Introduction
Colorectal cancer (CRC) is the third most commonly
diagnosed cancer among males and females (1). Surgical resection is an effective
treatment for early-stage CRC, while chemotherapy is the main
treatment for patients with advanced CRC. In recent years,
effective observation strategies following curative treatment for
CRC have helped improve the overall survival (OS) of patients, and
a number of CRC patients are eligible for such curative treatment
(2–4). The fecal immunochemical test (FIT),
a simple and easy method, has been widely used in CRC screening
programs (5–7). However, the 5-year survival rate
significantly declines when cancer cells spread to adjacent organs
or lymph nodes (8–10). Due to the genetic heterogeneity of
CRC, studying the tumor initiation and progression at a molecular
level can help to uncover the pathogenesis of CRC. During the past
decades, researchers have identified various important gene
biomarkers, including tumor oncogenes (such as KRAS, BRAF and
PIK3CA) and suppressor genes (such as A00PC, TP53 and PTEN). These
genes are critical for the genesis and development of CRC,
demonstrating the potential clinical significance for the treatment
of CRC (11,12).
As accumulating evidence has suggested inflammation
is a tumor-promoting hallmark that contributes to tumor initiation
and progression (13). CRC has
been reported to be associated with chronic bowel inflammation
(14–16), indicating the crucial roles of
inflammatory genes in the tumorigenesis and development of CRC.
Additionally, a number of studies have reported the importance of
single inflammatory genes in CRC. For instance, Cho et al
(17) revealed that genetic
variation in PPARGC1A affected the role of diet-associated
inflammation in CRC. Furthermore, Li et al (18) identified that the inflammatory
molecule PSGL-1, involved in the nuclear factor-κB signaling
pathway, can promote CRC growth by activating macrophages. However,
the study of inflammatory genes in the context of functional
interaction networks remains unclear, which drives us to predict
novel network-based inflammation-associated signatures. In
addition, a variety of public gene expression profiling datasets
and protein-protein interactions (PPIs) make it possible to study
the network of inflammatory genes further in order to identify
novel biomarkers for CRC. Chuang et al (19) used a protein-network-based
approach to identify biomarkers as sub-networks. Similarly, by
developing a network-based method, Li et al (20) identified cancer prognostic
biomarkers based on microarray and network datasets.
The present study aimed to investigate how
inflammatory genes functioned in the context of
inflammation-associated networks and their prognostic value in CRC,
and two inflammation-associated networks were constructed.
Topological properties were used to measure the network structure,
as well as the crucial positions of the inflammatory and
inflammation-associated genes in the two networks in CRC.
Furthermore, a 14-gene module was identified in the IRDN, and
notably, all 14 genes in this module were inflammatory genes. The
prognostic significance of this module comprising 14 inflammatory
genes was validated by three independent datasets. The current
study findings highlighted the novel role of the
inflammation-associated network in CRC, providing an insight into
the inflammation-mediated mechanisms involved in this disease.
Materials and methods
Data resources
CRC patients with whole-genome gene expression
profiles (Affymetrix Human Exon 1.0 ST Array) were downloaded from
the publicly available Gene Expression Omnibus (GEO) database
(https://www.ncbi.nlm.nih.gov/geo/). A
total of 90 specimens were included in the study of network
construction, including 77 tumor and 13 normal samples from the
study by Sveen et al (21)
with the accession number GSE24550 (Table I). In addition, another three GEO
datasets, namely GSE24549, GSE24551 and GSE103479 (Table I) (22,23), were downloaded for validation
analyses. The GSE24550 dataset, which is a subset of GSE24551, was
selected as a training set. Furthermore, CRC patients' stage
information was obtained for these four datasets and other clinical
features (age, gender, BRAF/KRAS/P53 mutations) for the GSE103479
dataset.
 | Table IClinical features of all CRC patients
included in this study. |
Table I
Clinical features of all CRC patients
included in this study.
| Characteristic | GSE24550
(n=77) | GSE24549
(n=83) | GSE24551
(n=160) | GSE102479
(n=155) |
|---|
| Stage, no. (%) | | | | |
| II | 44 (57.1) | 46 (55.4) | 90 (56.2) | 83 (53.5) |
| III | 33 (42.9) | 37 (44.6) | 70 (43.8) | 72 (46.5) |
| Age, no. (%) | | | | |
| ≤65 | n/a | n/a | n/a | 51 (32.9) |
| >65 | n/a | n/a | n/a | 102 (56.8) |
| Gender, no.
(%) | | | | |
| Male | n/a | n/a | n/a | 87 (56.1) |
| Female | n/a | n/a | n/a | 68 (43.9) |
| BRAF mutation | | | | |
| WT | n/a | n/a | n/a | 122 (78.7) |
| MT | n/a | n/a | n/a | 15 (9.7) |
| KRAS mutation | | | | |
| WT | n/a | n/a | n/a | 83 (53.5) |
| MT | n/a | n/a | n/a | 54 (34.8) |
| P53 mutation | | | | |
| WT | n/a | n/a | n/a | 59 (38.1) |
| MT | n/a | n/a | n/a | 78 (50.3) |
Gene Ontology (GO) (24) terms associated with the
inflammatory response were obtained from the study by Plaisier
et al (25). The genes
annotated to these inflammation-associated GO terms were obtained
from the AmiGO2 tool (26) of the
GO Consortium. Finally, 909 inflammatory genes were collected for
subsequent analyses.
The protein-protein interaction (PPI) data were
downloaded from the Human Protein Reference Database (HPRD) release
9 (http://www.hprd.org/) (27). It contained >42,000 manually
curated interactions between 9,826 human genes.
Differentially expressed gene (DEG)
analysis
The raw array data (.CEL files) of samples in the
four GEO datasets were uniformly pre-processed using the Robust
Multichip Average algorithm for background correction, quantified
normalization and log2-transformation (28). To account for the heterogeneity of
multiple microarray datasets in systematic measurements, each
dataset was standardized independently by the Z-score
transformation to balance the expression intensities of each probe
(29). DEGs were identified using
a two-tailed t-test for GSE24550. Genes with a false discovery rate
(FDR) cutoff value of 0.001 following adjustment of the P-value
were considered as DEGs. The unsupervised hierarchical clustering
of CRC samples and DEGs was performed with R software (https://www.r-project.org/) using the Euclidean
distance and complete linkage method.
Network construction and analysis
Cytoscape version 3.2.0 (30) was used for the construction of the
networks in the current study. The first network was established
according to the following procedure: The human PPI network was
initially downloaded from the HPRD, all the inflammatory gene
symbols were acquired from AmiGO2, and all the inflammatory genes
were mapped to the human PPI network. Subsequently, a sub-network
including inflammatory genes and their direct interacting genes in
the network (referred to as inflammatory neighbor genes) was
selected. Finally, the sub-network was termed the
inflammation-related neighbor network (IRNN). The second
sub-network was constructed as follows: DEGs of CRC were mapped to
IRNN, and then a sub-network including DEGs and their direct
interacting genes in the IRNN (referred to as DEG neighbor genes)
was selected. The second sub-network was termed the
inflammation-related driver network (IRDN) for subsequent
analyses.
In order to explore the topological properties of
the inflammatory genes in the IRNN and IRDN, the following features
were analyzed: Degree, betweenness centrality (BC) and closeness
centrality (CC), which were used to decipher the structure of the
sub-networks and to identify specific vital molecules. Degree was
used to determine the number of neighbors for each node. BC
represented the key role of a node in communication and information
diffusion (31). Node CC measured
the local cohesiveness, such as how close a node is to other nodes.
The IRNN and IRDN visualization and topological properties were
analyzed using Cytoscape version 3.2.0 and the R software.
Identifying prognostic modules in
IRDN
The cFinder algorithm (32) is a classic method used to identify
modules from a network, in which the identification and
visualization of overlapping dense groups of nodes was conducted
using the Clique Percolation Method (33). Modules were identified through
this method in the present study by locating the k-clique
percolation clusters of the network. A k-clique percolation cluster
includes the following two parts: i) all nodes that can be reached
via chains of adjacent k-cliques from each other; and ii) the links
in these cliques. Larger values of k correspond to a higher
stringency during the identification of dense groups and provide
smaller groups with a higher density of links inside them.
Functional enrichment analysis
Genes were functionally annotated to identify
enriched pathways based on DAVID Bioinformatics Resources (version
6.7; http://david.abcc.ncifcrf.gov/)
(34). The DAVID enrichment
analyses were limited to Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathways and GO-FAT biological process (BP) terms, with the
whole human genome as background. Functional categories were
visualized and clustered using the Enrichment Map plugin (35) in Cytoscape version 3.2.0.
Survival analysis
In order to validate whether the module identified
in the aforementioned step was associated with CRC patient
survival, the expression of mRNAs in the module was extracted.
Next, Cox proportional hazard analysis was used to obtain the
regression coefficient of each gene associated with patient
survival. The classifier was built as the linear combination of the
gene expression values of the selected genes using the standardized
Cox regression coefficient as the weight. A risk score formula for
each patient was established by including the expression values of
each selected gene weighted by their estimated regression
coefficients in the univariate Cox regression analysis. As a
result, patients were divided into the high-risk and low-risk
groups (36) using the median
value of the risk score as the threshold. Kaplan-Meier survival
plots and log-rank tests were used to assess the differences in
disease-free survivalb (DFS) duration between the high-risk and
low-risk patients. In addition, the sensitivity and specificity of
the module for survival prediction was evaluated using receiver
operating characteristic (ROC) curve analysis, and the area under
the ROC curve (AUC) was calculated.
Statistical analysis
In the construction of IRNN, two-tailed t-test and
FDR adjustion were used to identify CRC-related DEGs. Furthermore,
univariate and multivariate Cox regression analyses and log-rank
test were applied in survival analysis.
Results
Inflammatory genes serve a crucial role
in CRC
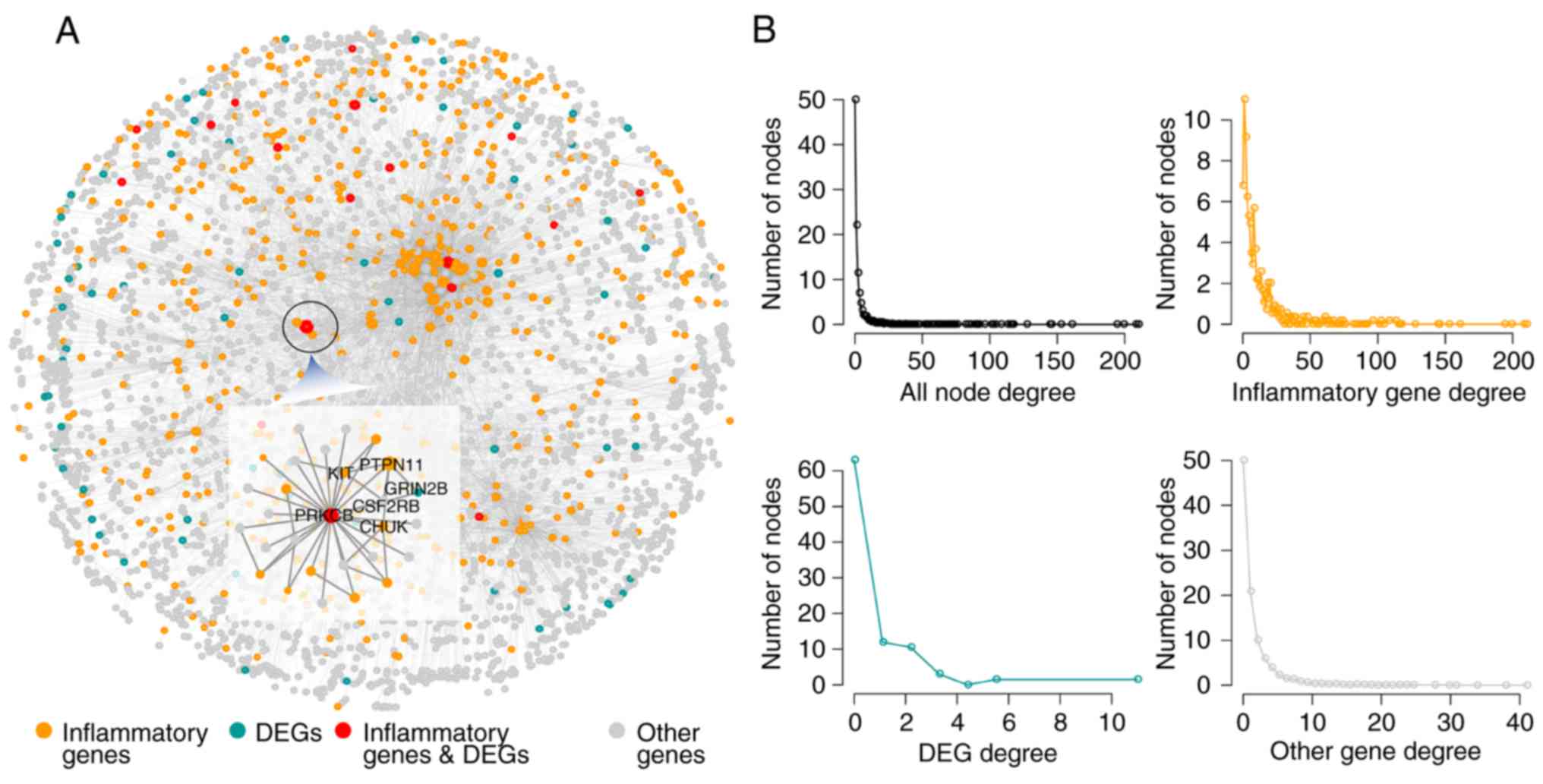
Based on the PPI network, an IRNN was constructed,
which included 9,293 interactions of 3,526 nodes (575 inflammatory
genes and their 2,928 neighbors; Fig.
1A). In the IRNN, there were 17 inflammatory genes (such as
CCL19, CD36, COL1A1, CXCL13, ITK and KIT) that were DEGs, as well
as 558 inflammatory genes (including PRKACA, TRAF2, PIK3R1, SHC1,
PTPN11, LCK and GRB2) that were associated with the DEGs.
In order to explore the construction and features of
the IRNN, network topology analysis was performed. As observed, the
global degree distribution of nodes in the IRNN closely followed
the power law distribution (Fig.
1B). Furthermore, the inflammatory genes, DEGs and other nodes
exhibited a small-world network organization (Fig. 1B), which suggested that the IRNN
constructed was biologically significant.
Inflammatory genes serve critical hub
roles by interacting with DEGs in CRC
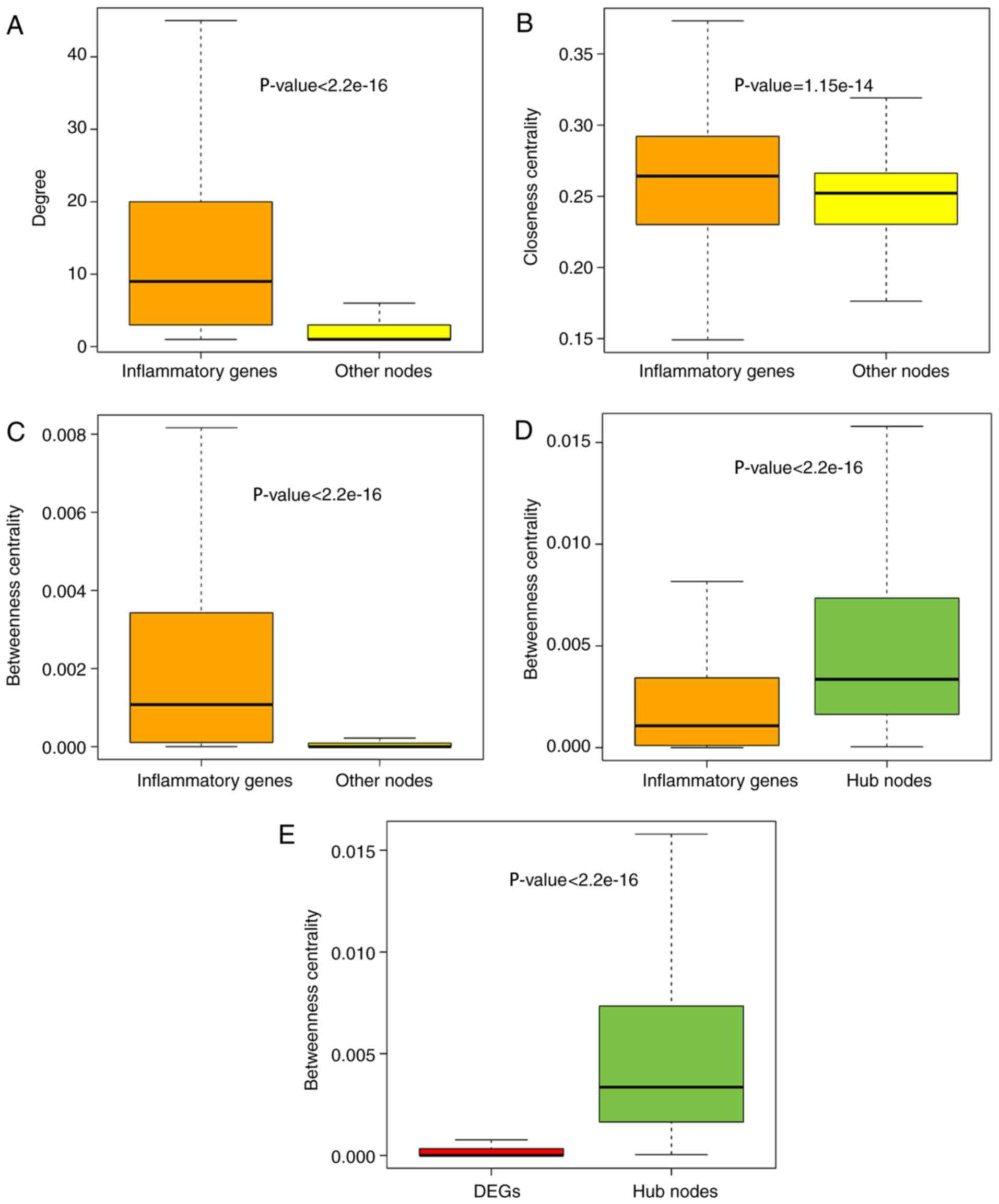
To further explore the role of inflammatory genes in
the IRNN, analysis of the topological features, including the
degree, CCs and BCs, was performed. The results demonstrated that
inflammatory genes in the IRNN had a higher degree, CCs and BCs
compared with their neighbors that were non-inflammatory genes in
the IRNN (P<2.2×10−16 for degree;
P=1.15×10−14 for CC; P<2.2×10−16 for BC;
Wilcoxon rank sum test; Fig.
2A–C, respectively), indicating that inflammatory genes were
central within the IRNN.
As observed earlier, inflammatory genes tended to be
located in the hub node positions in the IRNN; therefore, the
present study further deciphered these inflammatory hub genes in
detail. Previously, hubs were typically defined as the top 10–20%
of nodes in the networks based on their degree (37,38). Thus, the top 10% of nodes were
selected as the hub components based on the highest degrees,
identifying a total of 347 hub nodes. Furthermore, all the hub
nodes were found to belong to inflammatory genes with a
significantly higher BC compared with that of all the inflammatory
genes in the IRNN (P<2.2×10−16; Fig. 2D). These findings indicated that
certain inflammatory genes had critical hub roles in the IRNN.
However, the BCs of hub genes were higher in comparison with those
of DEGs (P<2.2×10−16; Fig. 2E), indicating that these DEGs
served critical roles in the communication with their neighboring
nodes in the IRNN. The KIT gene, a DEG in the IRNN sub-network,
encodes c-kit tyrosine kinase whose inhibitor STI571 reportedly
exhibits a substantial therapeutic activity in patients with CRC
(39). KIT, which interacted with
two hub inflammatory genes (FYN and SRC) in the IRNN, is inhibited
by dasatinib, which is studied in solid tumors such as CRC by
combining with capecitabine and oxaliplatin (40). These observations demonstrated
that hub inflammatory gene-associated DEGs were more likely to be
essential for CRC development and progression.
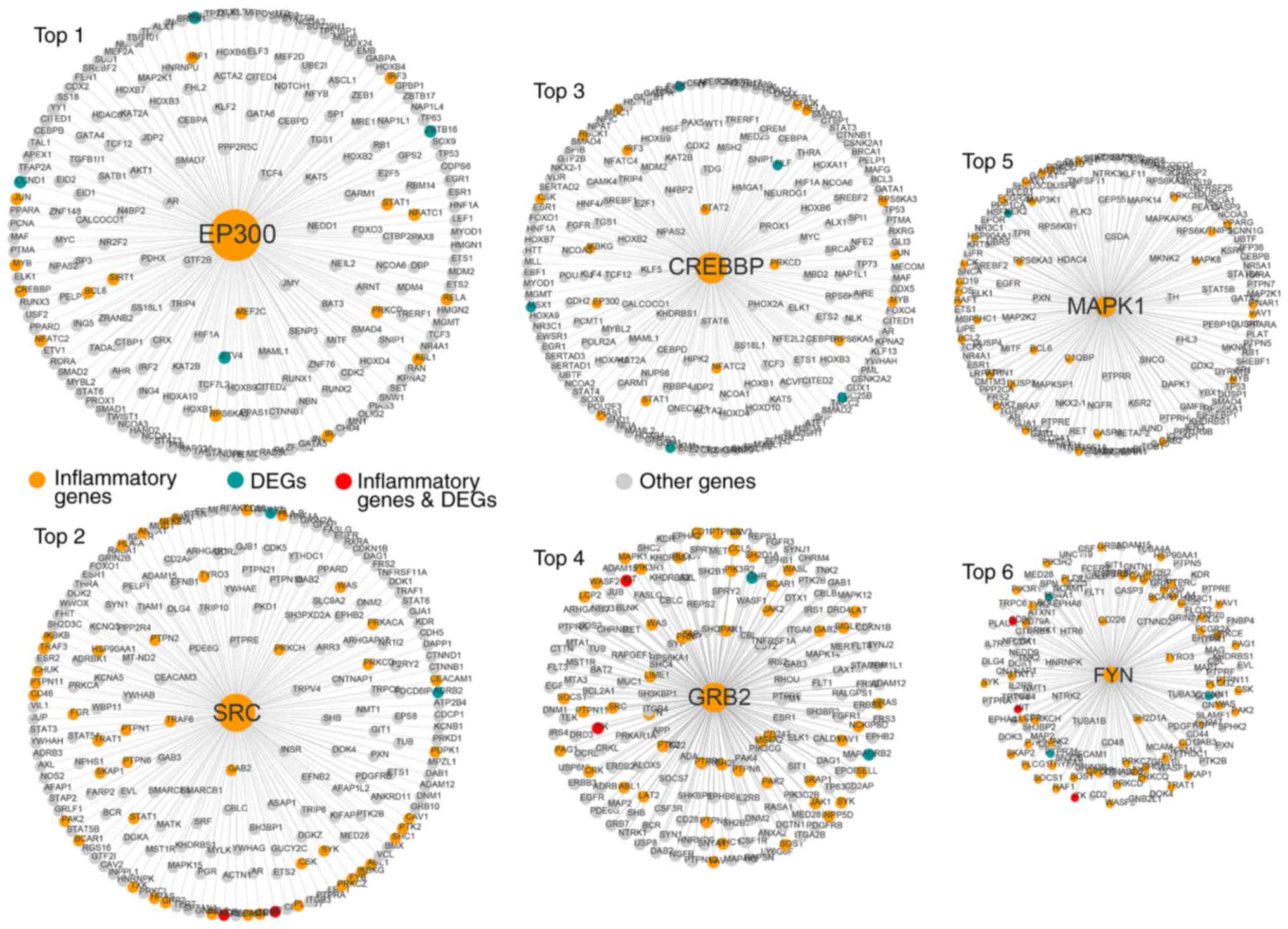
The top six inflammatory genes (EP300, SRC, CREBBP,
GRB2, MAPK1 and FYN) in terms of the node degree had a direct
connection in the network, indicating that inflammatory genes
function as hubs in the sub-networks (Fig. 3).
Inflammatory genes directly interact with
CRC-associated DEGs
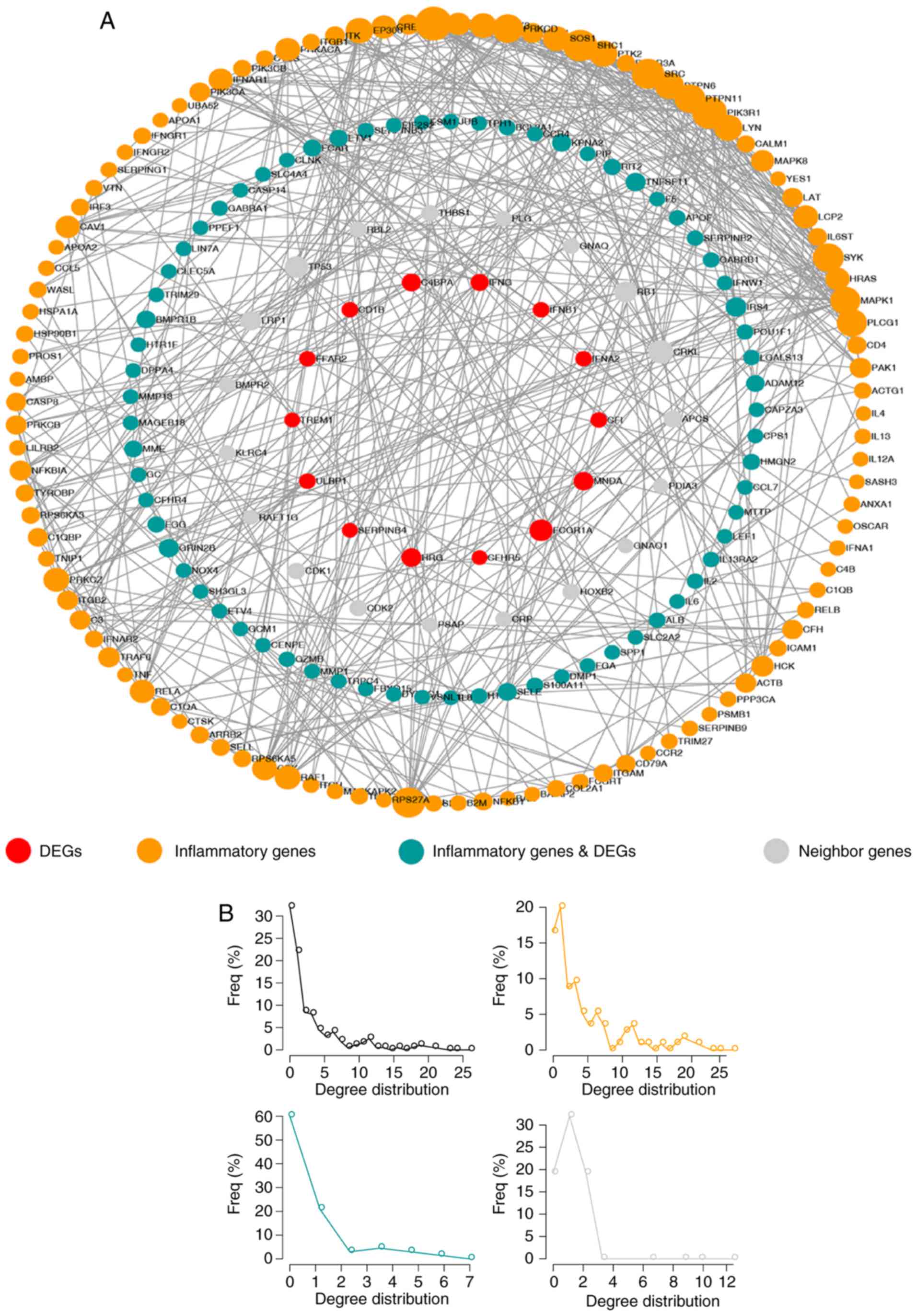
To further explore the association between
inflammatory genes and DEGs, a network termed IRDN was constructed,
which included DEGs and their neighbors extracted from the IRNN
(Fig. 4A). The IRDN included 209
genes, of which 83 were DEGs. In total, 121 of the genes in IRDN
were inflammatory genes, and 17 of these were also DEGs. Similar to
the nodes in IRNN, the nodes in the IRDN also exhibited a
small-world network organization (Fig. 4B), which suggested that the IRDN
was also biologically significant. These results suggested that the
IRDN obtained from IRNN may serve a crucial role in the biogenesis
of CRC.
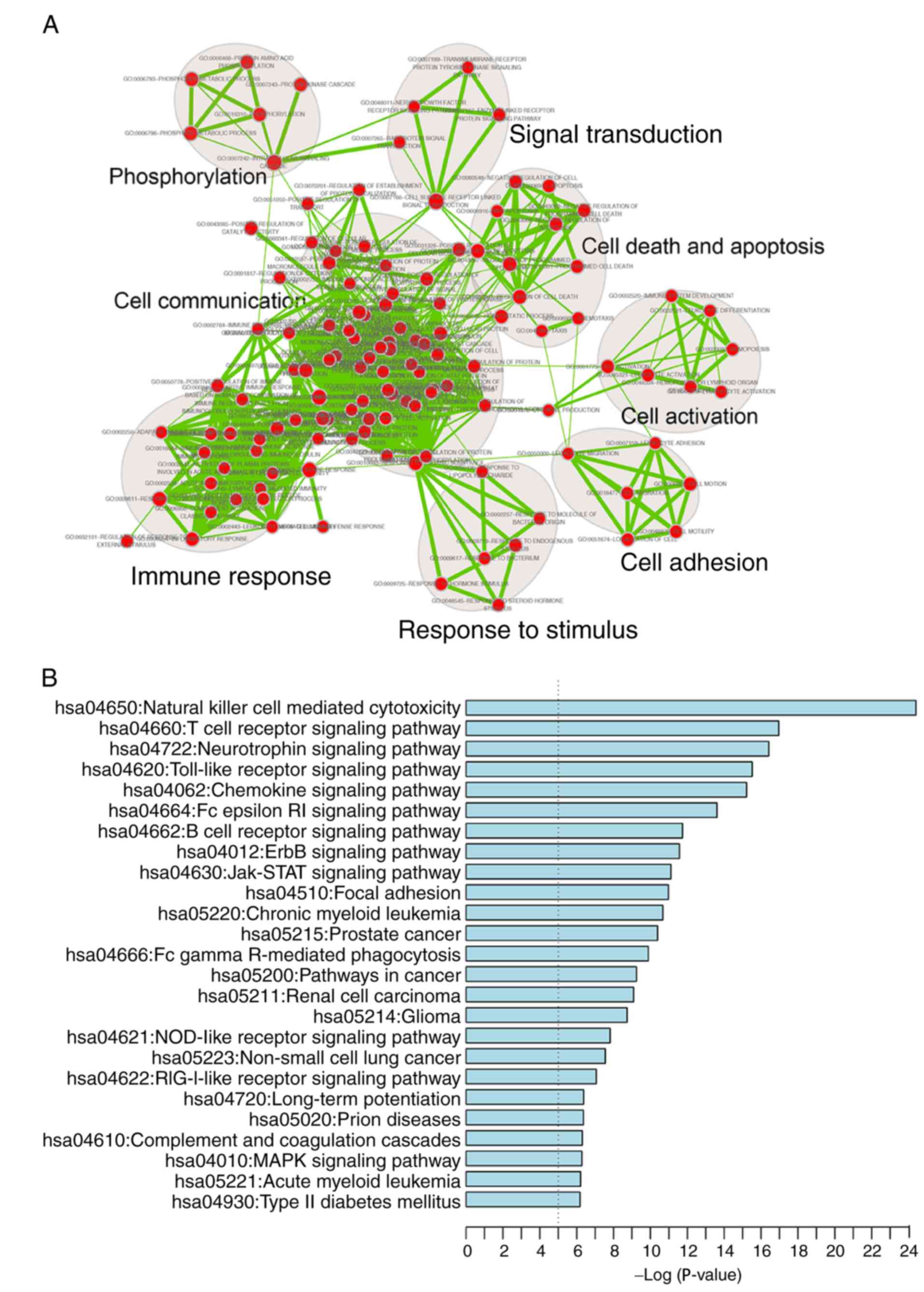
A functional enrichment analysis of all genes in
IRDN was also performed based on GO and KEGG pathway analyses. IRDN
mRNAs were enriched in 200 GO_BP_FAT terms (FDR<0.001) mainly in
eight functional clusters, including cell adhesion, cell
communication, cell activation, immune response, cell death and
apoptosis, signal transduction, response to stimulus and
phosphorylation (Fig. 5A). In
addition, 25 KEGG pathways (FDR<0.01) were involved, including
cancer, focal adhesion and several signaling pathways (Fig. 5B). All enriched signaling
pathways, including ErbB, Jak-STAT, MAPK and NOD-like receptor
signaling, are known to be contributors to CRC pathogenesis
(41–44).
Identifying inflammatory network-based
biomarkers in CRC
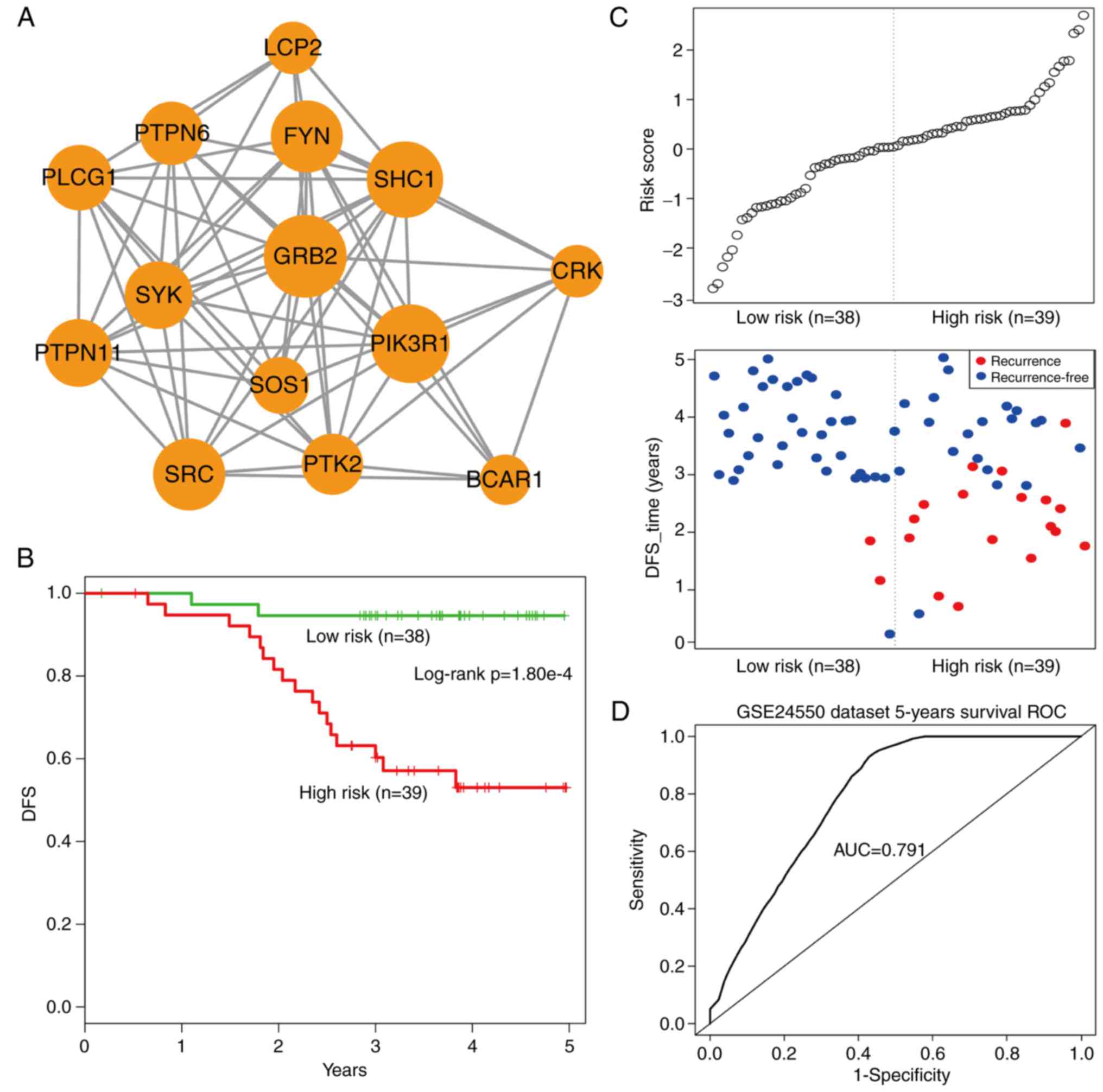
Using the cFindeR software, two
inflammation-associated modules in IRDN with k-clique ≥5 were
identified. Next, the study investigated whether these two
inflammatory gene-associated modules have a prognostic potential in
CRC. Using the training dataset GSE24550 (Table II), a risk-score formula was
created according to the expression of these two module genes to
generate DFS prediction. Using the median risk score of the
training series as the cutoff point, the risk scores of the module
genes were calculated for each patient and then patients were
ranked according to their risk score. The patients were grouped
into the high-risk or low-risk categories using the median risk
score of the training series as the cutoff point. As a result, the
module comprising 14 inflammatory genes with k-clique=6 (Fig. 6A) was able to divide patients into
the high- and low-risk groups with a significantly reduced DFS
observed in the high-risk group (log-rank test,
P=1.80×10−4; Fig. 6B).
Additionally, the distribution of gene risk score and the survival
status of the training dataset are shown in Fig. 6C. Furthermore, a time-dependent
ROC curve analysis was performed to evaluate the sensitivity and
specificity for module survival prediction. The module achieved an
AUC value of 0.791 (Fig. 6D),
suggesting a substantially effective performance.
| Table IIGene Expression Omnibus microarray
datasets used in the present study. |
Table II
Gene Expression Omnibus microarray
datasets used in the present study.
| Datasets | Platform | Number of
patients | Overall type | No. of tumor
samples | No. of normal
samples |
|---|
| GSE24550 | HuEx-1_0-st | 90 | DFS | 77 | 13 |
| GSE24549 | HuEx-1_0-st | 83 | DFS | 83 | 0 |
| GSE24551 | HuEx-1_0-st | 173 | DFS | 160 | 13 |
| GSE103497 | ADXECv1a520743 | 156 | OS | 156 | 0 |
Validating the prognostic potential of
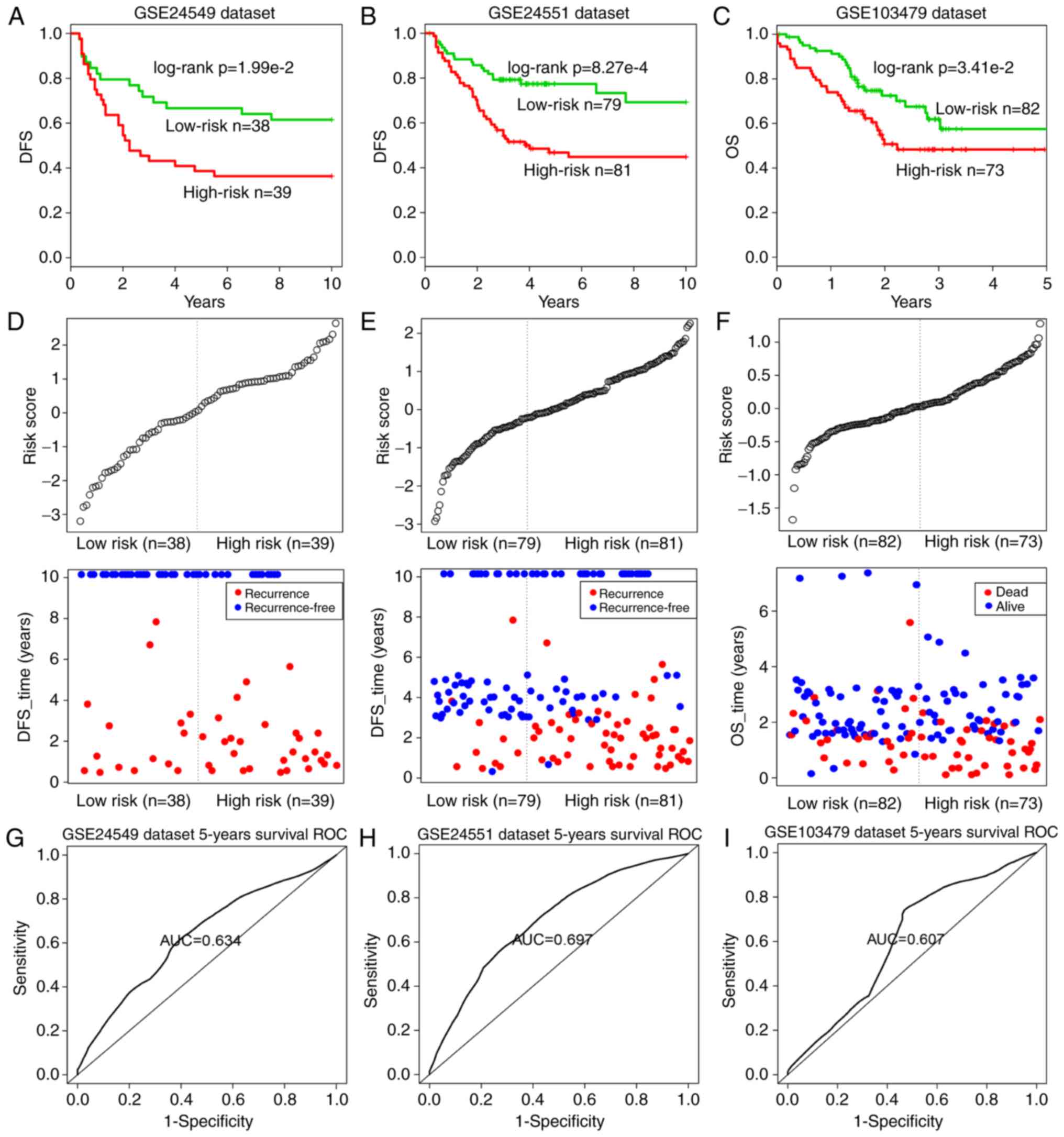
14-inflammatory-gene-module in independent datasets
To validate the prognostic performance of the
14-inflammatory-gene-module, three independent GEO datasets were
used in the subsequent analyses (Table II). Using the same risk score
formula as earlier, patients in each dataset were classified into
the high-risk and low-risk groups using the median score of the
training series as the cutoff point. Consistent with the previous
findings, patients in the high-risk group had significantly shorter
median DFS or OS when compared with those in the low-risk group
(GSE24549 patients: Log-rank test P=1.99×10−2, Fig. 7A; GSE24551 patients: Log-rank test
P=8.27×10−4, Fig. 7B;
GSE103479 patients: Log-rank test P=3.41×10−2, Fig. 7C). Similarly, the distribution of
gene risk scores and the survival statuses of the three datasets
are displayed in Fig. 7D–F.
Patients with high-risk scores tended to present poorer clinical
outcomes compared with patients with low-risk scores. The
time-dependent ROC curve analysis was performed to evaluate the
sensitivity and specificity for module survival prediction in these
three GEO datasets. The module achieved AUC values of 0.634, 0.697
and 0.607 in the GSE24549, GSE24551 and GSE103479 datasets,
respectively, indicating that high prediction performance (Fig. 7G–I).
Subsequent to further adjusting for other clinical
markers univariate analysis indicated that the
14-inflammatory-gene-module was significantly associated with the
DFS of CRC patients from the GSE24549 [hazard ratio (HR)=1.28; 95%
confidence interval (CI)=0.99–1.66; P=0.052] and GSE24551 datasets
(HR=1.88; 95% CI=1.42–2.47; P<0.001), as well as the OS of
patients from the GSE103479 dataset (HR=2.13; 95% CI=1.24–3.64;
P=0.006), as an independent risk factor (Table III). Subsequently, when
multivariate analysis was performed to investigate the independence
of the module to other clinical factors, the module was not
independent of the stage in GSE24549 (HR=1.93; 95% CI=1.05–3.56;
P=0.034), and GSE103479 (HR=2.37; 95% CI=1.34–4.21; P=0.003)
datasets (Table III).
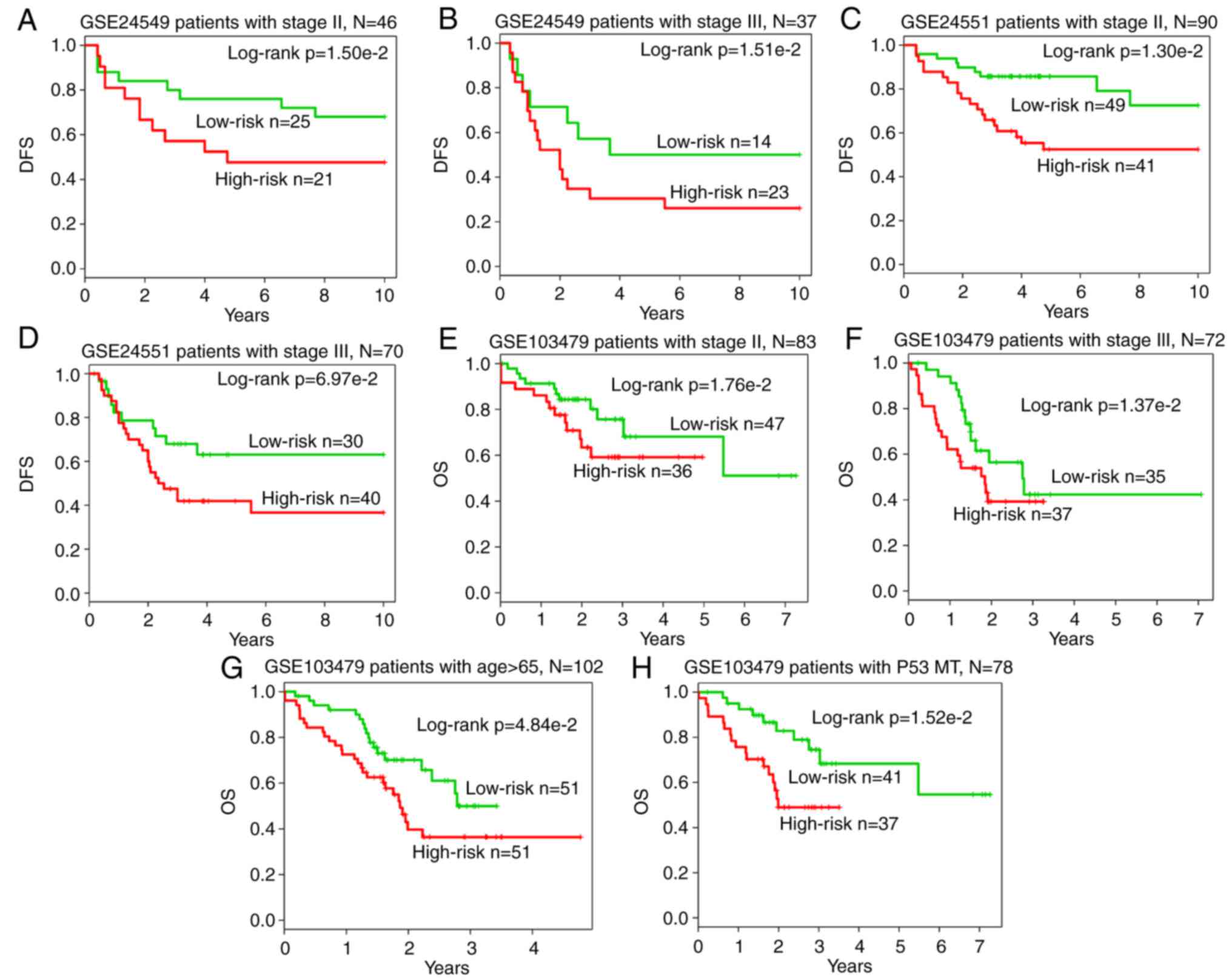
Furthermore, data stratification analysis on GSE24549 patients
indicated that the module was independent of stage
(P=1.50×10−2 for stage II group; P=1.51×10−2
for stage III group; Fig. 8A and
B), as well as the GSE24551 patients (P=1.30×10−2
for stage II group; P= 6.97×10−2 for stage III group;
Fig. 8C and D) and GSE103479
patients (P=1.76×10−2 for stage II group;
P=1.37×10−2 for stage III group; Fig. 8E and F). In addition, in terms of
OS survival in GSE103479 patients, the module was independent of
the patient age and the mutation of P53 (P=4.84×10−2 for
patients aged >65 years; P=1.52×10−2 for the P53
mutated group; Fig. 8G and H).
Based on these data, it is concluded that the
14-inflammatory-gene-module is a strong prognostic indicator for
CRC.
| Table IIIUnivariate and multivariate Cox
regression analysis of the module gene signature and disease-free
survival of colorectal cancer patients in the training and test
Gene Expression Omnibus datasets. |
Table III
Univariate and multivariate Cox
regression analysis of the module gene signature and disease-free
survival of colorectal cancer patients in the training and test
Gene Expression Omnibus datasets.
| Variables | Univariate model
| Multivariate model
|
|---|
| HR | 95% CI of HR | P-value | HR | 95% CI of HR | P-value |
|---|
| GSE24550 (DFS) | | | | | | |
| Module risk
score | 2.17 | 1.42–3.31 | <0.001 | 2.13 | 1.36–3.33 | 0.001 |
| Stage | 1.77 | 0.72–4.35 | 0.216 | 1.12 | 0.43–2.90 | 0.817 |
| GSE24549 (DFS) | | | | | | |
| Module risk
score | 1.28 | 0.99–1.66 | 0.052 | 1.24 | 0.97–1.57 | 0.080 |
| Stage | 2.02 | 1.11–3.71 | 0.022 | 1.93 | 1.05–3.56 | 0.034 |
| GSE24551 (DFS) | | | | | | |
| Module risk
score | 1.88 | 1.42–2.47 | <0.001 | 1.76 | 1.34–2.32 | <0.001 |
| Stage | 1.97 | 1.19–3.25 | 0.008 | 1.61 | 0.96–2.70 | 0.069 |
| GSE103479 (OS) | | | | | | |
| Module risk
score | 2.13 | 1.24–3.64 | 0.006 | 1.58 | 0.92–2.70 | 0.094 |
| Stage | 2.18 | 1.30–3.67 | 0.003 | 2.37 | 1.34–4.21 | 0.003 |
| Age | 1.06 | 1.03–1.09 | <0.001 | 1.05 | 1.02–1.09 | 0.001 |
| Gender | 0.97 | 0.59–1.62 | 0.919 | 1.26 | 0.71–2.26 | 0.428 |
| BRAF mutation | 1.91 | 0.90–4.07 | 0.091 | 1.17 | 0.50–2.78 | 0.714 |
| KRAS mutation | 0.87 | 0.51–1.51 | 0.629 | 0.97 | 0.53–1.77 | 0.913 |
| P53 mutation | 0.57 | 0.33–0.98 | 0.041 | 0.68 | 0.39–1.19 | 0.173 |
Discussion
Inflammatory genes serve a key role in cell-cell
communication, and dysfunctional inflammatory response can cause
various diseases, including cancer. Thus, deciphering the
inflammation-mediated mechanisms in CRC can provide opportunities
for the early detection and treatment of CRC. A variety of
inflammation-associated genes in CRC have been previously studied.
For instance, Hickish et al (45) revealed that MABp1 was
associated with an antitumor activity and relief of debilitating
symptoms in patients with advanced CRC. Foersch et al
(46) reported that inhibition of
VEGFR2 signaling led to senescence of human CRC, which may improve
the CRC patient survival in clinical practice. Zhu et al
(47) also suggested that
STING mediated protection against colorectal tumorigenesis
by participating in intestinal inflammation.
In the present study, a systematical analysis on the
inflammatory genes in CRC was performed, developing an integrated
network-based strategy to construct and identify the
inflammation-associated networks and modules. The current study
built two networks, namely IRNN and IRDN, with inflammatory genes
in the IRNN closely interacting with cancer genes, and genes in the
IRDN enriched in numerous cancer-associated functions and pathways.
These inflammatory genes may modulate the communication between
cancer and normal cells. In addition, the current study highlighted
the importance of inflammatory genes in the prognosis of CRC
through the identification of a 14-inflammatory-gene-module, which
was further validated by three independent datasets, providing a
novel insight for CRC diagnosis and therapy.
Notably, the top six hub nodes in IRNN were all
inflammatory genes, including EP300, which encodes the E1A-binding
protein p300 and is important in the processes of cell
proliferation and differentiation. The frame-shift mutations of
EP300 have been reported to contribute to cancer pathogenesis by
deregulating E1A-binding protein p300-mediated functions (48). Furthermore, the tyrosine kinases
of the SRC family participate in oncogenic signaling in advanced
CRC, which is crucial in the progression of CRC (49). The CBP protein (encoded by
CREBBP), together with p300, may influence colonic cell physiology
by affecting Wnt signaling, as well as affect colonic tumorigenesis
and stem cell pluripotency (50).
In addition, Ding et al (51) observed that Grb2-associated binder
2 (Gab2) promoted intestinal tumor metastasis by inducing
epithelial-to-mesenchymal transition through the MEK/ERK/MMP
pathway, which indicated that Gab2 may function as a novel
prognostic factor for CRC patients. Additionally, with the
immunohistochemistry study, researchers have found that MAPK is
highly expressed and extremely active in colorectal cells and can
be a therapeutically relevant target in inflammatory bowel disease
(52,53).
In conclusion, although the inflammation-associated
genes and modules should be confirmed by further experimental
validations, the present study indeed provided a new direction for
the exploration of the genesis and development of CRC using two
inflammation-mediated gene networks. Furthermore, a
14-inflammatory-gene-module with prognostic potential was
identified.
Acknowledgments
Not applicable.
References
|
1
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kishiki T, Lapin B, Matsuoka H, Watanabe
T, Takayasu K, Kojima K, Sugihara K and Masaki T: Optimal
surveillance protocols after curative resection in patients with
stage IV colorectal cancer: A multicenter retrospective study. Dis
Colon Rectum. 61:51–57. 2018.
|
|
3
|
van der Stok EP, Spaander MCW, Grünhagen
DJ, Verhoef C and Kuipers EJ: Surveillance after curative treatment
for colorectal cancer. Nat Rev Clin Oncol. 14:297–315. 2017.
View Article : Google Scholar
|
|
4
|
Kahi CJ, Boland CR, Dominitz JA,
Giardiello FM, Johnson DA, Kaltenbach T, Lieberman D, Levin TR,
Robertson DJ and Rex DK; United States Multi-Society Task Force on
Colorectal Cancer: Colonoscopy surveillance after colorectal cancer
resection: Recommendations of the US Multi-society task force on
colorectal cancer. Gastroenterology. 150:758–768.e11. 2016.
View Article : Google Scholar
|
|
5
|
Cooper JA, Parsons N, Stinton C, Mathews
C, Smith S, Halloran SP, Moss S and Taylor-Phillips S:
Risk-adjusted colorectal cancer screening using the FIT and routine
screening data: Development of a risk prediction model. Br J
Cancer. 118:285–293. 2018. View Article : Google Scholar :
|
|
6
|
Westwood M, Lang S, Armstrong N, van
Turenhout S, Cubiella J, Stirk L, Ramos IC, Luyendijk M, Zaim R,
Kleijnen J and Fraser CG: Faecal immunochemical tests (FIT) can
help to rule out colorectal cancer in patients presenting in
primary care with lower abdominal symptoms: A systematic review
conducted to inform new NICE DG30 diagnostic guidance. BMC Med.
15:1892017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Passamonti B, Malaspina M, Fraser CG,
Tintori B, Carlani A, D'Angelo V, Galeazzi P, Di Dato E, Mariotti
L, Bulletti S, et al: A comparative effectiveness trial of two
faecal immunochemical tests for haemoglobin (FIT). Assessment of
test performance and adherence in a single round of a
population-based screening programme for colorectal cancer. Gut.
67:485–496. 2018. View Article : Google Scholar
|
|
8
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
11
|
van Geel RM, Beijnen JH, Bernards R and
Schellens JH: Treatment individualization in colorectal cancer.
Curr Colorectal Cancer Rep. 11:335–344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Markowitz SD and Bertagnolli MM: Molecular
origins of cancer: Molecular basis of colorectal cancer. N Engl J
Med. 361:2449–2460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sakurai T, Kashida H, Watanabe T, Hagiwara
S, Mizushima T, Iijima H, Nishida N, Higashitsuji H, Fujita J and
Kudo M: Stress response protein cirp links inflammation and
tumorigenesis in colitis-associated cancer. Cancer Res.
74:6119–6128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Waldner MJ and Neurath MF: Master
regulator of intestinal disease: IL-6 in chronic inflammation and
cancer development. Semin Immunol. 26:75–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Z, Cao AT and Cong Y: Microbiota
regulation of inflammatory bowel disease and colorectal cancer.
Semin Cancer Biol. 23:543–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho YA, Lee J, Oh JH, Chang HJ, Sohn DK,
Shin A and Kim J: Genetic variation in PARGC1A may affect the role
of diet-associated inflammation in colorectal carcinogenesis.
Oncotarget. 8:8550–8558. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li J, Zhou Z, Zhang X, Zheng L, He D, Ye
Y, Zhang QQ, Qi CL, He XD, Yu C, et al: Inflammatory molecule,
PSGL-1, deficiency activates macrophages to promote colorectal
cancer growth through NFκB signaling. Mol Cancer Res. 15:467–477.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chuang HY, Lee E, Liu YT, Lee D and Ideker
T: Network-based classification of breast cancer metastasis. Mol
Syst Biol. 3:1402007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li J, Lenferink AE, Deng Y, Collins C, Cui
Q, Purisima EO, O'Connor-McCourt MD and Wang E: Identification of
high-quality cancer prognostic markers and metastasis network
modules. Nat Commun. 1:342010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sveen A, Agesen TH, Nesbakken A, Rognum
TO, Lothe RA and Skotheim RI: Transcriptome instability in
colorectal cancer identified by exon microarray analyses:
Associations with splicing factor expression levels and patient
survival. Genome Med. 3:322011. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sveen A, Ågesen TH, Nesbakken A, Meling
GI, Rognum TO, Liestøl K, Skotheim RI and Lothe RA: ColoGuidePro: A
prognostic 7-gene expression signature for stage III colorectal
cancer patients. Clin Cancer Res. 18:6001–6010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Agesen TH, Sveen A, Merok MA, Lind GE,
Nesbakken A, Skotheim RI and Lothe RA: ColoGuideEx: A robust gene
classifier specific for stage II colorectal cancer prognosis. Gut.
61:1560–1567. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Plaisier CL, Pan M and Baliga NS: A
miRNA-regulatory network explains how dysregulated miRNAs perturb
oncogenic processes across diverse cancers. Genome Res.
22:2302–2314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carbon S, Ireland A, Mungall CJ, Shu S,
Marshall B and Lewis S; AmiGO Hub; Web Presence Working Group:
AmiGO: Online access to ontology and annotation data.
Bioinformatics. 25:288–289. 2009. View Article : Google Scholar :
|
|
27
|
Peri S, Navarro JD, Kristiansen TZ,
Amanchy R, Surendranath V, Muthusamy B, Gandhi TK, Chandrika KN,
Deshpande N, Suresh S, et al: Human protein reference database as a
discovery resource for proteomics. Nucleic Acids Res. 32:D497–D501.
2004. View Article : Google Scholar :
|
|
28
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheadle C, Vawter MP, Freed WJ and Becker
KG: Analysis of microarray data using Z score transformation. J Mol
Diagn. 5:73–81. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang C, Jiang W, Li W, Lian B, Chen X, Hua
L, Lin H, Li D, Li X and Liu Z: Topological properties of the drug
targets regulated by microRNA in human protein-protein interaction
network. J Drug Target. 19:354–364. 2011. View Article : Google Scholar
|
|
32
|
Adamcsek B, Palla G, Farkas IJ, Derényi I
and Vicsek T: CFinder: Locating cliques and overlapping modules in
biological networks. Bioinformatics. 22:1021–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Palla G, Derényi I, Farkas I and Vicsek T:
Uncovering the overlapping community structure of complex networks
in nature and society. Nature. 435:814–818. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar
|
|
35
|
Merico D, Isserlin R, Stueker O, Emili A
and Bader GD: Enrichment map: A network-based method for gene-set
enrichment visualization and interpretation. PLoS One.
5:e139842010. View Article : Google Scholar :
|
|
36
|
Alizadeh AA, Gentles AJ, Alencar AJ, Liu
CL, Kohrt HE, Houot R, Goldstein MJ, Zhao S, Natkunam Y, Advani RH,
et al: Prediction of survival in diffuse large B-cell lymphoma
based on the expression of 2 genes reflecting tumor and
microenvironment. Blood. 118:1350–1358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu J, Li Y, Lu J, Pan T, Ding N, Wang Z,
Shao T, Zhang J, Wang L and Li X: The mRNA related ceRNA-ceRNA
landscape and significance across 20 major cancer types. Nucleic
Acids Res. 43:8169–8182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu SL, Chen HY, Chang GC, Chen CY, Chen
HW, Singh S, Cheng CL, Yu CJ, Lee YC, Chen HS, et al: MicroRNA
signature predicts survival and relapse in lung cancer. Cancer
Cell. 13:48–57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Attoub S, Rivat C, Rodrigues S, Van
Bocxlaer S, Bedin M, Bruyneel E, Louvet C, Kornprobst M, André T,
Mareel M, et al: The c-kit tyrosine kinase inhibitor STI571 for
colorectal cancer therapy. Cancer Res. 62:4879–4883.
2002.PubMed/NCBI
|
|
40
|
Montero JC, Seoane S, Ocaña A and
Pandiella A: Inhibition of SRC family kinases and receptor tyrosine
kinases by dasatinib: Possible combinations in solid tumors. Clin
Cancer Res. 17:5546–5552. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Blaj C, Schmidt EM, Lamprecht S, Hermeking
H, Jung A, Kirchner T and Horst D: Oncogenic effects of high MAPK
activity in colorectal cancer mark progenitor cells and persist
irrespective of RAS mutations. Cancer Res. 77:1763–1774. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Barry GS, Cheang MC, Chang HL and Kennecke
HF: Genomic markers of panitumumab resistance including ERBB2/HER2
in a phase II study of KRAS wild-type (wt) metastatic colorectal
cancer (mCRC). Oncotarget. 7:18953–18964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang SW, Hu J, Guo QH, Zhao Y, Cheng JJ,
Zhang DS, Fei Q, Li J and Sun YM: AZD1480, a JAK inhibitor,
inhibits cell growth and survival of colorectal cancer via
modulating the JAK2/STAT3 signaling pathway. Oncol Rep.
32:1991–1998. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xiong H, Du W, Zhang YJ, Hong J, Su WY,
Tang JT, Wang YC, Lu R and Fang JY: Trichostatin A, a histone
deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing
the promoter-associated histone acetylation of SOCS1 and SOCS3 in
human colorectal cancer cells. Mol Carcinog. 51:174–184. 2012.
View Article : Google Scholar
|
|
45
|
Hickish T, Andre T, Wyrwicz L, Saunders M,
Sarosiek T, Kocsis J, Nemecek R, Rogowski W, Lesniewski-Kmak K,
Petruzelka L, et al: MABp1 as a novel antibody treatment for
advanced colorectal cancer: A randomised, double-blind,
placebo-controlled, phase 3 study. Lancet Oncol. 18:192–201. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Foersch S, Sperka T, Lindner C, Taut A,
Rudolph KL, Breier G, Boxberger F, Rau TT, Hartmann A, Stürzl M, et
al: VEGFR2 signaling prevents colorectal cancer cell senescence to
promote tumorigenesis in mice with colitis. Gastroenterology.
149:177–189.e10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu Q, Man SM, Gurung P, Liu Z, Vogel P,
Lamkanfi M and Kanneganti TD: Cutting edge: STING mediates
protection against colorectal tumorigenesis by governing the
magnitude of intestinal inflammation. J Immunol. 193:4779–4782.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim MS and Lee SH, Yoo NJ and Lee SH:
Frameshift mutations of tumor suppressor gene EP300 in gastric and
colorectal cancers with high microsatellite instability. Hum
Pathol. 44:2064–2070. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sirvent A, Benistant C and Roche S:
Oncogenic signaling by tyrosine kinases of the SRC family in
advanced colorectal cancer. Am J Cancer Res. 2:357–371.
2012.PubMed/NCBI
|
|
50
|
Bordonaro M and Lazarova DL: CREB-binding
protein, p300 butyrate, and Wnt signaling in colorectal cancer.
World J Gastroenterol. 21:8238–8248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ding C, Luo J, Yu W, Gao S, Yang L, Chen C
and Feng J: Gab2 is a novel prognostic factor for colorectal cancer
patients. Int J Clin Exp Pathol. 8:2779–2786. 2015.PubMed/NCBI
|
|
52
|
Lowenberg M, Verhaar A, van den Blink B,
Ten Kate F, van Deventer S, Peppelenbosch M and Hommes D: Specific
inhibition of c-Raf activity by semapimod induces clinical
remission in severe Crohn's disease. J Immunol. 175:2293–2300.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hardwick JC, van den Brink GR, Offerhaus
GJ, van Deventer SJ and Peppelenbosch MP: NF-kappaB, p38 MAPK and
JNK are highly expressed and active in the stroma of human colonic
adenomatous polyps. Oncogene. 20:819–827. 2001. View Article : Google Scholar : PubMed/NCBI
|