Introduction
Benign tumors of adipose tissue are often exhibited
in humans with rare phosphatase and tensin homolog
(PTEN) Hamartoma Tumor Syndrome (PHTS), which is caused by
heterozygous germ line mutations within the tumor suppressor
PTEN gene (1). Although
noncancerous, these lipomas frequently develop to form extended
fatty tissue tumors (lipomatosis), which may infiltrate other
tissues and cause numerous side effects, including organ
obstruction, resulting in loss of function and chronic pain
(2). A previous study assessed a
child with a severe PHTS phenotype who exhibited abdominal
lipomatosis (3). It was
demonstrated that an individual treatment attempt using the
mammalian target of rapamycin complex (mTOR)1 inhibitor sirolimus
led to an improvement in the patient's general condition and a
decrease in thymus size; however, lipomatosis regression was not
observed (3). At present, no
targeted therapies for the treatment of PHTS-associated lipomatosis
exist. Surgical excision of the tumor and symptomatic treatment
remain the only form of therapy available (4). There is therefore an urgent need to
uncover novel therapeutic approaches for patients with PHTS and
non-resectable adipose tissue tumors.
Statins, also known as
3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, are widely
utilized to decrease serum cholesterol levels (5). Previous studies have demonstrated
that statins elicit numerous non-lipid modifying effects, which
have been revealed to exhibit anti-tumor effects in vitro,
in vivo and in epidemiological studies (6-9).
In particular, simvastatin was demonstrated to increase PTEN
protein levels and activity, thus downregulating the activation of
the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)
signaling pathway (10-13). Furthermore, a case report of a
54-year-old man described a dramatic reduction of an isolated
subcutaneous lipoma associated with simvastatin therapy (14).
The aim of the present study was to assess the
effects of simvastatin on three lipoma cell lines, termed lipoma
PTEN-deficient 1-3 (LipPD1-3), established from the resected
lipoma tissue of pediatric patients with heterozygous germ line
PTEN deletion or mutations. It was hypothesized that
simv-astatin treatment may lead to attenuated cell growth or the
induction of apoptosis in these PTEN haploinsufficient
lipoma cells by increasing levels of the PTEN protein.
Materials and methods
Informed consent
Written informed consent was provided by the parents
of all patients enrolled in the present study. A total of 3
patients (2 male, 1 female) and 2 healthy control patients (1 male,
1 female), aged 0-18 years were enrolled in the present study. All
patients were admitted to the Hospital for Child and Adolescent
Medicine (Leipzig University, Leipzig, Germany) and samples were
collected between July 2007 and December 2016. Patients with
morbidities other than PHTS were excluded. One patients received
sirolimus following lipoma resection (3). All LipPD cells used in this study
were obtained from lipomatous adipose tissue that was resected for
diagnostic and therapeutic reasons. Control primary pre-adipocytes
were removed from adipose tissue obtained from pediatric or young
adult patients during routine surgery. Ethical approval for these
studies was obtained from the Ethics Committee of the University of
Leipzig (ref. no. 425-12-171220; Leipzig, Germany).
Cell lines, cell culture and
treatments
Genetic analyses of PTEN revealed a
heterozygous deletion affecting exons 2-9 of 9 of PTEN in
LipPD1 cells (3,15). A heterozygous PTEN mutation
was detected in LipPD2 (c.404T>A, p.I135K) and LipPD3 (exon 1,
c.76A>C, p.T26P). A schematic presentation of the genetic
changes in PTEN is presented in Fig. 1A. The following cells were used as
PTEN wild-type controls: Simpson-Golabi-Behmel (SGBS, kindly
supplied by Dr M. Wabitsch, University Hospital, Ulm, Germany)
(16,17) cells and normal primary
pre-adipocytes obtained from pediatric or healthy young adult
adipose tissue. All cell strains were established according to
protocols as described previously (3,17).
Cells were maintained in Dulbecco's Modified Eagle's medium
(DMEM)/F12 medium supplemented with 10% fetal calf serum, glutamine
(2 mM), biotin (33 mM) and pantothenic acid (17 mM; all from
Biochrom, Ltd., Cambridge, UK) at 37°C in a humidified atmosphere
containing 5% CO2. InSolution™ simvastatin sodium salt
was purchased from EMD Millipore (Billerica, MA, USA). Medium was
replaced with serum-free DMEM 24 h prior to cell stimulation with
0, 0.1, 1 and 10 µM simvastatin for 6, 12, 24 or 48 h.
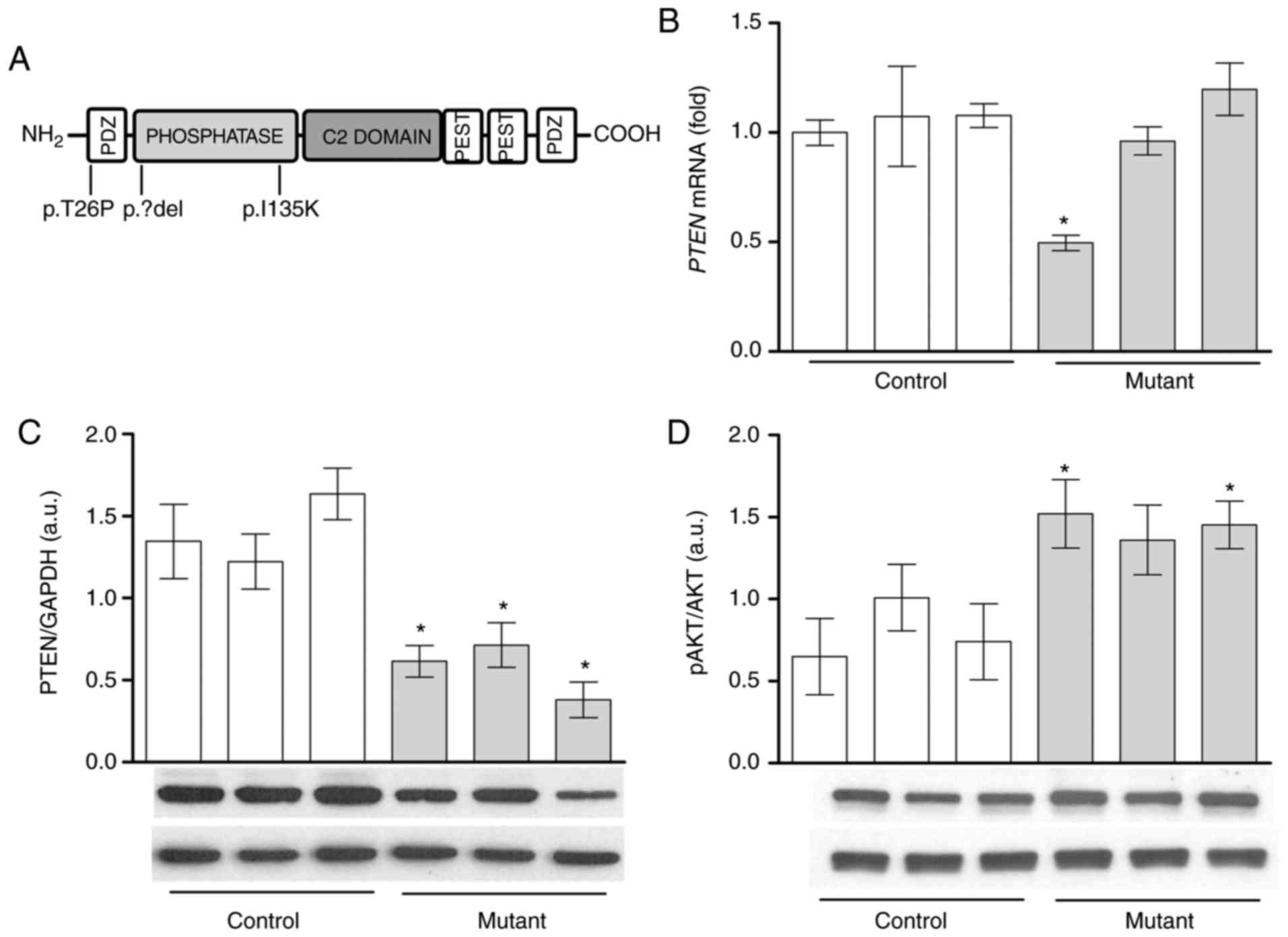 | Figure 1AKT phosphorylation is increased with
lower PTEN protein levels in lipoma cells compared with
PTEN wild-type pre-adipocytes. (A) Schematic representation
of the PTEN protein domains and mutation sites. Heterozygous
deletion (LipPD1) or mutations (LipPD2 and LipPD3) of the
PTEN gene are depicted. PTEN (B) mRNA and (C) protein
expression in mutant lipoma cells (LipPD1-3) in comparison with
PTEN wild-type control (SGBS, normal primary)
pre-adipocytes. mRNA data were normalized to the Tata box
binding protein and Glyceraldehyde-3-phosphate dehydrogenase,
respectively. (D) Phosphorylation of AKT (threonine 308) in mutant
lipoma cells (LipPD1-3) compared with wild-type control cells.
Phosphorylation of AKT was normalized to total AKT levels. All data
are presented as the mean ± standard error of the mean. Statistical
analysis was performed using a Student's t-test.
*P<0.05 vs. the mean of all three PTEN
wild-type cells. AKT, protein kinase B; PTEN, phosphatase
and tensin homolog; LipPD1-3, lipoma PTEN-deficient 1-3;
p, phosphorylated; PDZ, Post synaptic density protein (PSD95),
Drosophila disc large tumor suppressor (Dlg1) and zonula
occludens-1 protein (ZO-1), PEST, proline (P), glutamic acid (E),
serine (S) and threonine (T). |
Cell viability and apoptosis
The effect of simvastatin on proliferation was
measured using a Cell Proliferation Reagent WST-1 (Roche
Diagnostics GmbH, Mannheim, Germany) according to the
manufacturer's protocol. Cells were seeded in 96-well plates at a
density of 10,000 cells/cm2 and incubated for 48 h with
0, 0.1, 1 and 10 µM simvastatin. Apoptosis was determined
using Annexin V-fluorescein isothiocyanate (FITC; BD Pharmingen; BD
Bioscience, San Jose, CA, USA)/propidium iodide (PI) staining
according to manufacturer's protocol. Briefly, cells were seeded at
a density of 5,000 cells/cm2 and incubated for 48 h with
0, 1 or 10 µM simvastatin. Cells were trypsinized and
incubated with Annexin V-FITC/PI for 10 min at 4°C in the dark.
Annexin V-FITC-positive cells represent early apoptosis and Annexin
V-FITC/PI double positive cells indicate late apoptotic cells.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
For qPCR analysis mRNA was extracted from LipPD1
cells using the RNeasy mini kit (Qiagen GmbH, Hilden, Germany)
according to the manufacturer's protocol. A total of 1 µg
mRNA was reverse transcribed at 39°C for 1 h into cDNA using M-MLV
Reverse Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). PCR reactions were performed using Taqman
Mastermix FAST qPCR Master Mix Plus Low ROX (Eurogentec, Liege,
Belgium) or Absolute qPCR SYBR-Green Low ROX Mix (Thermo Fisher
Scientific, Inc.) and the Applied Biosystems 7500 Real-Time PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Primers and probes were designed using the PrimerExpress software
version 3.0 (Applied Biosystems; Thermo Fisher Scientific, Inc.)
and sequences are presented in Table
I. The following thermocycling conditions were used: Activation
at 95°C for 15 min, 40 cycles of denaturation at 95°C for 15 sec,
annealing at 60°C for 30 sec, elongation at 72°C for 30 sec. A
standard curve of serial dilutions of plasmid DNA of the respective
target gene was included on each plate. The copy number of each
sample was calculated from the standard curve and normalised to the
mean of the two housekeeping genes Tata box binding protein
(TBP) or hypoxanthine phosphoribosyltransferase
(HPRT) expression (18).
 | Table IPrimer and probe sequences. |
Table I
Primer and probe sequences.
| Gene | Direction | Sequence |
|---|
| PTEN | Forward |
GTTTACCGGCTCAAAT |
| Reverse CCCCC | ACTTTCACAGT |
| PPARγ | Forward |
GATCCAGTGGTTGCAGATTACAA |
| Reverse |
GAGGGAGTTGGAAGGCTCTTC |
| Probe |
TGACCTGAAACTTCAAGAGTACCAAAGTGCAA |
| TBP | Forward |
TTGTAAACTTGACCTAAAGACCATTGC |
| Reverse |
TTCGTGGCTCTCTTATCCTCATG |
| Probe |
AACGCCGAATATAATCCCAAGCGGTTTG |
| HPRT | Forward |
GGCAGTATAATCCAAAGATGGTCAA |
| Reverse |
GTCTGGCTTATATCCAACACTTCGT |
| Probe C |
AAGCTTGCTGGTGAAAAGGACCCC |
Western blot analysis
For protein analysis, cells were lysed with modified
radioimmunoprecipitation assay buffer [50 mM Tris HCl; pH 7.4; 1%
NP-40; 0.25% sodium deoxycholate; 1× Roche complete proteases
inhibitor cocktail (Roche Diagnostics GmbH); 1 mM EDTA; 1 mM sodium
orthovana-date; and 1 mM sodium fluoride], protein concentrations
were determined using a DC protein assay (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and proteins (20 µg/lane) were
separated by 10% SDS-PAGE. Following semi-dry transfer onto
nitrocellulose membranes, membranes were blocked using 5% non-fat
dry milk in TBS buffer containing 0.1% Tween-20 (TBST) for 1 h at
room temperature. Blots were then incubated overnight at 4°C with
the appropriate primary antibody. The antibodies utilized in
western blotting are summarized in Table II. Blots were washed three times
for 5 min with TBST and incubated with horseradish peroxidase
(HRP)-goat anti rabbit (P-0448) or HRP-goat anti mouse (P-0447
antibodies (Dako; Agilent Technologies, Inc., Santa Clara, CA, USA)
at a dilution of 1:2,000 for 2 h at room temperature. Protein bands
were detected using Luminata Classico Western horseradish
peroxidase Substrate (EMD Millipore) or Amersham ECL Prime Western
Blotting Detection Reagent (GE Healthcare, Chicago, IL, USA).
Glyceraldehyde-3-phosphate dehydrogenase (EMD Millipore) was used
as a loading control. ImageJ 1.41 was used for densitometric
analysis (National Institutes of Health, Bethesda, MD, USA).
| Table IIPrimary antibodies utilized in
western blotting. |
Table II
Primary antibodies utilized in
western blotting.
| Antibody | Supplier | Cat. no. | Dilution |
|---|
| 4E-BP1 | NEB | 9644 | 1:1,000 |
| Phospho-4E-BP1
(Threonine 37/46) | NEB | 2855 | 1:1,000 |
| AKT | NEB | 9272 | 1:1,000 |
| Phospho-AKT
(Threonine 308) | NEB | 4056 | 1:1,000 |
| Phospho-AKT (Serine
473) | NEB | 4060 | 1:1,000 |
| GAPDH | Merck KGaA | MAB374 | 1:100,000 |
| p44/42 MAPK
(ERK1/2) | NEB | 9102 | 1:1,000 |
| Phospho-p44/42
mitogen activated protein | NEB | 9101 | 1:1,000 |
| kinase (ERK1/2)
(Threonine 202/Tyrosine 204) | | | |
| mTOR | NEB | 2983 | 1:1,000 |
| Phospho-mTOR
(Serine 2448) | NEB | 2971 | 1:1,000 |
| Peroxisome
proliferator-activated receptor γ | NEB | 2443 | 1:1,000 |
| Phosphatase and
tensin homolog | NEB | 9559 | 1:1,000 |
Small interfering (si)RNA-mediated
knockdown of PPARγ
LipPD1 cells were microporated using the Neon
Transfection System 100 µl kit (Invitrogen; Thermo Fisher
Scientific, Inc.) with the PPARγ ON-TARGETplus SMARTpool siRNA
reagent and the respective negative control (ON-TARGETplus control
pool; GE Healthcare Dharmacon, Inc., Lafayette, CO, USA) as
previously described (18). A
total of 106 cells were transfected with siRNA at a
final concentration of 500 nmol/l, seeded at a density of
105 cells/well in 6 well plates and cultured for 24 h.
The following day, the culture medium was replaced with serum-free
DMEM for 24 h. Cells were then stimulated using simvastatin (10
µM) as above.
Statistical analysis
Data are presented as the mean ± standard error of
the mean of at least three independent experiments. Significant
differences were determined using GraphPad Prism 6 software
(GraphPad Software, Inc., La Jolla, CA, USA) and the unpaired
Student's t-test or one-way analysis of variance followed by a post
hoc Bonferroni multiple comparison test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Decreased PTEN protein levels and
enhanced AKT in PTEN deficient lipoma cells
Basal PTEN mRNA and protein levels were
analyzed to assess the impact of the heterozygous PTEN
deletions or mutations (Fig. 1A)
on lipoma cells. Lipoma cells with a large heterozygous PTEN
deletion (3) possessed a
significantly lower PTEN mRNA expression compared with PTEN
wild-type control pre-adipocytes (P<0.05; Fig. 1B). PTEN mRNA expression was
similar in lipoma cells with PTEN point mutations and
non-mutant cells (Fig. 1B).
However, in all three lipoma cell cultures, PTEN protein
levels were lower compared with PTEN wild-type control cells
(P<0.05; Fig. 1C). It was then
assessed whether reduced PTEN protein levels led to enhanced
AKT phosphorylation. Mutation-positive cells exhibited
significantly higher levels of phosphorylated AKT (Thr308) compared
with control cells (LipPD1 and LipPD3; P<0.05) (Fig. 1D).
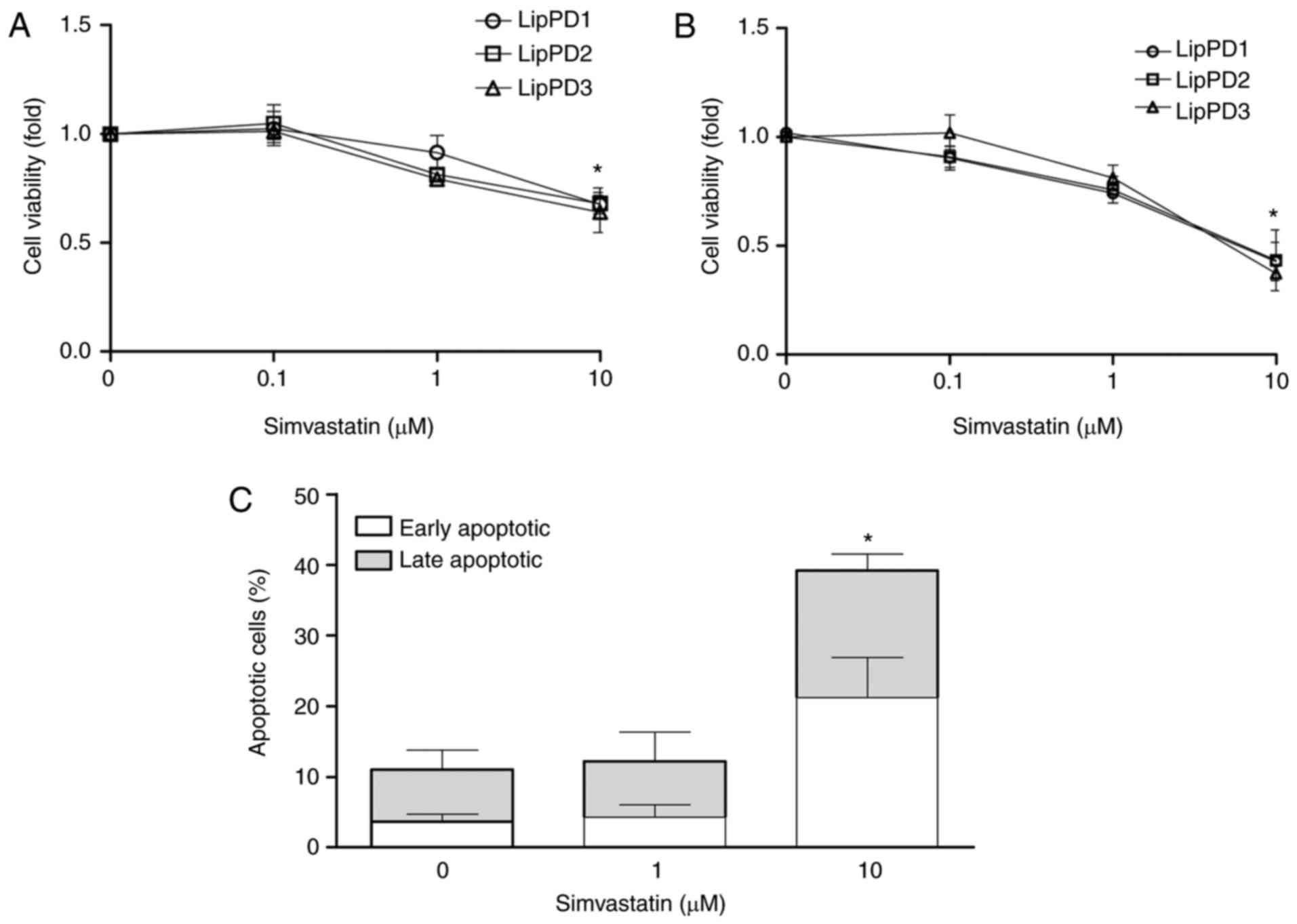
Simvastatin treatment induces apoptosis
in lipoma cells
The effect of simvastatin on the viability of lipoma
cells was determined following incubation for 24, 48 and 72 h
(Fig. 2A). Simvastatin at 10
µM significantly decreased the viability of LipPD1, LipPD2
and LipPD3 cells to 67.5±8.3, 68.0±14.3 and 64.0±9.2% at 48 h and
to 42.3±8.8%, 43.3±14.0% and 37.2±6.9% at 72 h incubation,
(P<0.05; Fig. 2A and B). At
the same dose and incubation time, simvastatin induced apoptosis by
28% in LipPD1 compared with LipPD1 cells incubated without
simvastatin (Fig. 2C). Following
incubation with 10 µM simvastatin, cells were equally
distributed between early and late apoptotic phases. No effect on
cell viability was observed following an incubation of 24 h (data
not shown).
Simvastatin treatment decreases AKT/mTOR
activation
Previous studies have demonstrated that simvastatin
upregulates PTEN transcription (8-10),
and so PTEN mRNA expression in lipoma cells was assessed
following incubation with simvastatin. Stimulation with 10
µM simvastatin resulted in a significant increase in the
expression of PTEN mRNA to 1.4±0.1-fold in LipPD1 compared
with control cells following a 6 h incubation (P<0.05; data not
shown). The effect of simvastatin on the PTEN protein and
AKT/mTOR pathway activation following 6, 12, 24 and 48 h of
incubation was then assessed. Simvastatin (10 µM) resulted
in an increase in phosphorylated and total PTEN protein
levels in LipPD1 cells when incubated for 24 h (Fig. 3A–C). In addition, a downregulation
in AKT phosphorylation at T308 and S473 (Fig. 3A, D and E) was observed following
24 and 48 h incubation. The activation of mTOR, a downstream target
of AKT, was then examined. The results demonstrated that 48 h
incubation with 10 µM simvastatin significantly decreased
mTOR1 phosphorylation at Ser2448 by 48.8±1.2% (P<0.05; Fig. 3F and G) and phosphorylation of the
mTOR target 4E-binding protein-1 (4EBP-1) by 64.9±1.2% (P<0.05;
Fig. 3F and H). Increased PTEN
protein levels were also observed in LipPD2 and LipPD3 cells
following 6 h of incubation with 10 µM simvastatin (both
P<0.05; data not shown). These results indicate that simvastatin
incubation affects PTEN protein levels and AKT/mTOR
activation in a time-dependent manner.
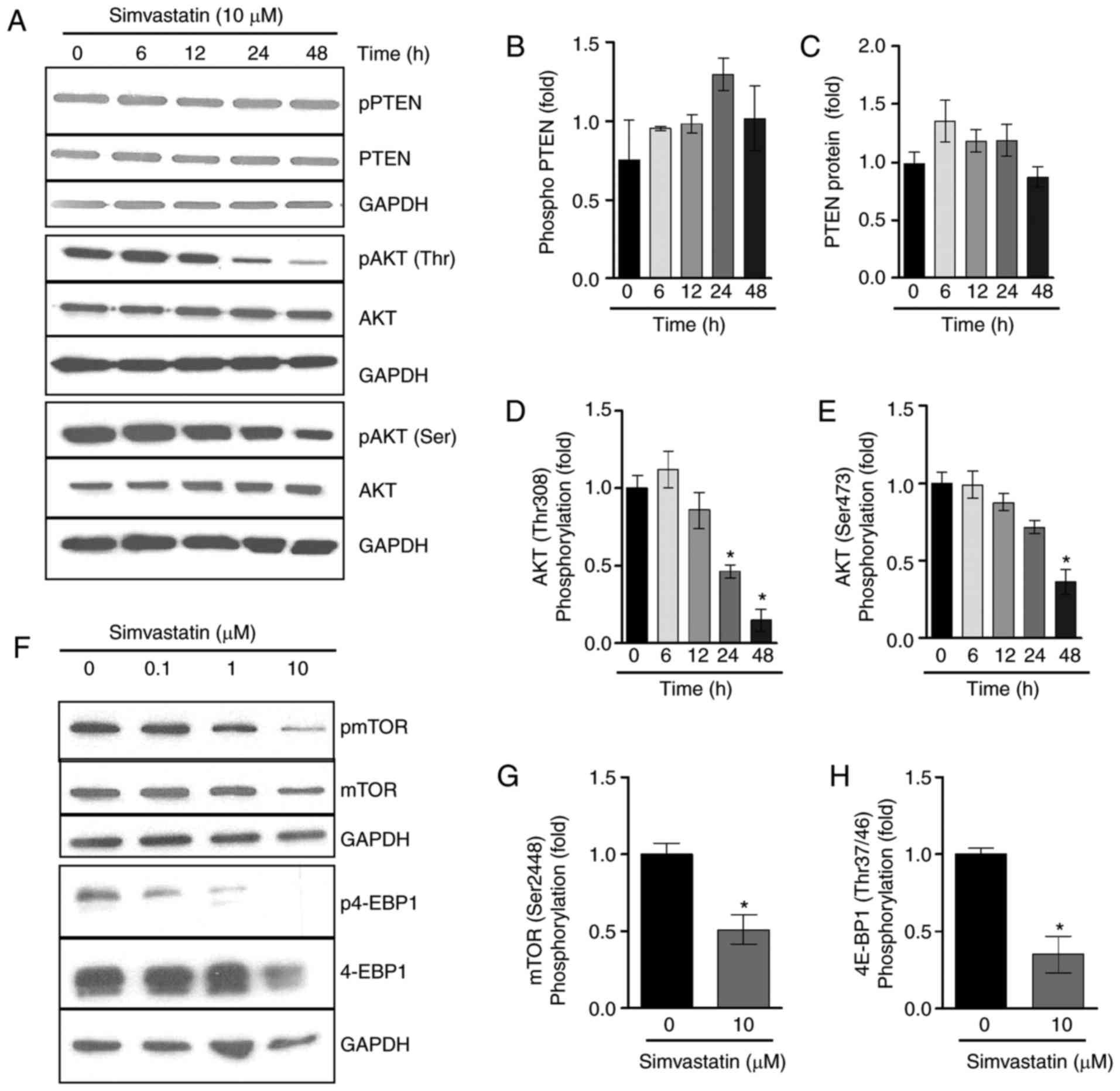 | Figure 3Simvastatin treatment decreased
AKT/mTOR activation following prolonged treatment. (A) LipPD1 cells
were stimulated with 10 µM simvastatin in culture medium for
6, 12, 24 or 48 h. PTEN mRNA expression was quantified using
reverse transcriptase polymerase chain reactions. Data were
normalized to TBP and then compared with the expression
level exhibited in the untreated controls. Relative expression of
(B) phospho PTEN, (C) PTEN, (D) phospho AKT and (E)
total AKT. (F) LipPD1 cells were stimulated with 10 µM
simvastatin in culture medium for 48 h. Relative expression of (G)
mTOR (Ser2448) and (H) 4E-BP1 (Thr37/46) were calculated. All data
are presented as the mean ± standard error of the mean. Statistical
analysis was performed using a Student's t-test or one-way analysis
of variance with a post hoc Bonferroni test. *P<0.05
vs. untreated cells. AKT, protein kinase B; mTOR, mammalian target
of rapamycin; PTEN, phosphatase and tensin homolog;
phospho, phosphorylated; LipPD1-3, lipoma PTEN-deficient
1-3; TBP, Tata box binding protein; Ser, Serine; Thr,
threonine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; pAKT,
phosphorylated AKT; 4E-BP1, 4E-binding protein-1; pmTOR,
phosphorylated mTOR; p4E-BP1, phosphorylated 4E-BP1. |
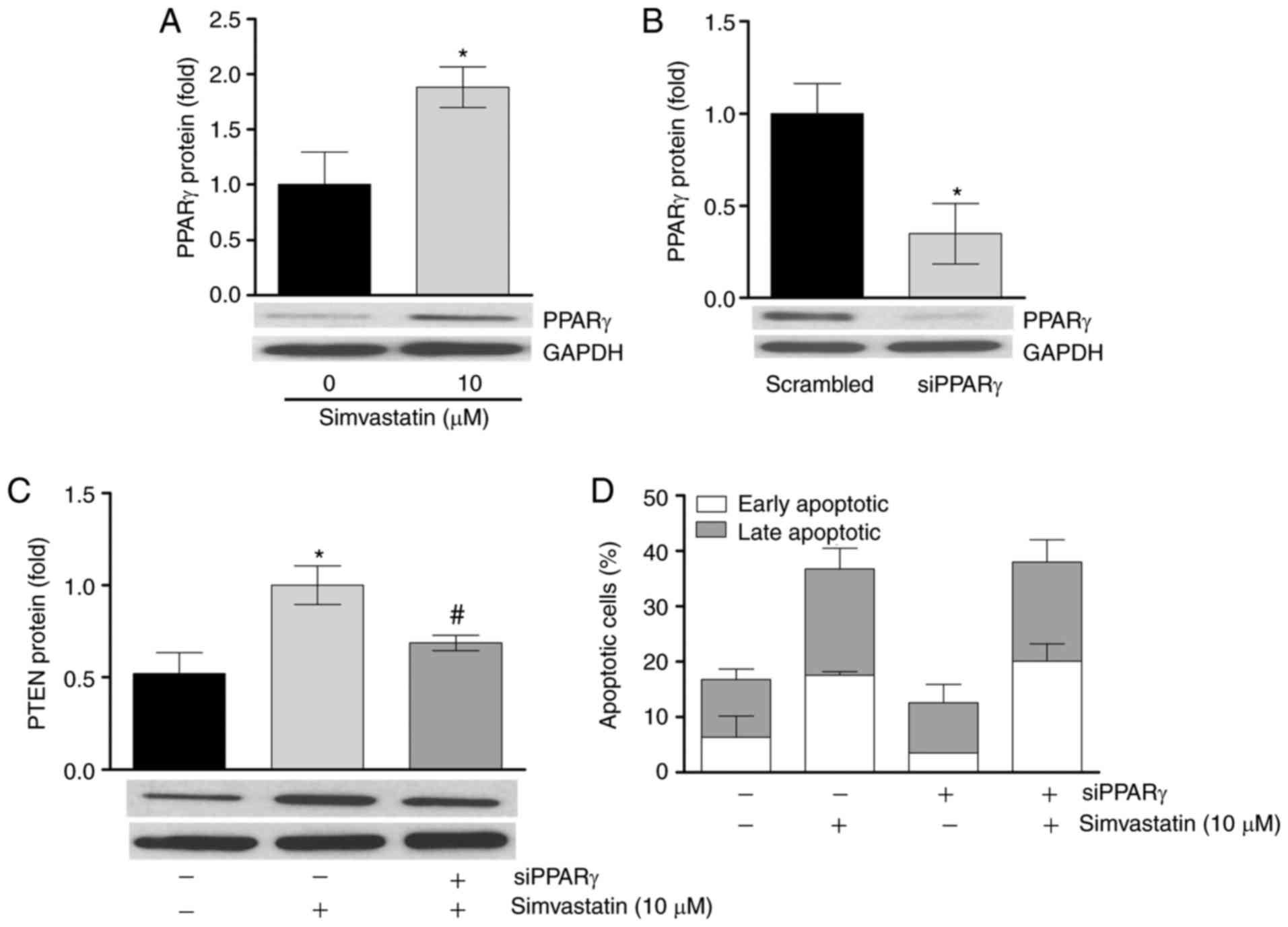
PPARγ mediates simvastatin action on PTEN
protein expression, but not on apoptosis
The present study aimed to identify the mediators of
simvastatin. Previous studies have demonstrated that simvastatin
acts through the activation of nuclear factor (NF)κB (9,13,19). However, in the present study, no
significant effect on NF-κB phosphorylation was observed following
incubation with simvastatin (data not shown). Additionally, no
significant upregulation of the NF-κB target protein B-cell
lymphoma-2 (13,20,21) was detected (data not shown).
Previous studies have indicated the ability of the transcriptional
regulator PPARγ to induce PTEN mRNA (22-25). The results of the present study
demonstrated that a 6 h simvastatin incubation significantly
upregulated LipPD1 cell PPARγ protein expression by 1.88±0.3-fold
(P<0.05; Fig. 4A). It was then
determined whether knockdown of PPARγ abrogated the stimulatory
effect of simvastatin on PTEN levels. siRNA-mediated
knockdown of PPARγ to 34.8±1.2% was confirmed at the protein level
(Fig. 4B) and this knockdown
significantly attenuated the upregulation of PTEN mRNA and
protein induced by simvastatin (P<0.05; Fig. 4C). These results indicate that
simvastatin induces PTEN expression in a PPARγ-dependent
manner in LipPD1 cells. Furthermore, the present study assessed
whether PPARγ knockdown would effect simvastatin-induced apoptosis.
No significant differences in apoptosis induction or in the
distribution of cells between early and late apoptotic phases were
identified in PPARγ-knockdown cells compared with
control-transfected cells following stimulation with simvastatin
(Fig. 4D).
Discussion
At present, treatment options for patients with
non-resectable PHTS-associated lipomatosis are limited. Sirolimus,
an inhibitor of mTOR1, was successfully used to treat patients with
other PHTS-associated complications (23-25). However, it was not effective at
reducing lipoma growth in a patient with PHTS due to the
heterozygous germ line deletion of PTEN (3). The aim of the present study was to
test the effects of simvastatin on the growth of human lipoma cells
derived from tumors with heterozygous mutations or deletions of
PTEN. Simvastatin was selected for use as it has been
demonstrated to exert anti-proliferative and growth-inhibitory
actions on a number of cancer cell lines and tumor animal models
(6,7,10,18,22,26-30). In accordance with these results,
the present study demonstrated that simvastatin reduced the
viability of PTEN mutant lipoma cells by inducing apoptosis
in a time- and dose-dependent manner.
A potential mechanism of simvastatin action is the
increase of PTEN protein levels and consequent suppression of
AKT/mTOR signaling. This has been addressed by previous studies in
various tissues, cancer cell lines and cancer xenograft animal
models (8-10). In accordance with previous studies
using tissues harvested from patients with PHTS (28,34), the results of the present study
demonstrated that PTEN protein levels are reduced and AKT
activation is increased in PTEN mutant lipoma cell cultures
compared with PTEN wild-type cells. Furthermore, decreased
PTEN mRNA levels were only identified in lipoma cells with a
large heterozygous PTEN deletion (LipPD1 cells) and not in
lipoma cells with heterozygous PTEN mutations (LipPD2 and
LipPD3 cells). This contrast between normal mRNA expression and low
protein levels may indicate the occurrence of a
post-transcriptional event, such as increased protein degradation,
due to mutations in PTEN.
Simvastatin treatment resulted in a transient
upregulation of PTEN mRNA and protein levels in lipoma
cells. In the present study, it was investigated by which mechanism
simvastatin mediated this effect. The transcriptional regulator
PPARγ has previously been reported to upregulate the transcription
of PTEN and thereby influence AKT phosphorylation in
non-malignant and cancer cells (10,19-22). The results of the present study
demonstrate that PPARγ exhibits a similar expression pattern to
PTEN following simvastatin stimulation of lipoma cells.
However, the transient knockdown of PPARγ abrogated the increase in
lipoma cell PTEN protein levels following simvastatin
treatment. Additionally, PPARγ knockdown had no effect on
simvastatin-mediated induction of apoptosis. This is in contrast to
previous studies, which demonstrated that the induction of
apoptosis by PPARγ agonists occurs via the upregulation of
PTEN (10,19,22,32-34).
Previous studies have reported various pathways by
which simvastatin affects cell viability that are dependent on the
inhibition of the cholesterol biosynthesis pathway, including the
attenuation of NF-κB activation by a decrease in intracellular
isoprenoid concentrations followed by impaired inner membrane
attachment and Ras, Rac or Rho protein function (32,33). However, the lipoma cells used in
the present study did not exhibit a decrease in NF-κB
phosphorylation following incubation with simvastatin. The action
of simvas-tatin in prostate cancer cells may be associated with
decreasing cellular cholesterol levels, leading to altered membrane
lipid raft structures, causing a reduction in AKT phosphorylation
and thus resulting in apoptosis (38). This effect of simvastatin on
caveolar raft structures has also been demonstrated in adipocytes
(39). In accordance with this,
the present study detected a decrease in AKT, mTOR and the mTOR
target 4EBP-1 phosphorylation following prolonged treatment with
simvastatin.
In conclusion, the results of the present study
support the hypothesis that simvastatin treatment reduces the
growth of lipoma cells and may be a promising candidate for the
treatment of lipomatosis associated with PTEN
haploinsufficiency. However, the effects of simvastatin are not
mediated by PPARγ-facilitated upregulation of PTEN in lipoma
cells. Further studies in suitable animal models are required to
evaluate the use of simvastatin treatment in patients with
lipomatosis associated with PTEN haploinsufficiency.
Acknowledgments
Not applicable.
Abbreviations:
|
4E-BP-1
|
4E-binding protein
|
|
mTOR
|
mammalian target of rapamycin
|
|
PTEN
|
phosphatase and tensin homologue
|
|
PHTS
|
PTEN hamartoma tumor syndrome
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
PPARγ
|
peroxisome proliferator-activated
receptor gamma
|
|
siRNA
|
small interfering RNA
|
Notes
[1]
Funding
The present study was supported by the
Mitteldeutsche Kinderkrebsforschung (to FK), the European Union's
Horizon 2020 research and innovation program under the Marie
Sklodowska-Curie agreement (grant no. 705869; to AG) and from
Deutsche Forschungsgemeinschaft/Collaborative Research Centre
SFB1052/B10 (to AG and WK).
[2] Availability
of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
AG and NH conceived the study. AG, NH and FK
designed the study and provided a major contribution to writing the
manuscript. AG, NH, FK, TS and MP analysed data. WK interpreted
patient data and discussed results. KL and AK provided expertise on
adipocyte culture and knockdown experiments. FK, TS and SR
performed experiments. All authors read and approved the final
manuscript.
[4] Ethics
approval and consent to participate
Ethical approval for these studies was obtained from
the Ethics Committee of the University of Leipzig (ref. no.
425-12-171220; Leipzig, Germany). Written informed consent was
provided by the parents of all patients enrolled in the present
study.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Eng C: PTEN: One gene, many syndromes. Hum
Mutat. 22:183–98. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tan MH, Mester JL, Ngeow J, Rybicki LA,
Orloff MS and Eng C: Lifetime cancer risks in individuals with
germline PTEN mutations. Clin Cancer Res. 18:400–407. 2012.
View Article : Google Scholar :
|
|
3
|
Schmid GL, Kässner F, Uhlig HH, Körner A,
Kratzsch J, Händel N, Zepp FP, Kowalzik F, Laner A, Starke S, et
al: Sirolimus treatment of severe PTEN hamartoma tumor syndrome:
Case report and in vitro studies. Pediatr Res. 75:527–534. 2014.
View Article : Google Scholar
|
|
4
|
Hollander MC, Blumenthal GM and Dennis PA:
PTEN loss in the continuum of common cancers, rare syndromes and
mouse models. Nat Rev Cancer. 11:289–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mauro VF and MacDonald JL: Simvastatin: A
review of its pharmacology and clinical use. DICP. 25:257–64. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Desai CS, Martin SS and Blumenthal RS:
Non-cardiovascular effects associated with statins. BMJ.
349:g37432014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhong S, Zhang X, Chen L, Ma T, Tang J and
Zhao J: Statin use and mortality in cancer patients: Systematic
review and meta-analysis of observational studies. Cancer Treat
Rev. 41:554–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nielsen SF, Nordestgaard BG and Bojesen
SE: Statin use and reduced cancer-related mortality. N Engl J Med.
367:1792–1802. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Campbell MJ, Esserman LJ, Zhou Y,
Shoemaker M, Lobo M, Borman E, Baehner F, Kumar AS, Adduci K, Marx
C, et al: Breast cancer growth prevention by statins. Cancer Res.
66:8707–8714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang Z, Tang Y, Fang J, Zhou Z, Xing Z,
Guo Z, Guo X, Wang W, Jiao W, Xu Z and Liu Z: Simvastatin inhibits
renal cancer cell growth and metastasis via AKT/mTOR, ERK and
JAK2/STAT3 pathway. PLoS One. 8:e628232013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang T, Seah S, Loh X, Chan CW, Hartman M,
Goh BC and Lee SC: Simvastatin-induced breast cancer cell death and
deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by
metabolic products of the mevalonate pathway. Oncotarget.
7:2532–2544. 2016.
|
|
12
|
Chen YQ, Zhao LY, Zhang WZ and Li T:
Simvastatin reverses cardiomyocyte hypertrophy via the upregulation
of phosphatase and tensin homolog expression. Exp Ther Med.
10:797–803. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ghosh-Choudhury N, Mandal CC,
Ghosh-Choudhury N and Ghosh Choudhury G: Simvastatin induces
derepression of PTEN expression via NFkappaB to inhibit breast
cancer cell growth. Cell Signal. 22:749–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Self TH and Akins D: Dramatic reduction in
lipoma associated with statin therapy. J Am Acad Dermatol. 58(2
Suppl): S30–S31. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wilhelm F, Kässner F, Schmid G, Kratzsch
J, Laner A, Wabitsch M, Körner A, Kiess W and Garten A:
Phosphatidylinositol 3-kinase (PI3K) signalling regulates
insulin-like-growth factor binding protein-2 (IGFBP-2) production
in human adipocytes. Growth Horm IGF Res. 25:115–120. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fischer-Posovszky P, Newell FS, Wabitsch M
and Tornqvist HE: Human SGBS cells-a unique tool for studies of
human fat cell biology. Obes Facts. 1:184–189. 2008. View Article : Google Scholar
|
|
17
|
Wabitsch M, Brenner RE, Melzner I, Braun
M, Möller P, Heinze E, Debatin KM and Hauner H: Characterization of
a human preadipocyte cell strain with high capacity for adipose
differentiation. Int J Obes Relat Metab Disord. 25:8–15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bernhard F, Landgraf K, Klöting N,
Berthold A, Büttner P, Friebe D, Kiess W, Kovacs P, Blüher M and
Körner A: Functional relevance of genes implicated by obesity
genome-wide association study signals for human adipocyte biology.
Diabetologia. 56:311–322. 2013. View Article : Google Scholar
|
|
19
|
Tu J, Li W, Zhang Y, Wu X, Song Y, Kang L,
Liu W, Wang K, Li S, Hua W and Yang C: Simvastatin inhibits
IL-1β-induced apoptosis and extracellular matrix degradation by
suppressing the NF-κB and MAPK pathways in nucleus pulposus cells.
Inflammation. 40:725–734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Spampanato C, De Maria S, Sarnataro M,
Giordano E, Zanfardino M, Baiano S, Cartenì M and Morelli F:
Simvastatin inhibits cancer cell growth by inducing apoptosis
correlated to activation of Bax and downregulation of BCL-2 gene
expression. Int J Oncol. 40:935–941. 2012. View Article : Google Scholar
|
|
21
|
Åberg M, Wickström M and Siegbahn A:
Simvastatin induces apoptosis in human breast cancer cells in a
NFκB-dependent manner and abolishes the anti-apoptotic signaling of
TF/FVIIa and TF/FVIIa/FXa. Thromb Res. 122:191–202. 2008.
View Article : Google Scholar
|
|
22
|
Pi WF, Guo XJ, Su LP and Xu WG:
Troglitazone upregulates PTEN expression and induces the apoptosis
of pulmonary artery smooth muscle cells under hypoxic conditions.
Int J Mol Med. 32:1101–1109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Farrow B and Evers BM: Activation of
PPARgamma increases PTEN expression in pancreatic cancer cells.
Biochem Biophys Res Commun. 301:50–53. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vella V, Nicolosi ML, Giuliano S, Bellomo
M, Belfiore A and Malaguarnera R: PPAR-γ agonists as antineoplastic
agents in cancers with dysregulated IGF axis. Front Endocrinol
(Lausanne). 8:312017.
|
|
25
|
Wang G, Cao R, Wang Y, Qian G, Dan HC,
Jiang W, Ju L, Wu M, Xiao Y and Wang X: Simvastatin induces cell
cycle arrest and inhibits proliferation of bladder cancer cells via
PPARγ signalling pathway. Sci Rep. 6:357832016. View Article : Google Scholar
|
|
26
|
Marsh DJ, Trahair TN, Martin JL, Chee WY,
Walker J, Kirk EP, Baxter RC and Marshall GM: Rapamycin treatment
for a child with germline PTEN mutation. Nat Clin Pract Oncol.
5:357–361. 2008.PubMed/NCBI
|
|
27
|
Iacobas I, Burrows PE, Adams DM, Sutton
VR, Hollier LH and Chintagumpala MM: Oral rapamycin in the
treatment of patients with hamartoma syndromes and PTEN mutation.
Pediatr Blood Cancer. 57:321–323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heindl M, Händel N, Ngeow J, Kionke J,
Wittekind C, Kamprad M, Rensing-Ehl A, Ehl S, Reifenberger J,
Loddenkemper C, et al: Autoimmunity, intestinal lymphoid
hyperplasia, and defects in mucosal B-cell homeostasis in patients
with PTEN hamartoma tumor syndrome. Gastroenterology.
142:1093–1096.e6. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang Z, Li W, Liu J, Li J, He F, Jiang Y,
Yang L, Li P, Wang B, Wang Y, et al: Simvastatin suppresses the DNA
replication licensing factor MCM7 and inhibits the growth of
tamoxifen-resistant breast cancer cells. Sci Rep. 7:417762017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang ST, Ho HJ, Lin JT, Shieh JJ and Wu
CY: Simvastatin-induced cell cycle arrest through inhibition of
STAT3/SKP2 axis and activation of AMPK to promote 27 and 21
accumulation in hepatocellular carcinoma cells. Cell Death Dis.
8:e26262017. View Article : Google Scholar
|
|
32
|
Cafforio P, Dammacco F, Gernone A and
Silvestris F: Statins activate the mitochondrial pathway of
apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis.
26:883–891. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Denoyelle C, Vasse M, Körner M, Mishal Z,
Ganné F, Vannier JP, Soria J and Soria C: Cerivastatin, an
inhibitor of HMG-CoA reductase, inhibits the signaling pathways
involved in the invasiveness and metastatic properties of highly
invasive breast cancer cell lines: An in vitro study.
Carcinogenesis. 22:1139–1148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen HH, Händel N, Ngeow J, Muller J, Hühn
M, Yang HT, Heindl M, Berbers RM, Hegazy AN, Kionke J, et al:
Immune dysregulation in patients with PTEN hamartoma tumor
syndrome: Analysis of FOXP3 regulatory T cells. J Allergy Clin
Immunol. 139:607–620.e15. 2017. View Article : Google Scholar
|
|
35
|
Lin CF, Young KC, Bai CH, Yu BC, Ma CT,
Chien YC, Chiang CL, Liao CS, Lai HW and Tsao CW: Rosiglitazone
regulates anti-inflammation and growth inhibition via PTEN. Biomed
Res Int. 2014:7879242014.PubMed/NCBI
|
|
36
|
Aiello A, Pandini G, Frasca F, Conte E,
Murabito A, Sacco A, Genua M, Vigneri R and Belfiore A: Peroxisomal
proliferator-activated receptor-gamma agonists induce partial
reversion of epithelial-mesenchymal transition in anaplastic
thyroid cancer cells. Endocrinology. 147:4463–4475. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han S and Roman J: Rosiglitazone
suppresses human lung carcinoma cell growth through
PPARgamma-dependent and PPARgamma-independent signal pathways. Mol
Cancer Ther. 5:430–437. 2006. View Article : Google Scholar
|
|
38
|
Zhuang L, Kim J, Adam RM, Solomon KR and
Freeman MR: Cholesterol targeting alters lipid raft composition and
cell survival in prostate cancer cells and xenografts. J Clin
Invest. 115:959–968. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Khan T, Hamilton MP, Mundy DI, Chua SC and
Scherer PE: Impact of simvastatin on adipose tissue: Pleiotropic
effects in vivo. Endocrinology. 150:5262–5272. 2009. View Article : Google Scholar : PubMed/NCBI
|