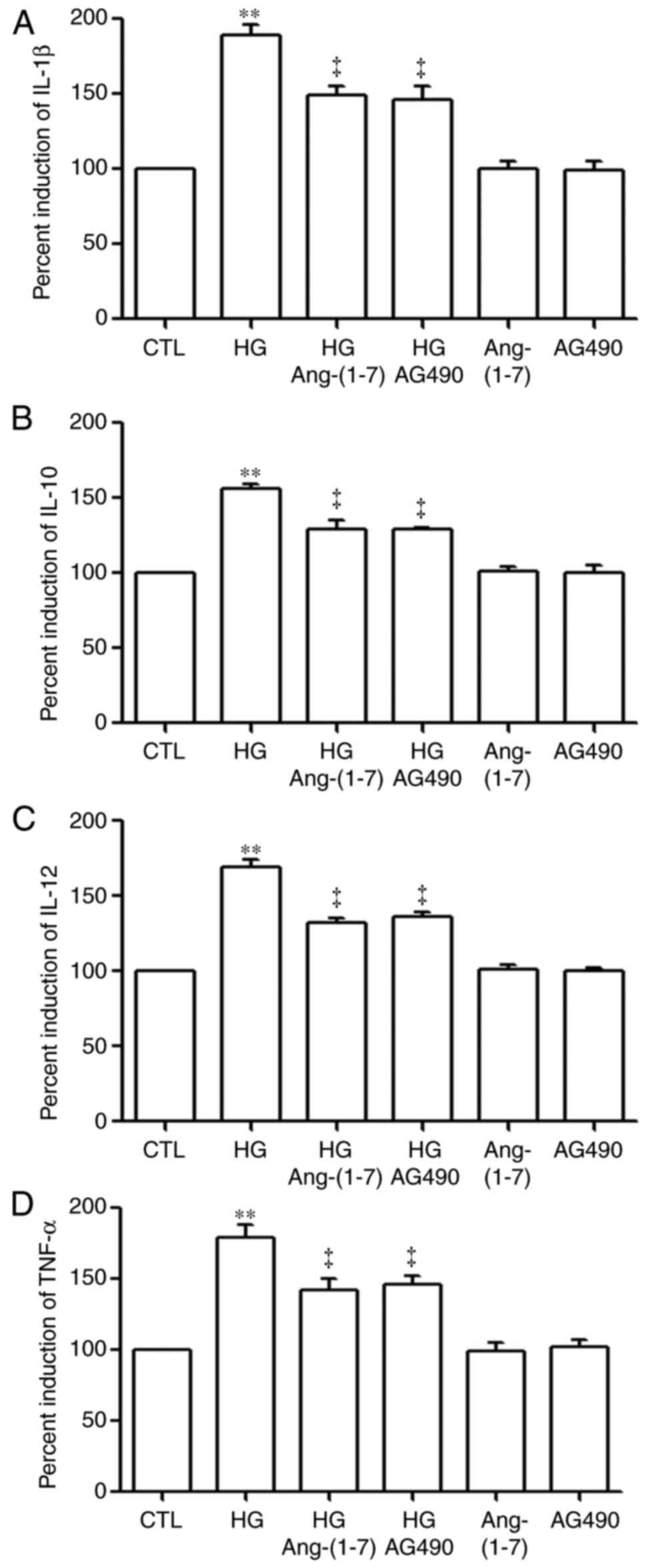 | Figure 11Ang-(1-7) and AG490 alleviate
HG-induced production of IL-1β, IL-10, IL-12 and TNF-α in human
umbilical vein endothelial cells. The cells were treated with HG
for 24 h with or without co-conditioning with 2 µmol/l
Ang-(1-7) or 20 µmol/l AG490 (inhibitor of the Janus kinase
2/signal transducer and activator of transcription 3 pathway) for
24 h. An enzyme-linked immunosorbent assay was performed to
determine the levels of (A) IL-1β, (B) IL-10, (C) IL-12 and (D)
TNF-α in cell supernatants. Data are shown as the mean ± standard
error of the mean (n=3). **P<0.01 vs. CTL group;
‡P<0.01, compared with the HG-treated group.
Ang-(1-7), angiotensin-(1-7);
HG, high glucose (40 mmol/l glucose). IL, interleukin; TNF-α, tumor
necrosis factor-α; CTL, control. |
Discussion
In the present study, the data revealed several
novel findings indicating that the JAK2/STAT3 signaling pathway is
relevant to the potential mechanisms responsible for HG-induced
HUVEC injury and inflammation, and the effect of Ang-(1-7) on
protecting vascular endothelium. First, in the HUVECs, the
JAK2/STAT3 signaling pathway was involved in the HG-induced HUVEC
injury and inflammation. Secondly, Ang-(1-7) exerted endothelial
protection against HG-induced injury and inflammation. Finally, the
protective effects on the vascular endothelium by Ang-(1-7) were
associated with inhibition of the JAK2/STAT3 pathway.
The data obtained demonstrated that the exposure of
HUVECs to HG for 24 h led to injury and inflammation, as
characterized by an increase in apoptotic cells, expression levels
of caspase-3 (a death effector domain), oxidative stress
(demonstrated by increased ROS production), decreased activation of
SOD, increased expression of Nox4 (an important component of the
NADPH oxidase family), increased expression level of eNOS,
decreased cell viability and dissipation of MMP, and the
upregulation of secretion of inflammatory cytokines (IL-1β, ll-6,
IL-12 and TNF-α). These results are consistent with those of
previous studies (5,6-9,29-31) and demonstrated that the damage in
HG-induced HUVEC injury and inflammation was extensive. However,
the associated mechanism remains to be fully elucidated.
The JAK2/STAT3 pathway is known to mediate survival
signals, which contribute to cell proliferation, differentiation,
growth and apoptosis (14-16).
Similarly, the effects of the JAK2/STAT3 pathway on the
cardiovascular system are also important and have been widely
examined. Accumulating evidence has demonstrated that the
JAK2/STAT3 pathway is involved in the progress of various
stimulation-induced cardiovascular complication (32-36), including apoptosis (32,33), reticulum stress (32), ROS (35,36), contractile dysfunction (36) and inflammation (37). Currently, the potential roles of
the JAK2/STAT3 pathway in hyperglycemia-induced cardiovascular
complication remain to be fully elucidated. Fiaschi et al
identified a novel role for STAT3 as a crucial signaling molecule
of collagen I production in cardiac fibroblasts induced by a
diabetic environment (38). In
addition, in streptozotocin (STZ)-induced diabetic rats, wortmannin
(an inhibitor of PI3K) and AG490 (an inhibitor of JAK2),
synergistically mitigate myocardial ischemia reperfusion injuries
(39). This indicates that
PI3K/Akt and JAK2/STAT3 are synergistically involved in myocardial
injury in diabetes.
As the effects of the JAK2/STAT3 pathway in
HG-induced HUVEC injury and inflammation remain to be fully
elucidated, the present study aimed to elucidate the mechanism.
Firstly, the role of HG on activation of the JAK2/STAT3 pathway in
HUVECs was investigated. The results showed that exposure of the
HUVECs to HG upregulated the expression levels of p-JAK2 and
p-STAT3, indicating that HG activated the JAK2/STAT3 pathway in
HUVECs. Secondly, the associated roles of JAK2/STAT3 pathway
activation were examined in HG-stimulated injury. The data
indicated that co-treatment of the HUVECs with HG and AG490, an
inhibitor of the JAK2 pathway, significantly alleviated HG-induced
injuries, including apoptosis, cytotoxicity, mitochondrial damage
and oxidative stress, as evidenced by a decrease in the number of
apoptotic cells, decreased expression levels of caspase-3 and Nox4,
and an increase in the activation of SOD, cell viability,
expression of eNOS and dissipation of MMP. These results suggested
that JAK2/STAT3 activation was involved in HG-stimulated injury in
HUVECs. In addition, as it has been demonstrated that hyperglycemia
is involved in vascular endothelium inflammation in vitro
and in vivo (29,40-43), the present study further examined
the role of JAK2/STAT3 activation on the HG-induced inflammatory
response in HUVECs. Similar to the results of previous studies
(29,40-43), it was found that exposure of the
HUVECs to HG promoted inflammatory responses, as indicated by the
upregulated production of IL-1β, IL-6, IL-12 and TNF-α. However,
the increased production of IL-1β, IL-6, IL-12 and TNF-α was
decreased by AG490. This suggested that the JAK2/STAT3 pathway was
involved in the HG-induced production of pro-inflammatory factors
(IL-1β, IL-6, IL-12 and TNF-α). The above data provide definitive
and novel evidence that activation of the JAK2/STAT3 pathway
contributed to HG-induced injury and inflammation in HUVECs.
An important finding of the present study relates to
the various endothelial protective effects of Ang-(1-7) on
HG-induced injury and inflammation in HUVECs. Firstly, it was found
that Ang-(1-7) markedly alleviated HG-induced cytotoxicity, as
characterized by increased cell viability. These results are
supported by previous findings that toxicity induced by various
factors, including lipopolysaccharide, long-term hypoxia and
STZ-induced diabetes is alleviated by Ang-(1-7) (44-46). Secondly, Ang-(1-7) can exert
anti-apoptotic effects against HG-induced apoptosis in HUVECs.
According to previous studies, the anti-apoptotic effect of
Ang-(1-7) is widely recognized, and is associated with various
systems and organs, including the endocrine system (46), reproductive system (47), circulatory system (48), respiratory system (49), urinary system (50) and motor system (51). These data demonstrate that
Ang-(1-7) protects cells against the injury induced by various
factors by exerting anti-apoptotic effects. In the present study,
data indicated that Ang-(1-7) protected the HUVECs against
HG-induced apoptosis, as indicated by decreases in cell apoptosis
and the expression of caspase-3. Ang-(1-7) is also involved in the
oxidative stress induced by HG in HUVECs. In the circulatory
system, Ang-(1-7) exerts cardiovascular protective effects by
attenuating oxidative stress in cardiac (53), cardiomyocyte autophagy (52), hypertension (53) and vascular remodeling (54). Additionally, AVE 0991, an analog
of Ang-(1-7), attenuates cardiac hypertrophy via inhibiting
oxidative stress (55). Ang-(1-7)
also mediates the endothelial protection of signaling by reducing
the oxidative stress induced by diabetes (56). Consistent with these previous
findings, the results of the present study demonstrated that
Ang-(1-7) protected HUVECs against HG-induced apoptosis by reducing
oxidative stress, as characterized by a decrease in the production
of ROS and expression level of Nox4, an important component of the
NADPH oxidase family, and an increase in the activation of SOD. The
findings of the present study also revealed that Ang-(1-7) had
mitochondrial protective effects against HG-induced mitochondrial
injury (a loss of MMP), which was comparable with a previous study
showing that Ang-(1-7) alleviated the loss of MMP during
H2O2-induced in pancreatic β cells (57). The endothelial protective effect
of Ang-(1-7) is also associated with its anti-inflammatory effect.
In previous studies, Ang-(1-7) exerted anti-inflammatory effects in
cardiomyocytes (58) and
pulmonary microvascular endothelial cells (59) of the circulatory system.
Similarly, the anti-inflammatory effect of Ang-(1-7) has been shown
to contribute to endothelial protection in endothelial cells
(60,61). However, whether Ang-(1-7) inhibits
HG-induced inflammation in HUVECs remains to be elucidated. In the
present study, Ang-(1-7) significantly inhibited the HG-induced
expression of inflammatory factors (IL-1β, IL-6, IL-12 and TNF-α).
This indicates that the endothelial protective effect of Ang-(1-7)
was involved in its anti-inflammatory effect. It was also found
that Ang-(1-7) ameliorated the expression level of eNOS induced by
HG in the HUVECs. Exposure of the HUVECs to HG for 24 h upregulated
the expression level of eNOS. These data were supported by previous
studies (62,63). Co-treatment of the HUVECs with HG
and Ang-(1-7) considerably elevated the expression level of eNOS.
However, the association between Ang-(1-7) and eNOS remains to be
elucidated and requires further investigation in vitro and
in vivo.
Another important finding of the present study
involves the effects of inhibition of the JAK2/STAT3 pathway on the
endothelial protective effects of exogenous Ang-(1-7) against
HG-induced multifarious endothelial cell injury and inflammation.
Several studies have reported that Ang-(1-7) exerts endothelial
protective effects (44,47,55,58,59). The present study examined whether
Ang-(1-7) protected HUVECs against HG-induced injury and
inflammation by suppressing the JAK2/STAT3 pathway. The results
demonstrated that exogenous Ang-(1-7) widely antagonized JAK2/STAT3
pathway activation and inflammatory factors (IL-1β, IL-6, IL-12 and
TNF-α). In addition, similar to the inhibitory effects of AG490, an
inhibitor of the JAK2 pathway, as indicated above, co-treatment of
the HUVECs with HG and Ang-(1-7) mitigated HG-induced endothelial
cell injury and inflammation. These results showed that the
inhibitory effect of the JAK2/STAT3 pathway may be a crucial
mechanism responsible for the endothelial protective effects of
exogenous Ang-(1-7) against HG-induced endothelial cell injury and
inflammation. Similarly, previous evidence demonstrates that
exogenous Ang-(1-7) reduced Ang II-induced (25) or monocrotaline-induced (64) cell injury through inhibition of
the JAK2/STAT3 pathway. Previous findings have also indicated that
Ang-(1-7) exerts cardioprotective protection against HG-induced
injury (65). These studies
support the results of the present study.
In conclusion, the present study provided novel
evidence that the JAK2/STAT3 pathway contributed to HG-induced
endothelial cell injury and inflammation, and that exogenous
Ang-(1-7) exerted endothelial protection against HG-induced
endothelial cell injury and inflammation via the inhibitory effect
on the JAK2/STAT3 pathways. These findings may assist in the
development of novel therapeutic methods for the prevention and
treatment of hyperglycemia-associated endothelial cell injury and
inflammation.
Acknowledgments
The present study was supported by grants from the
Guangdong Medical Research Foundation (grant no. A2012172), the
Technology Planning Project of Huangpu District (grant no.
201544-01), the Science and Technology Planning Project of
Guangdong Province, China (grant nos. 2012B031800358,
2012B031800365 and 2010B08071044), Medical Scientific Research
Foundation of Guangdong Province (A2015287) and the Guangdong
Natural Science Foundation (grant no. S2011010004381).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Shaw JE, Sicree RA and Zimmet PZ: Global
estimates of the prevalence of diabetes for 2010 and 2030. Diabetes
Res Clin Pract. 87:4–14. 2010. View Article : Google Scholar
|
|
2
|
Guariguata L, Whiting DR, Hambleton I,
Beagley J, Linnenkamp U and Shaw JE: Global estimates of diabetes
prevalence for 2013 and projections for 2035. Diabetes Res Clin
Pract. 103:137–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bachmann KN and Wang TJ: Biomarkers of
cardiovascular disease: Contributions to risk prediction in
individuals with diabetes. Diabetologia. Sep 28–2017.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Capellini VK, Celotto AC, Baldo CF, Olivon
VC, Viaro F, Rodrigues AJ and Evora PR: Diabetes and vascular
disease: Basic concepts of nitric oxide physiology, endothelial
dysfunction, oxidative stress and therapeutic possibilities. Curr
Vasc Pharmacol. 8:526–544. 2010. View Article : Google Scholar
|
|
5
|
Taguchi K, Sakata K, Ohashi W, Imaizumi T,
Imura J and Hattori Y: Tonic inhibition by G protein-coupled
receptor kinase 2 of Akt/endothelial nitric-oxide synthase
signaling in human vascular endothelial cells under conditions of
hyperglycemia with high insulin levels. J Pharmacol Exp Ther.
349:199–208. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao H, Ma T, Fan B, Han C, Luo J and Kong
L: Protective effect of trans-δ-viniferin against high
glucose-induced oxidative stress in human umbilical vein
endothelial cells through the SIRT1 pathway. Free Radic Res.
50:68–83. 2016. View Article : Google Scholar
|
|
7
|
Zhang Y, Liu T, Chen Y, Dong Z, Zhang J,
Sun Y, Jin B, Gao F, Guo S and Zhuang R: CD226 reduces endothelial
cell glucose uptake under hyperglycemic conditions with
inflammation in type 2 diabetes mellitus. Oncotarget.
7:12010–12023. 2016.PubMed/NCBI
|
|
8
|
Bhatt MP, Lim YC, Hwang J, Na S, Kim YM
and Ha KS: C-peptide prevents hyperglycemia-induced endothelial
apoptosis through inhibition of reactive oxygen species-mediated
transglutaminase 2 activation. Diabetes. 62:243–253. 2013.
View Article : Google Scholar
|
|
9
|
Surico D, Farruggio S, Marotta P, Raina G,
Mary D, Surico N, Vacca G and Grossini E: Human chorionic
gonadotropin protects vascular endothelial cells from oxidative
stress by apoptosis inhibition, cell survival signalling activation
and mitochondrial function protection. Cell Physiol Biochem.
36:2108–2120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Samanta A, Perazzona B, Chakraborty S, Sun
X, Modi H, Bhatia R, Priebe W and Arlinghaus R: Janus kinase 2
regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia.
25:463–472. 2011. View Article : Google Scholar
|
|
11
|
Kiu H and Nicholson SE: Biology and
significance of the JAK/STAT signalling pathways. Growth Factors.
30:88–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Copf T, Goguel V, Lampin-Saint-Amaux A,
Scaplehorn N and Preat T: Cytokine signaling through the JAK/STAT
pathway is required for long-term memory in Drosophila. Proc Natl
Acad Sci USA. 108:8059–8064. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hiroi T, Wajima T, Kaneko Y, Kiuchi Y and
Shimizu S: An important role of increase intetrahydrobiopterin via
H2O2-JAK2 signaling pathway in late phase of
ischaemic preconditioning. Exp Physiol. 95:609–621. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qi QR and Yang ZM: Regulation and function
of signal transducer and activator of transcription 3. World J Biol
Chem. 5:231–239. 2014.PubMed/NCBI
|
|
15
|
Corcoran RB, Contino G, Deshpande V,
Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA
and Bardeesy N: STAT3 plays a critical role in KRAS-induced
pancreatic tumorigenesis. Cancer Res. 71:5020–5029. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aittomäki S and Pesu M: Therapeutic
targeting of the Jak/STAT pathway. Basic Clin Pharmacol Toxicol.
114:18–23. 2014. View Article : Google Scholar
|
|
17
|
Manea SA, Manea A and Heltianu C:
Inhibition of JAK/STAT signaling pathway prevents
high-glucose-induced increase in endothelin-1 synthesis in human
endothelial cells. Cell Tissue Res. 340:71–79. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Amiri F, Venema VJ, Wang X, Ju H, Venema
RC and Marrero MB: Hyperglycemia enhances angiotensin II-induced
janus-activated kinase/STAT signaling in vascular smooth muscle
cells. J Biol Chem. 274:32382–32386. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hao PP, Chen YG, Liu YP, Zhang MX, Yang
JM, Gao F, Zhang Y and Zhang C: Association of plasma
angiotensin-(1-7) level and left ventricular function in patients
with type 2 diabetes mellitus. PLoS One. 8:e627882013. View Article : Google Scholar
|
|
20
|
Reich HN, Oudit GY, Penninger JM, Scholey
JW and Herzenberg AM: Decreased glomerular and tubular expression
of ACE2 in patients with type 2 diabetes and kidney disease. Kidney
Int. 74:1610–1616. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng
JB, Wang R, Wang XP, Dong B, Gao F, et al: Angiotensin-converting
enzyme (ACE) 2 overexpression ameliorates glomerular injury in a
rat model of diabetic nephropathy: A comparison with ACE
inhibition. Mol Med. 17:59–69. 2011.
|
|
22
|
Benter IF, Yousif MH, Cojocel C,
Al-Maghrebi M and Diz DI: Angiotensin-(1-7) prevents
diabetes-induced cardiovascular dysfunction. Am J Physiol Heart
Circ Physiol. 292:H666–H672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yousif MH, Dhaunsi GS, Makki BM, Qabazard
BA, Akhtar S and Benter IF: Characterization of Angiotensin-(1-7)
effects on the cardiovascular system in an experimental model of
type-1 diabetes. Pharmacol Res. 66:269–275. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hao PP, Yang JM, Zhang MX, Zhang K, Chen
YG, Zhang C and Zhang Y: Angiotensin-(1-7) treatment mitigates
right ventricular fibrosis as a distinctive feature of diabetic
cardiomyopathy. Am J Physiol Heart Circ Physiol. 308:H1007–H1019.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kandalam U and Clark MA: Angiotensin II
activates JAK2/STAT3 pathway and induces interleukin-6 production
in cultured rat brainstem astrocytes. Regul Pept. 159:110–106.
2010. View Article : Google Scholar
|
|
26
|
Mori J, Patel VB, Ramprasath T, Alrob OA,
DesAulniers J, Scholey JW, Lopaschuk GD and Oudit GY: Angiotensin
1-7 mediates renoprotection against diabetic nephropathy by
reducing oxidative stress, inflammation, and lipotoxicity. Am J
Physiol Renal Physiol. 306:F812–F821. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Huanga Z, Zhuanga X, Xieb C, Hua X, Donga
X, Guoa Y, Lic S and Liao X: Exogenous hydrogen sulfide attenuates
high glucose-induced cardiotoxicity by inhibiting NLRP3
inflammasome activation by suppressing TLR4/NF-κB pathway in H9c2
Cells. Cell Physiol Biochem. 40:v1578–v1590. 2016. View Article : Google Scholar
|
|
29
|
Ku SK and Bae JS: Baicalin, baicalein and
wogonin inhibits high glucose-induced vascular inflammation in
vitro and in vivo. BMB Rep. 48:519–524. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chu P, Han G, Ahsan A, Sun Z, Liu S, Zhang
Z, Sun B, Song Y, Lin Y, Peng J and Tang Z: Phosphocreatine
protects endothelial cells from Methylglyoxal induced oxidative
stress and apoptosis via the regulation of PI3K/Akt/eNOS and NF-κB
pathway. Vascul Pharmacol. 91:26–35. 2017. View Article : Google Scholar
|
|
31
|
Wang XM, Song SS, Xiao H, Gao P, Li XJ and
Si LY: Fibroblast growth factor 21 protects against high glucose
induced cellular damage and dysfunction of endothelial nitric-oxide
synthase in endothelial cells. Cell Physiol Biochem. 34:658–671.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cui ZT, Liu JP and Wei WL: The effects of
tanshinone IIA on hypoxia/reoxygenation-induced myocardial
microvascular endothelial cell apoptosis in rats via the JAK2/STAT3
signaling pathway. Biomed Pharmacother. 83:1116–1126. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang X, Guo CX, Zeng XJ, Li HH, Chen BX
and Du FH: A soluble receptor for advanced glycation end-products
inhibits myocardial apoptosis induced by ischemia/reperfusion via
the JAK2/STAT3 pathway. Apoptosis. 20:1033–1047. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao GL, Yu LM, Gao WL, Duan WX, Jiang B,
Liu XD, Zhang B, Liu ZH, Zhai ME, Jin ZX, et al: Berberine protects
rat heart from ischemia/reperfusion injury via activating
JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress.
Acta Pharmacol Sin. 37:354–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li L, Li M, Li Y, Sun W, Wang Y, Bai S, Li
H, Wu B, Yang G, Wang R, et al: Exogenous H2S
contributes to recovery of isch-emic post-conditioning-induced
cardioprotection by decrease of ROS level via down-regulation of
NF-κB and JAK2-STAT3 pathways in the aging cardiomyocytes. Cell
Biosci. 6:262016. View Article : Google Scholar
|
|
36
|
Wu L, Tan JL, Wang ZH, Chen YX, Gao L, Liu
JL, Shi YH, Endoh M and Yang HT: ROS generated during early
reperfusion contribute to intermittent hypobaric hypoxia-afforded
cardioprotection against postischemia-induced Ca2+
overload and contractile dysfunction via the JAK2/STAT3 pathway. J
Mol Cell Cardiol. 81:150–161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ni CW, Hsieh HJ, Chao YJ and Wang DL:
Interleukin-6-induced JAK2/STAT3 signaling pathway in endothelial
cells is suppressed by hemodynamic flow. Am J Physiol Cell Physiol.
287:C771–C780. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fiaschi T, Magherini F, Gamberi T,
Lucchese G2, Faggian G2, Modesti A and Modesti PA: Hyperglycemia
and angiotensin II cooperate to enhance collagen I deposition by
cardiac fibroblasts through a ROS-STAT3-dependent mechanism.
Biochim Biophys Acta. 1843:2603–2610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang T, Mao X, Li H, Qiao S, Xu A, Wang J,
Lei S, Liu Z, Ng KF, Wong GT, et al: N-Acetylcysteine and
allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via
adiponectin and attenuated myocardial postischemic injury in
diabetes. Free Radic Biol Med. 63:291–303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hein TW, Xu W, Xu X and Kuo L: Acute and
chronic hyperglycemia elicit JIP1/JNK-mediated endothelial
vasodilator dysfunction of retinal arterioles. Invest Ophthalmol
Vis Sci. 57:4333–4340. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu N, Shen H, Liu H, Wang Y, Bai Y and Han
P: Acute blood glucose fluctuation enhances rat aorta endothelial
cell apoptosis, oxidative stress and pro-inflammatory cytokine
expression in vivo. Cardiovasc Diabetol. 15:1092016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pollack RM, Donath MY, LeRoith D and
Leibowitz G: Anti-inflammatory agents in the treatment of diabetes
and its vascular complications. Diabetes Care. 39(Suppl 2):
S244–S252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Halvorsen B, Santilli F, Scholz H,
Sahraoui A, Gulseth HL, Wium C, Lattanzio S, Formoso G, Di Fulvio
P, Otterdal K, et al: LIGHT/TNFSF14 is increased in patients with
type 2 diabetes mellitus and promotes islet cell dysfunction and
endothelial cell inflammation in vitro. Diabetologia. 59:2134–2144.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li Y, Zeng Z, Li Y, Huang W, Zhou M, Zhang
X and Jiang W: Angiotensin-converting enzyme inhibition attenuates
lipopoly-saccharide-induced lung injury by regulating the balance
between angiotensin-converting enzyme and angiotensin-converting
enzyme 2 and inhibiting mitogen-activated protein kinase
activation. Shock. 43:395–404. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gopallawa I and Uhal BD:
Angiotensin-(1-7)/mas inhibits apoptosis in alveolar epithelial
cells through upregulation of MAP kinase phosphatase-2. Am J
Physiol Lung Cell Mol Physiol. 310:L240–L248. 2016. View Article : Google Scholar
|
|
46
|
He J, Yang Z, Yang H, Wang L, Wu H, Fan Y,
Wang W, Fan X and Li X: Regulation of insulin sensitivity, insulin
production, and pancreatic β cell survival by angiotensin-(1-7) in
a rat model of streptozotocin-induced diabetes mellitus. Peptides.
64:49–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Al-Maghrebi M and Renno WM: The
ACE/Angiotensin (1-7)/mas axis protects against testicular ischemia
reperfusion injury. Urology. 94:312.e1–8. 2016. View Article : Google Scholar
|
|
48
|
Yang HY, Bian YF, Zhang HP, Gao F, Xiao
CS, Liang B, Li J, Zhang NN and Yang ZM: Angiotensin-(1-7)
treatment ameliorates angiotensin II-induced apoptosis of human
umbilical vein endothelial cells. Clin Exp Pharmacol Physiol.
39:1004–1010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ma X, Xu D, Ai Y, Zhao S, Zhang L, Ming G
and Liu Z: Angiotensin-(1-7)/mas signaling inhibits
lipopolysaccharide-induced ADAM17 shedding activity andapoptosis in
alveolar epithelial cells. Pharmacology. 97:63–71. 2016. View Article : Google Scholar
|
|
50
|
Kim CS, Kim IJ, Bae EH, Ma SK, Lee J and
Kim SW: Angiotensin-(1-7) attenuates kidney injury due to
obstructive nephropathy in rats. PLoS One. 10:e01426642015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Meneses C, Morales MG, Abrigo J, Simon F,
Brandan E and Cabello-Verrugio C: The angiotensin-(1-7)/Mas axis
reduces myonuclear apoptosis during recovery from angiotensin
II-induced skeletal muscle atrophy in mice. Pflugers Arch.
46:1975–1984. 2015. View Article : Google Scholar
|
|
52
|
Lin L, Liu X, Xu J, Weng L, Ren J, Ge J
and Zou Y: Mas receptor mediates cardioprotection of
angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte
autophagy and cardiac remodelling through inhibition of oxidative
stress. J Cell Mol Med. 20:48–57. 2016. View Article : Google Scholar
|
|
53
|
Shi Y, Lo CS, Padda R, Abdo S, Chenier I,
Filep JG, Ingelfinger JR, Zhang SL and Chan JS: Angiotensin-(1-7)
prevents systemic hypertension, attenuates oxidative stress and
tubulointerstitial fibrosis, and normalizes renal
angiotensin-converting enzyme 2 and Mas receptor expression in
diabetic mice. Clin Sci. 128:649–663. 2015. View Article : Google Scholar
|
|
54
|
McKinney CA, Fattah C, Loughrey CM,
Milligan G and Nicklin SA: Angiotensin-(1-7) and angiotensin-(1-9):
Function in cardiac and vascular remodelling. Clin Sci.
126:815–827. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ma Y, Huang H, Jiang J, Wu L, Lin C, Tang
A, Dai G, He J and Chen Y: AVE 0991 attenuates cardiac hypertrophy
through reducing oxidative stress. Biochem Biophys Res Commun.
474:621–625. 2016. View Article : Google Scholar
|
|
56
|
Zhang Y, Liu J, Luo JY, Tian XY, Cheang
WS, Xu J, Lau CW, Wang L, Wong WT, Wong CM, et al: Upregulation of
angiotensin (1-7)-mediated signaling preserves endothelial function
through reducing oxidative stress in diabetes. Antioxid Redox
Signal. 23:880–892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang F, Liu C, Wang L, Cao X, Wang YY and
Yang JK: Antioxidant effect of angiotensin (1-7) in the protection
of pancreatic β cell function. Mol Med Rep. 14:1963–1969. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Papinska AM, Mordwinkin NM, Meeks CJ,
Jadhav SS1 and Rodgers KE: Angiotensin-(1-7) administration
benefits cardiac, renal and progenitor cell function in db/db mice.
Br J Pharmacol. Jun 15–2015.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li Y, Cao Y, Zeng Z, Liang M, Xue Y, Xi C,
Zhou M and Jiang W: Angiotensin-converting enzyme
2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced
apoptosis of pulmonary microvascular endothelial cells by
inhibiting JNK/NF-κB pathways. Sci Rep. 5:82092015. View Article : Google Scholar
|
|
60
|
Zhang YH, Zhang YH, Dong XF, Hao QQ, Zhou
XM, Yu QT, Li SY, Chen X, Tengbeh AF, Dong B and Zhang Y: ACE2 and
Ang-(1-7) protect endothelial cell function and prevent early
atherosclerosis by inhibiting inflammatory response. Inflamm Res.
64:253–260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang L, Hu X, Zhang W and Tian F:
Angiotensin (1-7) ameliorates angiotensin II-induced inflammation
by inhibiting LOX-1 expression. Inflamm Res. 62:219–228. 2013.
View Article : Google Scholar
|
|
62
|
Yu JW, Deng YP, Han X, Ren GF, Cai J and
Jiang GJ: Metformin improves the angiogenic functions of
endothelial progenitor cells via activating AMPK/eNOS pathway in
diabetic mice. Cardiovasc Diabetol. 15:882016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bretón-Romero R, Feng B, Holbrook M, Farb
MG, Fetterman JL, Linder EA, Berk BD, Masaki N, Weisbrod RM,
Inagaki E, et al: Endothelial dysfunction in human diabetes is
mediated by Wnt5a-JNK signaling. Arterioscler Thromb Vasc Biol.
36:561–569. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Haga S, Tsuchiya H, Hirai T, Hamano T,
Mimori A and Ishizaka Y: A novel ACE2 activator reduces
monocrotaline-induced pulmonary hypertension by suppressing the
JAK/STAT and TGF-β cascades with restored caveolin-1 expression.
Exp Lung Res. 41:21–31. 2015. View Article : Google Scholar
|
|
65
|
Lei Y, Xu Q, Zeng B, Zhang W, Zhen Y, Zhai
Y, Cheng F, Mei W, Zheng D, Feng J, et al: Angiotensin-(1-7)
protects cardiomyo-cytes against high glucose-induced injuries
through inhibiting reactive oxygen species-activated leptin-p38
mitogen-activated protein kinase/extracellular signal-regulated
protein kinase 1/2 pathways, but not the leptin-c-Jun N-terminal
kinase pathway in vitro. J Diabetes Investig. 8:434–445. 2017.
View Article : Google Scholar :
|