Introduction
Affecting ~12-13% of the general population,
neuropathic pain due to disease or lesions affecting the
somatosensory nervous system is a prevalent condition, for which
currently there is no effective treatment (1,2).
It is a condition that is often refractory to opioids or requires
larger doses that possess unacceptable side effects (3). Therefore, it is important to
understand the pathophysiology of neuropathic pain and the
underlying molecular mechanisms, which will facilitate development
of novel therapies.
Neuropathic pain is closely associated with
hyperresponsiveness of sensory neurons in the dorsal root ganglion
(DRG) and the spinal cord dorsal horn (4). Previous studies have shown that
elevated levels of proinflammatory cytokine tumor necrosis factor-α
(TNF-α) in DRG neurons is critical for neuropathic pain processing
(5). TNF-α antibodies have been
shown to reduce pain-associated behaviors in animal models of
neuropathic pain (5), suggesting
a key role of TNF-α in neuropathic pain and nociception. TNF-α
receptor TNFR1 reportedly is expressed on most cells as a major
mediator of the cytotoxicity of TNF-α (6).
Nitric oxide (NO) is involved in sensitization of
sensory neurons after noxious stimuli and is linked with the
development and maintenance of nociception (7,8).
The production of NO mainly depends on the expression of nitric
oxide synthases (NOSs) and their activities (7). Neuronal NOS (nNOS) is localized in
neurons and is dynamically regulated after peripheral inflammation,
while inducible NOS (iNOS) and endothelial NOS (eNOS) is normally
absent in neural tissues under normal conditions (7,8).
Previous studies have shown beneficial effects of selective
blockers for NOS in reducing neuropathic pain (7,8).
Accumulating evidence supports a role of
cannabinoids in modulating neuropathic pain (9,10).
Endogenous cannabinoids and their receptors have been found to be
expressed in key areas associated with pain processing and markedly
increase in these areas in models of chronic pain (11-15). Cannabinoids mainly function
through cannabinoid receptor type 1 (CB1) and CB2 (16). The CB1 receptor is expressed
primarily in the nervous system, whereas the CB2 receptor is mainly
expressed in immune cells and is also detectable in brainstem
neurons and spinal cord (17).
Recent report has shown that both CB1 and CB2 are expressed in DRG
neurons (18). Previous studies
have shown significant antinociceptive effects of cannabinoid
receptor agonists in animal models of spontaneous, inflammatory and
neuropathic pain (19).
In the present study, we explored the effects and
the underlying mechanisms of crosstalk between TNF-α and
cannabinoid on the expression/activity of NOS in DRG neurons.
Materials and methods
Cell culture and treatment
Rat DRG neurons (cat. no. R-DRG-505) and primary
neuron medium (cat. no. CC-3256) were purchased from Lonza Inc.
(Houston, TX, USA). The cells were suspensions of high quality
sensory neurons prepared by standardized methods and were ready for
immediate culture. The cells were treated with recombinant human
TNF-α (cat. no. T6674; Sigma-Aldrich, Beijing, China) in different
concentrations (5, 10, 20, 30 and 40 ng/ml) for 1, 5, 10, 15, 20
and 25 h. For kinase inhibitor or TNFR1 inhibitor SPD304 (cat. no.
S1697; Sigma-Aldrich) treatment, DRG neurons were pretreated with a
kinase inhibitor or SPD304 for 30 min, and then incubated with
TNF-α (40 ng/ml) and the kinase inhibitor or SPD304 for 25 h. For
the synthetic cannabinoid WIN-55,212-2 mesylate (WIN-55) (cat. no.
1038; Tocris Bioscience, Bristol, UK) treatment, DRG neurons were
treated with WIN-55 (100, 200, 300 and 400 ng/ml) in the presence
of TNF-α (40 ng/ml) for 1, 5, 10, 15, 20 and 25 h with or without
selective CB1 antagonist MJ15 (cat. no. 4063) or selective CB2
antagonist JTE907 (cat. no. 2479) (both from Tocris Bioscience).
For p38 mitogen-activated protein kinase (MAPK) knockdown, p38 MAPK
siRNA (cat. no. sc-156091; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) was transfected into DRG neurons using Lipofectamine 3000
transfection reagent (cat. no. L3000008; Thermo Fisher Scientific,
Beijing, China) by the manufacturer's protocol; the cells were
subjected to subsequent experiments 24 h after the
transfection.
NOS activity assay
The NOS activity was measured with a NOS activity
assay kit (cat. no. KA1345; Abnova, Taipei, Taiwan) according to
the manufacturer's protocol. This assay is based on the conversion
of [14C]-L-arginine to [14C]-L-citrulline by
NOS. NOS activity was calculated as follows: NOS activity
(pmol/µg/min) = [14C]-radioactivity in the
flow-through (CPM)/total protein amount (µg)/specific
radioactivity of [14C]-L-arginine (Bq/pmol). Each
experiment was repeated for three independent times in
duplicates.
Real-time quantitative RT-PCR and
measurement of nNOS mRNA stability
RNA was prepared from cells using TRIzol reagent
(Thermo Fisher Scientific) followed by purification with TURBO
DNA-free system (Ambion, Austin, TX, USA). cDNA was synthe sized
using SuperScript II reverse transcriptase and random hexamer
primers (both from Thermo Fisher Scientific). RT-qPCR was performed
using an ABI-PRISM 7700 Sequence detection system (Applied
Biosystems; Thermo Fisher Scientific) and the fluorescent dye
SYBR-Green Master Mix (Thermo Fisher Scientific) as described by
the manufacturer. The primers used were as follows: forward,
5′-CACGTGGTCCTCATTCTGAG-3′ and reverse, 5′-CAGATCGACGGCTTTGGT-3′
for nNOS; forward, 5′-TCGTACTTGGGATGCTCCATGG-3′ and reverse,
5′-TCCTGCAGGCTCACGGTCAA-3′ for iNOS; forward,
5′-CCCTGTAATTGGAATGAGTCCAC-3′ and reverse, 5′-GCTGGAATTACCGCGGCT-3′
for 18S rRNA. The PCR amplification condition was: 20 sec at 95°C
followed by 40 cycles of 3 sec at 95°C and 30 sec at 60°C. Relative
quantification of the nNOS or iNOS level was determined using the
2−ΔΔCt method (20)
and normalized against that of 18S rRNA in the same sample. Each
experiment was repeated for three independent times in duplicates.
For measurement of the nNOS mRNA stability, DRG neurons were
treated with or without TNF-α in the presence or absence of WIN-55,
p38 MAPK-siRNA or scramble control siRNA (Scr) for 25 h. Then the
cells were cultured in media containing transcription inhibitor
actinomycin D (1 mg/ml) for 1, 2 and 4 h. The mRNA levels of nNOS
were determined with real-time quantitative RT-PCR at 1, 2 and 4 h
of actinomycin D treatment.
Western blot analysis
Whole cell lysates were extracted by incubating the
DRG neurons with lysis buffer (50 mM Tris/HCl pH 7.2, 150 mM NaCl,
l% (v/v) Triton X-100, 1 mM sodium orthovanadate, 50 mM sodium
pyrophosphate, 100 mM sodium fluoride, 0.01% (v/v) aprotinin, 4
µg/ml pepstatin A, 10 µg/ml leupeptin and 1 mM
phenylmeth anesulfonyl fluoride; all purchased from Sigma-Aldrich)
on ice for 30 min and removing cell debris by centrifugation at
2,000 × g for 15 min at 4°C. Equal amount of proteins for each
sample were separated by 10% sodium dodecyl sulfate
(SDS)-polyacrylamide gel and blotted onto a polyvinylidene
difluoride microporous membrane (Millipore, Billerica, MA, USA).
The membranes were blocked with 5% skim milk powder in TBS-T for 2
h and incubated for 1 h with a 1:1,000 dilution of rabbit anti-rat
nNOS (NOS1) polyclonal antibody (cat. no. sc-648) or mouse
anti-rabbit glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
monoclonal antibody (cat. no. sc-32233), and then washed and
revealed using bovine anti-rabbit (cat. no. sc-2370) or bovine
anti-mouse (cat. no. sc-2371) (all from Santa Cruz Biotechnology,
Inc.) secondary antibody (1:5,000, 1 h). Peroxidase was revealed
with an ECL kit purchased from GE Healthcare (Shanghai, China).
Three independent experiments were performed.
p38 MAPK activity assay
p38 MAPK activity was measured with a p38 MAPK assay
kit (cat. no. 9820; Cell Signaling Technology, Beverly, MA, USA)
according to the manufacturer's protocol (21). Briefly, cell lysates were
sonicated and centrifuged at 15,000 rpm for 10 min at 4°C. The
supernatant containing equivalent amounts of protein (200
µg) was incubated by gentle rocking with 20 µl of
immobilized phospho-p38-MAPK monoclonal antibody for 16 h at 4°C.
The immunoprecipitates were washed twice with the lysing buffer and
pelleted by centrifugation. The p38 MAPK assay was carried out
using ATF2 fusion protein (2 µg) as a substrate in the
presence of 200 µM ATP and 1X kinase buffer following the
manufacturer's recommendations. Samples were resolved on a 12%
SDS-PAGE gel and visualized by autoradiography.
Statistical analysis
Statistical analyses were performed with SPSS for
Windows 10.0 (SPSS, Inc., Chicago, IL, USA). All values were
expressed as mean ± SD. Comparisons of means among multiple groups
were performed with one-way ANOVA followed by post hoc pairwise
comparisons using Tukey's tests. p<0.05 was considered
statistically significant in this study.
Results
TNF-α-induced expression of nNOS in DRG
neurons by a p38 MAPK-dependent mechanism
To examine the effect of TNF-α on the expression and
activity of NOS in DRG neurons, we treated DRG neurons with TNF-α
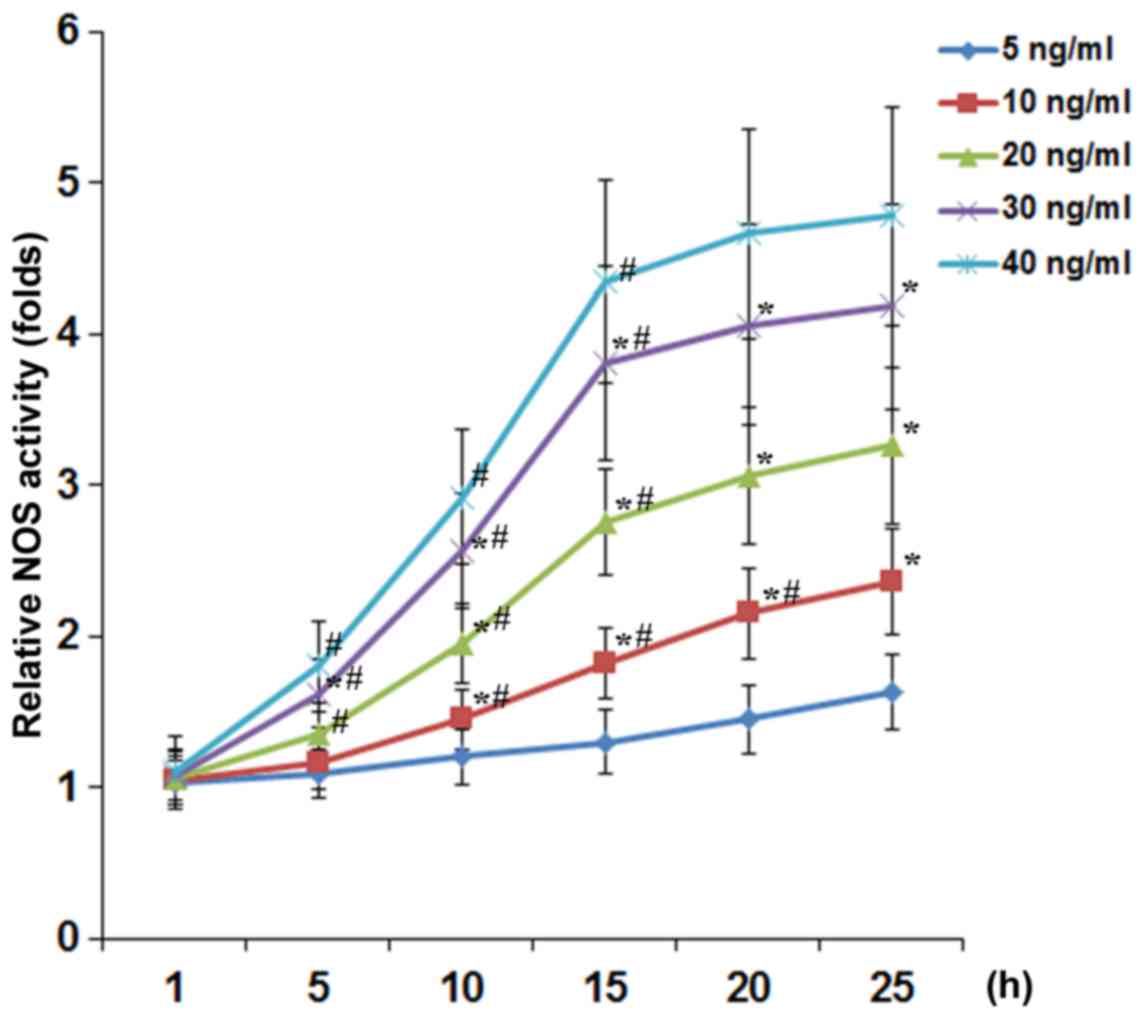
(5, 10, 20, 30 and 40 ng/ml) for 1, 5, 10, 15, 20 and 25 h. As show
in Fig. 1, TNF-α concentration-
and time-dependently increased the NOS activity in DRG neurons. At
TNF-α concentrations ≥20 ng/ml, the inducing effect of TNF-α on the
NOS activity reached a plateau after 15 h of treatment. As shown in
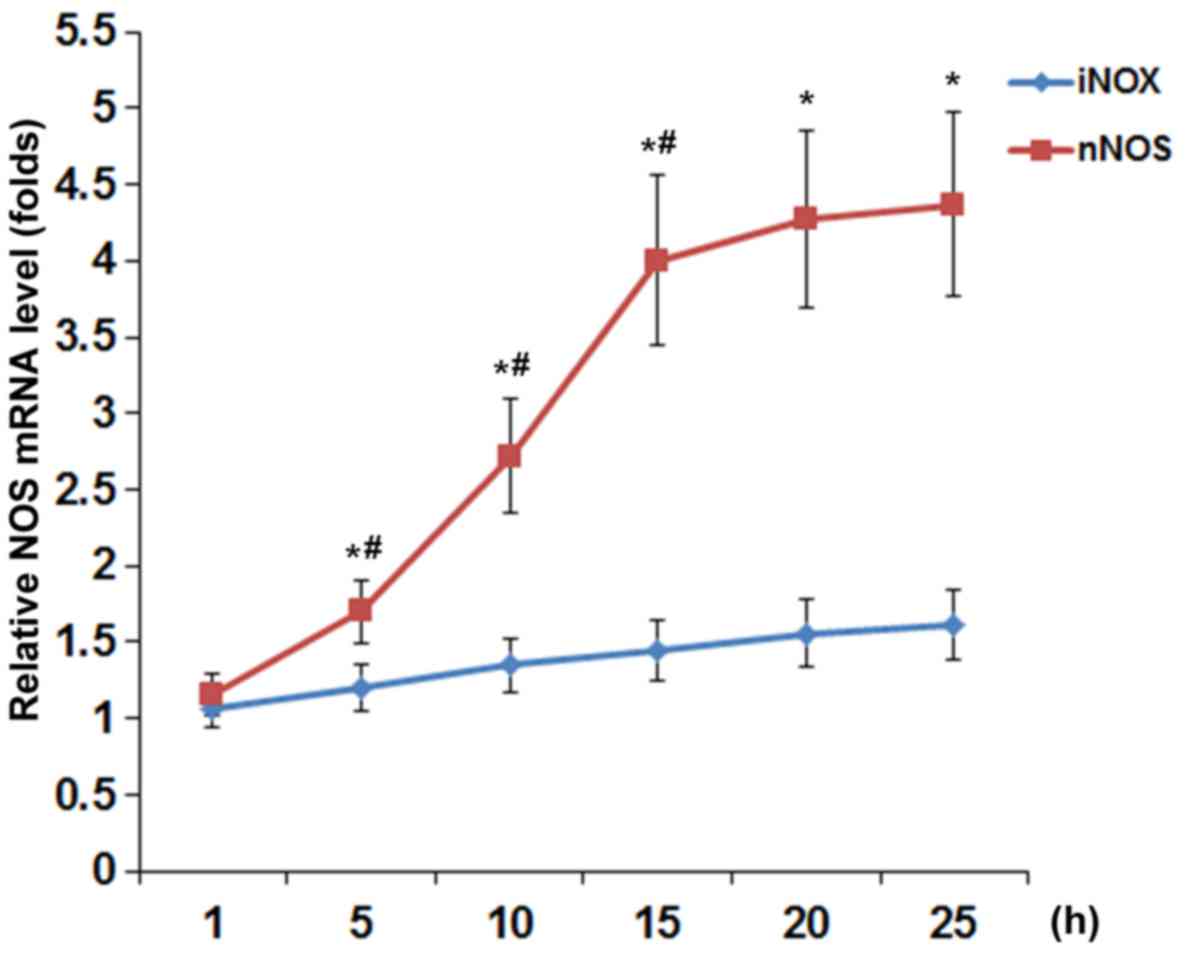
Fig. 2, TNF-α (40 ng/ml)
time-dependently increased the mRNA levels of nNOS but not iNOS or
eNOS (barely detectable; data not shown) in DRG neurons, with the
nNOS mRNA level showing similar data trend as the NOS activity in
Fig. 1.
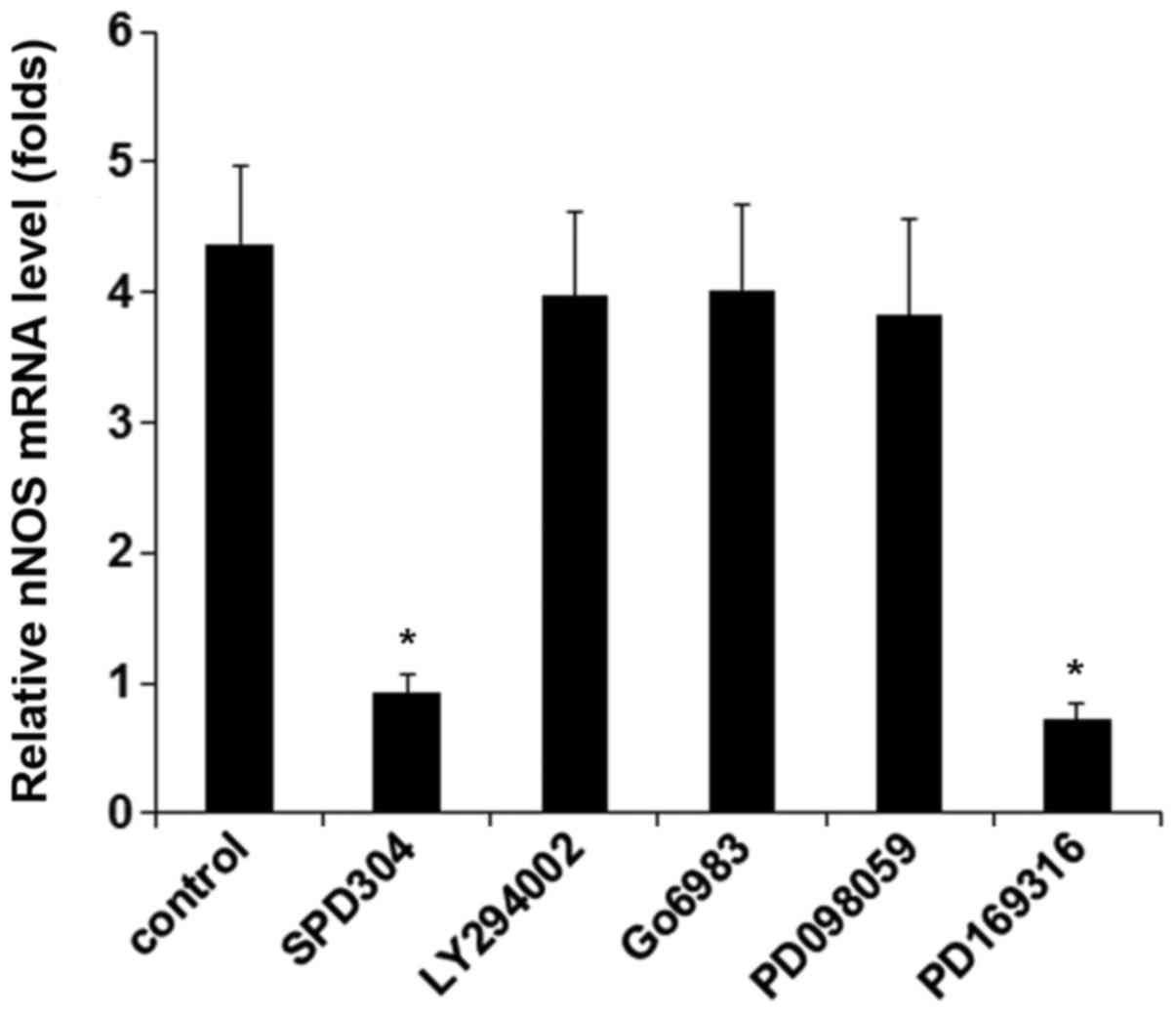
In DRG neurons treated with TNF-α (40 ng/ml for 25
h), inhibiting TNFR1 by SPD304 or inhibiting p38 MAPK by PD169316
completely abolished TNF-α-induced expression of nNOS, while
inhibition of phosphatidylinositol-3 kinase, protein kinase C, and
mitogen-activated protein kinase showed no significant effect
(Fig. 3). The results suggested
that TNF-α induced the expression/activity of nNOS in DRG neurons
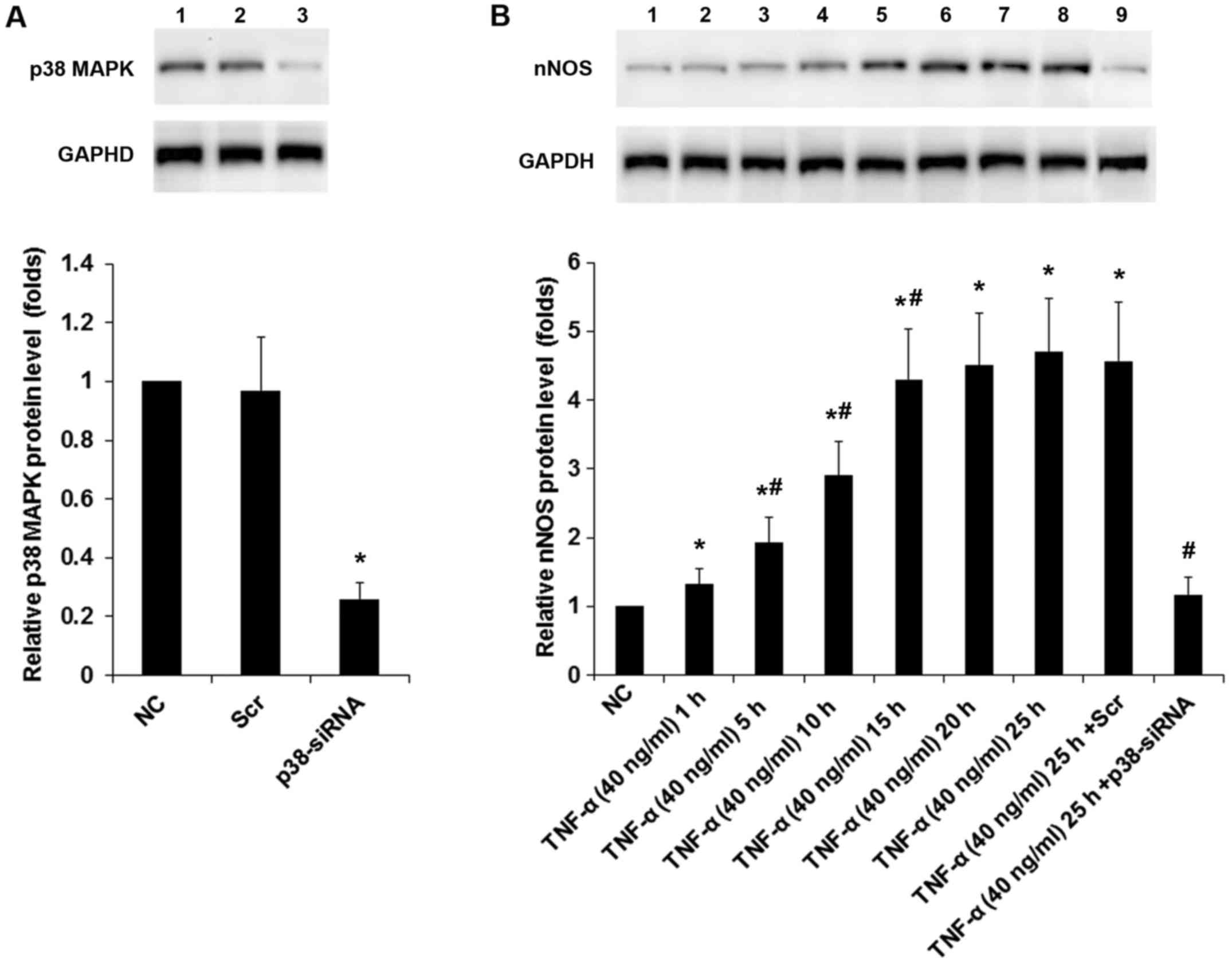
via TNFR1 by a p38 MAPK-dependent mechanism. We next knocked down
p38 MAPK in DRG neurons. As shown in Fig. 4A, the constitutive expression of
p38 MAPK was knocked down by specific siRNA by ~75% compared with
the controls. Western blot analyses confirmed that TNF-α (40 ng/ml)
time-dependently increased the protein levels of nNOS in DRG
neurons; this effect was largely abolished by p38 MAPK-siRNA
(Fig. 4B).
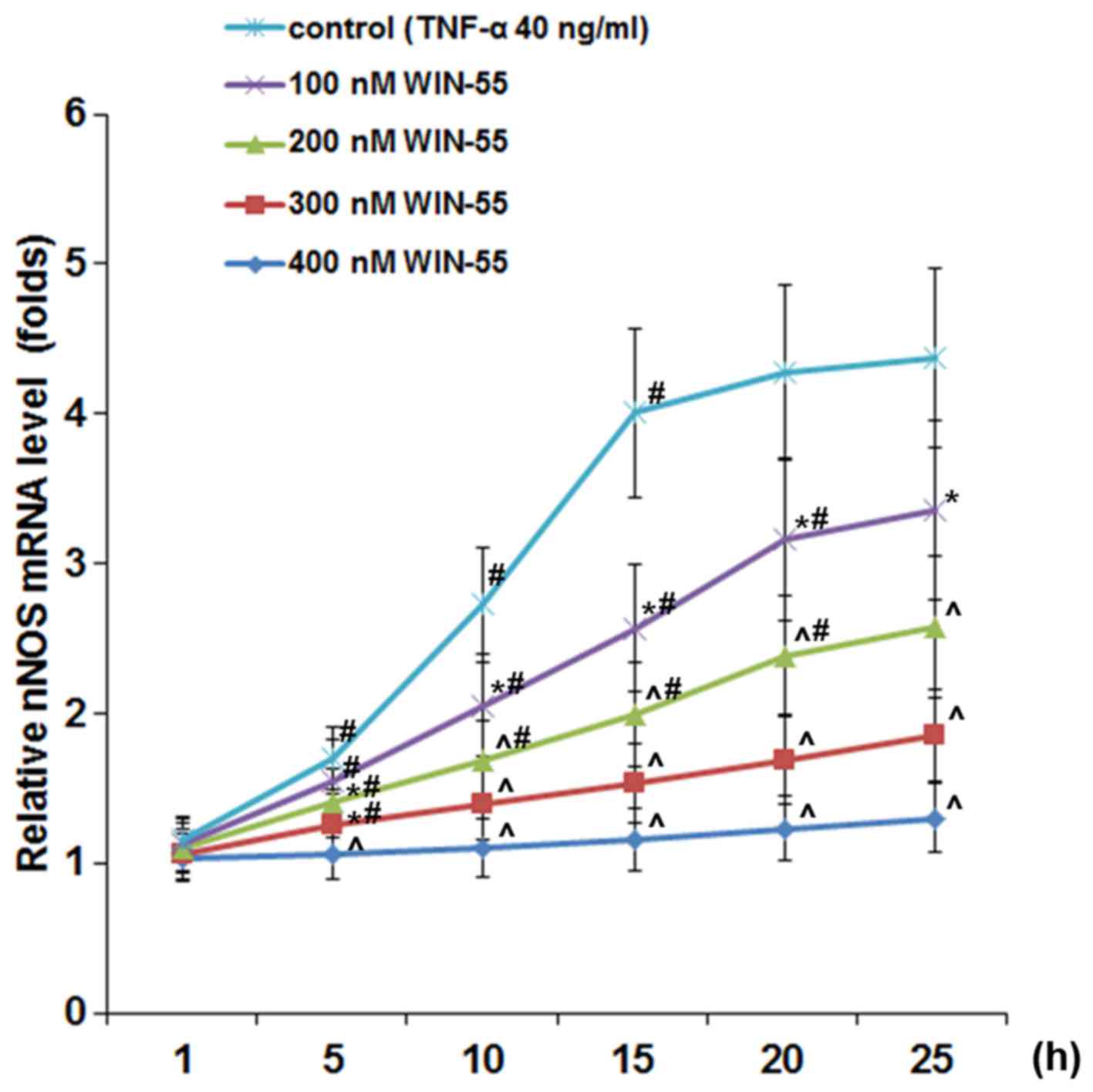
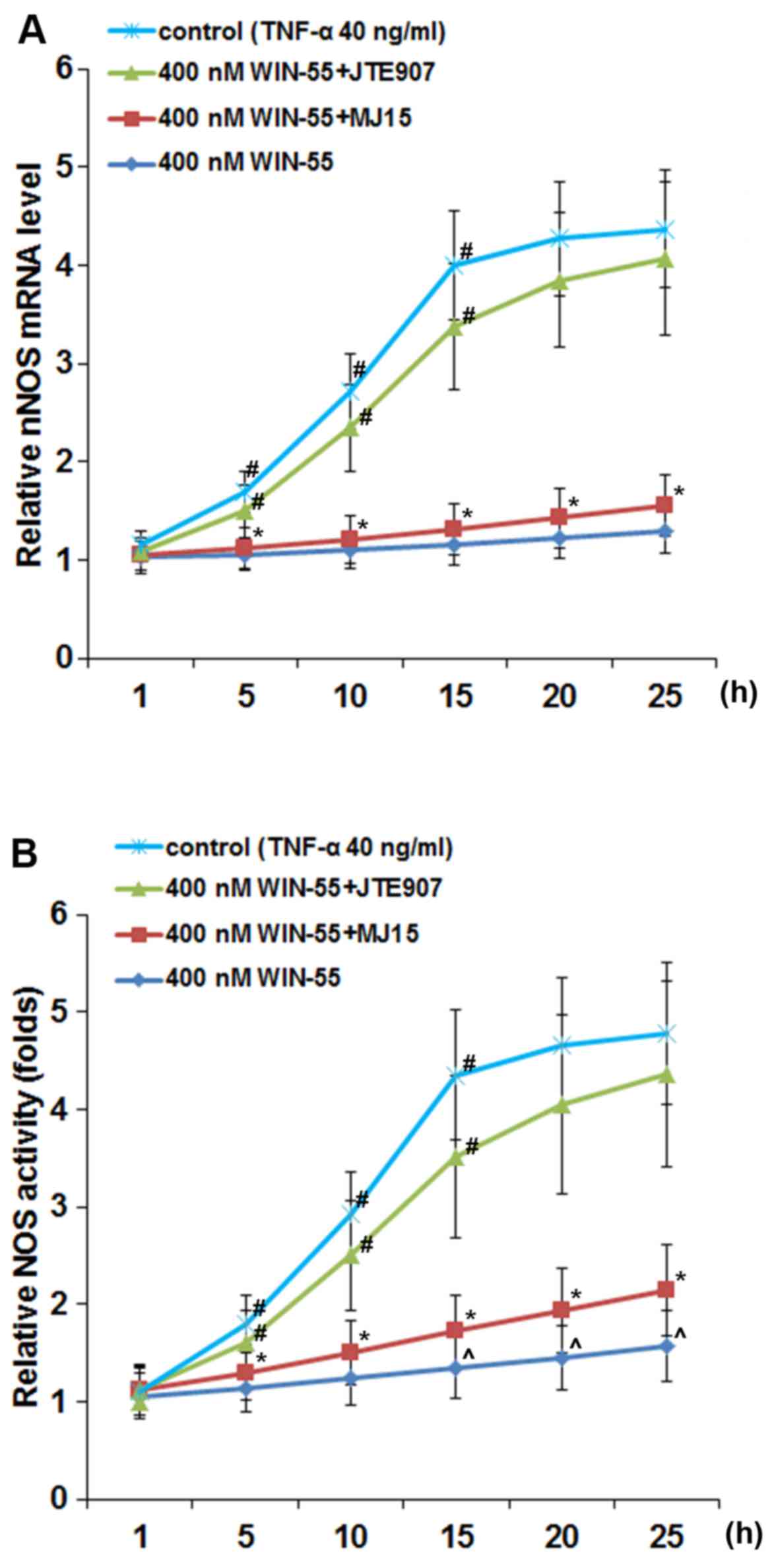
WIN-55 inhibits TNF-α-induced expression
of nNOS via CB2 in DRG neurons
As shown in Fig.
5, in DRG neurons treated with TNF-α (40 ng/ml), synthetic
cannabinoid WIN-55 (100, 200, 300 and 400 ng/ml) concentration- and
time-dependently abolished TNF-α-induced expression of nNOS, with
WIN-55 at 400 ng/ml completely abolishing the effect of TNF-α. In
DRG neurons treated with TNF-α (40 ng/ml) in the presence of WIN-55
(400 ng/ml), selective CB2 antagonist JTE907 but not selective CB1
antagonist MJ15 largely blocked the inhibitory effect of WIN-55 on
TNF-α-induced expression of nNOS (Fig. 6A) and NOS activity (Fig. 6B).
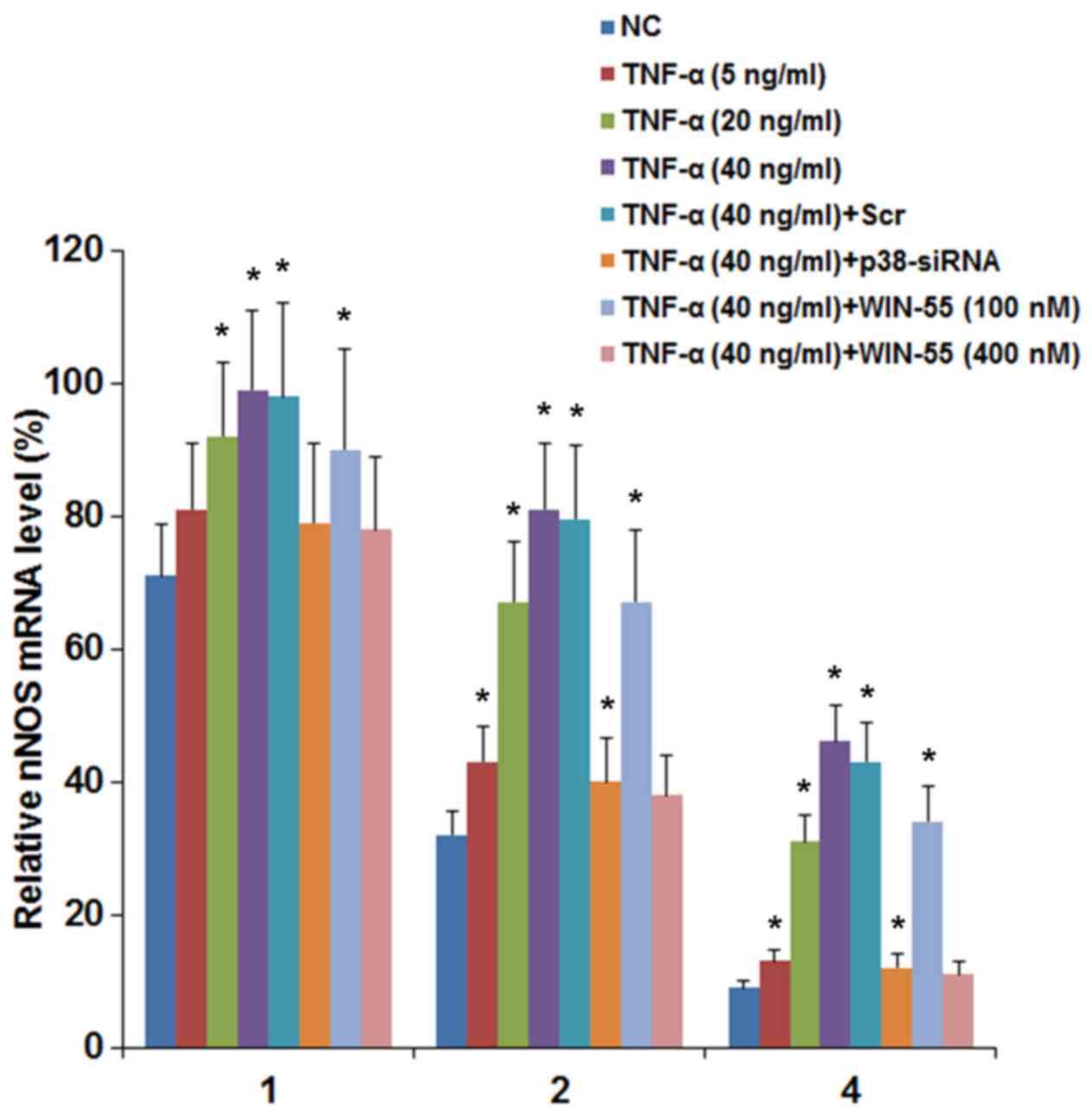
WIN-55 inhibits TNF-α-induced increase of
nNOS mRNA stability in DRG neurons
The above findings suggested that TNF-α induced the
expression of nNOS in DRG neurons by a p38 MAPK-dependent
mechanism; WIN-55 effectively inhibited this effect mainly via CB2.
Since TNF-α and WIN-55 showed no significant effect on the nNOS
gene promoter (data not shown), we next examined the effect of
TNF-α and WIN-55 on the stability of nNOS mRNA. DRG neurons were
treated with TNF-α (5, 20 and 40 ng/ml) for 25 h in the presence of
WIN-55 (100 or 400 nM). Then the cells were cultured in media
containing transcription inhibitor actinomycin D for 1, 2 and 4 h.
The mRNA levels of nNOS were determined at 1, 2 and 4 h of
actinomycin D treatment and expressed as percentages of that
immediately before actinomycin D treatment in each experimental
group. As shown in Fig. 7, the
relative nNOS mRNA levels in the control cells at 1, 2 and 4 h of
actinomycin D treatment were 71, 32 and 9%, respectively. TNF-α
concentration-dependently elevated the relative nNOS mRNA levels,
up to 99, 81 and 46% by 40 ng/ml of TNF-α at 1, 2 and 4 h of
actinomycin D treatment, respectively (Fig. 7). On the other hand, WIN-55
concentration-dependently inhibited the effect of TNF-α.
Particularly, in the presence of 40 ng/ml of TNF-α, WIN-55 at 400
nM decreased the relative nNOS mRNA levels to 78, 38 and 11% at 1,
2 and 4 h of actinomycin D treatment, respectively, nearly down to
the control level (Fig. 7). In
addition, in the presence of 40 ng/ml of TNF-α, siRNA-mediated
knockdown of p38 MAPK decreased the relative nNOS mRNA levels down
to 81, 41 and 12% at 1, 2 and 4 h of actinomycin D treatment,
respectively (Fig. 7). The
findings suggested that TNF-α increased the mRNA stability of nNOS
in DRG neurons by a p38 MAPK-dependent mechanism; WIN-55 inhibited
this effect.
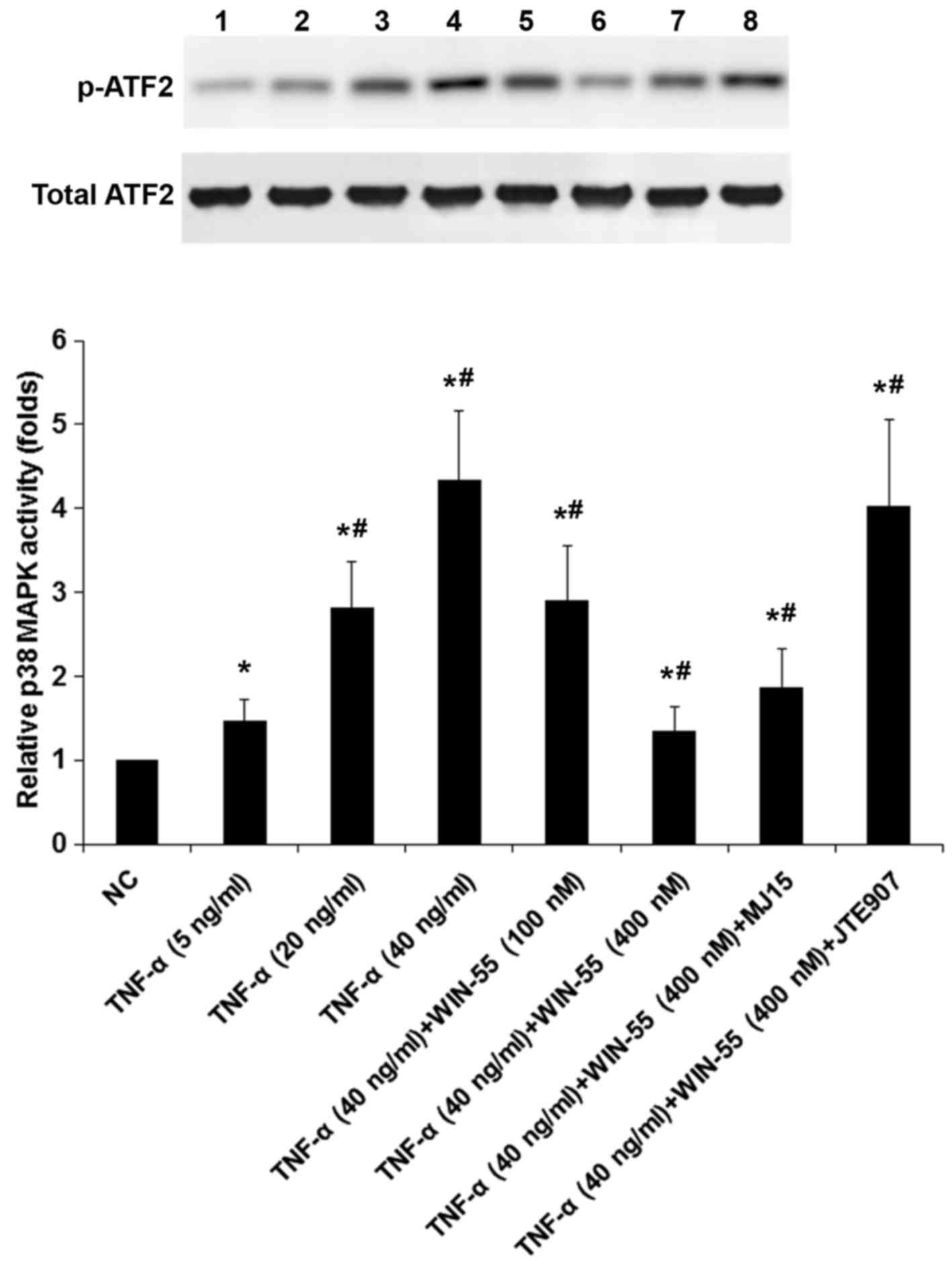
WIN-55 inhibits TNF-α-induced p38 MAPK
activity in DRG neurons
To explore potential functional interaction of TNF-α
with WIN-55 on p38 MAPK signaling, we measured p38 MAPK activities
in DRG neurons treated with TNF-α (5, 20 and 40 ng/ml) in the
presence of WIN-55 (100 or 400 ng/ml) for 25 h with or without MJ15
or JTE907 (Fig. 8). As evidenced
by increased levels of phosphorylated ATF2, TNF-α
concentration-dependently induced the p38 MAPK activity, which was
concentration-dependently decreased by WIN-55; the inhibitory
effect of WIN-55 was largely blocked by JTE907 but not MJ15
(Fig. 8). The findings suggested
that WIN-55 inhibited TNF-α-induced p38 MAPK activity in DRG
neurons mainly via CB2.
Acknowledgments
Not applicable.
Discussion
TNF-α is an established pain modulator in the
peripheral nervous system following peripheral nerve injury
(5). Elevated levels of TNF-α in
DRG neurons reportedly is critical for neuropathic pain processing
(5). NO plays an important role
in the development and maintenance of nociception (7). It has been shown that the regulation
of NO production mainly depends on the expression of NOSs and their
activities (7). Previous studies
have shown beneficial effects of selective blockers for NOS in
reducing neuropathic pain (7,8).
Accumulating evidence also support an important role of
cannabinoids in the modulation of neuropathic pain (9,10).
Significant antinociceptive effects of cannabinoid receptor
agonists have been demonstrated in animal models of neuropathic
pain (19). In the present study,
we provide the first evidence that cannabinoids inhibits
TNF-α-induced expression/activity of NOS in DRG neurons, thereby
linking the functions of TNF-α, NOS and cannabinoid in DRG
neurons.
We found that TNF-α significantly increased the
expression of nNOS but not iNOS and eNOS in DRG neurons via TNFR1.
This is in agreement with previous studies showing that only nNOS
is increased in mouse spinal cord after inflammation induction
(7) and that TNFR1 is expressed
on most cells as a major mediator of the cytotoxicity of TNF-α
(6). TNF-α in the concentration
range of 5-40 ng/ml increasingly elevated the NOS activity in DRG
neurons, until reaching a plateau at 30-40 ng/ml. In addition, at
TNF-α concentrations ≥20 ng/ml, the inducing effect of TNF-α on the
NOS activity reached a plateau after 15 h of treatment. The
findings suggest that TNF-α is an effective fast inducer of the NOS
activity, which is in line with its strong and fast induction of
nNOS in DRG neurons.
Jia et al showed that TNF-α could
phosphorylate and activate p38 MAPK (22). Wang et al showed that a
specific TNF-α inhibitor significantly reduced phosphorylated p38
MAPK levels induced by chronic constriction injury in DRG neurons
(23). Xu et al suggested
that p38 MAPK activation in DRG neurons is necessary for the
initiation and maintenance of neuropathic pain (24). These previous findings suggest
that TNF-α/p38 MAPK signaling is an important pathway for
neuropathic pain. This is in agreement with our findings that
TNF-α-induced p38 MAPK activity is required for TNF-α-induced mRNA
stability/expression/activity of nNOS, another key player in
neuropathic pain. How TNF-α-induced activation of p38 MAPK leads to
increased mRNA stability of nNOS in DRG neurons will be explored in
our future studies.
We found that synthetic cannabinoid WIN-55 potently
inhibited TNF-α-induced activation of p38 MAPK largely via CB2,
which explains for why WIN-55 inhibited TNF-α-p38 MAPK
signaling-induced expression of nNOS via CB2 in DRG neurons. This
is in agreement with previous studies showing that CB2 agonists
markedly inhibited the activation of p38 MAPK in DRG and
effectively controlled neuropathic pain in animal models (19,25,26). Taken together, our findings
suggest that cannabinoids are potent inhibitors of the TNF-α/p38
MAPK/NOS signaling axis in DRG neurons; the effect is mainly
mediated via CB2. This adds new insights into the molecular
mechanisms underlying the pharmacologic effects of cannabinoids on
neuropathic pain.
Endogenous cannabinoids and their receptors have
been found to be expressed in key areas associated with pain
processing and markedly increase in these areas in models of
chronic pain (11-15), suggesting that endogenous
cannabinoids are natural modulators of chronic pain. As endogenous
cannabinoids may well participate in regulating TNF-α/p38 MAPK/NOS
signaling during the development of neuropathic pain, our findings
also adds new insights into the pathophysiology of neuropathic pain
and other chronic pains, e.g. inflammatory pain, in which TNF-α/p38
MAPK/NOS signaling is very likely involved.
In conclusion, our findings suggest that TNF-α
induces expression/activity of nNOS in DRG neurons by increasing
its mRNA stability by a p38 MAPK-dependent mechanism; synthetic
cannabinoid WIN-55 inhibits this effect of TNF-α by inhibiting p38
MAPK via CB2.
Funding
This study was supported by the Hunan Provincial
Natural Science Foundation (grants no. 2015A537), Changsha, Hunan,
China.
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RT participated in the study design, collected the
data, carried out data analysis, and drafted the manuscript. LC
participated in the study design, carried out data analysis, and
performed data check and proofreading. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Saarto T and Wiffen PJ: Antidepressants
for neuropathic pain. Cochrane Database Syst Rev.
3:CD0054542005.
|
|
2
|
Lin HC, Huang YH, Chao TH, Lin WY, Sun WZ
and Yen CT: Gabapentin reverses central hypersensitivity and
suppresses medial prefrontal cortical glucose metabolism in rats
with neuropathic pain. Mol Pain. 10:632014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Przewlocki R and Przewlocka B: Opioids in
neuropathic pain. Curr Pharm Des. 11:3013–3025. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chu LW, Chen JY, Wu PC and Wu BN:
Atorvastatin prevents neuroinflammation in chronic constriction
injury rats through nuclear NFκB downregulation in the dorsal root
ganglion and spinal cord. ACS Chem Neurosci. 6:889–898. 2015.
View Article : Google Scholar
|
|
5
|
Yang Y, Wu H, Yan JQ, Song ZB and Guo QL:
Tumor necrosis factor-α inhibits angiotensin II receptor type 1
expression in dorsal root ganglion neurons via β-catenin signaling.
Neuroscience. 248:383–391. 2013. View Article : Google Scholar
|
|
6
|
Westacott CI, Atkins RM, Dieppe PA and
Elson CJ: Tumor necrosis factor-alpha receptor expression on
chondrocytes isolated from human articular cartilage. J Rheumatol.
21:1710–1715. 1994.PubMed/NCBI
|
|
7
|
Boettger MK, Uceyler N, Zelenka M, Schmitt
A, Reif A, Chen Y and Sommer C: Differences in inflammatory pain in
nNOS-, iNOS- and eNOS-deficient mice. Eur J Pain. 11:810–818. 2007.
View Article : Google Scholar
|
|
8
|
Tao F, Tao YX, Zhao C, Doré S, Liaw WJ,
Raja SN and Johns RA: Differential roles of neuronal and
endothelial nitric oxide synthases during carrageenan-induced
inflammatory hyperalgesia. Neuroscience. 128:421–430. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hama A and Sagen J: Antinociceptive effect
of cannabinoid agonist WIN 55,212-2 in rats with a spinal cord
injury. Exp Neurol. 204:454–457. 2007. View Article : Google Scholar
|
|
10
|
Rahn EJ and Hohmann AG: Cannabinoids as
pharmacotherapies for neuropathic pain: From the bench to the
bedside. Neurotherapeutics. 6:713–737. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hegyi Z, Kis G, Holló K, Ledent C and
Antal M: Neuronal and glial localization of the cannabinoid-1
receptor in the superficial spinal dorsal horn of the rodent spinal
cord. Eur J Neurosci. 30:251–262. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hsieh GC, Pai M, Chandran P, Hooker BA,
Zhu CZ, Salyers AK, Wensink EJ, Zhan C, Carroll WA, Dart MJ, et al:
Central and peripheral sites of action for CB2 receptor mediated
analgesic activity in chronic inflammatory and neuropathic pain
models in rats. Br J Pharmacol. 162:428–440. 2011. View Article : Google Scholar :
|
|
13
|
Lim G, Sung B, Ji RR and Mao J:
Upregulation of spinal cannabinoid-1-receptors following nerve
injury enhances the effects of Win 55,212-2 on neuropathic pain
behaviors in rats. Pain. 105:275–283. 2003. View Article : Google Scholar
|
|
14
|
Amaya F, Shimosato G, Kawasaki Y,
Hashimoto S, Tanaka Y, Ji RR and Tanaka M: Induction of CB1
cannabinoid receptor by inflammation in primary afferent neurons
facilitates antihyperalgesic effect of peripheral CB1 agonist.
Pain. 124:175–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fernández-Ruiz J, Pazos MR,
García-Arencibia M, Sagredo O and Ramos JA: Role of CB2 receptors
in neuroprotective effects of cannabinoids. Mol Cell Endocrinol.
286(Suppl 1): S91–S96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mbvundula EC, Bunning RA and Rainsford KD:
Arthritis and cannabinoids: HU-210 and Win-55,212-2 prevent
IL-1alpha-induced matrix degradation in bovine articular
chondrocytes in-vitro. J Pharm Pharmacol. 58:351–358. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Q, Bhat M, Bowen WD and Cheng J:
Signaling pathways from cannabinoid receptor-1 activation to
inhibition of N-methyl-D-aspartic acid mediated calcium influx and
neurotoxicity in dorsal root ganglion neurons. J Pharmacol Exp
Ther. 331:1062–1070. 2009. View Article : Google Scholar :
|
|
18
|
Anand U, Otto WR, Sanchez-Herrera D, Facer
P, Yiangou Y, Korchev Y, Birch R, Benham C, Bountra C, Chessell IP,
et al: Cannabinoid receptor CB2 localisation and agonist-mediated
inhibition of capsaicin responses in human sensory neurons. Pain.
138:667–680. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Paszcuk AF, Dutra RC, da Silva KA, Quintão
L, Campos MM and Calixto JB: Cannabinoid agonists inhibit
neuropathic pain induced by brachial plexus avulsion in mice by
affecting glial cells and MAP kinases. PLoS One. 6:e240342011.
View Article : Google Scholar :
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
21
|
Wang X, Wu H and Miller AH: Interleukin
1alpha (IL-1alpha) induced activation of p38 mitogen-activated
protein kinase inhibits glucocorticoid receptor function. Mol
Psychiatry. 9:65–75. 2004. View Article : Google Scholar
|
|
22
|
Jia P, Wang J, Wang L, Chen X, Chen Y, Li
WZ, Long R, Chen J, Shu YW, Liu K, et al: TNF-α upregulates Fgl2
expression in rat myocardial ischemia/reperfusion injury.
Microcirculation. 20:524–533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang RK, Zhang QQ, Pan YD and Guo QL:
Etanercept decreases HMGB1 expression in dorsal root ganglion
neuron cells in a rat chronic constriction injury model. Exp Ther
Med. 5:581–585. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu JT, Xin WJ, Wei XH, Wu CY, Ge YX, Liu
YL, Zang Y, Zhang T, Li YY and Liu XG: p38 activation in uninjured
primary afferent neurons and in spinal microglia contributes to the
development of neuropathic pain induced by selective motor fiber
injury. Exp Neurol. 204:355–365. 2007. View Article : Google Scholar
|
|
25
|
Wilkerson JL, Gentry KR, Dengler EC,
Wallace JA, Kerwin AA, Kuhn MN, Zvonok AM, Thakur GA, Makriyannis A
and Milligan ED: Immunofluorescent spectral analysis reveals the
intrathecal cannabinoid agonist, AM1241, produces spinal
anti-inflammatory cytokine responses in neuropathic rats exhibiting
relief from allodynia. Brain Behav. 2:155–177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wilkerson JL, Gentry KR, Dengler EC,
Wallace JA, Kerwin AA, Armijo LM, Kuhn MN, Thakur GA, Makriyannis A
and Milligan ED: Intrathecal cannabilactone CB(2)R agonist, AM1710,
controls pathological pain and restores basal cytokine levels.
Pain. 153:1091–1106. 2012. View Article : Google Scholar
|