Introduction
Osteoarthritis (OA) is characterized by the
progressive destruction and loss of the matrix of articular
cartilage due to an imbalance between the anabolic and catabolic
activities of chondrocytes (1).
Several studies have reported that inflammation serves an important
role in OA progression (2–5),
and it is widely accepted that the activation of inflammatory
cytokines, such as interleukin (IL)-1β, tumor necrosis factor-α
(TNF-α) and IL-6, is critical in this process (6). OA patients display elevated IL-1β
levels in the synovial fluid, synovial membrane, cartilage and
subchondral bone layer (7).
Similar to other inflammatory cytokines, IL-1β blocks the
production of cartilage structural proteins, including type II
collagen and aggrecan (8,9). Furthermore, IL-1β was reported to
upregulate the production of matrix metalloproteinases (MMPs) in
chondrocytes, specifically MMP-13, leading to increased destruction
of cartilage components (10). A
later study revealed that IL-1β stimulation of human chondrocytes
significantly upregulated the mRNA levels of both
MMP-13 and a disintegrin and metalloproteinase with
thrombospondin motifs 5 (ADAMTS-5) (11), which have been identified as the
major enzymes responsible for cartilage degradation during OA
progression (12,13).
Aquaporins (AQPs) are small integral membrane
proteins that constitute a family with 13 members thus far
identified in mammals (including AQP0 to 12), and are essential for
water transport across membranes (14). Certain AQPs are also known as
aquaglyceroporins since they participate in the transport of other
small molecules, such as glycerol, urea or ammonia. AQP9, which was
first identified in leukocytes (14), is an aquaglyceroporin that is
expressed in numerous organs, with high expression levels detected
in the liver, epididymis, testis, spleen and brain (15). There have been several studies on
the functions of AQPs in certain organs. Early death has been
observed in AQP11 knockout (KO) mice due to malfunction of the
kidney (16), while renal
dysfunction has also been reported in AQP1, 3 and 7 KO mice
(17,18).
In humans, AQPs have been implicated in several
inflammatory diseases. For instance, overexpression of AQP1 has
been detected in autoimmune and alcoholic pancreatitis (19), Alzheimer disease (20), and cases of rheumatoid and
psoriatic arthritis (21). AQP9
has also been connected to a number of diseases (22). Rojek et al (22) demonstrated that AQP9 is important
for hepatic glycerol metabolism and suggested that it may serve a
role in glycerol and glucose metabolism in diabetes mellitus. More
recently, Spegel et al (23) confirmed the importance of AQP9 in
maintaining appropriate blood glucose levels. Furthermore, AQP9
downregulation was reported to result in glaucomatous optic
neuropathy (24).
Notably, several studies have demonstrated that AQPs
also participate in signal transduction (25–27). Specifically, AQPs are implicated
in the phagocytic functions and the activation and migration of
immune cells (25,26), as well as in proinflammatory
cytokine secretion during inflammation (27). Based on these previous findings on
the signal transduction function of AQPs, as well as the reported
high expression of AQP9 in patients with hydrarthrosis and
synovitis (28), it can be
hypothesized that AQP9 may alter the inflammatory signal
transduction in chondrocytes and that it may serve an important
role in OA progression. Therefore, focusing on IL-1β-induced
inflammation, the present study attempted to determine the
mechanisms associated with AQP9 functions in chondrocytes.
Materials and methods
Cell culture
Normal human articular chondrocytes isolated from
the knee (NHAC-kn; Lonza Group, Ltd., Walkersville, MD, USA) were
cultured in 15 ml of Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (BioWhittaker; Lonza
Group, Ltd., Basel, Switzerland) and 100 units/ml of
penicillin-streptomycin at 37°C in a 5% CO2 atmosphere.
Cells at passages 3–5 were used in the experiments of the present
study and considered as ̔normal human chondrocytes̓.
Human cartilage samples
OA cartilage samples were obtained from the femoral
condyle of patients undergoing total knee arthroplasty for primary
OA (n=5). Normal cartilage samples were also obtained from patients
with femoral neck fractures who were undergoing femoral head
replacement surgeries (n=5). The mean age of the OA patients was
77.2 years, while the average age of patients with femoral neck
fractures was 83.9 years; however, there was no significant
difference in the age between the two groups (Table I). All samples were obtained in
accordance with the World Medical Association Declaration of
Helsinki Ethical Principles for Medical Research Involving Human
Subjects (29).
 | Table IClinical characteristics of
samples. |
Table I
Clinical characteristics of
samples.
| Parameter | Total | Osteoarthritis | Femoral neck
fracture |
|---|
| Number of
patients | 10 | 5 (50%) | 5 (50%) |
| Mean age
(years) | 80.2±5.6 | 77.2±6.1 | 83.9±4.8 |
| Sex | | | |
| Male | 2 | 1 | 1 |
| Female | 8 | 4 | 4 |
Normal and OA cartilage samples were fixed in 4%
para-formaldehyde, decalcified, embedded in paraffin wax and cut
into 10-µm slices. Cartilage destruction was evaluated by
safranin O (cat. no. S0145; Tokyo Chemical Industry, Tokyo, Japan)
and fastgreen (cat. no. 10720; Chroma-Gesellschaft; Thermo Fisher
Scientific, Inc.) staining. Histological evaluation was performed
using the grading system described by Mankin et al (30).
Detection of AQP expression by standard
reverse transcription-polymerase chain reaction (RT-PCR)
RT-PCR was conducted to detect the expression of
AQP mRNA in human chondrocytes (n=5). Briefly, chondrocytes
were plated into 6-well plates at a density of 1.0×105
cells/cm2 and cultured at 37°C for 24 h. Cells were
treated with 10 ng/ml recombinant human IL-1β (R&D Systems,
Inc., Minneapolis, MN, USA) for 24 h, followed by RNA extraction
using a QIAshredder and RNeasy mini kit (Qiagen, Hilden, Germany)
according to the manufacturer's protocol. Next, 1 µg of
total RNA was reverse-transcribed into first-strand cDNA using 1.25
µM oligo-dT primers in 40 µl PCR buffer II containing
2.5 mM MgCl2, 0.5 mM dNTP Mix, 0.5 units RNase
inhibitor, and 1.25 units MuLV reverse transcriptase (PerkinElmer,
Inc., Foster City, CA, USA) at 42°C for 60 min. Subsequently, the
cDNA solutions were used in PCR analysis for the detection of human
AQP1-12 expression. The cDNA amplification was
performed under the following PCR conditions: 94°C for 5 min;
followed by 35 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C
for 30 sec; and a final extension at 72°C for 2 min using AmpliTaq
Gold DNA Polymerase (cat. no. N8080241; Thermo Fisher Scientific,
Inc.) and specific primers (Table
II). The PCR products were electrophoresed on 3% agarose gels
at 100 V for 30 min, and the gels were visualized under ultraviolet
transillumination following the electrophoresis. The primer
sequences used for the detection of human AQP1-12 are
presented in Table I.
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as
a reference gene.
| Table IISpecific primers for reverse
transcription-polymerase chain reaction amplifications. |
Table II
Specific primers for reverse
transcription-polymerase chain reaction amplifications.
| Gene | Primer sequence
(5′-3′)
|
|---|
| Forward | Reverse |
|---|
| GAPDH |
GTTCGACAGTCAGCCGCATC |
GGAATTTGCATGGGTGGA |
| AQP1 |
TGGACACCTCCTGGCTATTG |
GGGCCAGGATGAAGTCGTAG |
| AQP2 C |
ACCCCTGCTCTCTCCATA |
GAAGACCCAGTGGTCATCAAAT |
| AQP3 |
GCTGTATTATGATGCAATCTGGC |
TAAGGGAGGCTGTGCCTATG |
| AQP4 |
GAAGGCATGAGTGACAGACC |
ATTCCGCTGTGACTGCTTTC |
| AQP5 |
GCCACCTTGTCGGAATCTAC |
TAAAGCATGGCAGCCAGGAC |
| AQP6 |
CACCTCATTGGGATCCACTT |
GTTGTAGATCAGTGAGGCCA |
| AQP7 |
ATCTCTGGAGCCCACATGAA |
GAAGGAGCCCAGGAACTG |
| AQP8 |
GTGCCTGTCGGTCATTGAG |
CAGGGTTGAAGTGTCCACC |
| AQP9 |
TCTCTGAGTTCTTGGGCACG |
GGTTGATGTGACCACCAGAG |
| AQP10 |
GATAGCCATCTACGTGGGTG |
CACAGAAAGCAGACAGCAAC |
| AQP11 |
TCCGAACCAAGCTTCGTATC |
TAGCGAAAGTGCCAAAGCTG |
| AQP12 |
ACTTGTTCTTCTGGCCGTAG |
CTTACTGGAGTACGTGCAGG |
| MMP-3 |
ATTCCATGGAGCCAGGCTTTC |
CATTTGGGTCAAACTCCAACTGTG |
| MMP-13 |
TGCTGCATTCTCCTTCAGGA |
ATGCATCCAGGGGTCCTGGC |
| ADAMTS5 |
TATGACAAGTGCGGACTATG |
TTCAGGGCTAAATAGGCAGT |
Immunohistochemical assay
Deparaffinized sections were digested with
proteinase K (Dako; Agilent Technologies, Inc., Santa Clara, CA,
USA) for 10 min and then treated with 3% hydrogen peroxide (Wako
Pure Chemical Industries, Inc., Osaka, Japan) to block any
endogenous peroxidase activity. Next, the sections were incubated
overnight at 4°C with the following primary antibodies at a 1:50
dilution: Anti-IL-1β (sc-7884) and anti-AQP9 (sc-74409) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA),
anti-phosphorylated-IκB kinase (p-IKK) (#2697) was from Cell
Signaling Technology (Danvers, MA, USA), while anti-MMP-3
(ab53015), anti-MMP-13 (ab39012) and anti-ADAMTS-5 (ab41037)
antibodies were obtained from Abcam (Cambridge, UK). Sections were
subsequently incubated with horseradish peroxidase-labeled mouse
anti-rabbit immunoglobulin G (Histofine Simple Stain MAX PO (R);
Nichirei Biosciences, Inc., Tokyo, Japan) at room temperature for
60 min. The peroxidase substrate 3,3′-diaminobenzidine (Histofine
Simple Stain DAB Solution; Nichirei Biosciences, Inc.) was added
and the peroxidase activity was detected by the production of a
brown reaction product. The sections were counterstained with
hematoxylin and then examined microscopically. Stained cells were
independently counted by three blinded observers in three areas of
high magnification fields, at both the superficial and deep zones
of the cartilage tissues.
Reverse transcription-quantitative (q)PCR
analysis
Chondrocytes (n=5) were plated into 6-well plates at
a density of 1.0×105 cells/cm2, cultured at
37°C for 24 h and then transfected with the following: Non-specific
small interfering RNA (siRNA), serving as the negative control (NC)
siRNA (cat. no. AM4613; Thermo Fisher Scientific, Inc.); 10 nM
AQP9-1 specific siRNA (siAQP9-1; cat. no. 4392420; Invitrogen;
Thermo Fisher Scientific, Inc.); or 10 nM AQP9-2 specific siRNA
(siAQP9-2; cat. no. 4392422; Invitrogen; Thermo Fisher Scientific,
Inc.). Transfection was performed for 24 h using the Lipofectamine
RMAiMAX transfection reagent (cat. no. 13778150; Invitrogen; Thermo
Fisher Scientific, Inc.). A control group which was treated only
with DMEM medium was used. Subsequent to changing medium, 10 ng/ml
recombinant human IL-1β was added for 24-h incubation. As
previously reported, anti-inflammatory drugs can affect OA
progression (31). Thus, a group
of cultures was subjected to 30-min pretreatment with 10 µM
BMS-345541 (cat. no. 095M4739V; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), a p-IKK inhibitor, prior to the addition of
IL-1β (32).
RNA extraction and RT were performed as described
earlier in this study. The relative mRNA levels of AQP9,
MMP-3, MMP-13 and
ADAMTS-5 were analyzed by SYBR Green Real-Time PCR
Master Mix on an ABI Prism 7500 sequence-detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The results were
quantified using the quantification cycle (Cq) method (33), with GAPDH serving as an
internal control gene. The difference between the mean Cq values of
the gene of interest and the internal control gene was denoted as
ΔCq, and the difference between ΔCq and the Cq value of the
calibrated sample was denoted as ΔΔCq. The log2(ΔΔCq)
provides the relative value of gene expression. Primer sequences
used for the detection of the aforementioned genes are presented in
Table II.
Western blot analysis
Chondrocytes (n=5) were plated into 6-well plates at
a density of 1.0×105 cells/cm2, cultured at
37°C, and transfected with non-specific siRNA or AQP9-specific
siRNA (10 nM) for 24 h using the Lipofectamine RMAiMAX transfection
reagent. Following, incubation with fresh medium for another 24 h
at 37°C, chondrocytes were cultured with or without 10 ng/ml
recombinant human IL-1β for 30 min, washed with Tris-buffered
saline with Tween-20 (TBST) and then lysed in a buffer containing
25 mM Tris, 1% Nonidet P-40, 150 mM NaCl, 1.5 mM EGTA and a
protease/phosphatase inhibitor mix (Roche Diagnostics, Basel,
Switzerland). Lysates were centrifuged at 4°C at 15,000 × g for 10
min to remove cellular debris. Next, the supernatants were
collected, mixed with 4× electrophoresis sample buffer,
electrophoresed on a 7.5–15% SDS-polyacrylamide gradient gel
(Biocraft, Tokyo, Japan) and electrically transferred onto a
polyvinylidene difluoride blotting membrane (GE Healthcare Life
Sciences, Little Chalfont, UK). Membranes were blocked with 5%
skimmed milk in TBST for 30 min at 25°C. Membranes were incubated
with antibodies against p38 mitogen-activated protein kinase (MAPK)
(#9212), phosphorylated (p)-p38 MAPK (#9211), extracellular
signal-regulated kinase (ERK) (#4695), p-ERK (#4370), c-Jun
N-terminal kinase (JNK) (#9252), p-JNK (#9251), IKK (#2682) or
p-IKK (#2697), all of which were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G antibody
was used as a secondary antibody, and proteins were subsequently
visualized using the ECL Plus reagent (GE Healthcare Life Sciences)
in a Chemilumino Analyzer LAS-3000 mini (Fujifilm, Tokyo, Japan).
Protein expression was determined by semiquantification of
digitally captured images using the National Institutes of Health
ImageJ software (http://imagej.nih.gov/ij/).
Ethics
The study protocol was approved by the Ethics
Committee of Kobe University Graduate School of Medicine (Kobe,
Japan) on March 19th, 2015 (no. 1721), and all patients provided
informed consent prior to participation.
Statistical analysis
Data are expressed as the mean ± standard deviation.
A three-way analysis of variance with the Tukey-Kramer post-hoc
test was used to assess differences in experimental groups. The
Mann-Whitney U test was used to perform comparisons between groups.
A value of P<0.05 was considered to indicate a difference that
was statistically significant. Statistical analyses were performed
using BellCurve a software (Social Survey Research Information Co.,
Ltd., Tokyo, Japan) add-on for Excel (Microsoft Corporation,
Redmond, WA, USA).
Results
AQP1, 3, 7, 9 and 11 are expressed in
human chondrocytes
Electrophoresis analysis with PCR products
demonstrated that AQP1, 3, 7, 9 and 11 were expressed
in chondrocytes obtained from OA patients (Fig. 1A) and in IL-1β-treated normal
human chondrocytes (Fig. 1B). By
contrast, non-stimulated normal human chondrocytes did not express
any of the AQP genes (data not shown). Therefore, all
subsequent experiments were performed using normal human
chondrocytes under IL-1β stimulation.
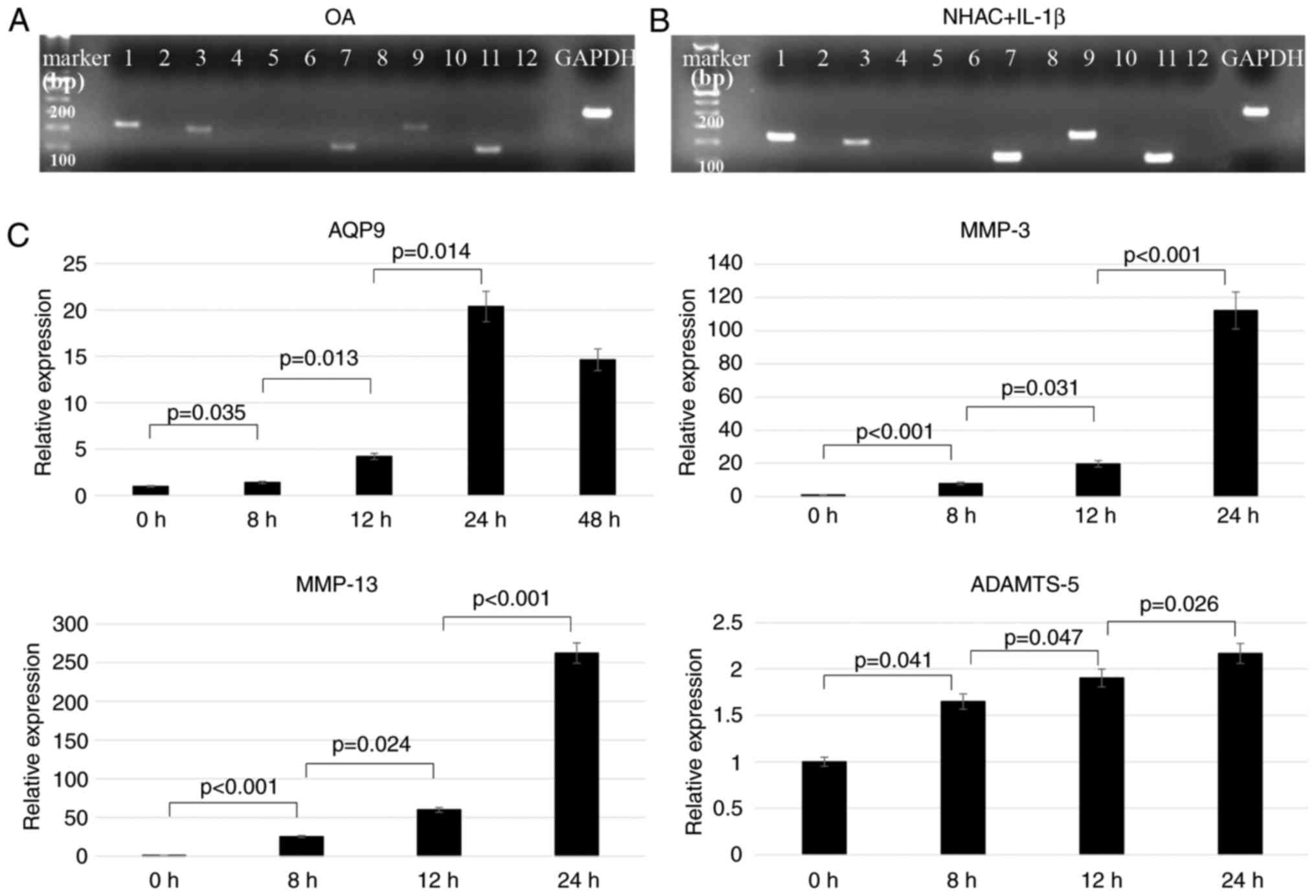 | Figure 1Expression of different AQPs and
catabolic genes in chondrocytes. Agarose gel electrophoresis of PCR
products obtained from (A) human osteoarthritis chondrocytes and
(B) IL-1β-treated normal human chondrocytes, using RT-PCR and
primers specific for each of the 12 human AQPs, namely
AQP1–12. (C) Chondrocytes were cultured with IL-1β
for 8, 12, 24 or 48 h, and the levels of AQP9,
MMP-3, MMP-13 and
ADAMTS-5 mRNA were quantified by qPCR (n=5). AQP,
aquaporin; MMP, matrix metalloproteinase; ADAMTS-5, a disintegrin
and metalloproteinase with thrombospondin motifs 5; IL-1β,
interleukin-1β; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction. |
IL-1β increases AQP9 mRNA expression in
chondrocytes
RT-qPCR demonstrated that the level of AQP9
mRNA was significantly increased with time upon exposure to 10
ng/ml IL-1β. The peak AQP9 mRNA expression occurred at 24 h
after IL-1β treatment. Increases were also observed in the mRNA
levels of MMP-3, MMP-13 and
ADAMTS-5 mRNA levels (Fig. 1C).
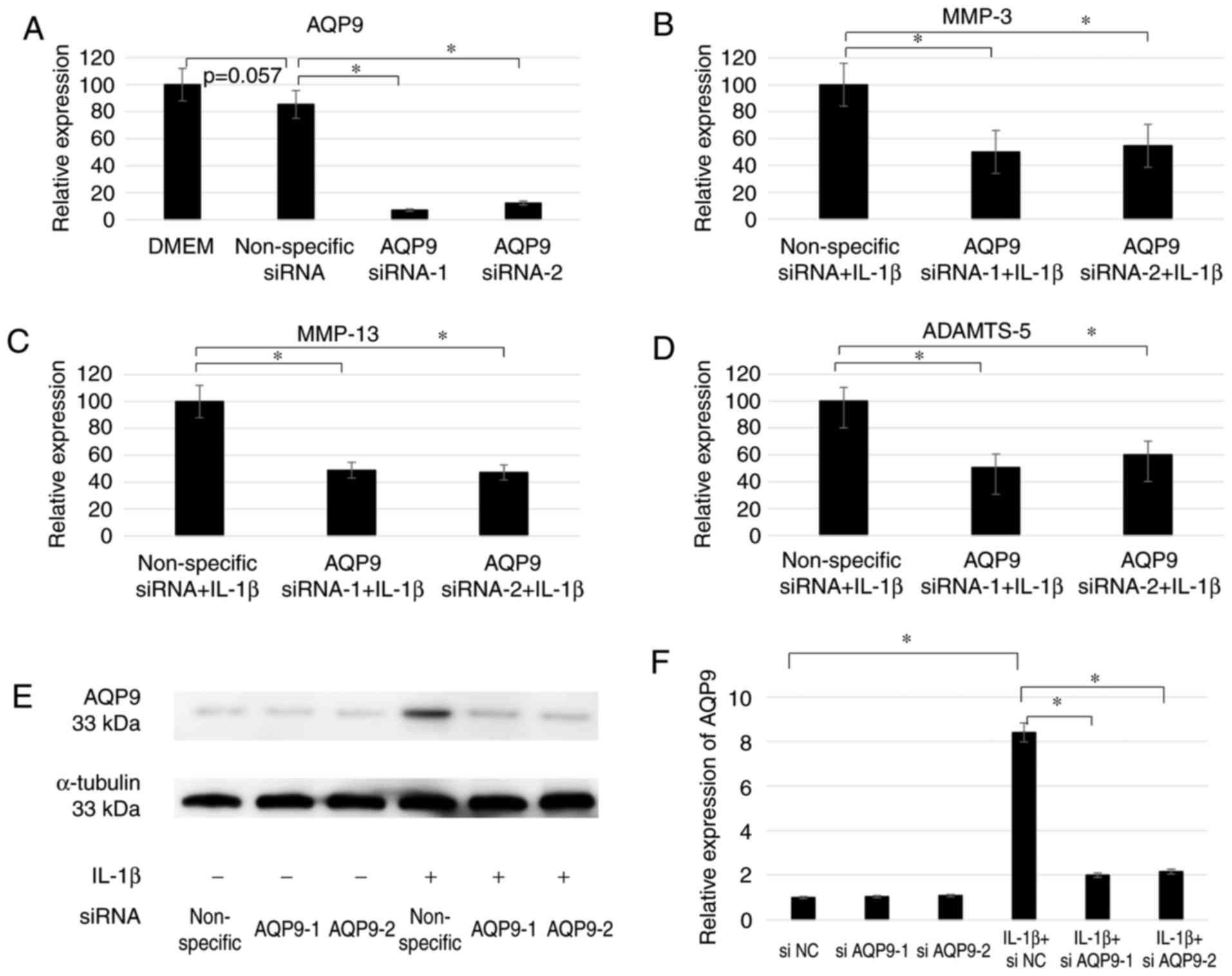
Downregulation of AQP9 expression
decreases catabolic gene expression in IL-1β-stimulated
chondrocytes
To investigate the functions of AQP9 in
chondrocytes, cells were transfected with two AQP9-specific siRNAs
and then treated with IL-1β. RT-qPCR demonstrated that the
knockdown was successfully achieved by siRNA transfection, as the
level of AQP9 mRNA in the siAQP9-1-transfected and siAQP9-2
transfected cells only amounted to 9.7 and 12.2%, respectively, of
the corresponding level in the cells transfected with non-specific
siRNA (Fig. 2A). The
downregulation of AQP9 expression also resulted in decreased
MMP-3, MMP-13 and
ADAMTS-5 mRNA levels (Fig. 2B–D). The level of
MMP-3 mRNA was significantly decreased to 49.9 and
54.5% using siAQP9-1 and siAQP9-2, respectively, under IL-1β
stimulation (both P<0.001; Fig.
2B). In addition, the level of MMP-13 mRNA was
decreased to 48.9 and 47.2% (both P<0.005; Fig. 2C), and the level of
ADAMTS-5 mRNA was decreased to 50.5 and 60.1% (both
P<0.005; Fig. 2D), following
transfection with siAQP9-1 and siAQP9-2, respectively. These
findings suggest that AQP9 positively regulates the expression
levels of MMP-3, MMP-13 and
ADAMTS-5. Furthermore, since IL-1β was previously
demonstrated to upregulate all four genes, it is likely that AQP9
mediates, at least partly, the ability of IL-1β to induce the
expression of the other three genes. Western blotting confirmed
that siAQP9-1 and siAQP9-2 significantly decreased AQP9 expression
at the protein level in chondrocytes with IL-1β stimulation in
comparison with the non-specific siRNA (Fig. 2E and F).
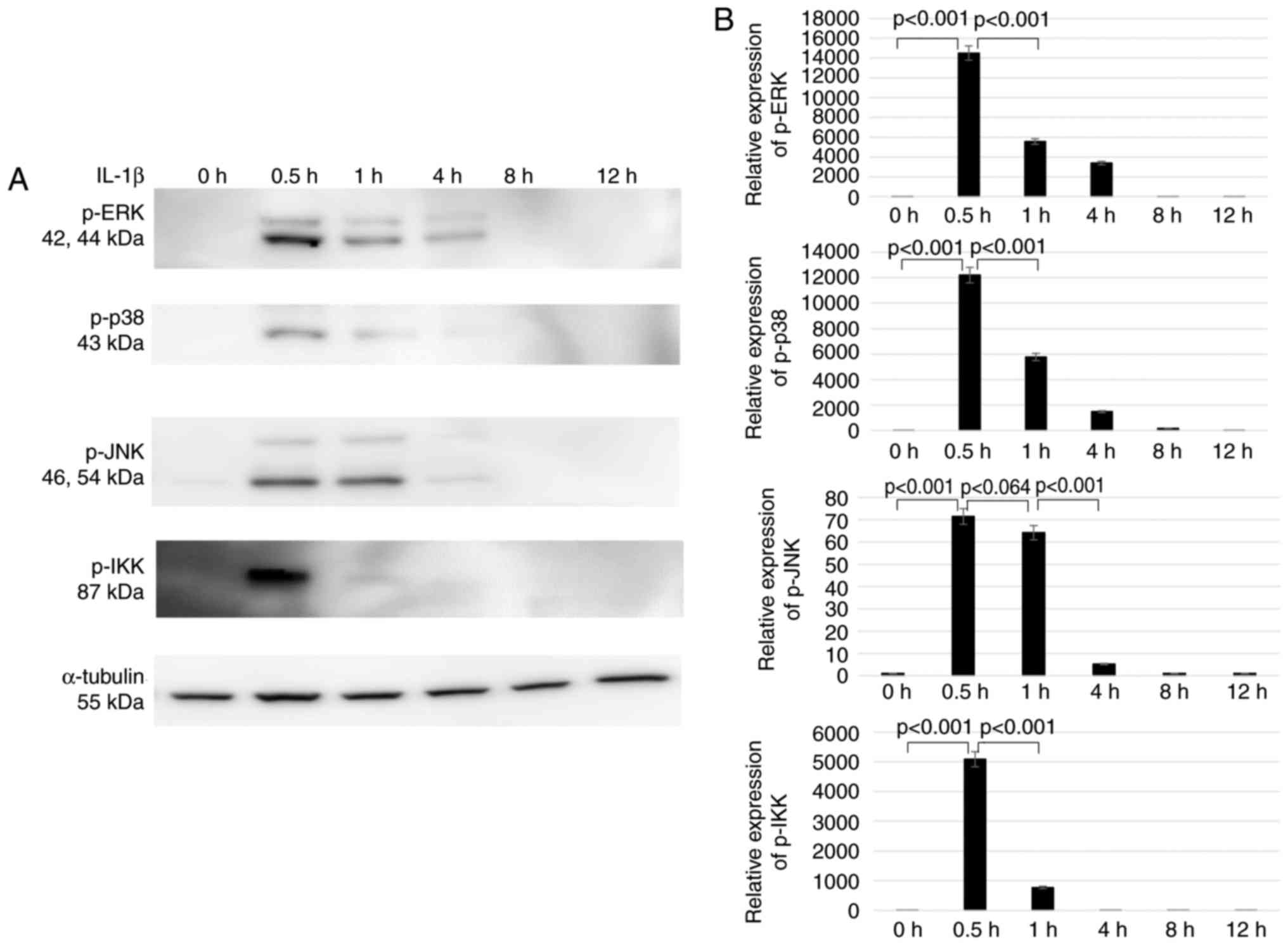
Downregulation of AQP9 expression
increases IKK phosphorylation in IL-1β-stimulated chondrocytes
To investigate the role of AQP9 in signal
transduction, the phosphorylation of ERK, p38 MAPK, JNK and IKK in
AQP9-knockdown chondrocytes treated with 10 ng/ml IL-1β was
assessed. The results confirmed the time-dependent change in
phosphorylation following IL-1β stimulation, and the
phosphorylation levels of ERK, p38 MAPK, JNK and IKK were markedly
elevated at 30 min (Fig. 3).
Therefore, the AQP9-knockdown (si AQP9-1) chondrocytes were treated
with IL-1β for 30 min in subsequent experiments. Next, it was
observed that the expression levels of ERK, p38 MAPK, JNK and IKK
proteins were not altered in comparison with those in cells treated
with the non-specific siRNA control (Fig. 4). However, the AQP9-knockdown
cells exhibited significantly lower IKK phosphorylation levels
compared with those in the control cells treated with non-specific
siRNA. This indicated that AQP9 positively regulates the
phosphorylation of IKK, which is a kinase necessary for the
activation of nuclear factor (NF)-κB. By contrast, the
phosphorylation levels of ERK, p38 MAPK and JNK were similar
between the knockdown and control cells (Fig. 4), indicating that AQP9 is not
involved in the activation of their corresponding signaling
pathways.
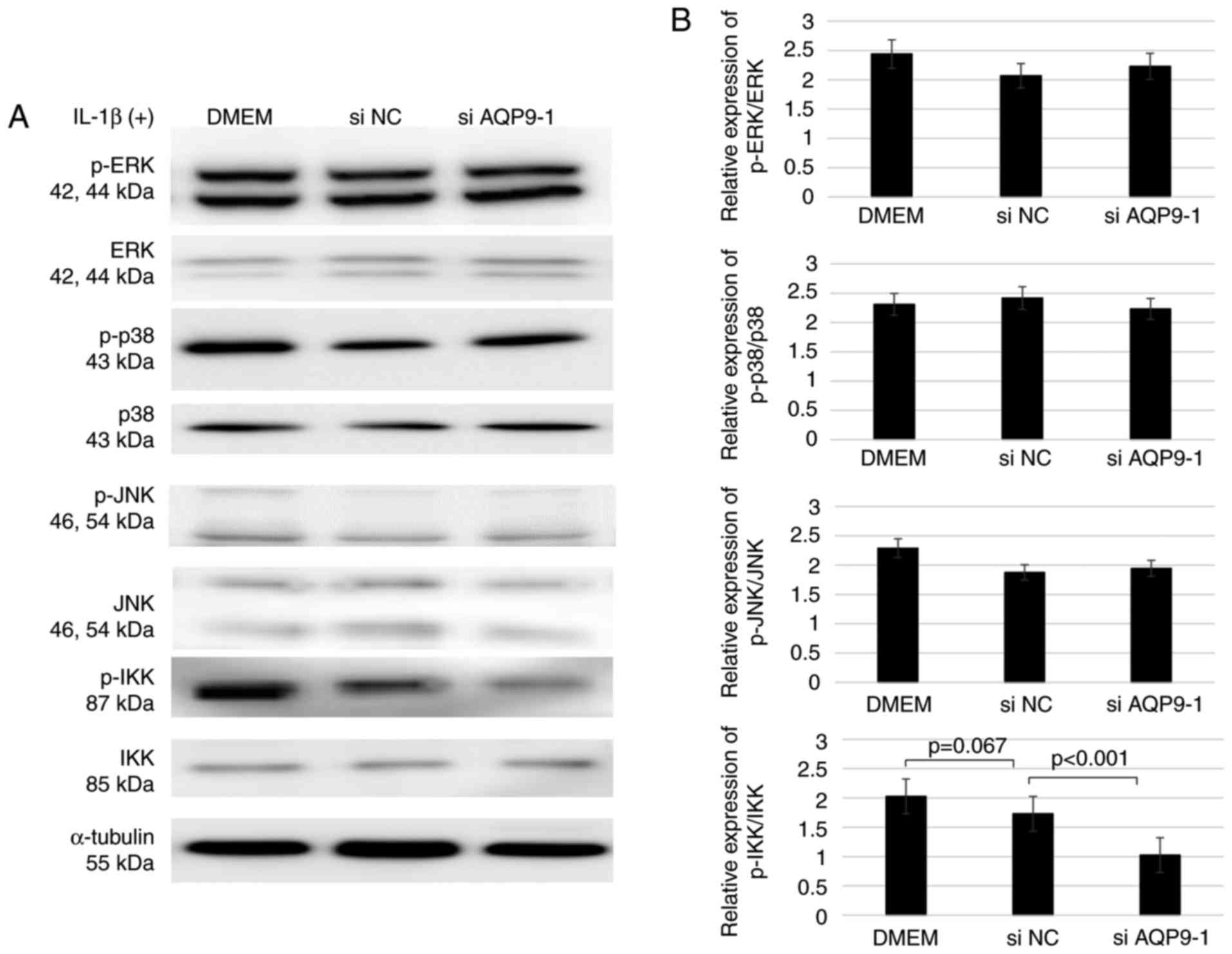 | Figure 4Levels of p-ERK, ERK, p-p38 MAPK, p38
MAPK, p-JNK, JNK, p-IKK and IKK following IL-1β stimulation of
chondrocytes transfected with siAQP9 or non-specific siNC, as
determined by western blotting. (A) Western blots and (B)
semiquantified results of protein expression levels are shown
(n=5). Each full length protein of ERK, p38MAPK, JNK and IKK was
used as the control to estimate the protein loading on the gel.
ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated
protein kinase; JNK, c-Jun N-terminal kinase; IKK, IκB kinase; p-,
phosphorylated; IL-1β, interleukin-1β; AQP9, aquaporin 9; si, small
interfering RNA; NC, negative control; DMEM, Dulbecco's modified
Eagle's medium. |
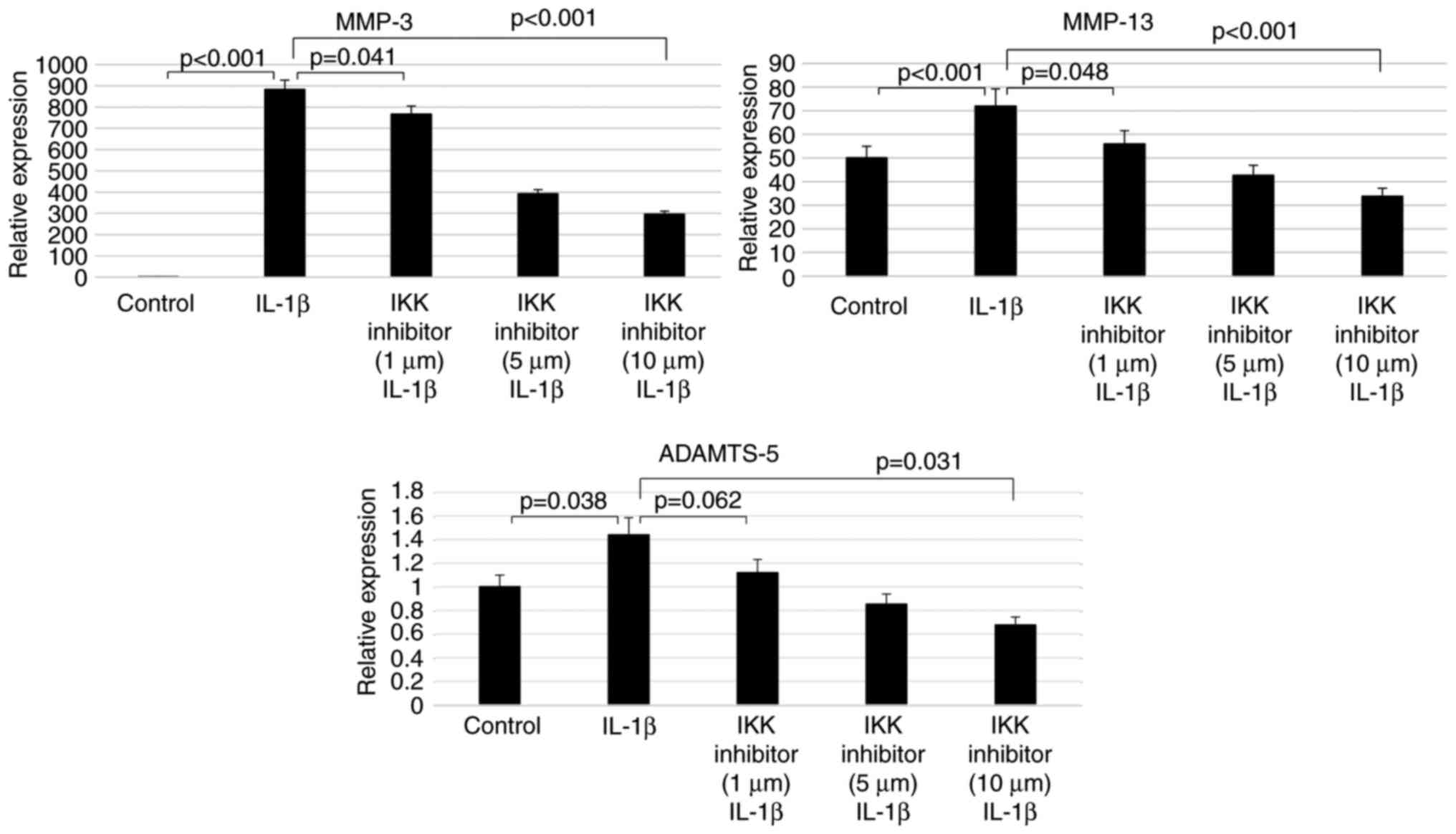
Inhibition of IKK phosphorylation
decreases catabolic gene expression in chondrocytes
To examine the involvement of the NF-κB signaling
pathway in the induction of MMP-3, MMP-13 and ADAMTS-5 expression
by IL-1β treatment, the chondrocytes were subsequently treated with
an inhibitor of IKK phosphorylation, BMS-345541. RT-qPCR
demonstrated that, in cells treated with 10 ng/ml IL-1β for 24 h,
the IKK inhibitor suppressed MMP-3,
MMP-13 and ADAMTS-5 mRNA levels in a
concentration-dependent manner. More specifically, 10 µM
BMS-345541 resulted in significantly lower MMP-3
(0.19%; P<0.001), MMP-13 (0.98%; P<0.001) and
ADAMTS-5 (24%; P=0.032) mRNA levels compared with
those in cells treated only with IL-1β (Fig. 5).
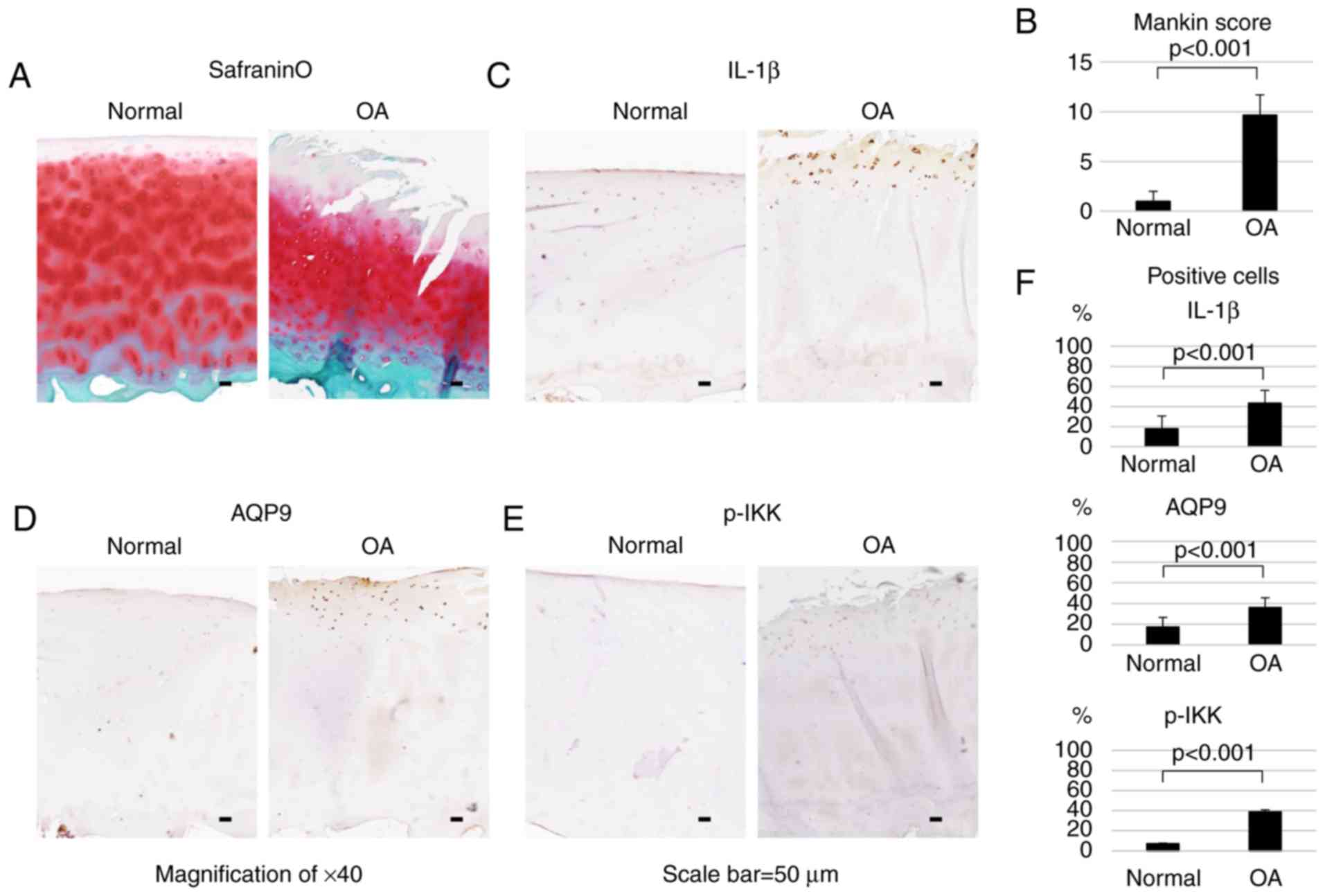
AQP9 and inflammatory markers are highly
expressed in OA cartilage
Subsequent to safranin O and fast green staining,
human OA cartilage exhibited mid-zone excavation and lower
proteoglycan content compared with the normal cartilage (Fig. 6A). Histological evaluation was
performed using the histopathology grading system by Mankin et
al (30), indicating that the
average score of the OA cartilage was significantly higher compared
with that of normal cartilage (Fig.
6B).
Immunohistological analysis demonstrated that IL-1β,
AQP9 and p-IKK were highly expressed in the surface layer of OA
cartilage (Fig. 6C–E). As shown
in Fig. 6F, the numbers of
IL-1β-, AQP9- and p-IKK-positive cells were significantly higher
(P<0.001) in OA cartilage as compared with those in normal
tissue. In OA and normal cells, the IL-1β-positive cell rates were
43.3 and 17.9%, respectively. Similarly, the AQP9-positive cell
rates were 36 and 17.1%, respectively, while the p-IKK-positive
cell rates were 38.8 and 7.14%, respectively.
Catabolic markers are highly expressed in
OA cartilage
To confirm the expression of catabolic markers in
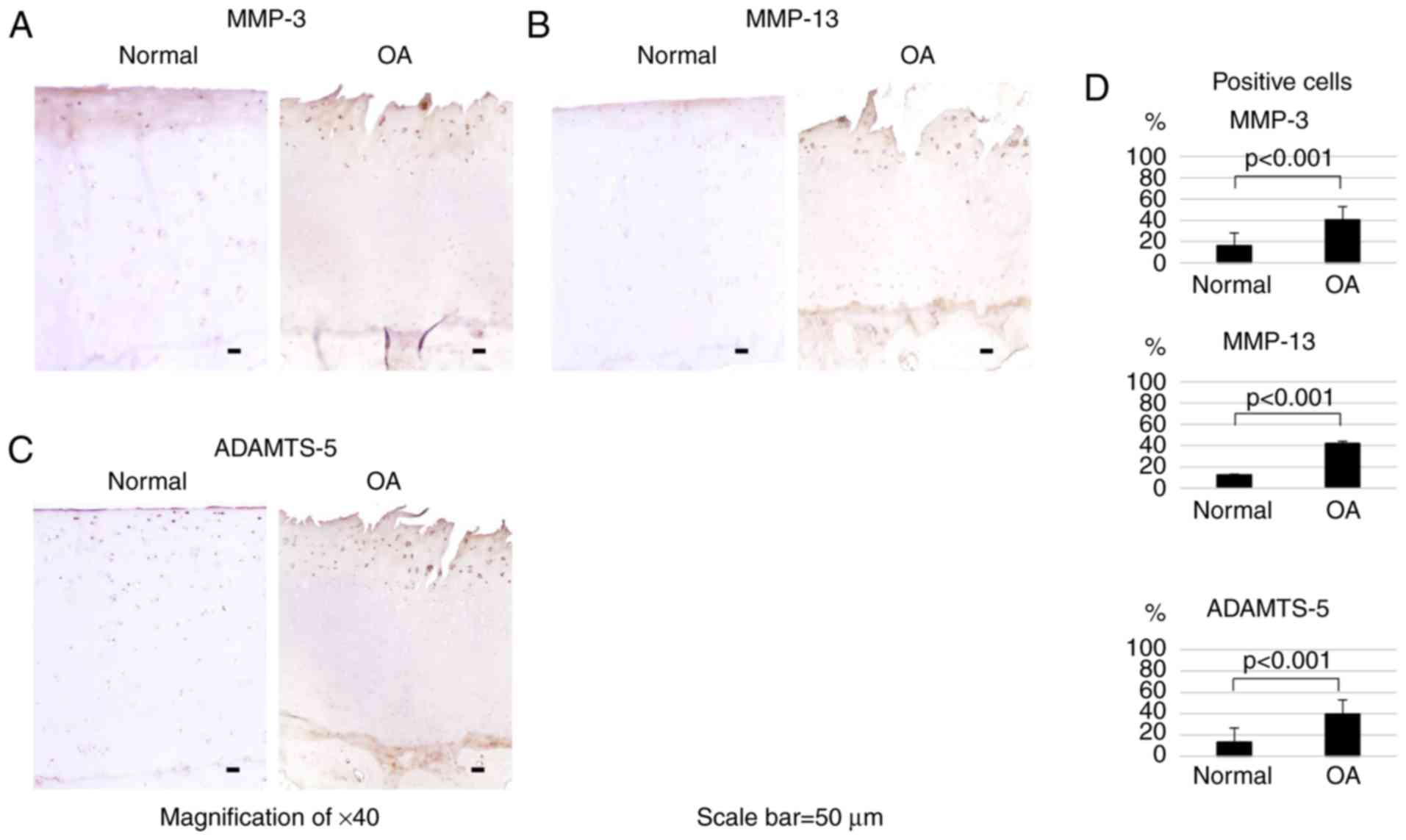
human OA cartilage tissue, immunohistochemical detection of MMP-3,
MMP-13 and ADAMTS-5 was then performed. The immunohistological
analysis revealed that all three proteins were highly expressed in
the surface layer of OA cartilage (Fig. 7A–C). As shown in Fig. 7D, the numbers of MMP-3-, MMP-13-
and ADAMTS-5-positive cells were significantly higher (P<0.001)
in OA cartilage compared with those in normal tissue. The
MMP-3-positive cell rates were 40.4 and 16% in OA and normal cells,
respectively, the MMP-13-positive cell rates were 41.9 and 12.5%,
and the ADAMTS-5-positive cell rates were 39.7 and 13.3%.
Discussion
The current study demonstrated that AQP9 and
inflammatory markers are highly expressed in OA cartilage tissue,
and that downregulation of AQP9 expression in normal human
chondrocytes reduced the IL-1β-induced expression of catabolic
genes. Matsushima et al (34) reported elevated AQP9 expression
levels in systemic inflammatory response syndrome patients and
suggested that AQP9 may be associated with F-actin polymerization,
which would implicate it in the morphologic and functional changes
observed in this syndrome. In addition, Mesko et al
(35) reported that changes in
AQP9 expression are universal markers of chronic inflammation and
may be one of the causes of this condition in autoimmune disease
patients. Nagahara et al (28) also reported the presence of
AQP9 mRNA in synovial tissues from patients with OA and
rheumatoid arthritis. The present study suggested that the elevated
expression levels of AQP9 in OA joints may increase the expression
of catabolic factors, such as MMPs and ADAMTSs. It is likely that
AQP9 mediates, at least partly, the IL-1β-induced expression of
MMP-3, MMP-13 and ADAMTS-5. Furthermore, the current findings
indicate that AQP9 positively regulates the phosphorylation of IKK,
which is known to activate NF-κB. Based on these results, it can be
hypothesized that IL-1β first upregulates AQP9, which then induces
the expression levels of MMP-3, MMP-13 and ADAMTS-5 by activating
the NF-κB cascade.
NF-κB is one of the transcription factors with a
central role in immunoreaction and is known to be activated by
various stimuli, including stress, cytokines and ultraviolet rays
(36). It is involved in numerous
physiological phenomena, such as acute and chronic inflammation,
cell proliferation and apoptosis. Defective control of NF-κB
activity leads to inflammatory diseases, such as rheumatoid
arthritis, inflammatory bowel disease, asthma and chronic
obstructive pulmonary disease (37,38), as well as cancer and septic shock
(39). Recently, Wang et
al (40) reported that
knockdown of AQP9 expression attenuated brain edema through the
inhibition of the NF-κB signaling pathway. To determine the
mechanism through which AQP9 affects catabolic gene expression in
IL-1β-stimulated cells, the present study investigated various
proteins associated with signaling, and the results revealed that
the depletion of AQP9 significantly reduced the abundance of p-IKK.
As the IKK complex serves a central role in regulating NF-κB
activity, the current results suggest that AQP9 functions as a
signal transduction regulator of NF-κB activity by positively
regulating the phosphorylation of IKK, which allows NF-κB to
mediate the ability of IL-1β to increase the levels of p-IKK and,
in turn, induce the expression of MMP-3, MMP-13 and ADAMTS-5. This
is supported by the observation that inhibition of IKK
phosphorylation reduced MMP-3, MMP-13 and ADAMTS-5 expression in
response to IL-1β stimulation. Notably, previous studies have
reported that certain AQPs function as inflammatory signal
potentiators, enhancing the release of inflammatory cytokines, such
as IL-1β, in response to increased activation of NF-κB (27,41). Together, these data suggest that
AQPs are involved in both triggering the inflammatory response and
mediating its effects on catabolic gene expression.
However, several limitations of the current study
require further elaboration. First, the correlation between the
upregulation of AQP9 and the NF-κB signaling pathway needs
clarification. Notably, it has been reported that inflammatory
factors increase AQP4 mRNA levels through the Toll-like
receptor 4 (TLR4) signaling pathway in the cortex and astrocytes,
which culminates in the activation of NF-κB, as is the case with
all TLR pathways (42). Thus,
there may be other factors linking AQP9 and NF-κB. Furthermore,
AQP9 overexpression models to confirm the results of inflammation
increase and in vivo experiments were not tested. In the
future, these experiments will be performed to confirm the
comprehensive functions of AQP9 in chondrocytes.
In conclusion, the data of the present study
demonstrated that AQP9 and inflammatory markers are highly
expressed in OA cartilage and that AQP9 downregulation results in
decreased catabolic gene expression in response to IL-1β
stimulation through NF-κB signaling. Thus, AQP9 may be an
attractive target for the treatment of inflammatory joint diseases,
such as OA.
Acknowledgments
The authors thank Mr. Takeshi Ueha, Ms. Kyoko
Tanaka, Ms. Minako Nagata and Ms. Maya Yasuda of Kobe University
Graduate School of Medicine (Kobe, Japan) for their technical
assistance.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KT conceived the study, participated in its design,
performed statistical analysis and data interpretation, and drafted
the manuscript. SHay conceived the study, participated in its
design and the acquisition of data, performed statistical analysis
and data interpretation, and drafted the manuscript. TM and SHas
participated in human sample collection and the drafting of the
manuscript. MH, NC and SKih conceived the study and participated in
its design and the acquisition of data. KT, SKir, YK and MT
contributed to data acquisition. KN and RK participated in
designing the study and helped revise the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of Kobe University Graduate School of Medicine (Kobe,
Japan) on March 19th, 2015 (no. 1721), and all patients provided
informed consent prior to participation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chevalier X, Groult N, Emod I and
Planchenault T: Proteoglycan-degrading activity associated with the
40 kDa collagen-binding fragment of fibronectin. Br J Rheumatol.
35:506–514. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Berenbaum F and van den Berg WB:
Inflammation in osteoarthritis: Changing views. Osteoarthritis
Cartilage. 23:1823–1824. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Berenbaum F: Osteoarthritis as an
inflammatory disease (osteoarthritis is not osteoarthrosis!).
Osteoarthritis Cartilage. 21:16–21. 2013. View Article : Google Scholar
|
|
4
|
Fernandes JC, Martel-Pelletier J and
Pelletier JP: The role of cytokines in osteoarthritis
pathophysiology. Biorheology. 39:237–246. 2002.PubMed/NCBI
|
|
5
|
Goldring MB: The role of cytokines as
inflammatory mediators in osteoarthritis: Lessons from animal
models. Connect Tissue Res. 40:1–11. 1999. View Article : Google Scholar
|
|
6
|
Loeser RF: Molecular mechanisms of
cartilage destruction: Mechanics, inflammatory mediators, and aging
collide. Arthritis Rheum. 54:1357–1360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wojdasiewicz P, Poniatowski LA and
Szukiewicz D: The role of inflammatory and anti-inflammatory
cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm.
2014:5614592014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shakibaei M, Schulze-Tanzil G, John T and
Mobasheri A: Curcumin protects human chondrocytes from
IL-l1beta-induced inhibition of collagen type II and beta1-integrin
expression and activation of caspase-3: An immunomorphological
study. Ann Anat. 187:487–497. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stove J, Huch K, Gunther KP and Scharf HP:
Interleukin-1beta induces different gene expression of stromelysin,
aggrecan and tumor-necrosis-factor-stimulated gene 6 in human
osteoarthritic chondrocytes in vitro. Pathobiology. 68:144–149.
2000. View Article : Google Scholar
|
|
10
|
Mengshol JA, Vincenti MP, Coon CI,
Barchowsky A and Brinckerhoff CE: Interleukin-1 induction of
collagenase 3 (matrix metalloproteinase 13) gene expression in
chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear
factor kappaB: Differential regulation of collagenase 1 and
collagenase 3. Arthritis Rheum. 43:801–811. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsushita T, Sasaki H, Takayama K, Ishida
K, Matsumoto T, Kubo S, Matsuzaki T, Nishida K, Kurosaka M and
Kuroda R: The overexpression of SIRT1 inhibited osteoarthritic gene
expression changes induced by interleukin-1β in human chondrocytes.
J Orthop Res. 31:531–537. 2013. View Article : Google Scholar
|
|
12
|
Wang M, Sampson ER, Jin H, Li J, Ke QH, Im
HJ and Chen D: MMP13 is a critical target gene during the
progression of osteoarthritis. Arthritis Res Ther. 15:R52013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miller RE, Tran PB, Ishihara S, Larkin J
and Malfait AM: Therapeutic effects of an anti-ADAMTS-5 antibody on
joint damage and mechanical allodynia in a murine model of
osteoarthritis. Osteoarthritis Cartilage. 24:299–306. 2016.
View Article : Google Scholar :
|
|
14
|
Ishibashi K, Hara S and Kondo S: Aquaporin
water channels in mammals. Clin Exp Nephrol. 13:107–117. 2009.
View Article : Google Scholar
|
|
15
|
Tsukaguchi H, Weremowicz S, Morton CC and
Hediger MA: Functional and molecular characterization of the human
neutral solute channel aquaporin-9. Am J Physiol. 277:F685–F696.
1999.PubMed/NCBI
|
|
16
|
Ishibashi K: New members of mammalian
aquaporins: AQP10-AQP12. Handb Exp Pharmacol. 251–262. 2009.
View Article : Google Scholar
|
|
17
|
Verkman AS: Roles of aquaporins in kidney
revealed by transgenic mice. Semin Nephrol. 26:200–208. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sohara E, Rai T, Sasaki S and Uchida S:
Physiological roles of AQP7 in the kidney: Lessons from AQP7
knockout mice. Biochim Biophys Acta. 1758:1106–1110. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ko SB, Mizuno N, Yatabe Y, Yoshikawa T,
Ishiguro H, Yamamoto A, Azuma S, Naruse S, Yamao K, Muallem S and
Goto H: Aquaporin 1 water channel is overexpressed in the plasma
membranes of pancreatic ducts in patients with autoimmune
pancreatitis. J Med Invest. 56(Suppl): 318–321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huysseune S, Kienlen-Campard P, Hebert S,
Tasiaux B, Leroy K, Devuyst O, Brion JP, De Strooper B and Octave
JN: Epigenetic control of aquaporin 1 expression by the amyloid
precursor protein. FASEB J. 23:4158–4167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mobasheri A, Moskaluk CA, Marples D and
Shakibaei M: Expression of aquaporin 1 (AQP1) in human synovitis.
Ann Anat. 192:116–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rojek AM, Skowronski MT, Fuchtbauer EM,
Füchtbauer AC, Fenton RA, Agre P, Frøkiaer J and Nielsen S:
Defective glycerol metabolism in aquaporin 9 (AQP9) knockout mice.
Proc Natl Acad Sci USA. 104:3609–3614. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Spegel P, Chawade A, Nielsen S, Kjellbom P
and Rutzler M: Deletion of glycerol channel aquaporin-9 (Aqp9)
impairs long-term blood glucose control in C57BL/6 leptin
receptor-deficient (db/db) obese mice. Physiol Rep. 3:e125382015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Naka M, Kanamori A, Negi A and Nakamura M:
Reduced expression of aquaporin-9 in rat optic nerve head and
retina following elevated intraocular pressure. Invest Ophthalmol
Vis Sci. 51:4618–4626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
de Baey A and Lanzavecchia A: The role of
aquaporins in dendritic cell macropinocytosis. J Exp Med.
191:743–748. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu N, Feng X, He C, Gao H, Yang L, Ma Q,
Guo L, Qiao Y, Yang H and Ma T: Defective macrophage function in
aquaporin-3 deficiency. FASEB J. 25:4233–4239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rabolli V, Wallemme L, Lo Re S,
Uwambayinema F, Palmai-Pallag M, Thomassen L, Tyteca D, Octave JN,
Marbaix E, Lison D, et al: Critical role of aquaporins in
interleukin 1β(IL-1β)-induced inflammation. J Biol Chem.
289:13937–13947. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nagahara M, Waguri-Nagaya Y, Yamagami T,
Aoyama M, Tada T, Inoue K, Asai K and Otsuka T: TNF-alpha-induced
aquaporin 9 in synoviocytes from patients with OA and RA.
Rheumatology (Oxford). 49:898–906. 2010. View Article : Google Scholar
|
|
29
|
General Assembly of the World Medical
Association: World medical association declaration of helsinki:
Ethical principles for medical research involving human subjects. J
Am Coll Dent. 81:14–18. 2014.
|
|
30
|
Mankin HJ, Dorfman H, Lippiello L and
Zarins A: Biochemical and metabolic abnormalities in articular
cartilage from osteo-arthritic human hips. II Correlation of
morphology with biochemical and metabolic data. J Bone Joint Surg
Am. 53:523–537. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pattoli MA, MacMaster JF, Gregor KR and
Burke JR: Collagen and aggrecan degradation is blocked in
interleukin-1-treated cartilage explants by an inhibitor of IkappaB
kinase through suppression of metalloproteinase expression. J
Pharmacol Exp Ther. 315:382–388. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kihara S, Hayashi S, Hashimoto S, Kanzaki
N, Takayama K, Matsumoto T, Chinzei N, Iwasa K, Haneda M, Takeuchi
K, et al: Cyclin-dependent kinase inhibitor-1-deficient mice are
susceptible to osteoarthritis associated with enhanced
inflammation. J Bone Miner Res. 32:991–1001. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Matsushima A, Ogura H, Koh T, Shimazu T
and Sugimoto H: Enhanced expression of aquaporin 9 in activated
polymor-phonuclear leukocytes in patients with systemic
inflammatory response syndrome. Shock. 42:322–326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mesko B, Poliska S, Szegedi A, Szekanecz
Z, Palatka K, Papp M and Nagy L: Peripheral blood gene expression
patterns discriminate among chronic inflammatory diseases and
healthy controls and identify novel targets. BMC Med Genomics.
3:152010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gilmore TD: The Rel/NF-kappaB signal
transduction pathway: Introduction. Oncogene. 18:6842–6844. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Holgate ST: Cytokine and anti-cytokine
therapy for the treatment of asthma and allergic disease. Cytokine.
28:152–157. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Williams RO, Paleolog E and Feldmann M:
Cytokine inhibitors in rheumatoid arthritis and other autoimmune
diseases. Curr Opin Pharmacol. 7:412–417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu SF and Malik AB: NF-kappa B activation
as a pathological mechanism of septic shock and inflammation. Am J
Physiol Lung Cell Mol Physiol. 290:L622–L645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang BF, Cui ZW, Zhong ZH, Sun YH, Sun QF,
Yang GY and Bian LG: Curcumin attenuates brain edema in mice with
intracerebral hemorrhage through inhibition of AQP4 and AQP9
expression. Acta Pharmacol Sin. 36:939–948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang M, Gao F, Liu H, Yu WH and Sun SQ:
Temporal changes in expression of aquaporin-3, -4, -5 and -8 in rat
brains after permanent focal cerebral ischemia. Brain Res.
1290:121–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Song TT, Bi YH, Gao YQ, Huang R, Hao K, Xu
G, Tang JW, Ma ZQ, Kong FP, Coote JH, et al: Systemic
pro-inflammatory response facilitates the development of cerebral
edema during short hypoxia. J Neuroinflammation. 13:632016.
View Article : Google Scholar : PubMed/NCBI
|