Introduction
Colorectal cancer (CRC) is one of the major types of
cancer and causes of mortality worldwide, and it remains the third
most common cause of cancer-associated mortality worldwide
(1,2). Oncogenes and tumor suppressor genes,
accompanied by deregulated gene expression owing to epigenetic
changes may be involved in the mutational events in the development
and progression of CRC (3).
MicroRNAs (miRNAs) are a class of small RNAs, ~22
nucleotides long, which are non-coding and control gene expression
through the repression of target messenger RNAs (mRNAs) (4,5).
The miRNAs negatively regulate gene expression by the degradation
or translational repression of a target mRNA by targeting the 3′
untranslational region (3′UTR) of specific mRNAs (6,7).
miRNAs are associated with the majority of pathophysiological
cellular processes, including development, differentiation, growth,
migration and apoptosis (8–10).
miR-125, a highly conserved miRNA, is transcribed from three
different clusters and has been shown to be dysregulated in
multiple malignancies through the suppression of multiple targets
(11). Martínez-Acuña et
al determined the expression of miR-125a-5p from nine cervical
cell lines by reverse transcription-polymerase chain reaction
(RT-PCR) analysis and found that miR-125a-5p was involved in the
migration of tumor cervical cancer cells by acting on
mitogen-activated protein kinase (MAPK)1 as a functional target
(12). miR-125 is also involved
in cancer inflammation by the regulating the expression of
cyclooxygenase-2 (COX-2), which is an important target in various
types of tumor (13).
It is known that the occurrence of CRC is associated
with multiple signaling pathways. miRNAs have shown promise in the
development of drugs targeting the specific novel pathways
associated with cancer. For example, anti-angiogenic therapies are
being used more frequently in the treatment of CRC. Vascular
endothelial growth factor (VEGF) is a number of the
platelet-derived growth factor family. It includes related
glycoproteins, including VEGF-A, VEGF-B, VEGF-C and VEGF-D, which
are coded by genes located on 6p23.1, 11q3.3, 4q34.3 and Xp22.31,
respectively, with differing functions and receptors (12). A previous study found that miR-126
offers therapeutic potential in CRC through targeting VEGF-A and
the subsequent regulation of angiogenesis (14). From results of a meta-analysis, it
has been found that the high expression of miR-125b may predict
poor survival rates in CRC, non-small cell lung cancer and prostate
cancer (15). Bi et al
(16) found that miR-125a
suppressed tumor cell proliferation by inhibiting the expression of
VEGF-A. The low expression of miR-125a in hepatocellular cancer was
offset by transfected cells with small interfering RNAs inhibiting
the expression of VEGF-A. However, whether miR-125 affects CRC
through the targeting of VEGF remains to be elucidated. In the
present study, a dual-luciferase reporter assay was used to detect
the level of miR-125 in order to assess whether miR-125 affects CRC
through the targeting of VEGF. The aim of the present study was to
investigate the role of miRNA-125 in CRC. Therefore, the effect of
miR-125 on the development of CRC was investigated by examining the
expression of VEGF, COX-2 and the MAPK signaling pathway.
Materials and methods
Materials
IMDM was obtained from Gibco; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Fetal bovine serum was
purchased from Zhejiang Tianhang Biological Technology, Co., Ltd.
(Zhejiang, China). miR-125 mimic and inhibitor were obtained from
Qiagen China Co., Ltd. (Shanghai, China).
Cell line and cell culture
The RKO cell line was obtained from American Type
Culture Collection (Manassas, VA, USA) and was cultured at 37°C in
IMDM (4,500 mg/l D-glucose, 110 mg/l sodium pyruvate, 25 mM HEPES
(5.958 g/l), 584 mg/l L-glutamine, 3.024 g/l NaHCO3, 15
mg/l-phenol red), which was supplemented with 10% fetal bovine
serum, 100 U/ml penicillin and 100 U/ml streptomycin in a 5%
CO2 incubator. The medium was replaced every 2 or 3
days.
Modulation of miR-125
An miR-125 mimic and antagomir were used to
overexpress or inhibit the expression of miR-125, respectively. A
scrambled oligonucleotide (GenePharm, Inc., Sunnyvale, CA, USA) was
used as a control. Transfection was performed using TransMessenger
transfection reagent (Qiagen GmbH, Hilden, Germany) following the
manufacturer’s protocol.
miR-125 expression assay
Total RNA form the RKO cells was extracted using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The
expression of miR-125 was determined using an miRNA plate assay kit
(Signosis, Inc., Santa Clara, CA, USA) in accordance with the
manufacturer’s protocol. The U6 snRNA was selected as an internal
control to normalized RNA content.
MTT assay
The cell cytotoxicity was evaluated using an MTT
assay. Briefly, the RKO cells, which had been transfected with the
miR-125 mimic or inhibitor or control miR-125, were seeded into
96-well culture plates (Costar; Corning Incorporated, Corning, NY,
USA) at a density of 1×106 cells/well and incubated with
5% CO2 at 37°C. The MTT solution (Sigma-Aldrich; EMD
Millipore, Billerica, MA, USA) was added into the 96-well plates
(final concentration of 5 mg/ml), and incubated at 37°C for 4 h.
Following incubation, 150 μl DMSO (Amresco, Inc., Solon, OH,
USA) was added into the 96-well plates to dissolve the crystal and
incubated for 15 min. Finally, the optical density values of the
obtained solution were examined using an enzyme-linked
immunosorbent assay (ELISA) reader (Thermo Fisher Scientific, Inc.)
at the wavelength of 490 nm.
Flow cytometry
The cells were fixed with 4% paraformaldehyde and
were permeabilized using 0.1% Triton X-100. Following washing with
PBS three times. The apoptosis assay was performed according to the
manufacturer’s protocol. Briefly, the cells were washed and
suspended in binding buffer and incubated with 10 μl of
Annexin V for 10 min and 10 μl of propidium iodide for 5
min. Following two washes in PBS, the cells were suspended in
binding buffer and analyzed immediately using the FACSCanto II flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed
using FlowJo software version 7.6.1 (Tree Star, Inc., Ashland, OR,
USA).
Caspase-3 activity measurement
RKO cell caspase-3 activity was measured using a
caspase-3 fluorescent assay kit (Enzo Life Sciences, Inc.,
Farmingdale, NY, USA).
Measurement of cellular DNA
fragmentation
The RKO cells were seeded at a density of
1×106 cells/well and incubated overnight at 37°C. Then,
the RKO cells were pre-labeled with BrdU (15 ng/well) and then
incubated with palmitate (500 μl/well) for 4 h at room
temperature. DNA fragmentation was measured in accordance with the
manufacturer’s protocol of the Cellular DNA Fragmentation ELISA kit
(Roche Applied Science, Penzberg, Germany).
Western blot analysis
The protein levels of VEGF, COX-2, PCNA and GAPDH
were determined using western blot analysis. The protein samples
were extracted from the cultured RKO cells which were treated with
a lysis buffer containing protease and phosphatase inhibitors (1 M
Tris-HCl, 5 M NaCl, 10% NP-40, 10% Na-deoxycholate, 100 mM EDTA,
0.1% aprotinin, 0.1% leupeptin, 0.1% pepstatin, 0.5% PMSF, 0.5%
Na3Vo4 and 0.5% NaF) and the protein
concentrations were determined using a BCA protein assay. The
quantity of protein loaded for separation was 1 mg/ml. Protein (20
μl) were separated by 12% SDS-PAGE gel and
electrophoretically transferred onto polyvinylidenefluoride
membranes. Following blocking with 5% non-fat dry milk for 2 h at a
room temperature, the membranes were washed with TBS-Tween-20 [150
mM Nacl, 50 mM Tris (pH 7.5) and 0.1% Tween-20] and incubated with
primary antibodies against VEGF (1:1,000; cat. no. 2463), COX-2
(1:1,000; cat. no. 4842), PCNA (1:1,000; cat. no. 13110) and GAPDH
(1:1,000; cat. no. 5174) (all from Cell Signaling Technology, Inc.,
Danvers, MA, USA) overnight at 4°C, and then incubated with the
horseradish peroxidase-conjugated secondary antibodies (1:5,000;
cat.no. ZB-2301; go at anti-rabbit; Bejing Zhongshan Jinqiao
Biotechnology Co., Ltd., Beijing, China) for 1 h at room
temperature. Proteinbands were visualized by enchanced
chemiluminescence using ECL reagents (Pierce; Thermo Fisher
Scientific, Inc.). GAPDH was used as the internal reference. Blots
were semi-quantified by densitometric analysis using the Image-Pro
Plus software version 6.0 (Media Cybernetics, Ine., Rockville, MD,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), the RNA was extracted from the RKO cells and was
reverse transcribed into complementary DNA using the TransScript
first-strand cDNA synthesis kit in accordance with the
manufacturer’s protocol (Qiagen GmbH). The following primers were
used: COX-2, forward, 5′-TTACAATGCTGACTATGGCTAC-3′ and reverse,
5′-CTGATGCGTGAAGTGCTG-3′; GAPDH, forward,
5′-GGAGCGAGATCCCTCCAAAAT-3′ and reverse,
5′-GGCTGTTGTCATACTTCTCATGG-3′. The total volume of PCR reaction was
20 μl:10 μl DNA Master SYBR green I (Finnzymes OY,
Espoo, Finland), 0.6 μl forward primer and 0.6 μl
reverse primer, 4 μl cDNA, 4.8 μl RNase-free water.
The following thermocycling conditions were used for the PCR:
Initial denaturation at 95°C for 15 min; 40 cycles of denaturation
at 95°C for 10 sec, annealing at 60°C for 30 sec extension at 72°C
for 30 sec. Using GraphPad Prism software version 5.01 (GraphPad
Software, Inc., La Jolla, CA, USA), relative gene expression was
quantified according to the comparative Cq method (17). With respect to the expression
levels of GAPDH, all results were normalized.
Luciferase assays
The wild-type luciferase vector (wt-Luc-VEGF)
contained has-miR-125 response elements in the 3′UTR of VEGF.
Putative binding sites located in the 3′UTR of VEGF were identified
using TargetScan version5 (www.targetscan.org/vert_5). The mutant (mu-Luc-VEGF)
vector with a mutation in the has-miR-125 response elements was
generated using site-directed gene mutagenesis. The reporter
vector, which consisted of a luciferase gene followed by the
miR-125 binding consensus sequence, was obtained from Signosis,
Inc. (Sunnyvale, CA, USA). The RKO cells were cultured for 24 h. To
treat cells with a miR-125 mimic or a control oligonucleotide, the
cells were transfected with 200 ng of plasmid DNA (wt-Luc-VEGF or
mu-Luc-VEGF) using Attractene transfection reagent (Qiagen GmbH) in
accordance with the manufacturer’s protocol. The pRL-CMV vector
served as an internal control by expressing Renilla
luciferase. At 24 h post-transfection, luciferase assays were
performed using the dual luciferase reporter assay system (Promega
Corporation, Madison, WI, USA).
Statistical analysis
The data are expressed as the mean ± standard
deviation. Two-way analysis of variance was used to assess the
statistical significance of the differences between groups followed
by Student-Neuman-Keuls test for multiple comparisons. P<0.05
was considered to indicate a statistically significant difference.
Statistical analysis was performed using SPSS software version 21.0
(IBM Corporation, Armonk, NY, USA).
Results
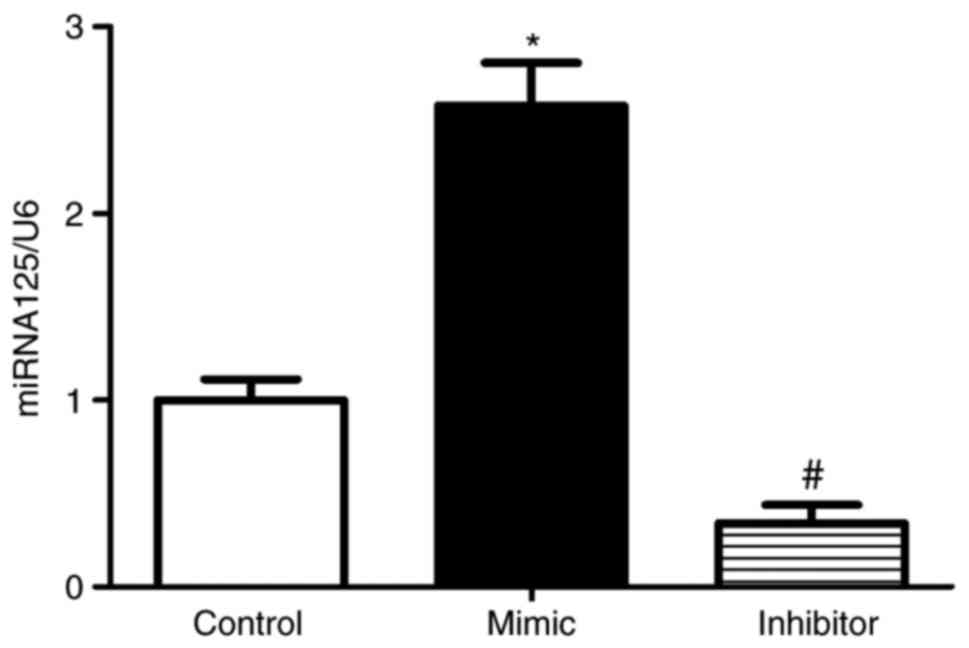
To assess the effect of miR-125 in RKO cells, the
RKOs cells were transfected with miR-125 mimic or inhibitor or
control miR. After 24 h, miR-125 was determined in the RKO cells
using the miRNA plate assay. The level of miR-125 was increased in
the RKO cells transfected with miR-125 mimic, compared with that in
the RKO cells. By contrast, the expression of miR-125 was
significantly downregulated in RKO cells transfected with miR-125
inhibitor (Fig. 1).
To determine the effect of miR-125 on the viability
of RKO cells, the RKO cells were transfected with miR-125 mimic or
inhibitor or control miR. After 0, 12, 24, 36, 48, 60 and 72 h, the
number and absorptivity of the cells were determined using MTT and
DMSO, respectively. The absorptivity of RKO cells transfected with
mimics was decreased and the absorptivity of RKO cells transfected
with inhibitor was increased, compared with the control (Fig. 2A). Following transfection of the
RKO cells with miR-125 mimics or inhibitors or control miR, the
expression of PCNA was determined by western blot analysis. The
protein level of PCNA was decreased in the RKO cells transfected
with mimics, compared with that in the control cells (Fig. 2B). Compared with the control,
whereas the protein levels of PCNA were increased in the RKO cells
transfected with inhibitors (Fig.
2C). This showed that miR-125 significantly altered the
expression of PCNA in the RKO cells. The growth of the RKO cells
was also measured by flow cytometry. The results showed that the
population of RKO cells transfected with mimics was decreased and
the population of RKO cells transfected with inhibitor was
increased, compared with the population in the control (Fig. 2D).
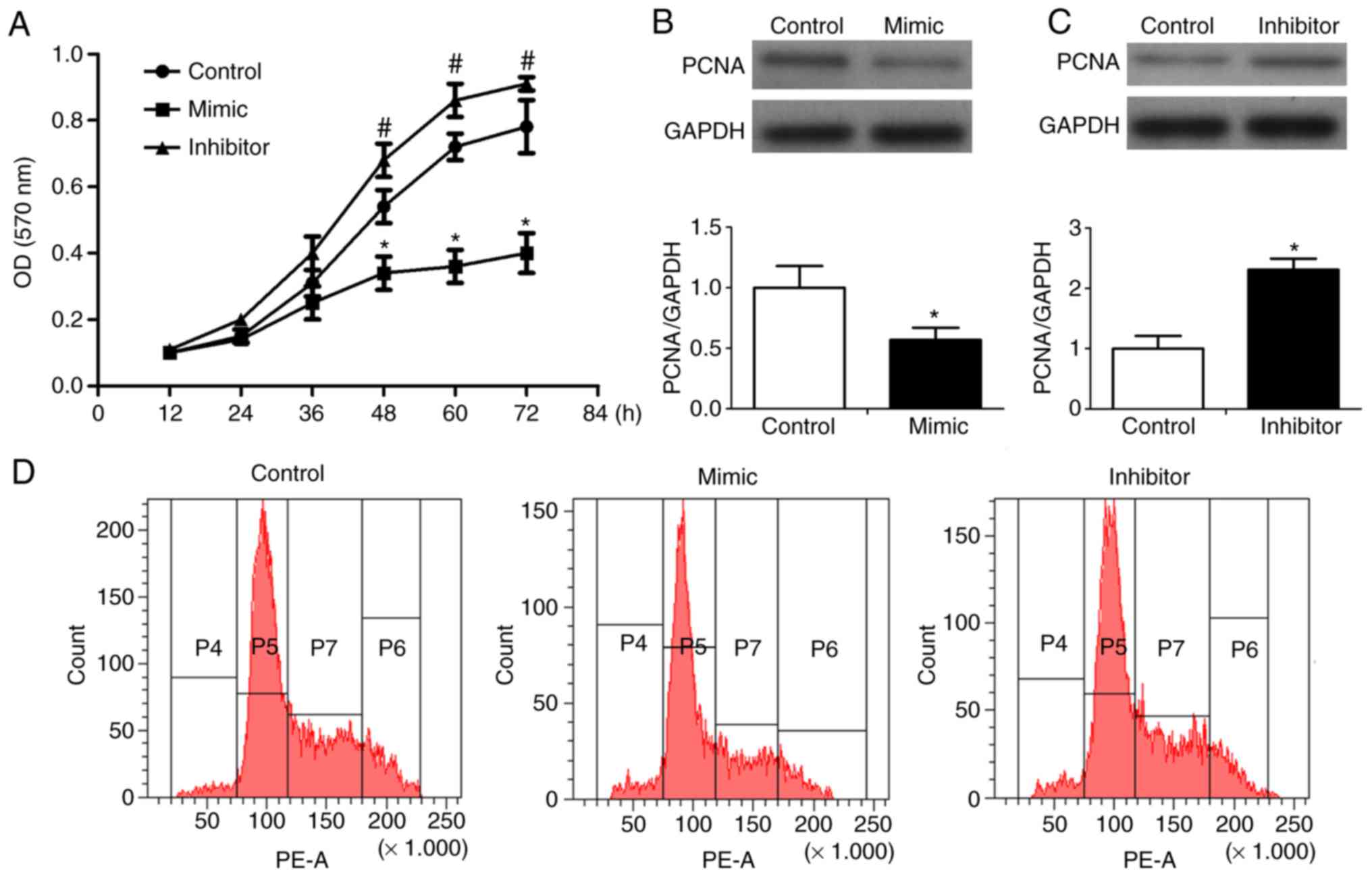 | Figure 2Effect of miR-125 on the cytotoxicity
of RKO cells. RKO cells were transfected with miR-125 mimic,
inhibitor, or miR negative control. (A) After 0, 12, 24, 36, 48, 60
and 72 h the number and absorptivity of cells were determined using
MTT and DMSO, respectively. (B) Western blot analysis was performed
to detect the expression and (C) levels of PCNA and GAPDH. (D)
Growth of RKO cells was measured by flow cytometry.
*P<0.05 vs. control; #P<0.05 vs.
control. miR, microRNA; PCNA, proliferating cell nuclear
antigen. |
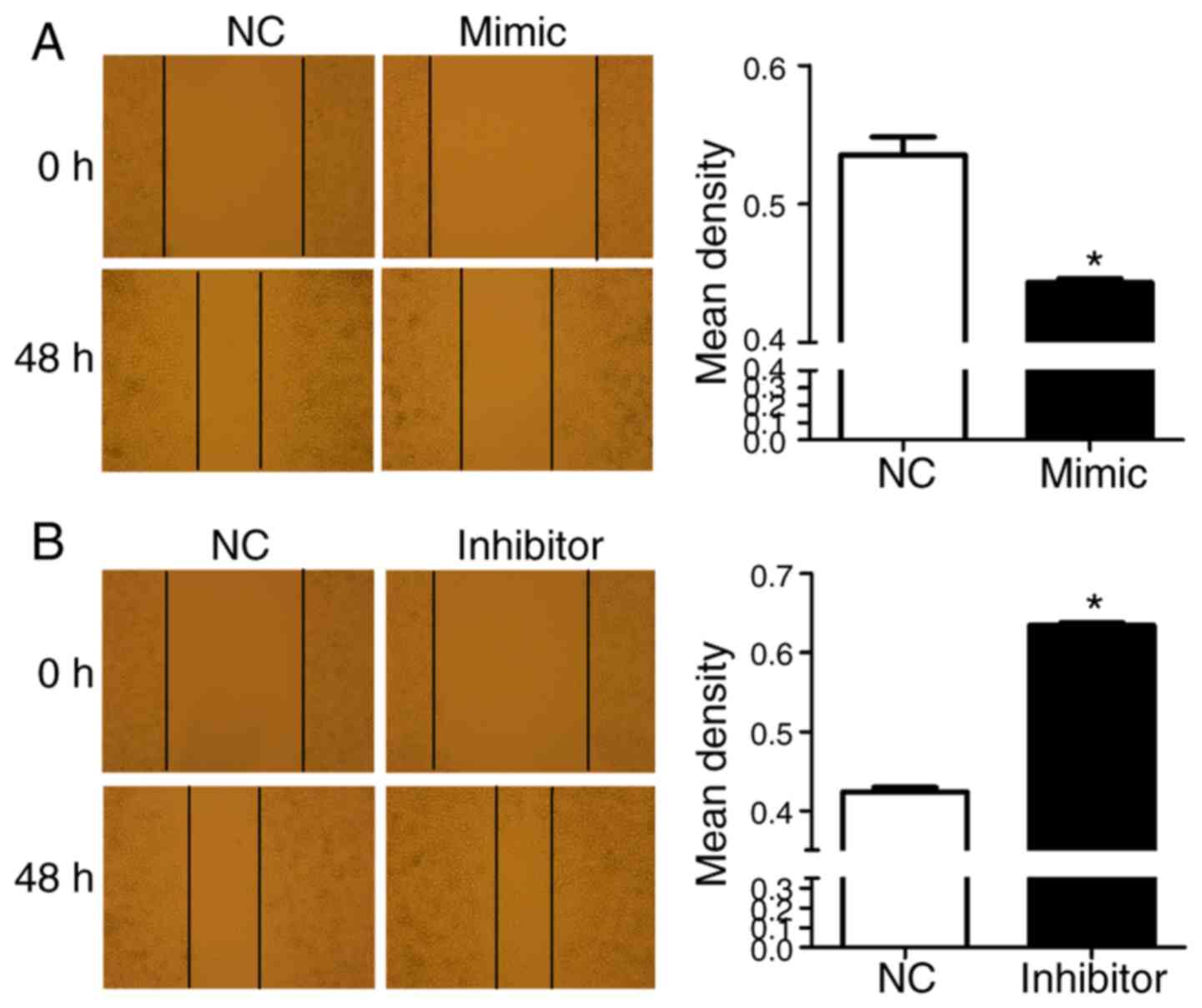
To investigate the role of miR-125 in RKO cell
migration, following transfection of the RKO cells with miR-125
mimics or inhibitors or control miR, cell scratches were observed
at 0 and 48 h, respectively. Compared with the negative control,
only a few cells migrated into the scratch wound in the miR-125
mimic group (Fig. 3A). By
contrast, a large number of cells migrated into the scratch wound
in the miR-125 inhibitor group (Fig.
3B).
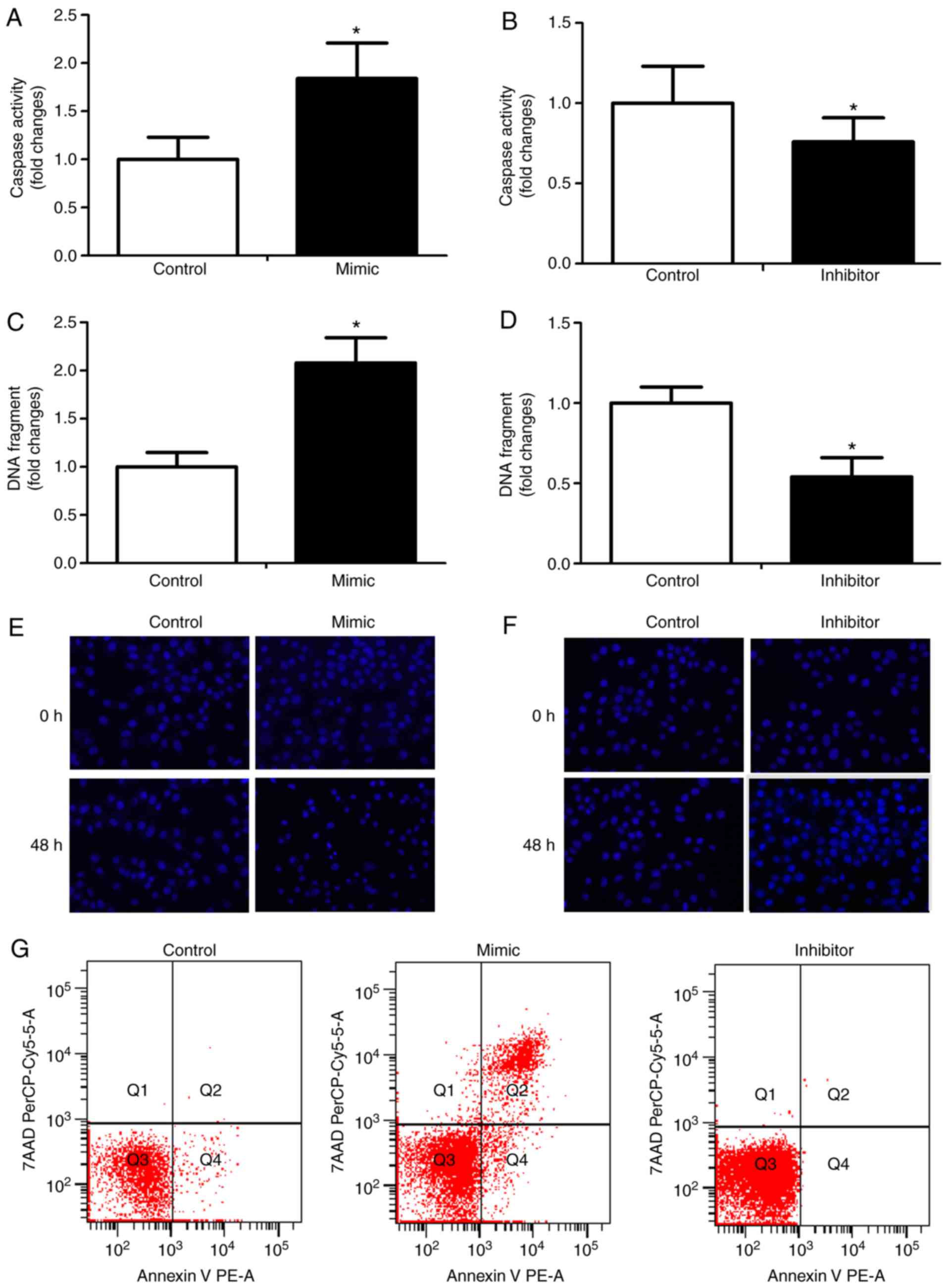
To assess the role of miR-125 in apoptosis, RKO
cells were transfected with miR-125 mimic or inhibitor or control
miR, and apoptosis was determined by caspase-3 activity, DNA
fragmentation and apoptotic morphology. The miR-125 mimic
significantly enhanced caspase-3 activity (Fig. 4A) and the inhibitor reduced
caspase-3 activity (Fig. 4B). The
miR-125 mimic increased DNA fragmentation (Fig. 4C), whereas the inhibitor decreased
DNA fragmentation (Fig. 4D). In
the miR-125 mimic group, marked morphological features of apoptosis
were shown (Fig. 4E). By
contrast, there were no notable morphological features of apoptosis
in the miR-125 inhibitor group (Fig.
4F). The apoptosis of RKO cells transfected with miR-125 mimic
or inhibitor or control miR was measured by flow cytometry. The
results showed that the apoptosis of RKO cells transfected with
mimics was increased and the population of RKO cells transfected
with inhibitor was decreased, compared with that in the control
(Fig. 4G).
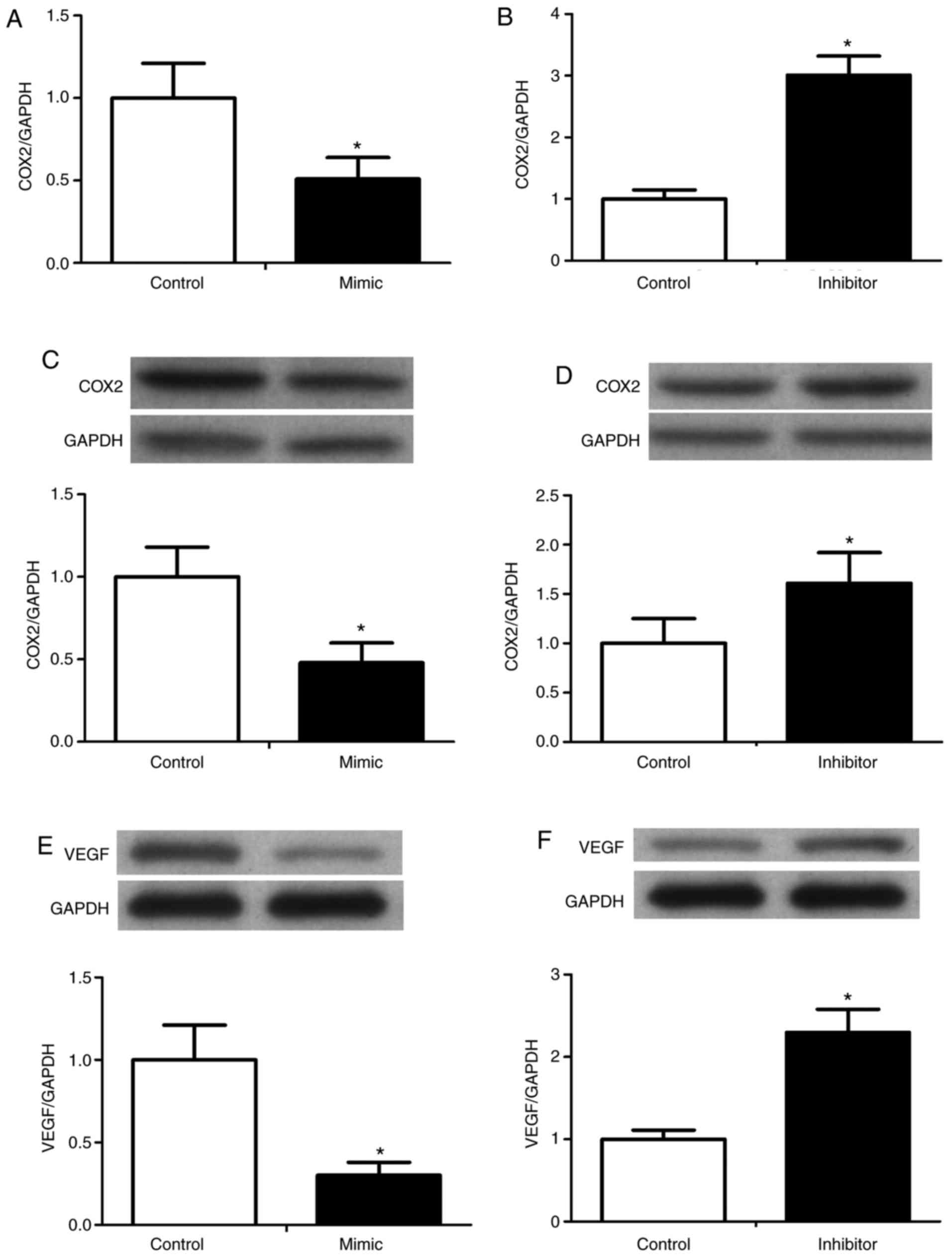
To determine the effect of miR-125 on the expression
of COX-2 and VEGF in RKO cells, the RNA expression of COX-2 was
determined using the RT-qPCR method, and the protein levels of
COX-2 and VEGF were determined by western blot analysis. The
preliminary experiment showed that VEGF was constitutively
expressed in the RKO cells (data not shown), therefore, the effect
of miR-125 on the expression of VEGF was investigated. The miR-125
mimics caused significant decreases in the expression of COX-2 and
VEGF, whereas the miR-125 inhibitors upregulated the expression of
COX-2 and VEGF (Fig. 5).
In the development of a primary tumor, uncontrolled
growth is a key component. The MAPK pathway is a major pathway
associated with uncontrolled growth in CRC, involving proteins
which include ERK, p38, JNK and RAS. Several miRNAs have been shown
to regulate the proteins involved in this pathway. For example,
Let7, miR-143, miR-18a* and miR-145 downregulate RAS and act as
tumor-suppressive miRNAs in CRC (18–21). In the present study, western blot
analysis was used to examine the effect of miR-125 on the
phosphorylation of ERK, p38 and JNK in RKO cells. The
phosphorylation of ERK, p38 and JNK was reduced by the miR-125
mimic, but were increased by the miR-125 inhibitor (Fig. 6).
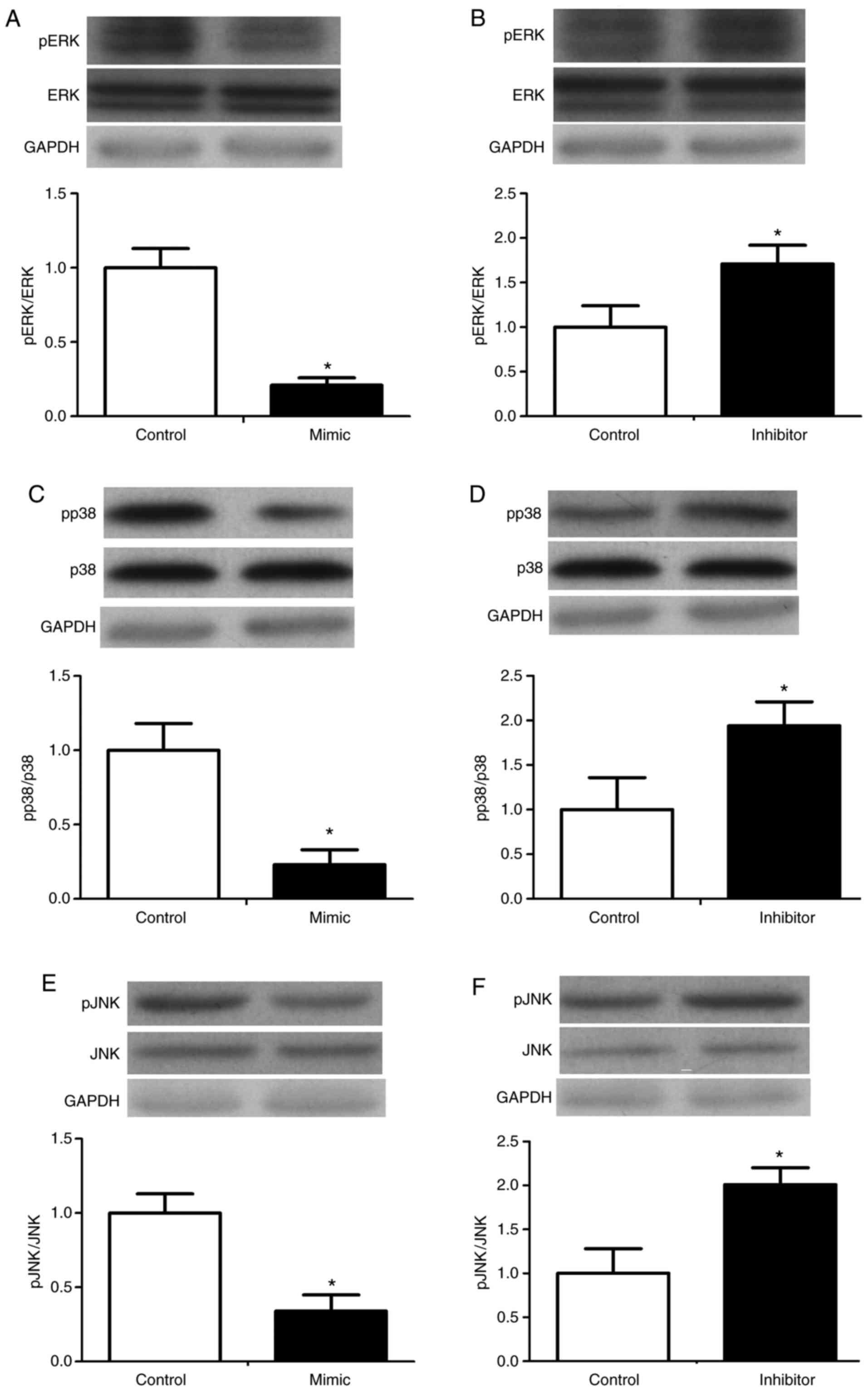 | Figure 6Effect of miR-125 on the
mitogen-activated protein kinase signaling pathway in RKO cells.
RKOs were transfected with miR-125 mimic, inhibitor, or a miR-125
negative control. Expression and phosphorylation of ERK in the (A)
mimic and (B) inhibitor groups, p38 in the (C) mimic and (D)
inhibitor groups, and JNK in the (E) mimic and (F) inhibitor groups
were determined by western blot analysis. *P<0.05 vs.
control. miR, microRNA; ERK, extracellular signal-regulated kinase;
JNK, c-Jun N-terminal kinase; pERK, phosphorylated ERK; pp38,
phosphorylated p38; pJNK, phosphorylated JNK. |
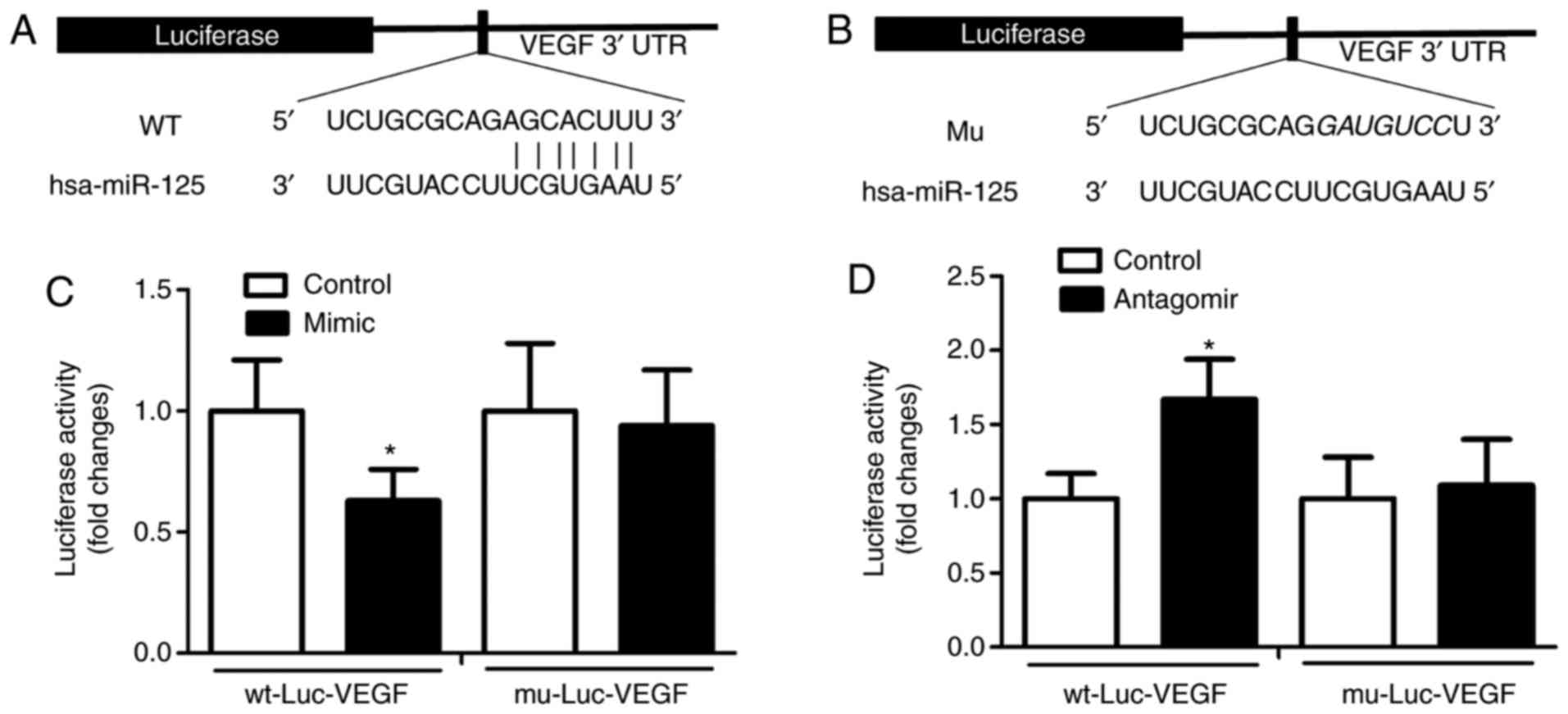
It was revealed by a dual-luciferase reporter assay
that miR-125 functionally interacted with the 3′UTR sequences of
VEGF. The alignments between miR-125 and the region within the
3′UTR of VEGF represent putative target sequences which can confer
inhibition of translation by miR-125. To assess whether VEGF is a
direct target of miR-125, the present study used a reporter vector,
which contained a luciferase gene followed by the 3′UTR of VEGF
mRNA (wt-Luc-VEGF). The luciferase activity in
wt-Luc-VEGF-transfected RKO cells was inhibited by the
overexpression of miR-125 by its mimic. To verify this, mutation of
the miR-125 putative binding sites on the 3′UTR of VEGF of
wt-Luc-VEGF was introduced by substitution (Fig. 7A and B). The mutation abrogated
the inhibitory effect of miR-125 on the luciferase activity of the
RKO cells (Fig. 7C). By contrast,
the inhibition of miR-125 by its inhibitors enhanced the luciferase
activity in the wt-Luc-VEGF-transfected RKO cells; the mutation of
wt-Luc-VEGF abrogated the enhanced effect of miR-125 on the
luciferase activity in RKO cells (Fig. 7D). These data demonstrated that
miR-125 directly targeted and inhibited the expression of VEGF in
RKO cells.
Discussion
The major finding of the present study was that
induction of the expression of miR-125 promoted the apoptosis of
RKO cells. miR-125 directly targeted VEGF and repressed the
expression of VEGF. Therefore, the results of the present study
suggested the importance of miR-125 in RKO cells by inducing cell
apoptosis.
The associations between miRNAs and malignancies
have been investigated in numerous studies, the results of which
have shown that miRNA deregulation is involved in all types of
cancer. In different types of cancer, different members of the
miR-125 family have been reported to have conflicting properties;
through acting as either tumor suppressors or oncogenes, they may
contribute to the initiation and progression of cancer (22–25).
The tumor-suppressor functions of miR-125 have been
shown in several types of cancer, including ovarian cancer
(23,26), bladder cancer (27), osteosarcoma (28), breast cancer (29–31), hepatocellular carcinoma (16,32,33), melanoma (34) and cutaneous squamous cell
carcinoma (35). Chen and Hu
(36) used PCR to investigate the
level and the methylation status of the miR-125 family in CRC
tissues and adjacent non-tumor tissues, and found that miR-125a and
miR-125b were significantly downregulated in CRC tissues, and the
methylation frequencies of miR-125a and miR-125b were higher in CRC
tissues. These results suggested the hypermethylation of miR-125 as
a potential biomarker for clinical outcome. In the present study,
it was shown that miR-125 also contributed to the apoptosis of CRC
cells. In response to CRC, miR-125 inhibitors prevented apoptosis,
whereas miR-125 mimics enhanced apoptosis. As VEGF stimulates
angiogenesis, proliferation and migration, it has been implicated
in tumor generation (37). In the
present study, the induction of miR-125 inhibited the expression of
VEGF, whereas the inhibition of miR-125 enhanced the expression of
VEGF. This confirmed that miR-125 directly targeted and inhibited
the expression of VEGF in RKO cells. Substantial data support the
use of targeted agents directed against the molecular signaling
pathways involved in angiogenesis, particularly the VEGF pathway
(38). MAPK is a type of VEGF
signaling pathway and MAPK has three signaling pathways: ERK, p38
and JNK. It is known that ERK, JNK and p38 transduce the signal
through phosphorylation. Martínez-Acuña et al determined the
expression of miR-125a-5p from nine cervical cell lines using
RT-PCR analysis and found that miR-125a-5p was involved in the
migration of cervical cancer tumor cells via MAPK1 as a functional
target (12). The present study
demonstrated that miR-125 mimics decreased the phosphorylation of
ERK, p38 and JNK, whereas miR-125 inhibitors increased this
phosphorylation. It is known that the majority of cases of
cancer-associated mortality are due to infection and inflammation.
COX-2 is a type of inflammatory maker. In previous years, several
studies have found that the miR-125 family is involved in cancer
inflammation by regulation of the RNA-binding protein HuR (39), and COX-2 is an important target in
various tumors (40,41). In the present study, it was shown
that miR-125 mimics significantly decreased the expression of
COX-2, whereas miR-125 inhibitors upregulated the expression of
COX-2.
Although the present study revealed interesting
results, there were a number of limitations. Firstly, the
investigation only used luciferase assays to investigate the
targeting association between miR-125 and VEGF, with no western
blot or RT-PCR assays performed to confirm the association.
Secondly, the levels of miR-125 were not examined in CRC tissues in
the present study, therefore, future investigations require the
levels of miRNA-125 to be detected in CRC tissues. Thirdly, the
levels of non-phosphorylated protein are expected to decrease
accordingly when phosphorylation occurs. However, there was no
change in the level of non-phosphorylated protein in the present
study, the reason for which remains to be elucidated, as does the
reason for miR-125 affecting the phosphorylation of MAPK.
Additionally, only in vitro experiments involving only one
cell line were performed, which is insufficient for a solid
conclusion. In subsequent investigations, the use of in vivo
experiments and animal models id required to confirm the
conclusion.
In conclusion, the present study confirmed that
miR-125 was important in RKO cells and VEGF was confirmed as a
direct target of miR-125. The promotion effect of miR-125 on
apoptosis was mediated through reducing the expression of VEGF.
Therefore, miR-125 may be a novel therapeutic target for CRC.
Acknowledgments
Not applicable.
References
|
1
|
Haggar FA and Boushey RP: Colorectal
cancer epidemiology: Incidence, mortality, survival, and risk
factors. Clin Colon Rectal Surg. 22:191–197. 2009. View Article : Google Scholar :
|
|
2
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dean M: Cancer as a complex developmental
disorder-nineteenth Cornelius P. Rhoads memorial award lecture.
Cancer Res. 58:5633–5636. 1998.PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meister G and Tuschl T: Mechanisms of gene
silencing by double-stranded RNA. Nature. 431:343–349. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumarswamy R and Thum T: Non-coding RNAs
in cardiac remodeling and heart failure. Circ Res. 113:676–689.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Quiat D and Olson EN: MicroRNAs in
cardiovascular disease: From pathogenesis to prevention and
treatment. J Clin Invest. 123:11–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huntzinger E and Izaurralde E: Gene
silencing by microRNAs: Contributions of translational repression
and mRNA decay. Nat Rev Genet. 12:99–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Winter J, Jung S, Keller S, Gregory RI and
Diederichs S: Many roads to maturity: MicroRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pillai RS, Bhattacharyya SN and Filipowicz
W: Repression of protein synthesis by miRNAs: How many mechanisms?
Trends Cell Biol. 17:118–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martínez-Acuña N, González-Torres A,
Tapia-Vieyra JV and Alvarez-Salas LM: MARK1 is a novel target for
miR-125a-5p: Implications for cell migration in cervical tumor
cells. Microrna. 2017.
|
|
13
|
Yin H, Sun Y, Wang X, Park J, Zhang Y, Li
M, Yin J, Liu Q and Wei M: Progress on the relationship between
miR-125 family and tumorigenesis. Exp Cell Res. 339:252–260. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aldebasi YH, Rahmani AH, Khan AA and Aly
SM: The effect of vascular endothelial growth factor in the
progression of bladder cancer and diabetic retinopathy. Int J Clin
Exp Med. 6:239–251. 2013.PubMed/NCBI
|
|
15
|
Stiegelbauer V, Perakis S, Deutsch A, Ling
H, Gerger A and Pichler M: MicroRNAs as novel predictive biomarkers
and therapeutic targets in colorectal cancer. World J
Gastroenterol. 20:11727–11735. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bi Q, Tang S, Xia L, Du R, Fan R, Gao L,
Jin J, Liang S, Chen Z, Xu G, et al: Ectopic expression of miR-125a
inhibits the proliferation and metastasis of hepatocellular
carcinoma by targeting MMP11 and VEGF. PLos One. 7:e401692012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-(delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Sun X, Zhang S and Ma X: Prognostic value
of MicroRNA-125 in various human malignant neoplasms: A
meta-analysis. Clin Lab. 61:1667–1674. 2015. View Article : Google Scholar
|
|
19
|
Johnson SM, Grosshans H, Shingara J, Byrom
M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D and Slack
FJ: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar
|
|
20
|
Tsang WP and Kwok TT: The miR-18a*
microRNA functions as a potential tumor suppressor by targeting on
K-Ras. Carcinogenesis. 30:953–959. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen X, Guo X, Zhang H, Xiang Y, Chen J,
Yin Y, Cai X, Wang K, Wang G, Ba Y, et al: Role of miR-143
targeting KRAS in colorectal tumorigenesis. Oncogene. 28:1385–1392.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin Y, Yan ZP, Lu NN, Xu Q, He J, Qian X,
Yu J, Guan X, Jiang BH and Liu LZ: Downregulation of miR-145
associated with cancer progression and VEGF transcriptional
activation by targeting N-RAS and IRS1. Biochim Biophys Acta.
1829:239–247. 2013. View Article : Google Scholar
|
|
23
|
Bousquet M, Harris MH, Zhou B and Lodish
HF: MicroRNA miR-125b causes leukemia. Proc Natl Acad Sci USA.
107:21558–21563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cowden Dahl KD, Dahl R, Kruichak JN and
Hudson LG: The epidermal growth factor receptor responsive miR-125a
represses mesenchymal morphology in ovarian cancer cells.
Neoplasia. 11:1208–1215. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang L, Huang Q, Zhang S, Zhang Q, Chang
J, Qiu X and Wang E: Hsa-miR- 125a-3p and hsa-miR-125a-5p are
downregulated in non-small cell lung cancer and have inverse
effects on invasion and migration of lung cancer cells. BMC Cancer.
10:3182010. View Article : Google Scholar
|
|
26
|
Jiang F, Liu T, He Y, Yan Q, Chen X, Wang
H and Wan X: MiR-125b promotes proliferation and migration of type
II endometrial carcinoma cells through targeting TP53INP1 tumor
suppressor in vitro and in vivo. BMC Cancer. 11:4252011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guan Y, Yao H, Zheng Z, Qiu G and Sun K:
MiR-125b targets BCL3 and suppresses ovarian cancer proliferation.
Int J Cancer. 128:2274–2283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang L, Luo J, Cai Q, Pan Q, Zeng H, Guo
Z, Dong W, Huang J and Lin T: MicroRNA-125b suppresses the
development of bladder cancer by targeting E2F3. Int J Cancer.
128:1758–1769. 2011. View Article : Google Scholar
|
|
29
|
Liu LH, Li H, Li JP, Zhong H, Zhang HC,
Chen J and Xiao T: MiR-125b suppresses the proliferation and
migration of osteosarcoma cells through down-regulation of STAT3.
Biochem Biophys Res Commun. 416:31–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li W, Duan R, Kooy F, Sherman SL, Zhou W
and Jin P: Germline mutation of microRNA-125a is associated with
breast cancer. J Med Genet. 46:358–360. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scott GK, Goga A, Bhaumik D, Berger CE,
Sullivan CS and Benz CC: Coordinate suppression of ERBB2 and ERBB3
by enforced expression of micro-RNA miR-125a or miR-125b. J Biol
Chem. 282:1479–1486. 2007. View Article : Google Scholar
|
|
32
|
Mattie MD, Benz CC, Bowers J, Sensinger K,
Wong L, Scott GK, Fedele V, Ginzinger D, Getts R and Haqq C:
Optimized high-throughput microRNA expression profiling provides
novel biomarker assessment of clinical prostate and breast cancer
biopsies. Mol Cancer. 5:242006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liang L, Wong CM, Ying Q, Fan DN, Huang S,
Ding J, Yao J, Yan M, Li J, Yao M, et al: MicroRNA-125b
suppressesed human liver cancer cell proliferation and metastasis
by directly targeting oncogene LIN28B2. Hepatology. 52:1731–1740.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jia HY, Wang YX, Yan WT, Li HY, Tian YZ,
Wang SM and Zhao HL: MicroRNA-125b functions as a tumor suppressor
in hepatocellular carcinoma cells. Int J Mol Sci. 13:8762–8774.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kappelmann M, Kuphal S, Meister G,
Vardimon L and Bosserhoff AK: MicroRNA miR-125b controls melanoma
progression by direct regulation of c-Jun protein expression.
Oncogene. 32:2984–2991. 2013. View Article : Google Scholar
|
|
36
|
Chen H and Hu Z:
Hypermethylation-associated silencing of miR-125a and miR-125b: A
potential marker in colorectal cancer. Dis Markers.
2015:3450802015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nagy JA, Dvorak AM and Dvorak HF: VEGF and
the induction of pathological angiogenesis. Annu Rev Pathol.
2:251–275. 2007. View Article : Google Scholar
|
|
38
|
Aragon-Ching JB and Dahut WL:
Anti-angiogenesis approach to genitourinary cancer treatment.
Update Cancer Ther. 3:182–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dormoy-Raclet V, Ménard I, Clair E, Kurban
G, Mazroui R, Di Marco S, von Roretz C, Pause A and Gallouzi IE:
The RNA-binding protein HuR promotes cell migration and cell
invasion by stabilizing the beta-actin mRNA in a
U-rich-element-dependent manner. Mol Cell Biol. 27:5365–5380. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Denkert C, Weichert W, Pest S, Koch I,
Licht D, Köbel M, Reles A, Sehouli J, Dietel M and Hauptmann S:
Overexpression of the embryonic-le-thal abnormal vision-like
protein HuR in ovarian carcinoma is a prognostic factor and is
associated with increased cyclooxygenase-2 expression. Cancer Res.
64:189–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mrena J, Wiksten JP, Thiel A, Kokkola A,
Pohjola L, Lundin J, Nordling S, Ristimäki A and Haglund C:
Cyclooxygenase-2 is an independent prognostic factor in gastric
cancer and its expression is regulated by the messenger RNA
stability factor HuR. Clin Cancer Res. 11:7362–7368. 2005.
View Article : Google Scholar : PubMed/NCBI
|