Introduction
Pulmonary arterial hypertension (PAH) is a vascular
remodeling disease of the lungs that results in right ventricular
failure and death within a few years of diagnosis (1). Chronic hypoxia-induced PAH, a common
type of PAH, is predominantly secondary to disorders of the
respiratory system, including chronic obstructive pulmonary disease
(COPD) and obstructive sleep apnea (2). Although the precise pathogenesis of
hypoxia-induced PAH remains unclear, it is thought that vascular
endothelial damage and dysfunction serve a crucial role in
triggering pathological vascular remodeling (3). Pulmonary endothelial cell
dysfunction in hypoxia-induced PAH has been demonstrated to be
associated with decreased production of nitric oxide (NO) and
increased production of endothelin-1 (ET-1), which participate in
the subsequent abnormal proliferation of pulmonary endothelial
cells and pulmonary smooth muscle cells (4,5).
In addition, experimental studies have suggested that endothelial
cell apoptosis in the pulmonary microvasculature causes arteriolar
occlusion and increases pulmonary vascular resistance, suggesting
that vascular endothelial cell apoptosis is closely associated with
the pathogenesis of PAH (6,7).
The mechanisms responsible for endothelial cell apoptosis in the
early stages of hypoxia-induced PAH are yet to be fully
elucidated.
In recent studies, endoplasmic reticulum stress
(ERS) has been demonstrated to have an important role in the
development of PAH. Hypoxia, toxicity, infection and perturbation
of Ca2+ homeostasis lead to the accumulation of unfolded
or misfolded proteins, which results in ERS, activating the
adaptive cellular response termed the unfolded protein response
(UPR) (8,9). Under mildly stressful conditions,
these pathways activate several transcription factors involved in
cell survival. However, under conditions of severe acute or chronic
stress, the ER UPR shifts from survival to cell death signaling
pathways (10). In previous
studies, it was identified that ERS participates in the development
of PAH in vitro and in vivo. Sutendra and Michelakis
(1) reported that, in response to
hypoxia-induced stress, the Nogo B protein from mice pulmonary
artery smooth muscle cells (PASMCs), which regulates ER structure
and has been implicated in vascular remodeling, serves a role in
the development of PAH. Koyama et al (11) demonstrated that activation of all
branches of the UPR and accompanying inflammation in human
pulmonary smooth muscle cells have a major role in the pathogenesis
of PAH, and that the chemical chaperones that inhibit ERS may be
potential therapeutic agents for PAH. Although studies have
indicated that ERS participates in the process of PASMC
proliferation in hypoxia-induced PAH, the connection between
endothelial dysfunction and ERS in the development of
hypoxia-induced PAH remains unclear.
Fan et al (12) demonstrated that ERS-associated
proteins and the ERS-dependent apoptotic protein caspase-12 were
upregulated and the number of pulmonary apoptotic cells was
markedly increased in hypoxia-induced PAH in rats, indicating that
ERS-induced apoptosis may be one of the mechanisms underlying
hypoxic pulmonary hypertension and pulmonary vascular wall
remodeling. The present study aimed to investigate whether ERS
participates in hypoxia-induced apoptosis and damage in human
pulmonary arterial endothelial cells (HPAECs) through in
vitro experiments, and to provide additional evidence on the
role of ERS in hypoxia-induced PAH.
Fibroblast growth factor (FGF)21, a novel member of
the FGF superfamily, is a crucial endogenous regulator of lipid
metabolism and systemic glucose. Circulating endogenous FGF21 is
elevated in obesity, type 2 diabetes and coronary artery disease.
Therefore, FGF21 is considered a stress hormone, mediating the
adaptive metabolic response to various stress conditions (13). In addition, FGF21 is reported to
be involved in various pathological conditions, including fatty
liver disease, ERS, and chronic inflammation (14). Recent studies have indicated that
both triglycerides and tunicamycin-induced ERS stimulate FGF21
expression in hepatocytes and increase serum levels of FGF21,
indicating that FGF21 has a crucial role in ERS (15,16). Guo et al (17) demonstrated that administration of
FGF21 restores insulin signaling via inhibiting ERS in the adipose
tissue of high-fat diet-induced obese mice. In addition, FGF21 has
been reported to protect cardiac endothelial cells from damage and
to suppress inflammatory responses (18). FGF21 may be a signal of injured
target tissue and may have physiological roles in improving
endothelial function at an early stage of atherosclerosis and in
halting the development of coronary heart disease (18). It is well known that there are
many similarities between the cardiovascular system and the
pulmonary vasculature. However, research regarding the associations
between FGF21 and PAH is limited. Whether FGF21 could protect
endothelial cells from damage, as is the case in the cardiovascular
system, remains poorly delineated. Considering the aforementioned
relationship between FGF21 and ERS, the present study hypothesized
that FGF21 could decrease hypoxia-induced apoptosis and repair
damage to HPAECs to improve endothelial function though alleviating
ERS.
Materials and methods
Reagents
Salubrinal (an ERS inhibitor; cat. no. SML0951) and
tunicamycin (an ERS agonist; cat. no. T7765) were obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Rabbit antibodies
against binding immunoglobulin protein (BiP), protein kinase R-like
endoplasmic reticulum kinase (PERK), B-cell lymphoma-2 (Bcl-2),
caspase-4 and GAPDH were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Rabbit antibodies against phosphorylated
(p-) PERK were purchased from Biorbyt (Cambridge, UK). Mouse
antibodies against cluster of differentiation 31 (CD31) and
transcription factor C/EBP homologous protein (CHOP) were purchased
from Abcam (Cambridge, UK) and Cell Signaling Technology, Inc.,
respectively. An anti-mouse immunoglobulin G (IgG) horseradish
peroxidase (HRP)-linked antibody and an anti-rabbit IgG HRP-linked
antibody were purchased from Cell Signaling Technology, Inc. FGF21
was obtained from the Wenzhou Medical College Pharmacy School
(Wenzhou, China). Endothelial cell medium (ECM; 1001), fetal bovine
serum (FBS) and an endothelial cell growth supplement (ECGS) were
purchased from ScienCell Research Laboratories, Inc. (San Diego,
CA, USA). Phosphate-buffered saline (PBS) was purchased from
Hyclone; GE Healthcare Life Sciences (Logan, UT, USA). The cell
counting kit-8 (CCK-8) was purchased from Dojindo Molecular
Technologies (Kumamoto, Japan). The ELISA kit was purchased from
Boyun Biotechnology Company (Shanghai, China). The terminal
deoxyribonucleotide transferase-mediated dUTP nick end-labelling
(TUNEL) kit was purchased from Roche Diagnostics (Basel,
Switzerland).
Cell lines and cell culture
HPAECs (cat. no. 3100) were purchased from ScienCell
Research Laboratories, Inc., and used as previously described
(19). The HPAECs were cultured
in ECM, supplemented with 5% FBS, 1% ECGS, 100 U/ml penicillin and
100 µg/ml streptomycin. The medium was changed every 2 days.
When the cells reached 80-90% confluence, they were washed with PBS
and treated with 0.05% trypsin-EDTA for passaging. The cells were
identified through microscopy and by positive staining for CD31
(Fig. 1A). For experimental use,
HPAECs from passages 3-5 were sub cultured and randomly divided
into 6 groups: Normoxia group (N); hypoxia group (H); hypoxia+FGF21
group (H+F); hypoxia+salubrinal (ERS inhibitor; 20 µM) group
(H+S), with the concentrations used determined from a previous
study (20); hypoxia+tunicamycin
(ERS agonist; 2.5 ng/ml) group (H+T) with the concentrations used
determined from a previous study (21); and hypoxia+tunicamycin+FGF21 group
(H+T+F). The H+F group was subdivided into 5 groups with different
concentrations of FGF21, as follows: H+F (100 ng/ml), H+F (200
ng/ml), H+F (400 ng/ml), H+F (800 ng/ml) and H+F (1,600 ng/ml), to
obtain the optimum concentration of FGF21. Hypoxia-treated HPAECs
were treated as aforementioned for 24 h. All hypoxia groups were
kept for 24 h in the hypoxia incubator at 37°C with 5%
CO2, 5% O2 and 90% N2, with these
conditions based on the results of previous studies (22,23), whereas the normoxia group was
maintained in a normal incubator at 37°C with 21% O2, 5%
CO2 and 74% N2.
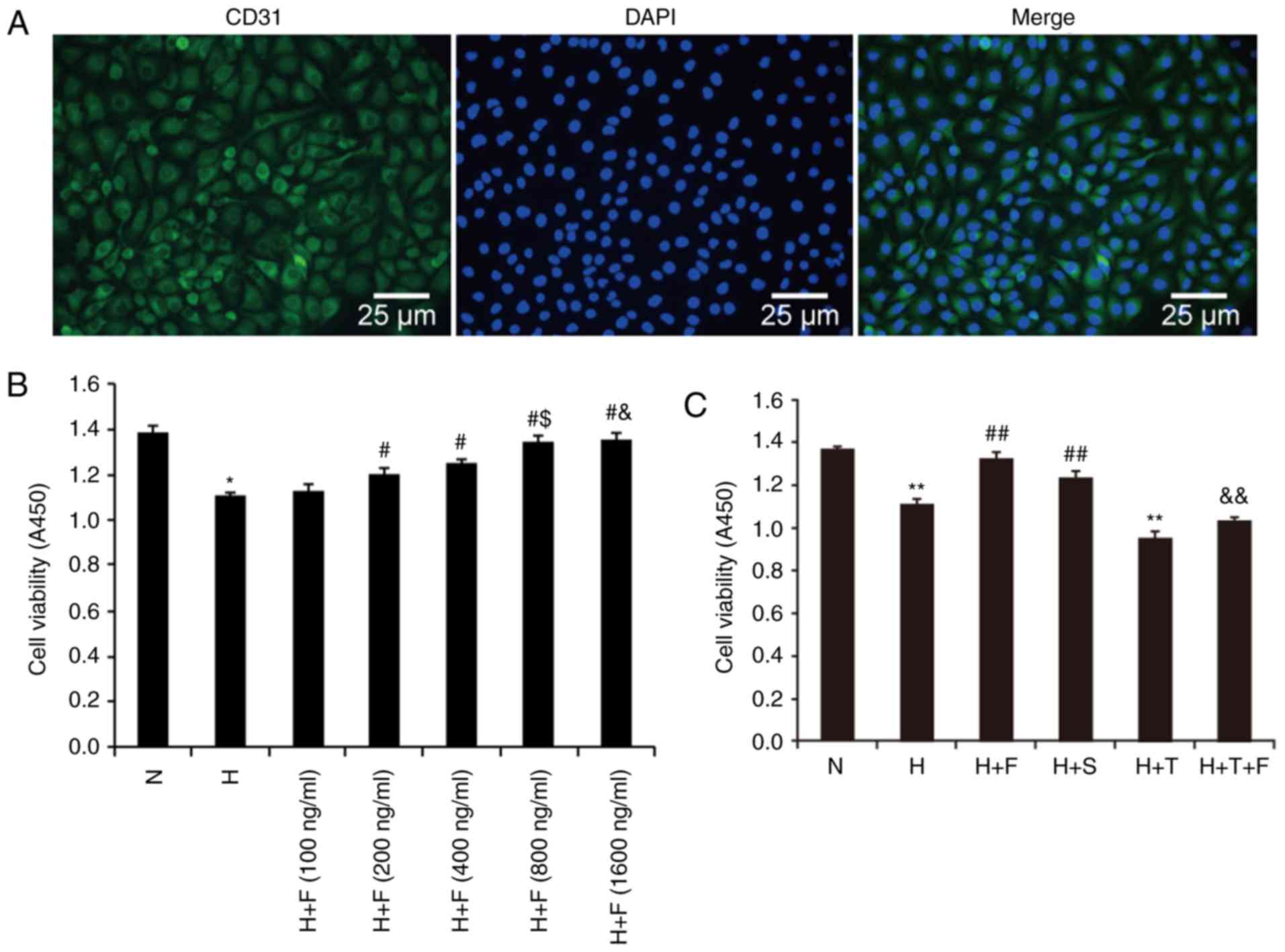 | Figure 1Characterizationof HPAECs and cell
viability in different experimental groups. (A) HPAECs were
identified by microscopy, based on positive staining for CD31
(magnification, ×100; scale bar, 25 µm). CD31 staining
appears green in the images, while nuclei are counterstained with
DAPI and appear blue. (B) HPAECs were exposed to hypoxia and
treated with different concentrations of FGF21 (100, 200, 400, 800
and 1,600 ng/ml). Absorbance of cell counting kit-8 at 450 nm in
HPAECs was used to determine the relative cell number and
viability. (C) HPAECs were exposed to hypoxia and treated with
FGF21 (800 ng/ml), ERS inhibitor salubrinal (20 µM), ERS
agonist tunicamycin (2.5 ng/ml) or tunicamycin plus FGF21. Cell
viability was analyzed via CCK-8 assays. The absorbance of CCK-8 at
450 nm was used to determine the relative cell numbers and
viability. Data are expressed as the mean ± standard deviation
(n=6). *P<0.05 vs. the N group;
**P<0.01 vs. the N group; #P<0.05 vs.
the H group; ##P<0.01 vs. the H group;
$P<0.05 vs. the H+F (400 ng/ml);
&P>0.05 vs. the H+T group (800 ng/ml);
&&P<0.01 vs. the H+T group. HPAECs, human
pulmonary arterial endothelial cells; FGF21, fibroblast growth
factor 21; ERS, endoplasmic reticulum stress; CCK-8, cell counting
kit-8; N, normoxia; H, hypoxia; F, FGF21; S, salubrinal; T,
tunicamycin. |
Cell viability assay
Cell viability was quantified through CCK-8 assays.
HPAECs were seeded at a density of 1×104 cells/well in
flat-bottomed 96-well plates. Following pre-incubation in complete
medium at 37°C and in 21% O2 and 5% CO2 for
24 h, HPAECs were pretreated with FGF21, salubrinal and tunicamyc
in prior to exposure to hypoxia. Cell viability was observed under
a microscope before the CCK-8 assay. Cell viability was observed
under a light microscope (magnification, ×100) prior to the CCK-8
assays. After 24 h of hypoxia, the CCK-8 reagent (10
µl/well) was added to each well. Following incubation at
37°C for 2 h, the absorbance of each well was determined using a
micro plate reader at 450 nm. Cell viability was determined based
on the absorbance of each well.
Detection of apoptosis
Cell apoptosis was evaluated using the TUNEL assay
(In Situ Cell Death Detection Kit, POD), according to the
manufacturer's protocol. HPAECs were exposed to hypoxia and treated
with or without the reagents indicated above. Following the
treatment, the cells adhered to cover slips and were fixed with 4%
paraformaldehyde in PBS (pH 7.4) for 1 h at 25°C. The cells were
then subjected to TUNEL assays, according to the manufacturer's
protocol. DAB was employed as the chromogen and hematoxylin as the
counterstain. Finally, the cover slips were observed via light
microscopy, and the % of TUNEL-positive cells was assessed in six
randomly selected fields from each cover slip.
Ultrastructural examination of the
endoplasmic reticulum of HPAECs
HPAECs were cultured in 25 cm2 flasks. At
the end of the hypoxia exposure period, the HPAECs were washed with
PBS and treated with 0.05% trypsin-EDTA to collect cells. Cell
aggregates were fixed with 2.5% glutaraldehyde and 1% osmic acid
for 1 hat 37°C, prior to being dehydrated with acetone and embedded
in epoxy resin 812. The fixed cell aggregates were subsequently cut
into ultrathin sections (the thickness of the sections was ~50 nm)
with an ultramicrotome and examined with a Hitachi H-7500
transmission electron microscope (Hitachi, Ltd., Tokyo, Japan)
following staining with uranyl acetate and lead citrate for 3-6 h
at 45°C and for 48 h at 65°C, respectively. Three fields were
obtained randomly.
Western blot analysis
Following treatment, HPAECs were lysed with ice-cold
RIPA lysis buffer containing PMSF for 30 min. Then, the lysates
were centrifuged at 16,000 × g for 30 min at 4°C, and the
supernatant was collected. The protein concentrations were
determined using a Pierce bicinchoninic acid protein assay kit.
Equal amounts of protein (20 µg) were then separated by 12%
SDS-PAGE, transferred onto polyvinylidene fluoride membranes,
blocked with 5% milk for 1 h at room temperature, and incubated
overnight with specific primary rabbit antibodies against BiP
(dilution, 1:1,000; 3183), PERK (dilution, 1:1,000; 3192), p-PERK
(dilution, 1:100; orb6693), Bcl-2 (dilution, 1:1,000; 2870),
caspase-4 (dilution, 1:1,000; ab22687) and GAPDH (dilution,
1:1,000; 5174S) and a mouse antibody against CHOP (dilution,
1:1,000; 2895) at 4°C. The membranes were subsequently washed with
TBS/0.1% Tween-20 (TBST) buffer and membranes that had been
incubated with anti-BiP, anti-PERK, anti-p-PERK, anti-Bcl-2,
anti-caspase-4 and anti-GAPDH antibodies were incubated with
anti-rabbit IgG HRP-linked antibody (dilution, 1:10,000; 7074),
while membranes that had been incubated with the anti-CHOP antibody
were incubated with an anti-mouse IgG HRP-linked antibody
(dilution, 1:10,000; 7076) for 1 h at room temperature. Super
Signal West Fem to Maximum Sensitivity substrate (cat. no. 3409;
Shanghai Boyun BioTech Co., Ltd., Shanghai, China) was used as the
visualization reagent. GAPDH was used as an internal control. The
optical density of the immune blots was quantified with Quantity
one-4.6.2 software (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Transwell migration chamber assay
The 24-well Transwell system (5 µm; Corning
Incorporated, Corning, NY, USA) was used to perform cell migration
assays. A total of 600 µl ECM, containing 5% FBS was added
to the lower chamber with the experimental treatments. Then, HPAECs
were suspended in ECM without serum, and 200 µl cell
suspension (at a density of 1.5×105/ml) was added to the
upper chambers of the Transwell plates. Following incubation at
37°C for 24 h, the non-migrated cells were removed using a cotton
swab. The migrated cells were then fixed with 4% paraformaldehyde
for 10 min, followed by staining with crystal violet for 30 min.
Finally, the number of migrated cells was counted under a
microscope (Nikon Corporation, Tokyo, Japan), six fields were
obtained randomly.
ELISA of NO and ET-1
The accumulated NO and ET-1 levels in the culture
medium were determined using ELISA kits (NO, cat. no. 30417H; ET-1,
cat. no. 30538H), according to the manufacturer's protocol.
Statistical analysis
All of the experiments were repeated at least three
times. The results are expressed as the mean ± standard deviation.
Statistical significance was determined by one-way analysis of
variance, followed by the least significant difference (LSD) test
(equal variances assumed) or Dunnett's T3 test (equal variances not
assumed). P<0.05 was considered to indicate a statistically
significant difference. All calculations were performed using SPSS
version 20.0 (IBM Corporation, Armonk, NY, USA).
Results
Hypoxia and the ERS agonist tunicamycin
reduce the viability of HPAECs, and this effect is improved by
FGF21 and the ERS inhibitor salubrinal
The absorbance measured at 450 nm in HPAECs
subjected to the CCK-8 assay was used to determine relative cell
numbers and viability. As indicated in Fig. 1B, the absorbance was reduced in
the H group compared with the N group (P<0.05). Under
cotreatment with FGF21, the absorbance was significantly enhanced
in the H+F (200 ng/ml), H+F (400 ng/ml), H+F (800 ng/ml) and H+F
(1,600 ng/ml) compared with the H group (P<0.05; Fig. 1B). The absorbance was
significantly increased in the H+F (800 ng/ml) group compared with
the H+F (400 ng/ml) group (P<0.05; Fig. 1B). Meanwhile, compared with the
H+F (800 ng/ml) group, there was no significance in the increase of
absorbance in the H+F (1,600 ng/ml) (P>0.05; Fig. 1B). We chose 800 ng/ml as the
optimum concentration of FGF21 in subsequent experiments.
As indicated in Fig.
1C, the absorbance was reduced in the H group and the H+T group
compared with that of the N group (P<0.01). Under co-treatment
with FGF21 or salubrinal, the absorbance was significantly enhanced
in the H+F and H+S groups compared with that of the H group
(P<0.01). The absorbance was increased in the H+T+F group
compared with that of the H+T group (P<0.01). The results
indicated that FGF21, as well as the ERS inhibitor salubrinal,
improved the viability of HPAECs under conditions of hypoxia.
Hypoxia and the ERS agonist tunicamycin
increase apoptosis, and this effect is reversed by FGF21 and the
ERS inhibitor salubrinal
The effects of FGF21 on the apoptosis of HPAECs were
evaluated by TUNEL assays. As illustrated in Fig. 2, the % of apoptotic HPAECs
(indicated by brown-stained cell nuclei) was significantly
increased in the H group compared with the N group (P<0.01).
FGF21 or salubrinal treatment decreased the % of apoptotic cells in
the H+F and H+S groups compared with the H group (P<0.01;
Fig. 2). There was a significant
increase in TUNEL-positive cells in the H+T group compared with the
N group (P<0.01; Fig. 2), but
cotreatment with FGF21 attenuated cell apoptosis in the H+T+F group
(P<0.01; Fig. 2). These
findings were consistent with the protective function of FGF21 in
cardiac endothelial cells (18),
suggesting that FGF21 could decrease the rate of apoptosis induced
by hypoxia and protect HPAECs from damage or stress.
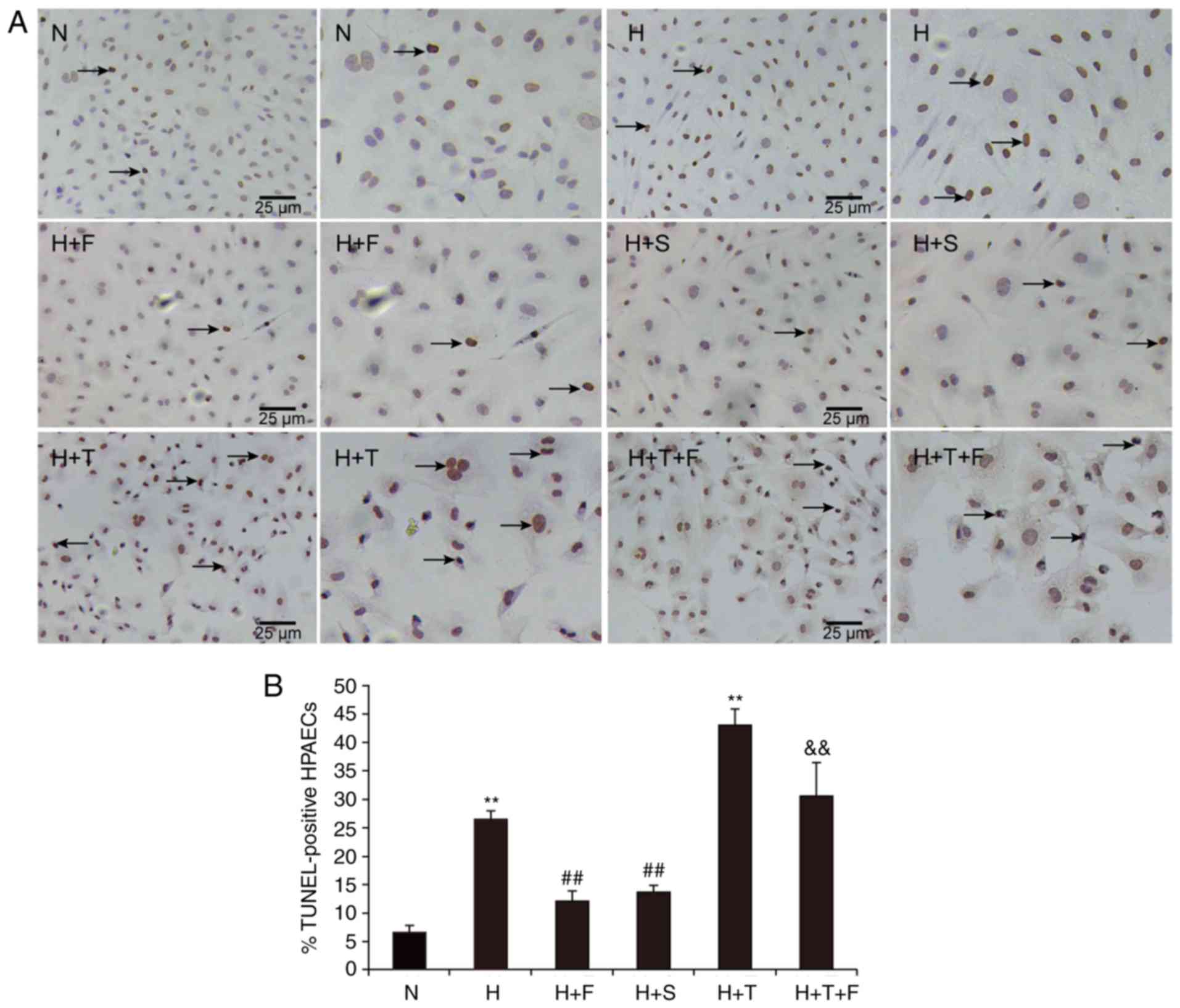 | Figure 2(A and B) Hypoxia increases the
apoptosis of HPAECs, and this is reversed by FGF21. The HPAECs from
each experimental group were stained with the TUNEL reagent and
analyzed by microscopy (magnification, ×100; scale bar, 25
µm). The arrows denote TUNEL-positive apoptotic HPAECs
(brown staining). Data are expressed as the mean ± standard
deviation (n=6). **P<0.01 vs. the N group;
##P<0.01 vs. the H group;
&&P<0.01 vs. the H+T group. HPAECs, human
pulmonary arterial endothelial cells; FGF21, fibroblast growth
factor 21; TUNEL, terminal deoxyribonucleotide transferase-mediated
dUTP nick end-labelling assay; N, normoxia; H, hypoxia; F, FGF21;
S, salubrinal; T, tunicamycin. |
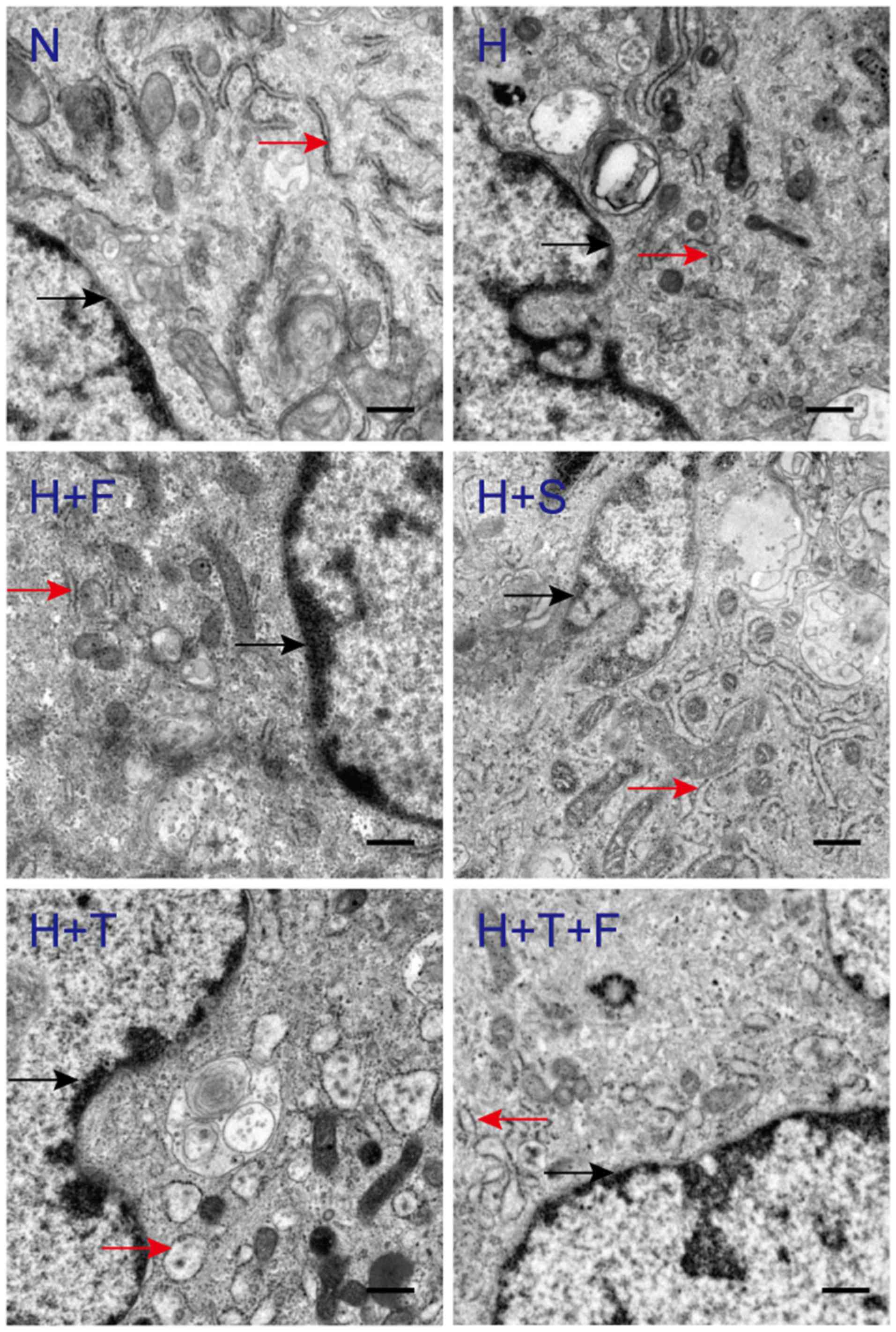
FGF21 reverses the hypoxia-induced
endoplasmic reticulum dilation of HPAECs
To determine whether FGF21 could protect HPAECs
against hypoxia-induced ERS, the cells were examined at the
ultrastructural level. Compared with the N group, the rough
endoplasmic reticulum lumens of the H group and the H+T group were
notably expanded, and ribosomes had become detached from the
endoplasmic reticulum (Fig. 3).
In the H+F and the H+S groups, the expansion of the endoplasmic
reticulum lumen was reversed by FGF21 or salubrinal, similar to the
normoxia group (Fig. 3). The
endoplasmic reticulum lumen of the H+T group was considerably
expanded, while the endoplasmic reticulum lumen of the H+T+F group
was not repaired well, and mild expansion of the endoplasmic
reticulum lumen continued to be observed in the H+T+F group
(Fig. 3). These results indicated
that FGF21 is a protective factor that could reverse the
hypoxia-induced endoplasmic reticulum dilation of HPAECs.
FGF21 alleviates hypoxia-induced ERS via
the PERK/CHOP signaling pathway and via inhibition of caspase-4
expression
In the present study, the roles of the PERK branch
and the ERS-dependent apoptosis factor caspase-4 were examined, as
regulators of ERS in hypoxia-induced HPAEC damage. To this effect,
the protein expression levels of these proteins were measured by
western blotting.
The expression of representative proteins in the
PERK branch of ERS, including BiP, p-PERK and CHOP, was
significantly increased by hypoxia in the H group compared with the
N group (P<0.01; Fig. 4A, B and
D). This result indicated that the PERK branch of ERS was
activated under hypoxic conditions in HPAECs. FGF21 or salubrinal
treatment reduced the expression of BiP, p-PERK and CHOP in the H+F
and H+S groups, respectively, compared the H group (P<0.01;
Fig. 4A, B and D). The expression
of BiP, p-PERK and CHOP was significantly increased in the H+T
group compared with the N group (P<0.01; Fig. 4A, B and D), but cotreatment with
FGF21 attenuated their expression in the H+T+F group (P<0.01;
Fig. 4A, B and D). By contrast,
the expression of PERK exhibited the opposite trend to the
expression of p-PERK (Fig. 4B).
The ratio of p-PERK/PERK exhibited a consistent trend with the
expression of p-PERK (Fig. 4B and
C).
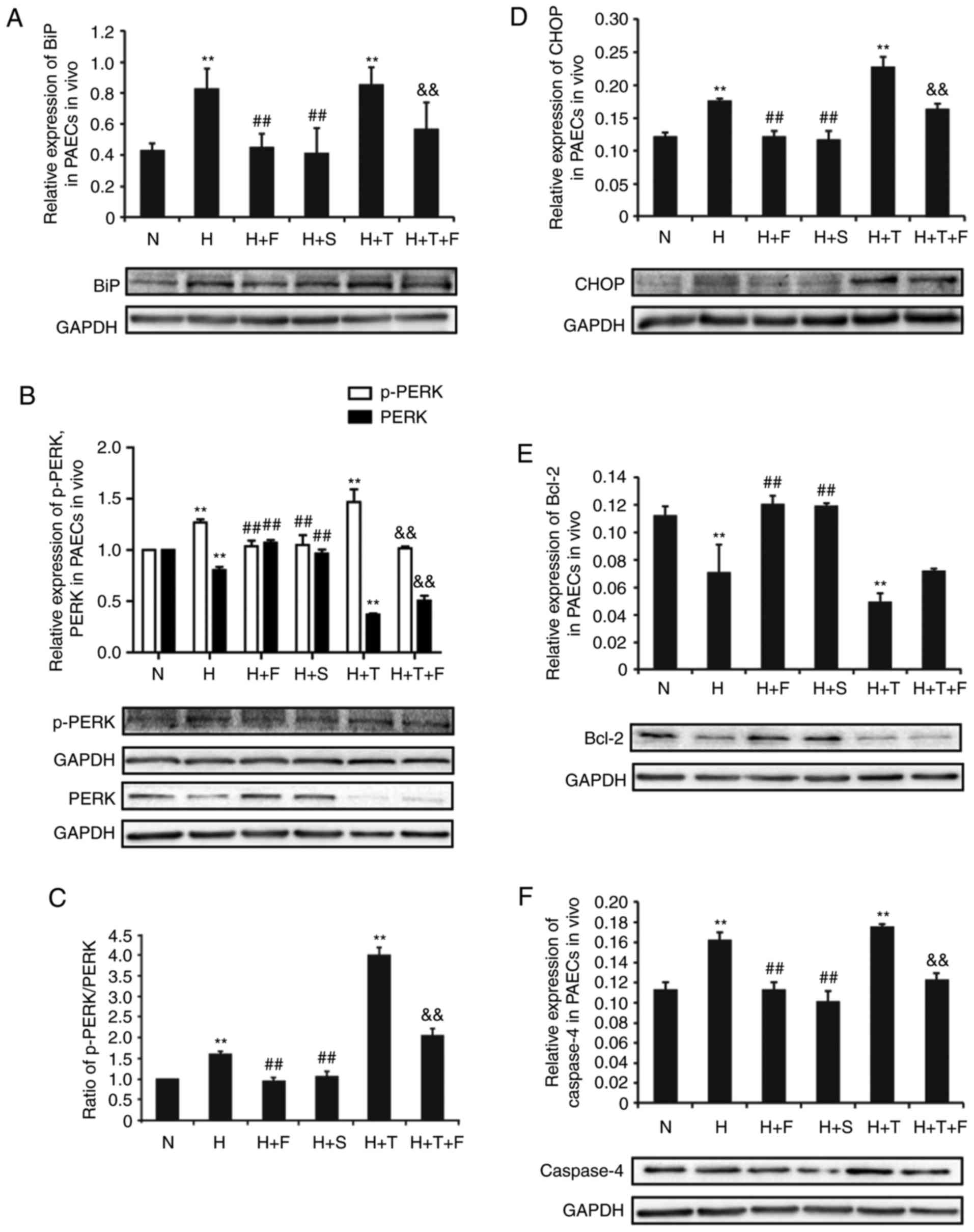 | Figure 4FGF21 alleviates the hypoxia-induced
ERS by modulating the expression of ERS-related proteins in HPAECs.
HPAECs were treated as indicated and protein expression levels were
examined by western blotting. Representative blots and
quantification are shown for (A) BiP, (B) p-PERK and PERK, (C) the
ratio of p-PERK/PERK, (D) CHOP, (E) Bcl-2. (F) Relative expression
of the ERS-dependent apoptotic protein caspase-4 in HPAECs was
analysed by western blotting. GAPDH was used as an internal
control. All experiments were performed in triplicate, and data are
expressed as the mean ± standard deviation (n=3).
**P<0.01 vs. N group; ##P<0.01 vs. the
H group; &&P<0.01 vs. the H+T group. FGF21,
fibroblast growth factor 21; ERS, endoplasmic reticulum stress;
HPAECs, human pulmonary arterial endothelial cells; BiP, binding
immunoglobulin protein; p-, phosphorylated; PERK, protein kinase
R-like endoplasmic reticulum kinase; CHOP, transcription factor
C/EBP homologous protein; Bcl-2, B cell lymphoma-2; N, normoxia; H,
hypoxia; F, FGF21; S, salubrinal; T, tunicamycin. |
The antiapoptotic mediator Bcl-2 is a downstream
target of CHOP, which can downregulate the expression of Bcl-2,
thereby increasing the rate of apoptosis. In the present study, the
expression of Bcl-2 was downregulated in the H group and the H+T
group compared with the N group (P<0.01; Fig. 4E). FGF21 or salubrinal treatment
increased the expression of Bcl-2 in the H+F and H+S groups
compared with the H group (P<0.01; Fig. 4E).
The expression of the ERS-induced apoptosis protein
caspase-4 was significantly elevated in the H group and the H+T
group compared with the N group (P<0.01; Fig. 4F). Under cotreatment with FGF21 or
salubrinal, the expression of caspase-4 was reduced in the H+F and
H+S groups, respectively, compared with the H group (P<0.01;
Fig. 4F). The expression of
caspase-4 was decreased in the H+T+F group compared with the H+T
group (P<0.01; Fig. 4F). In
summary, the present findings indicate, for the first time, that
ERS is one of the crucial mechanisms in hypoxia-induced HPAEC
apoptosis, which was reversed by FGF21 through modulation of the
PERK/CHOP signaling pathway and inhibition of caspase-4
expression.
Hypoxia and the ERS agonist tunicamycin
inhibit the migratory ability of HPAECs, and this effect is
improved by FGF21 and the ERS inhibitor salubrinal
As indicated in Fig.
5, the migratory ability of HPAECs was decreased in the H group
compared with the N group (P<0.01). The migratory ability of
HPAECs was significantly increased in the H+F and H+S groups
compared with the H group (P<0.01; Fig. 5). There was a significant
reduction in the number of migrated cells in the H+T group compared
with the N group (P<0.01; Fig.
5), but cotreatment with FGF21 increased the number of migrated
cells in the H+T+F group (P<0.01; Fig. 5).
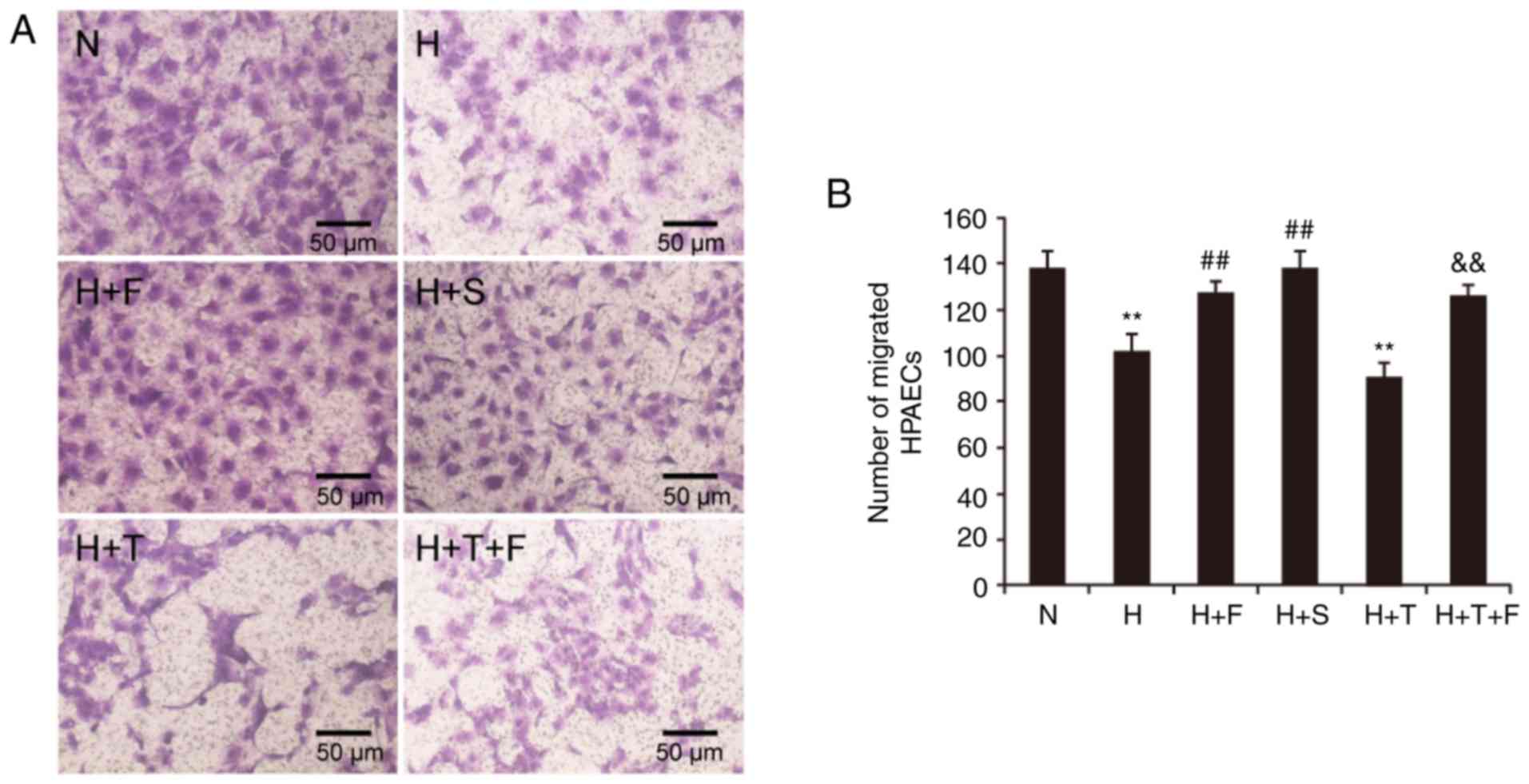 | Figure 5(A and B) Effect of FGF21 on the
migratory ability of HPAECs. HPAEC migration was assessed using
Transwell migration chambers. Representative images are shown for
the migrated cells in each experimental group (magnification, ×100;
scale bar, 50 µm), as well as quantification of the numbers
of migrated HPAECs. Data are expressed as the mean ± standard
deviation (n=6). **P<0.01 vs. the N group;
##P<0.01 vs. the H group;
&&P<0.01 vs. the H+T group. FGF21, fibroblast
growth factor 21; HPAECs, human pulmonary arterial endothelial
cells; N, normoxia; H, hypoxia; F, FGF21; S, salubrinal; T,
tunicamycin. |
Hypoxia and the ERS agonist tunicamycin
reduce the secretion of NO and increase the secretion of ET-1, and
these effects are reversed by FGF21 and the ERS inhibitor
salubrinal
As presented in Fig.
6A, the secretion of NO was decreased in the H group and the
H+T group compared with the N group (P<0.01). NO secretion was
enhanced under FGF21 or salubrinal treatment in the H+F and H+S
groups, respectively, compared with the H group (P<0.05;
Fig. 6A). NO secretion was
increased in the H+T+F group compared with the H+T group
(P<0.01; Fig. 6A). As far as
ET-1 secretion is concerned, ET-1 levels were upregulated by
hypoxia in the H group compared with the N group (P<0.01;
Fig. 6B). FGF21 or salubrinal
treatment decreased the secretion of ET-1 in the H+F and H+S
groups, respectively, compared with the H group (P<0.01;
Fig. 6B). The secretion of ET-1
was significantly increased in the H+T group compared with the N
group (P<0.01; Fig. 6B), but
cotreatment with FGF21 reduced the secretion of ET-1 in the H+T+F
group (P<0.01; Fig. 6B). The
balance of NO and ET-1 production has a key role in the function of
endothelial cells. Hence, the present-findings suggested that FGF21
could improve endothelial function and protect HPAECs from damage
by regulating the secretion of NO and ET-1.
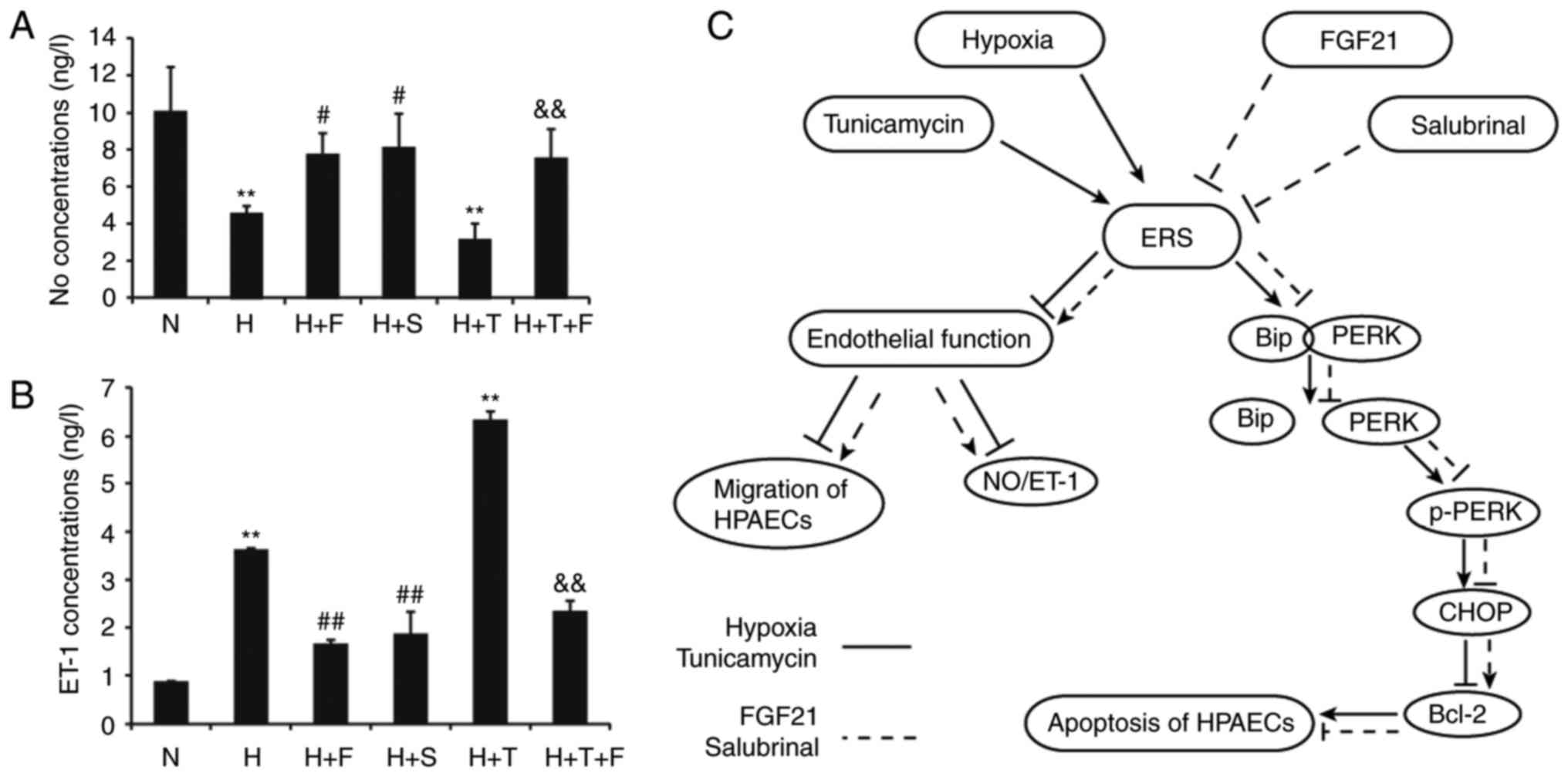 | Figure 6Effect of FGF21 on NO and ET-1
secretion in HPAECs. (A) HPAECs were treated as indicated and at
the end of treatment, the cell culture medium was collected and
assayed by ELISA for the levels of secreted NO and (B) the levels
of secreted ET-1. Data are expressed as the mean ± standard
deviation (n=3). **P<0.01 vs. the N group;
#P<0.05 and ##P<0.01 vs. the H group;
&&P<0.01 vs. the H+T group. (C) Schematic of
the proposed mechanism by which FGF21 attenuates hypoxia-induced
apoptosis and dysfunction by alleviating ERS in HPAECs. FGF21,
fibroblast growth factor 21; NO, nitric oxide; ET-1, endothelin-1;
HPAECs, human pulmonary arterial endothelial cells; ERS,
endoplasmic reticulum stress; N, normoxia; H, hypoxia; F, FGF21; S,
salubrinal; T, tunicamycin; BiP, binding immunoglobulin protein;
p-, phosphorylated; PERK, protein kinase R-like endoplasmic
reticulum kinase; CHOP, transcription factor C/EBP homologous
protein; Bcl-2, B cell lymphoma-2. |
Discussion
Abnormal growth of PASMCs, endothelial cell,
fibroblasts, and myofibroblastsis involved in the development of
vascular remodeling in hypoxia-induced PAH. The pathology of PAH is
also characterized by infiltration of activated inflammatory and
immune circulating cells. Among the cells types found within
remodeling pulmonary arteries, PASMCs have been the predominant
subject of scientific research, but pulmonary artery endothelial
cells (PAECs) are also increasingly studied (24). None of the drugs currently
available for treating PAH have been demonstrated to effectively
reverse the disease or significantly improve long-term survival
(1). The present study
demonstrated that hypoxia-stimulated ERS contributes towards
endothelial apoptosis and dysfunction, which is one of the
etiologies for hypoxia-induced PAH.
The data from the current study indicated that the
expression of ERS-associated proteins BiP, p-PERK, CHOP and
caspase-4 was significantly increased in HPAECs exposed to hypoxia.
This was accompanied by an increased rate of apoptosis and damaged
migratory and secretory abilities, indicating that hypoxic
conditions stimulate ERS, which in turn may induce endothelial
apoptosis and dysfunction in HPAECs. These results are consistent
with those of previous reports (12,25), where expression of the ERS marker
proteins glucose-regulated protein (GRP)78, GRP94 and caspase-12
were demonstrated to be upregulated in hypoxia-induced PAH in rats,
additionally, the number of pulmonary apoptotic cells in the
hypoxia group was markedly increased compared with the control
group (12,25), suggesting that ERS-induced
apoptosis may be one of the mechanisms underlying hypoxic pulmonary
hypertension and pulmonary vascular wall remodeling.
Under cellular stress, the ERS activates three
branches of the UPR: The double-stranded RNA-activated
PERK/eukaryotic translation initiation factor 2α (eIF2α) pathway;
the inositol-requiring enzyme 1 (IRE1)/X-box binding protein 1
(XBP1) pathway; and the activating transcription factor 6 (ATF6)
pathway (26).
PERK is a key ER resident transmembrane molecule
whose activation is thought to be the first event in sensing the
UPR (10). In resting cells under
physiological conditions, as one of the most highly expressed ER
chaperones, BiP (GRP78) binds to PERK. Under the effects of harmful
stress, PERK disassociates from BiP and is activated by
self-phosphorylation to initiate ERS (27-29). Chronic or severe ERS would
activate ERS-dependent apoptosis, including the CHOP and
caspase-mediated apoptotic pathways. The CHOP-mediated pathway is
associated with PERK/eIF2α mediated transcriptional activation of
the C/EBP-homologous protein CHOP, which then downregulates the
expression of the antiapoptotic mediator Bcl-2. Activation of the
caspase cascade is a well-known proapoptotic event that is also
involved in ERS-induced apoptosis (28). Several caspases are believed to
serve roles in this process, including caspase-2, caspase-4
(human), caspase-12 (rodents), caspase-7, caspase-3 and caspase-9.
It has been suggested that caspase-12 is specific to death signals
in ERS rather than to other cell death mechanisms (29,30). The present results demonstrated
that hypoxic conditions activated PERK/CHOP signaling and increased
the expression of human caspase-4, which may participate in the
apoptosis and damage of HPAECs.
Both in yeast and in mammalian cells, UPR signaling
can be initiated by the use of chemical agents that induce ERS,
such as the N-linked glycosylation inhibitor tunicamycin, the
calcium pump inhibitor thapsigargin, and the reducing agent
dithiothreitol (31). A known ERS
inducer tunicamycin mediates the degradation of ceramide synthase 6
(CerS6) and subsequent ATF-6 and caspase-3 activation. Senkal et
al (32) reported that
downregulation of CerS6 increased ATF-6 activation, as determined
by increased expression of its downstream target CHOP in UM-SCC-22A
cells. CerS6 knockdown also activated CHOP in human squamous lung
cancer H1650 cells, leading to cell death (32).
An increasing number of studies have reported that
ERS inhibitors protect cells under adverse stimuli. In the current
study, salubrinal was used as an ERS inhibitor. Salubrinal, a
selective inhibitor of cellular complexes, dephosphorylates eIF2α.
During ERS, PERK, an ER-resident transmembrane protein,
oligomerizes and phosphorylates eIF2α at serine 51 (33). Salubrinal induces rapid and robust
eIF2α phosphorylation and its downstream effects, including
downregulation of cyclin D1 and upregulation of growth arrest and
DNA damage inducible protein 34 and CHOP (34). The phosphorylation of eIF2a is
cytoprotective during ERS. In recent years, another ERS inhibitor,
4-phenylbutyric acid (4-PBA), has also been employed. 4-PBA can
inhibit ERS as a chemical chaperone by stabilizing peptide
structures, improving luminal folding capacity, and attenuating
cell damage. Wu et al (35) demonstrated that 4-PBA improved
pulmonary arterial remodeling and suppressed the expression of ERS
indicators, including GRP78, GRP94, ATF6, IRE-1, PERK, CHOP and
Bcl-2, in a monocrotaline-induced PAH rat model.
FGF21 is a member of the FGF family. In the adult
organism, FGFs are homeostatic factors and exhibit functions in
tissue repair and response to injury (36). FGF21 is an endocrine regulator
that participates in lipid and glucose metabolism, and has recently
been reported to protect cardiac endothelial cells from damage and
suppress inflammatory responses. In the present study, the role of
FGF21 in hypoxia-induced apoptosis and dysfunction was investigated
in HPAECs. Studies by Schaap et al (37) suggest that FGF21 expression is
regulated by ERS. The authors reported that FGF21 mRNA levels are
increased by triglyceride-induced ERS in rat H4IIE cells and rat
primary hepatocytes. Wan et al (8) demonstrated that FGF21 is the target
gene of CHOP, and that transcription and mRNA stabilization are
responsible for the CHOP-mediated induction of FGF21 expression in
ERS, indicating that ERS is the key mechanism of FGF21 regulation
in several metabolic diseases. In addition, Jiang et al
(38) indicated that
intraperitoneal administration of the ER stressor tunicamycin
activates ERS, which results in liver steatosis associated with
upregulated expression of hepatic FGF21 in the liver. Furthermore,
administration of recombinant FGF21 in mice alleviates PERK/CHOP
signaling in the liver, resulting in the reversal of ERS-induced
liver steatosis. In addition to the evidence presented above,
previous studies have indicated that FGF21 and ERS are closely
associated in metabolism. FGF21 may act as an inhibitor, similar to
salubrinal or 4-PBA, and attenuate ERS to protect cells from
damage. In the present study, the results following treatment of
HPAECs with hypoxia and the ERS agonist tunicamycin demonstrated
that administration of FGF21 ameliorated the hypoxia-induced
endothelial apoptosis and dysfunction, and improved the viability
of HPAECs. These observations are consistent with previous studies
on the cardiovascular system, indicating that FGF21 could prevent
damage to cardiac endothelial cells (18). In the present study, the results
revealed that FGF21 downregulated p-PERK and subsequently increased
the expression of CHOP, ultimately upregulating the expression of
the antiapoptotic mediator Bcl-2, and downregulating the expression
of caspase-4, which decreased the number of apoptotic cells and
enhanced the viability of HPAECs. Hypoxia and tunicamycin resulted
in an expanded shape of the ER in HPAECs, while this effect was
notably normalized following treatment with FGF21 and the ERS
inhibitor salubrinal. Furthermore, the results indicated that FGF21
alleviated hypoxia-induced endothelial dysfunction, by decreasing
the secretion of ET-1, increasing the secretion of NO and repairing
damage to the cell migration ability, processes that are involved
in the pathogenesis of hypoxia-induced PAH (39).
The balance between NO and ET-1 has a significant
role in the development of hypoxia-induced PAH. In patients with
PAH, changes in the expression of various endothelial vasoactive
mediators, including NO, ET-1, prostacyclin, serotonin and
thromboxane, have been increasingly recognized. Since most of these
mediators affect the growth of smooth muscle cells, this may
facilitate the development of pulmonary vascular hypertrophy. The
present study aimed to explore the mechanism of the apoptosis and
dysfunction of HPAECs under conditions of hypoxia through observing
the cell morphology and function, and protein level changes.
Endothelial function was investigated by assaying for the cell
migration ability, and the secretion levels of NO and ET-1.
Overproduction of eNOS, which stimulates the conversion of
L-arginine to citrulline, producing NO (40), prevents hypoxia-induced PAH in
transgenic mice (41). Several
previous studies suggest that NO protects against hypoxia-induced
vasoconstriction in the lungs, inhibits smooth muscle proliferation
and platelet aggregation, and decreases the secretion of ET-1
(39). ET-1 expression is
elevated in animal models of PH and in patients with PH (39). ET-1 receptor antagonists, such as
bosentan, improve the functional status of patients and other
indices of PH-related morbidity (42). The present results revealed that
hypoxia reduced the secretion of NO and increased the secretion of
ET-1 in HPAECs. In addition, FGF21 and the ERS inhibitor salubrinal
were demonstrated to improve the secretion of NO and ET-1 in HPAECs
under conditions of hypoxia.
In summary, the results of the present study
revealed that ERS is a key mechanism involved in the
hypoxia-induced apoptosis and dysfunction of HPAECs, which are
important processes in the initiation of hypoxia-induced PAH. FGF21
attenuated the hypoxia-induced dysfunction and apoptosis of HPAECs
through diminishing ERS, which was mediated by the PERK/CHOP
signaling pathway (as illustrated in Fig. 6C), and by the inhibition of
caspase-4 expression. Although FGF21 was demonstrated to alleviate
the main ERS-dependent death signaling pathway in HPAECs, the
mechanism by which FGF21 is associated with ERS has yet to be fully
elucidated. Additionally, the role of FGF21 in ERS and PAH in
vivo remains unclear and requires further research. However,
the present findings strongly support the notion that FGF21 may
have promising therapeutic potential for the treatment of
hypoxia-induced PAH.
Acknowledgments
Not applicable.
Abbreviations:
|
PAH
|
pulmonary arterial hypertension
|
|
ERS
|
endoplasmic reticulum stress
|
|
FGF21
|
fibroblast growth factor 21
|
|
HPAECs
|
human pulmonary arterial endothelial
cells
|
|
CCK-8
|
cell counting kit-8
|
|
TUNEL
|
terminal deoxyribonucleotide
transferase-mediated dUTP nick end-labelling assay
|
|
BiP
|
binding immunoglobulin protein
|
|
CHOP
|
transcription factor C/EBP homologous
protein
|
|
PERK
|
protein kinase R-like endoplasmic
reticulum kinase
|
|
Bcl-2
|
B cell lymphoma-2
|
|
NO
|
nitric oxide
|
|
ET-1
|
endothelin-1
|
|
COPD
|
chronic obstructive pulmonary
disease
|
|
UPR
|
unfolded protein response
|
|
PASMCs
|
pulmonary artery smooth muscle
cells
|
|
ECM
|
endothelial cell media
|
|
FBS
|
fetal bovine serum
|
|
ECGS
|
endothelial cell growth supplement
|
|
PBS
|
phosphate-buffered saline
|
|
eIF2α
|
eukaryotic translation initiation
factor 2α
|
|
IRE1
|
inositol-requiring enzyme 1
|
|
XBP1
|
X-box binding protein 1
|
|
ATF6
|
activating transcription factor 6
|
|
CerS6
|
ceramide synthase 6
|
|
4-PBA
|
4-phenylbutyric acid
|
Funding
The present study was supported by the Project of
Health Department of Zhejiang Province of China (grant no.
2016DTA005), the Natural Science Foundation Grants of Zhejiang
Province (grant no. Y17H010028) and the Chinese National Natural
Science Foundation Grants (grant no. 81473406).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
AC designed and performed the experiments, analysed
the data and wrote the paper; XH and LW designed the experiments
and helped to draft the manuscript; JL, JZ and XW performed the
experiments and collected data. ZX and MX performed the
experiments; ZC, ML, DY and ZH participated in the study design and
supervised the study; MC and PW collected data and performed the
analysis. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sutendra G and Michelakis ED: The
metabolic basis of pulmonary arterial hypertension. Cell Metab.
19:558–573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang X, Zou L, Yu X, Chen M, Guo R, Cai
H, Yao D, Xu X, Chen Y, Ding C, et al: Salidroside attenuates
chronic hypoxia-induced pulmonary hypertension via adenosine A2a
receptor related mitochondria-dependent apoptosis pathway. J Mol
Cell Cardiol. 82:153–166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Voelkel NF and Cool C: Pathology of
pulmonary hypertension. Cardiol Clin. 22:343–351. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eddahibi S, Guignabert C, Barlier-Mur AM,
Dewachter L, Fadel E, Dartevelle P, Humbert M, Simonneau G, Hanoun
N, Saurini F, et al: Cross talk between endothelial and smooth
muscle cells in pulmonary hypertension: Critical role for
serotonin-induced smooth muscle hyperplasia. Circulation.
113:1857–1864. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Humbert M, Montani D, Perros F, Dorfmüller
P, Adnot S and Eddahibi S: Endothelial cell dysfunction and cross
talk between endothelium and smooth muscle cells in pulmonary
arterial hypertension. Vascul Pharmacol. 49:113–118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sahara M, Sata M, Morita T, Hirata Y and
Nagai R: Nicorandil attenuates monocrotaline-induced vascular
endothelial damage and pulmonary arterial hypertension. PLoS One.
7:e333672012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Farkas L, Farkas D, Ask K, Möller A,
Gauldie J, Margetts P, Inman M and Kolb M: VEGF ameliorates
pulmonary hypertension through inhibition of endothelial apoptosis
in experimental lung fibrosis in rats. J Clin Invest.
119:1298–1311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wan XS, Lu XH, Xiao YC, Lin Y, Zhu H, Ding
T, Yang Y, Huang Y, Zhang Y, Liu YL, et al: ATF4- and
CHOP-dependent induction of FGF21 through endoplasmic reticulum
stress. Biomed Res Int. 2014:8078742014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Muñoz JP and Zorzano A: Endoplasmic
reticulum stress enters a Nogo Zone. Sci Transl Med. 3:88ps262011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koyama M, Furuhashi M, Ishimura S, Mita T,
Fuseya T, Okazaki Y, Yoshida H, Tsuchihashi K and Miura T:
Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid
prevents the development of hypoxia-induced pulmonary arterial
hypertension. Am J Physiol Heart Circ Physiol. 306:H1314–H1323.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan XF, Li WJ, Chen ZQ, Wang XR, Kong XX,
Mao SZ, Hu LG and Gong YS: Changes of endoplasmic reticulum
stress-induced apoptosis in pulmonary tissue of rats with hypoxic
pulmonary hypertension. Zhongguo Ying Yong Sheng Li Xue Za Zhi.
27:270–274. 2011.In Chinese. PubMed/NCBI
|
|
13
|
Ost M, Coleman V, Kasch J and Klaus S:
Regulation of myokine expression: Role of exercise and cellular
stress. Free Radic Biol Med. 98:78–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Inagaki T: Research perspectives on the
regulation and physiological functions of FGF21 and its association
with NAFLD. Front Endocrinol (Lausanne). 6:1472015.
|
|
15
|
Kim SH, Kim KH, Kim HK, Kim MJ, Back SH,
Konishi M, Itoh N and Lee MS: Fibroblast growth factor 21
participates in adaptation to endoplasmic reticulum stress and
attenuates obesity-induced hepatic metabolic stress. Diabetologia.
58:809–818. 2015. View Article : Google Scholar
|
|
16
|
Shimizu M, Morimoto H, Maruyama R, Inoue J
and Sato R: Selective regulation of FGF19 and FGF21 expression by
cellular and nutritional stress. J Nutr Sci Vitaminol (Tokyo).
61:154–160. 2015. View Article : Google Scholar
|
|
17
|
Guo Q, Xu L, Liu J, Li H, Sun H, Wu S and
Zhou B: Fibroblast growth factor 21 reverses suppression of
adiponectin expression via inhibiting endoplasmic reticulum stress
in adipose tissue of obese mice. Exp Biol Med (Maywood).
242:441–447. 2017. View Article : Google Scholar
|
|
18
|
Lü Y, Liu JH, Zhang LK, DU J, Zeng XJ, Hao
G, Huang J, Zhao DH, Wang GZ and Zhang YC: Fibroblast growth factor
21 as a possible endogenous factor inhibits apoptosis in cardiac
endothelial cells. Chin Med J (Engl). 123:3417–3421. 2010.
|
|
19
|
Li J, Zhou J, Zhang D, Song Y, She J and
Bai C: Bone marrow-derived mesenchymal stem cells enhance autophagy
via PI3K/AKT signalling to reduce the severity of
ischaemia/reper-fusion-induced lung injury. J Cell Mol Med.
19:2341–2351. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gong T, Wang Q, Lin Z, Chen ML and Sun GZ:
Endoplasmic reticulum (ER) stress inhibitor salubrinal protects
against ceramide-induced SH-SY5Y cell death. Biochem Biophys Res
Commun. 427:461–465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guoliang H, Kunlun H, Li F, Xin L and
Ruijun L: Salubrinal protects endoplasmic reticulum of cardiac
muscle cells against stress-associated apoptosis. Jun Yi Jin Xiu
Xue Yuan Xue Bao. 31:483–485. 2010.In Chinese.
|
|
22
|
Wang H, Zuo X, Wang Q, Yu Y, Xie L, Wang
H, Wu H and Xie W: Nicorandil inhibits hypoxia-induced apoptosis in
human pulmonary artery endothelial cells through activation of
mito-KATP and regulation of eNOS and the NF-κB pathway. Int J Mol
Med. 32:187–194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morecroft I, White K, Caruso P, Nilsen M,
Loughlin L, Alba R, Reynolds PN, Danilov SM, Baker AH and Maclean
MR: Gene therapy by targeted adenovirus-mediated knockdown of
pulmonary endothelial Tph1 attenuates hypoxia-induced pulmonary
hypertension. Mol Ther. 20:1516–1528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paulin R and Michelakis ED: The metabolic
theory of pulmonary arterial hypertension. Circ Res. 115:148–164.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mao SZ, Fan XF, Xue F, Chen R, Chen XY,
Yuan GS, Hu LG, Liu SF and Gong YS: Intermedin modulates hypoxic
pulmonary vascular remodeling by inhibiting pulmonary artery smooth
muscle cell proliferation. Pulm Pharmacol Ther. 27:1–9. 2014.
View Article : Google Scholar
|
|
26
|
Wang S and Kaufman RJ: The impact of the
unfolded protein response on human disease. J Cell Biol.
197:857–867. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Z, Lv Y, Zhao N, Guan G and Wang J:
Protein kinase R-like ER kinase and its role in endoplasmic
reticulum stress-decided cell fate. Cell Death Dis. 6:e18222015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vannuvel K, Renard P, Raes M and Arnould
T: Functional and morphological impact of ER stress on
mitochondria. J Cell Physiol. 228:1802–1818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rutkowski DT and Kaufman RJ: A trip to the
ER: Coping with stress. Trends Cell Biol. 14:20–28. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar
|
|
31
|
Richardson CE, Kinkel S and Kim DH:
Physiological IRE-1-XBP-1 and PEK-1 signaling in caenorhabditis
elegans larval development and immunity. PLoS Genet.
7:e10023912011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Senkal CE, Ponnusamy S, Manevich Y,
Meyers-Needham M, Saddoughi SA, Mukhopadyay A, Dent P, Bielawski J
and Ogretmen B: Alteration of ceramide synthase 6/C16-ceramide
induces activating transcription factor 6-mediated endoplasmic
reticulum (ER) stress and apoptosis via perturbation of cellular
Ca2 and ER/Golgi membrane network. J Biol Chem.
286:42446–42458. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schröder M and Kaufman RJ: ER stress and
the unfolded protein response. Mutat Res. 569:29–63. 2005.
View Article : Google Scholar
|
|
34
|
Boyce M, Bryant KF, Jousse C, Long K,
Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D and Yuan
J: A Selective Inhibitor of eIF2alpha dephosphorylation protects
cells from ER stress. Science. 307:935–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu Y, Adi D, Long M, Wang J, Liu F, Gai
MT, Aierken A, Li MY, Li Q, Wu LQ, et al: 4-Phenylbutyric acid
induces protection against pulmonary arterial hypertension in rats.
PLoS One. 11:e01575382016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ornitz DM and Itoh N: Fibroblast growth
factors. Genome Biol. 2:REVIEWS3005. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schaap FG, Kremer AE, Lamers WH, Jansen PL
and Gaemers IC: Fibroblast growth factor 21 is induced by
endoplasmic reticulum stress. Biochimie. 95:692–699. 2013.
View Article : Google Scholar
|
|
38
|
Jiang S, Yan C, Fang QC, Shao ML, Zhang
YL, Liu Y, Deng YP, Shan B, Liu JQ, Li HT, et al: Fibroblast growth
factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded
protein response and counteracts endoplasmic reticulum
stress-induced hepatic steatosis. J Biol Chem. 289:29751–29765.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Budhiraja R, Tuder RM and Hassoun PM:
Endothelial dysfunction in pulmonary hypertension. Circulation.
109:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marletta MA: Nitric oxide synthase
structure and mechanism. J Biol Chem. 268:12231–12234.
1993.PubMed/NCBI
|
|
41
|
Ozaki M, Kawashima S, Yamashita T, Ohashi
Y, Rikitake Y, Inoue N, Hirata KI, Hayashi Y, Itoh H and Yokoyama
M: Reduced hypoxic pulmonary vascular remodeling by nitric oxide
from the endothelium. Hypertension. 37:322–327. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rubin LJ, Badesch DB, Barst RJ, Galie N,
Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, et al:
Bosentan therapy for pulmonary arterial hypertension. N Engl J Med.
346:896–903. 2002. View Article : Google Scholar : PubMed/NCBI
|