Introduction
Neuropathic pain is a relatively common occurrence,
observed in ~0.6–1.5% of the US population, and it is a chronic
severely adynamic state initiated by a primary injury or the
dysfunction of the nervous system (1). There are several similarities
between pain and other neurobiological processes, such as learning
and memory (2). Two particularly
aggravating and prominent symptoms in different types of
neuropathic pain, caused by abnormal sensory perception of pain or
independent stimulating or persistent pain, are hyperalgesia and
allodynia (3). However, the
underlying potential cellular and molecular mechanisms are
relatively obscure, and current pain treatment remains
unsatisfactory. Previous studies have demonstrated that astrocytes
are activated during persistent neuropathic pain; in particular,
the late participation of astrocytic activation has been correlated
with nociception maintenance (4-7).
With rapid and sensitive response to various noxious or
physiological stimuli, astrocytes can increase the expression of
glial fibrillary acidic protein (GFAP) (8). Kim et al observed that the
lack of GFAP resulted in brief persistent allodynia following nerve
ligation (9). These findings
indicate that peripheral nerve damage-induced GFAP activity not
only serves as a marker for astrocyte hypertrophy, but also plays
an important role during the process of neuropathic pain. Moreover,
GFAP plays a key role in regulating astrocyte motility (10) and may be involved in synaptic
modulation in the central nervous system (CNS) (11-13).
Autophagy, which is a process of self-digestion, is
a dynamic regulated process consisting of autophagosomal induction,
formation and autophagosome-lysosome fusion, for the recycling of
damaged or malfunctioning proteins and organelles, such as injured
mitochondria (14). Following
exposure to various stressors, the double-membrane vesicle
encircles the degraded cytoplasm and organelles and fuses with the
lysosomal membrane to form an autolysosome. The autophagosomes are
then degraded and recycled to remodel new protein or organelles for
cell survival. Failure of the autophagosome-lysosome fusion may
lead to the accumulation of autophagosomes (15). Therefore, a simple assessment of
autophagy may be misleading, due to the increased autophagosomal
formation. It is also necessary to investigate the mechanisms
underlying autophagosomal induction, formation and
autophagosome-lysosome fusion (16). A number of studies have clearly
demonstrated high levels of autophagy-related proteins and/or
numbers of autophagosomes in a model of CNS injury due to
hypoxia/ischemia or trauma (17
and refs. therein). Accumulating evidence indicates a close
association between autophagy and neuropathic pain (18-20). Piao et al (21) reported that lack of the p62
autophagic protein plays a key role in the pathophysiology of
neuropathic pain. Autophagy inducers have been shown to protect
neurons during traumatic brain injury in mice and to also relieved
brain damage in a model of neonatal hypoxia/ischemia (22,23). Furthermore, autophagy regulates
the expression GFAP in astrocytes and has been associated with
allodynia and hyperalgesia in different diseases (24,25).
However, whether the autophagic-lysosomal pathway is
activated and whether autophagy regulates GFAP activity, allodynia
and hyperalgesia in neuropathic pain induced by chronic
constriction injury (CCI), remains unknown. Thus, the aim of the
present study was to discuss the entire autophagy process, from
autophagosomal formation to autophagosome-lysosome fusion, in the
setting of neuropathic pain. In addition, autophagy inhibitors and
inducers were used to examine the effects of autophagy on GFAP
activity, allodynia and hyperalgesia.
Materials and methods
Animals
A total of 192 Adult male Sprague-Dawley rats (8–10
weeks old, weighing 200–250 g) were used in all the experiments.
The rats were housed in a temperature-controlled (25°C) room with
an alternating 12-h light/dark cycle under specific pathogen-free
conditions; water and chow were provided ad libitum until
the commencement of the experiments. The rats were obtained from
the Laboratory Animal Center of the Military Medical Science
Academy of the Chinese People's Liberation Army. All experimental
procedures were approved by the Institutional Animal Care and Use
Committee of Tianjin Medical University and were performed in
accordance with the National Institutes of Health Guide for Care
and Use of Laboratory Animals. All efforts were made to minimize
animal suffering and the number of animals used.
Neuropathic pain induced by CCI
Neuropathic pain was induced by CCI of the sciatic
nerve according to the model previously described by Bennett and
Xie (26). The rats were
anesthetized with sevoflurane (induction with 3.0%; maintenance
with 1.5%) mixed with air through a nose mask under sterile
conditions. The left sciatic nerve was exposed and 3 loose
ligatures with 5-0 silk suture thread were made on the nerve at
1.0–1.5 mm intervals. The muscle and skin were sutured after
complete hemostasis. The rats were divided into 8 groups according
to the ligation/medicinal intervention as follows: The control
(Con) (n=40), CCI (n=48), CCI + 3-methyladenine (3-MA) (n=16), CCI
+ rapamycin (Rap) (n=24), CCI + chloroquine (CQ) (n=16), CCI +
bafilomycin (Baf) (n=16), CCI + Rap + CQ (n=16) and CCI + Rap + Baf
(n=16) groups in all the experiments. In the Con group, the sciatic
nerve was exposed but without ligation; the rats in all the other
groups underwent both exposure and ligation of the left sciatic
nerve. At the end of the experiments, the rats were euthanized by
CO2 exposure (CO2 displacement rate
equivalent to 20% of the chamber volume/min) and cervical
dislocation.
Experimental protocol
The experimental protocol was as follows:
Experiment 1: Autophagy-related
protein expression in rats with CCI-induced neuropathic pain
Neuropathic pain was induced by CCI of the sciatic
nerve and the L4-L6 spinal cord was collected for further detection
of autophagy-related proteins. Part of the spinal cord was used to
test for LC3II, Beclin 1 and p62 by western blot analysis prior to
(Con; Fig. 1) and at 1, 3, 7 and
14 days after CCI. In addition, the expression of GFAP, another
autophagy-related protein, was detected by western blot analysis
(as described below) at 1, 3, 7 and 14 days after CCI (n=8 for Con
and each point time). The remaining part of the L4-L6 spinal cord
was immediately stored in 4% paraformaldehyde for the measurement
of GFAP expression by immunofluorescence assay (as described below)
at 1, 3 and 7 days after CCI.
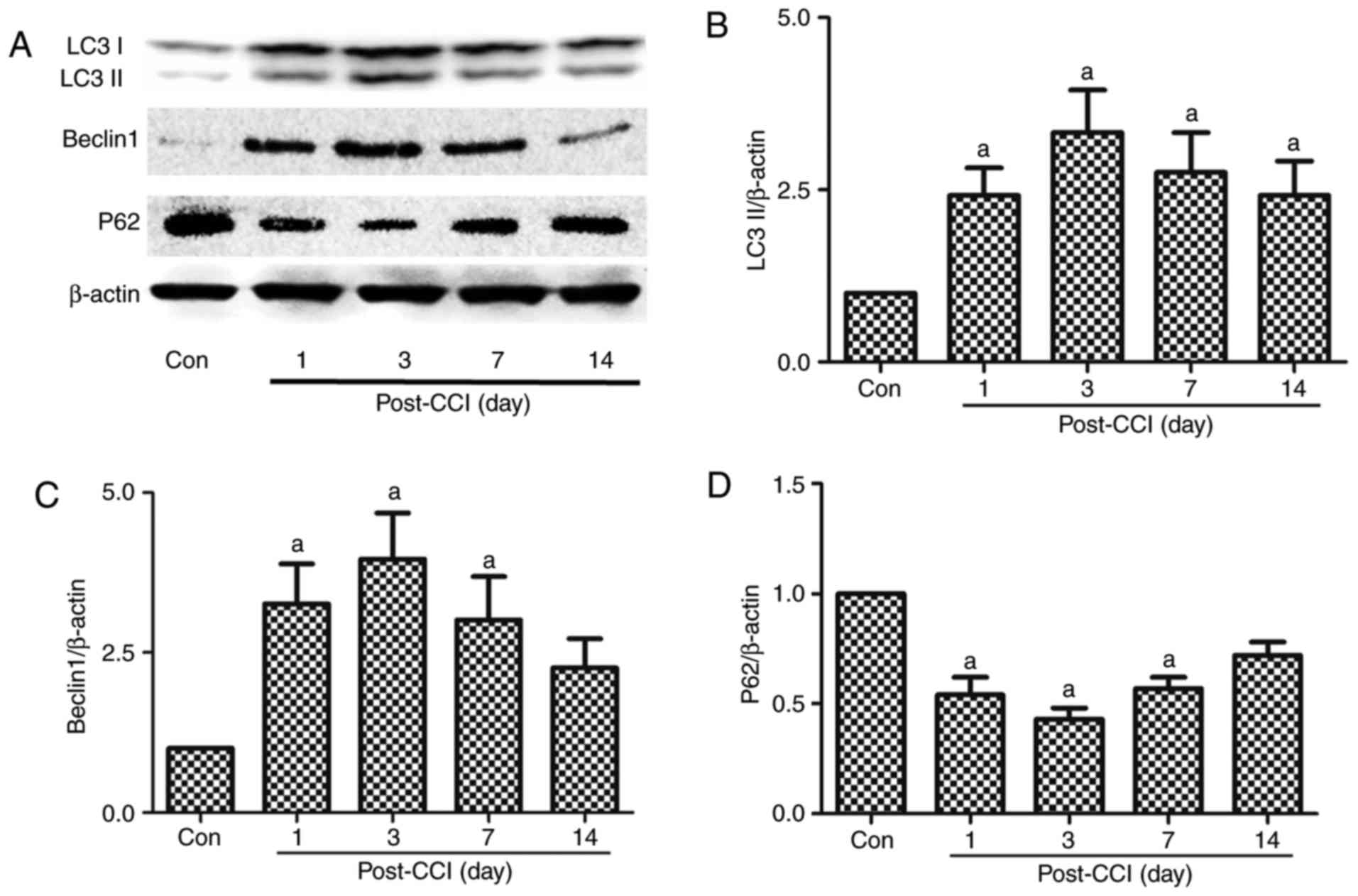 | Figure 1Expression of the autophagy-related
proteins, LC3II, Beclin 1 and p62, in the spinal dorsal horn of
rats with neuropathic pain. A part of the L4-L5 spinal cord was
collected for the detection of the autophagy-related proteins (A
and B) LC3II, (A and C) Beclin 1, and (A and D) p62 by western blot
analysis prior to and at 1, 3, 7 and 14 days after CCI. The value
in the Con group was set as 1, and other relative values in the CCI
groups were calculated by comparison. The values are expressed as
the means ± standard deviation (n=8 per group). Statistical
analysis was performed with one-way analysis of variance followed
by Tukey's test. aP<0.05 vs. Con group. Con, control;
CCI, chronic constriction injury. |
Experiment 2: Effect of autophagy
induction and inhibition on the expression of the autophagy-related
proteins, LC3II, Beclin 1 and p62, in rats with CCI-induced
neuropathic pain
The rats were randomly divided into 4 groups, namely
the Con (n=8 from Fig. 1), CCI
(n=8 from Fig. 1), 3-MA (n=8) and
Rap (n=8) groups. According to previous studies (14,22,23), 3-MA and Rap (Bio Vision, Mountain
View, CA, USA) were administered by intraperitoneal injection at a
dose of 15 and 10 mg/kg, respectively, 1 h prior to the CCI
operation. At 7 days after CCI, the autophagy-related protein
expression of LC3II, Beclin 1 and p62 in the spinal cord was
detected by western blot analysis in all 4 groups.
Experiment 3: Evaluation of the
effects of autophagy inhibition and induction on allodynia,
hyperalgesia and GFAP expression in CCI-induced neuropathic pain by
western blot analysis and immunofluorescence
Grouping was performed as described above in
Experiment 2. Mechanical allodynia and thermal hyperalgesia were
tested prior to (0 days) and at 1, 3, 7 and 14 days after CCI. At 7
days after CCI, GFAP expression in the spinal cord was detected by
western blot analysis and immunofluorescence assay in all 4 groups
(n=8 from Fig. 1).
Experiment 4: Lysosome-related protein
expression in rats with CCI-induced neuropathic pain
Neuropathic pain was induced by CCI (n=8) of the
sciatic nerve. A part of the L4-L6 spinal cord was collected for
the detection of lysosome-related proteins, lysosomal-associated
membrane protein type 2 (LAMP2) and Ras-related protein Rab-7a
(RAB7), by western blot analysis prior to (Con; Fig. 5) and at 1, 3, 7 and 14 days after
CCI (n=8 from Fig. 1).
Experiment 5: Effect of autophagy
inhibition and induction on lysosome-related protein expression,
allodynia and hyperalgesia in rats with CCI-induced neuropathic
pain
The rats were randomly divided into 7 groups as
follows: The Con (n=8 from Fig.
1), CCI (n=8 from Fig. 1),
CCI + chloroquine (CQ) (n=8), CCI + bafilomycin (Baf) (n=8), CCI +
Rap (n=8 from Fig. 3), CCI + Rap
+ CQ (n=8) and CCI + Rap + Baf (n=8) groups. According to previous
studies (15,27,28), Rap (Bio Vision), Baf (LC
Laboratories, Woburn, MA, USA) and CQ (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) were administered by intraperitoneal injection
at a dose of 10, 1 and 60 mg/kg, respectively, 1 h prior to the CCI
operation. At 7 days after CCI, the expression levels of the
autophagy-related proteins, LC3II and Beclin 1, and those of the
lysosome-related proteins, LAMP2 and RAB7 in the spinal cord were
detected by western blot analysis in all 7 groups. Mechanical
allodynia and thermal hyperalgesia were tested before and at 1, 3,
7 and 14 days after CCI.
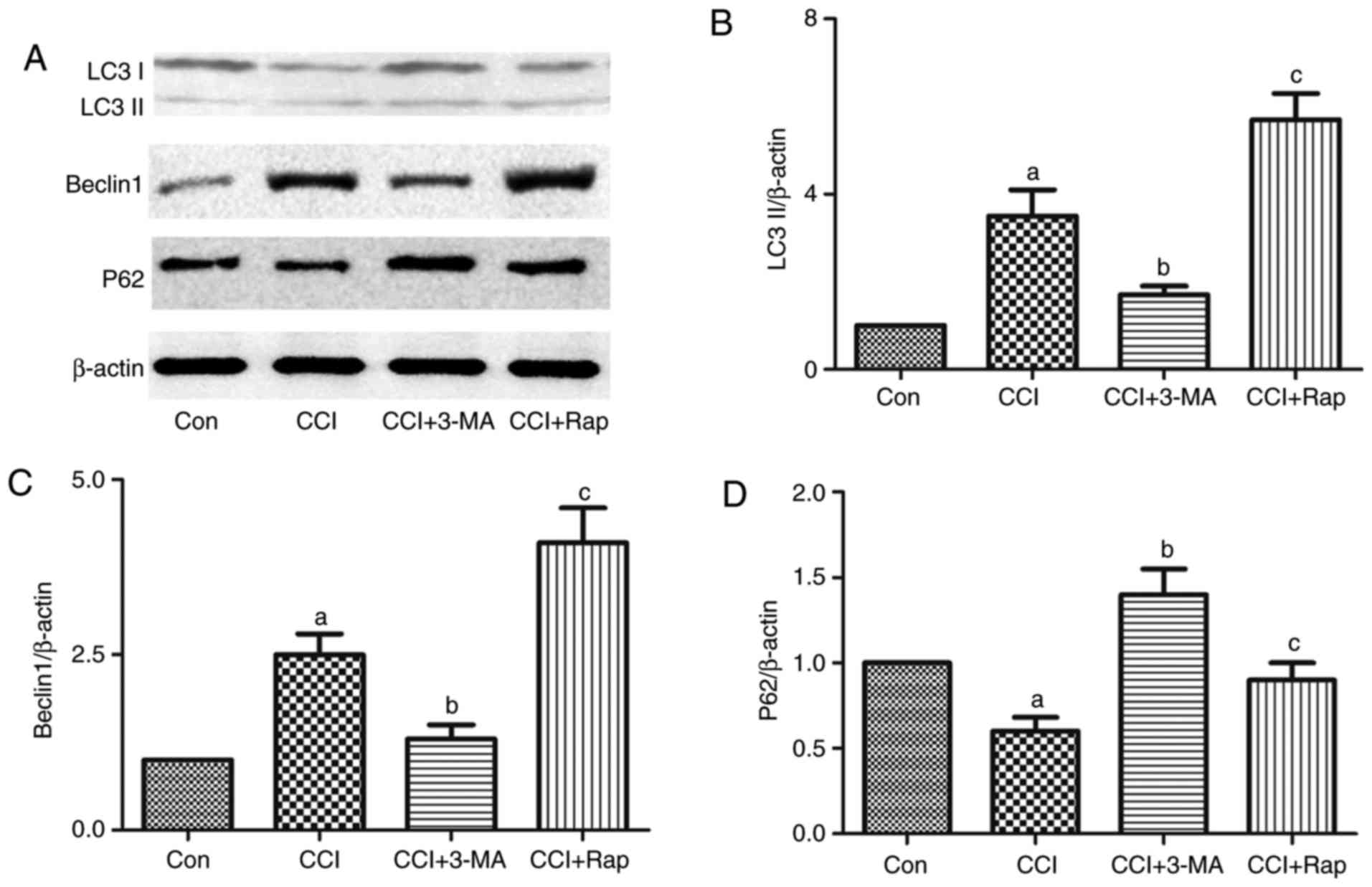 | Figure 3Effect of the autophagic inducer,
rapamycin, and inhibitor, 3-MA, on the expression of the
autophagy-related proteins, LC3II, Beclin 1 and p62, in the spinal
dorsal horn of rats with neuropathic pain. 3-MA and rapamycin were
administered by intraperitoneal injection at a dose of 15 and 10
mg/kg body weight, respectively, 1 h prior to the CCI operation.
L4-L5 spinal dorsal horn tissue was collected for the detection of
(A and B) LC3II, (A and C) Beclin 1 and (A and D) p62 7 days after
CCI operation by western blot analysis. The value in the Con group
was set as 1 and other relative values in the CCI groups were
calculated by comparison. The values are expressed as the means ±
standard deviation (n=8 per group). Statistical analysis was
performed with one-way analysis of variance followed by Tukey's
test. aP<0.05 vs. Con group. bP<0.05
vs. CCI group. cP<0.05 vs. CCI group and CCI + 3-MA
group. Con, control; CCI, chronic constriction injury; 3-MA,
3-methyladenine. |
Nociceptive behavioral assessment
To evaluate mechanical hyperalgesia, Von Frey
filaments were used to assess the presence of mechanical
hypersensitivity by measuring paw withdrawal threshold (BSEVF3,
Harvard Apparatus Co., Holliston, MA, USA). The procedure was as
follows: The rats were placed individually in transparent Perspex
boxes with wire mesh walls and floor for 30 min of habituation time
prior to behavioral tests. The filaments were individually applied
vertically to the plantar side of the right hind paw in ascending
order and repeated 3 times at 10-min intervals at each time point
per paw. A positive response was defined as the minimal force that
caused at least 2 withdrawals observed out of 3 consecutive trials
(29). A maximal cut-off value of
60 g was used to prevent tissue damage.
Thermal hyperalgesia was measured as previously
described by Bianchi et al (30). The rats were placed on a hot plate
(a round heated surface surrounded by Plexiglas) to acclimate to
the device (YLS-6B, Zhenghua Biological Instrument Equipment Co.,
Huaibei, China). The temperature was increased by a heat source
under the plantar surface of the hind paw. Clear paw withdrawal,
shaking and/or licking were considered nociceptive-like responses.
The nociceptive threshold was recorded in sec and repeated 3 times
at 10-min intervals at each time point. A cut-off time of 30 sec
was used to avoid tissue damage (4).
Western blot analysis
The L4-L6 spinal cord segments were rapidly removed
and homogenized in ice-cold sodium dodecyl sulfate (SDS) sample
buffer containing protease inhibitors (Sigma-Aldrich; Merck KGaA).
The lysate was centrifuged and the supernatant was removed as the
total protein. Total protein homogenates were separated on a 10%
SDS-polyacrylamide gel and blotted onto polyvinylidene difluoride
membranes by semidry electrophoretic transfer at 15 V for 60 min.
The membranes were blocked with Tris-buffered saline containing 5%
non-fat milk in Tris-Tween buffer saline for 1 h at room
temperature, and were first incubated overnight at 4°C with primary
antibodies against rat LC3II (dilution 1:1,000; Cat. no. ab48394,
Abcam, Cambridge, UK), Beclin 1 (dilution 1:1,000; Cat. no.
ab62557, Abcam), p62 (dilution 1:500; Cat. no. ab91526, Abcam),
GFAP (dilution 1:2,000; Cat. no. ab7260, Abcam), RAB7 (dilution
1:1,000; Cat. no. ab229647, Abcam) and LAMP2 (dilution 1:1,000;
Cat. no. ab203224, Abcam). Subsequently, the membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies (anti-mouse, sc-2005; 1:5,000; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA and anti-rabbit, sc-2357, 1:5,000; Santa Cruz
Biotechnology, Inc.) at room temperature for 1 h. The bands were
visualized by exposing the blots to Kodak Biomax MR Films and
quantified with the Bio-Rad Gel Doc 2000 system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Immunohistochemistry
The spinal cord was post-fixed and infiltrated, and
subsequently sliced into 10-µm-thick sections,
permeabilized, blocked and incubated with GFAP primary antibodies
(dilution 1:500, Abcam). The following day, the sections were
rewarmed to room temperature, washed and incubated with the
secondary antibody (dilution 1:500; Cat. no. ab150075, Abcam),
counterstained with DAPI (D-21490, Thermo Fisher Scientific, Inc.),
and washed for 30 min with PBS. Fluorescent images were captured
using a fluorescence microscope (TCS SP2; Leica, Wetzlar,
Germany).
Statistical analysis
All the results were analyzed using SPSS version
18.0 (SPSS, Inc., Chicago, IL, USA) and Prism® version
5.0 (GraphPad Software, San Diego, CA, USA) software. Statistical
differences between more than 2 groups were analyzed with one-way
analysis of variance, followed by Tukey's post hoc test. All data
are expressed as the means ± standard deviation. Statistical
significance was set at P<0.05.
Results
Activation of autophagy in the spinal
dorsal horn of rats with neuropathic pain induced by CCI
LC3 is a mammalian autophagic protein that localizes
to the autophagosome membrane in the cytosol. After autophagy is
activated, LC3I from the cytosol is converted into LC3II in the
autophagosome membrane. Beclin 1 is distributed in the plasma
membrane, cytoplasm and nucleus, and is vital for the localization
of autophagic proteins to a pre-autophagosomal structure (PAS),
depending on the interaction with the class III type
phosphoinositide 3-kinase (PI3KC3)/Vps34 (31). p62/SQSTM1 (p62) is a selective
autophagy receptor and is degraded by autophagy; thus, increased
levels of p62 reflect the inhibition of autophagy (32). In this experiment, spinal dorsal
horn tissues were collected for the detection of
autophagy-regulated proteins at 1, 3, 7 and 14 days after CCI. The
expression levels of LC3II and Beclin 1 increased significantly
from day 1 to 7 after CCI compared with the Con group, being the
highest on day 3 after CCI; no significant differences were
observed in these levels compared to the Con group at 14 days after
CCI (Fig. 1A–C). By contrast, p62
exhibited an opposite trend in the neuropathic pain model compared
with LC3II and Beclin 1. The p62 levels decreased from day 1 to 14,
with the lowest expression observed at 3 days after CCI (Fig. 1A and D). Autophagy was thus
activated after CCI, and there was a negative association between
LC3II, Beclin 1 and p62.
Astrocyte activation at different time
points in the model of neuropathic pain
The activation of astrocytes, which are
characterized by hyperplasia and hypertrophy with increased GFAP
expression, has been previously described in this pain model
(33,34). To specifically detect the
regulatory effect of neuropathic pain on astrocyte function, the
expression of GFAP was tested at various time points after CCI by
immunohistochemistry and western blot analysis. Compared with GFAP
staining and expression in the Con group, there was a significant
increase from day 1 to 7 after CCI (Fig. 2). Therefore, pain appeared to
activate astrocytes.
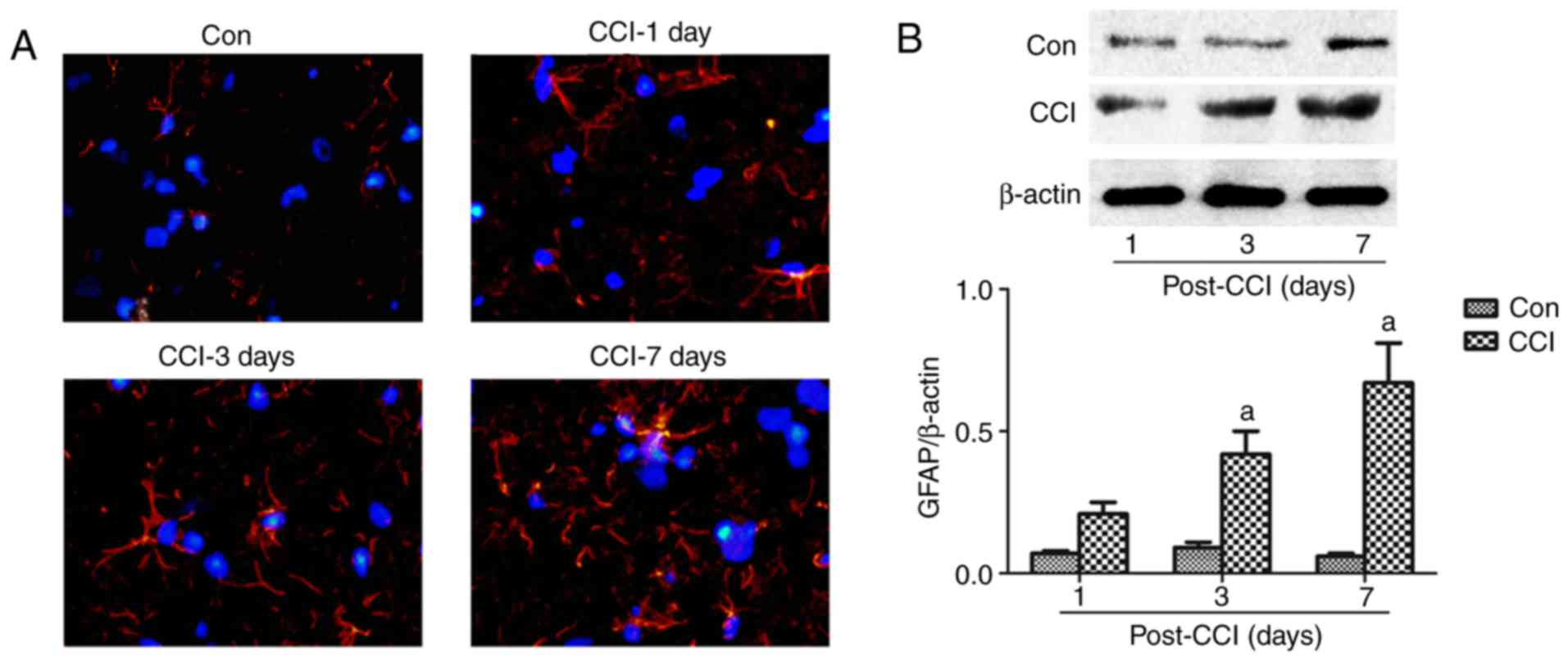 | Figure 2Astrocyte changes in the spinal
dorsal horn of rats with neuropathic pain. (A) A part of the L4-L6
spinal cord was obtained for the measurement of GFAP by
immunofluorescence assay at 1, 3 and 7 days after CCI. Red, GFAP;
blue, DAPI; magnification, ×40). (B) The L4-L5 spinal cord was
collected for the detection of GFAP expression by western blot
analysis at 1, 3, 7 and 14 days after CCI. The values are expressed
as the means ± standard deviation (n=8 per group). Statistical
analysis was performed with one-way analysis of variance followed
by Tukey's test. aP<0.05 vs. Con group. Con, control;
CCI, chronic constriction injury; GFAP, glial fibrillary acidic
protein; DAPI, 4′,6-diamidino-2-phenylindole. |
Effect of autophagy induction and
inhibition on LC3, Beclin 1 and p62 expression in the spinal dorsal
horn of rats with neuropathic pain
CCI induced the expression of the autophagy markers,
LC3 and Beclin 1, and inhibited the expression of the autophagy
adaptor, p62 (Figs. 1 and
3). Compared with the CCI group,
the use of the autophagy inhibitor, 3-MA, reduced the expression of
LC3 and Beclin 1, and increased the expression of p62 (Fig. 3), whereas the use of the autophagy
inducer, Rap, increased the expression of LC3 and Beclin 1, and
decreased the expression of p62 in this neuropathic pain model
(Fig. 3).
Effect of autophagy on allodynia,
hyperalgesia and GFAP expression in a rat model of neuropathic
pain
To determine whether autophagy regulates allodynia,
hyperalgesia and GFAP expression in a rat model of neuropathic pain
induced by CCI of the sciatic nerve, the rats were treated with the
autophagy inducer, Rap, and the autophagy inhibitor, 3-MA.
Neuropathic pain induced a rapid decrease in the threshold of
mechanical allodynia and thermal hyperalgesia compared with the Con
group (Fig. 4A and B). The
threshold of mechanical allodynia and thermal hyperalgesia markedly
decreased 1 day after CCI and remained stable until at least day 14
(Fig. 4A and B). Compared with
the CCI group, the use of the autophagy inhibitor, 3-MA,
significantly aggravated the decline of the threshold of mechanical
and thermal hyperalgesia in the CCI + 3-MA group (Fig. 4A and B). By contrast, the
threshold of mechanical and thermal hyperalgesia was markedly
increased following treatment with the autophagy inducer, Rap, in
the CCI + Rap group compared with the CCI group (Fig. 4A and B).
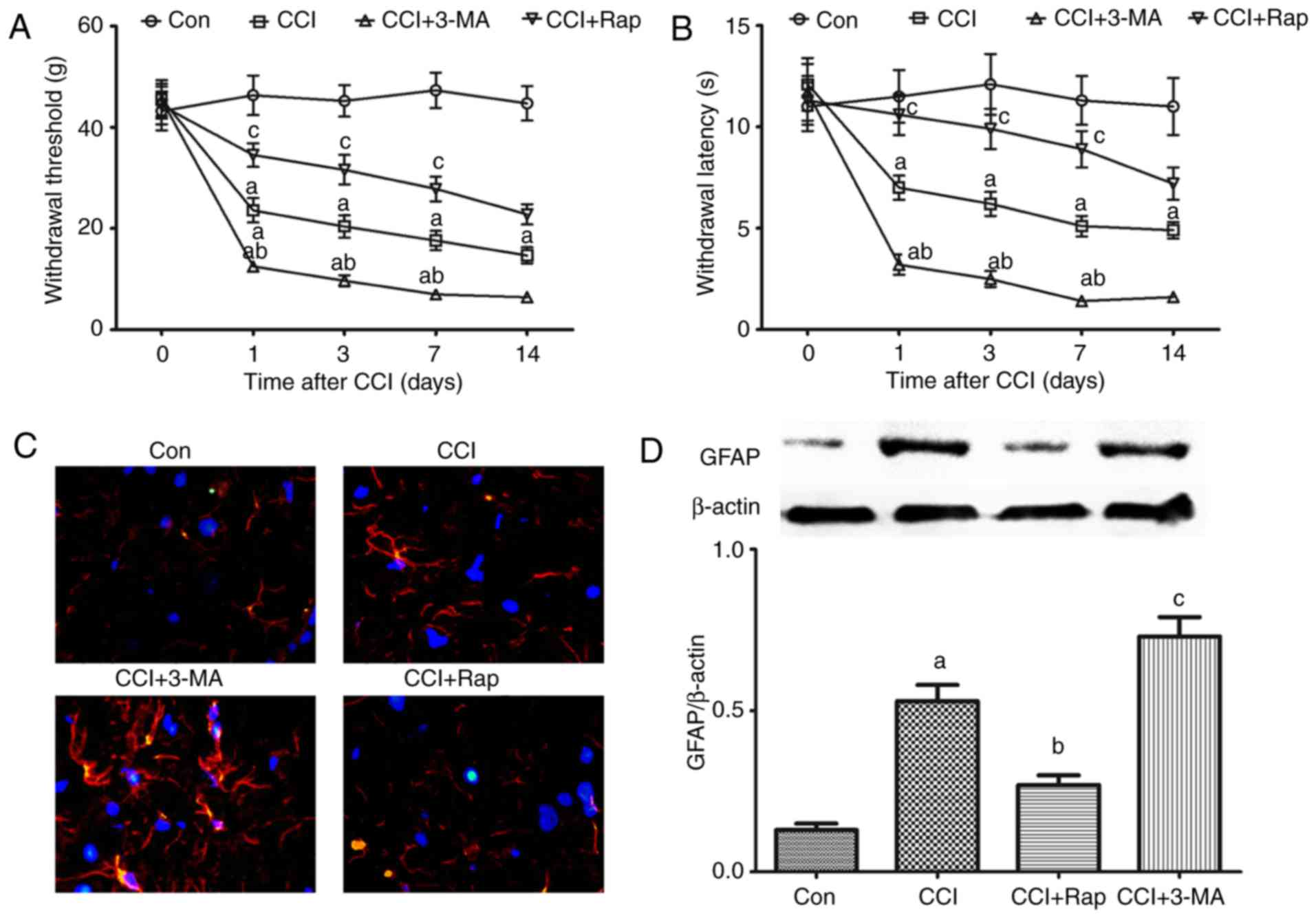 | Figure 4Effect of autophagy on allodynia,
hyperalgesia and astrocyte activation in CCI-induced neuropathic
pain. 3-MA and rapamycin were administered by intraperitoneal
injection at a dose of 15 and 10 mg/kg body weight, respectively, 1
h prior to the CCI operation. (A) Mechanical allodynia and (B)
thermal hyperalgesia were examined prior to and at 1, 3, 7 and 14
days after CCI. At 7 days after CCI, the spinal cord was collected
to detect the expression of GFAP by (C) immunofluorescence and (D)
western blot analysis. The values are expressed as the means ±
standard error of the mean (n=8 per group). Statistical analysis
was performed with one-way analysis of variance followed by Tukey's
test. aP<0.05 vs. Con group. bP<0.05
vs. CCI group. cP<0.05 vs. CCI group and CCI + 3-MA
group. Con, control; CCI, chronic constriction injury; 3-MA,
3-methyladenine; GFAP, GFAP, glial fibrillary acidic protein. |
Compared with the Con group, CCI induced GFAP
expression, and GFAP expression exhibited an increase at 7 days
after CCI surgery. Following treatment with 3-MA, GFAP expression
further increased in the CCI + 3-MA group compared with the CCI
group (Fig. 4C and D). By
contrast, the autophagy inducer, Rap, significantly inhibited the
increase in GFAP expression in the CCI + Rap group (Fig. 4C and D). These results indicate
that autophagy markedly inhibits mechanical allodynia, thermal
hyperalgesia and GFAP expression induced by CCI, whereas 3-MA
reverses the inhibitory effects of autophagy on mechanical
allodynia, thermal hyperalgesia and GFAP expression.
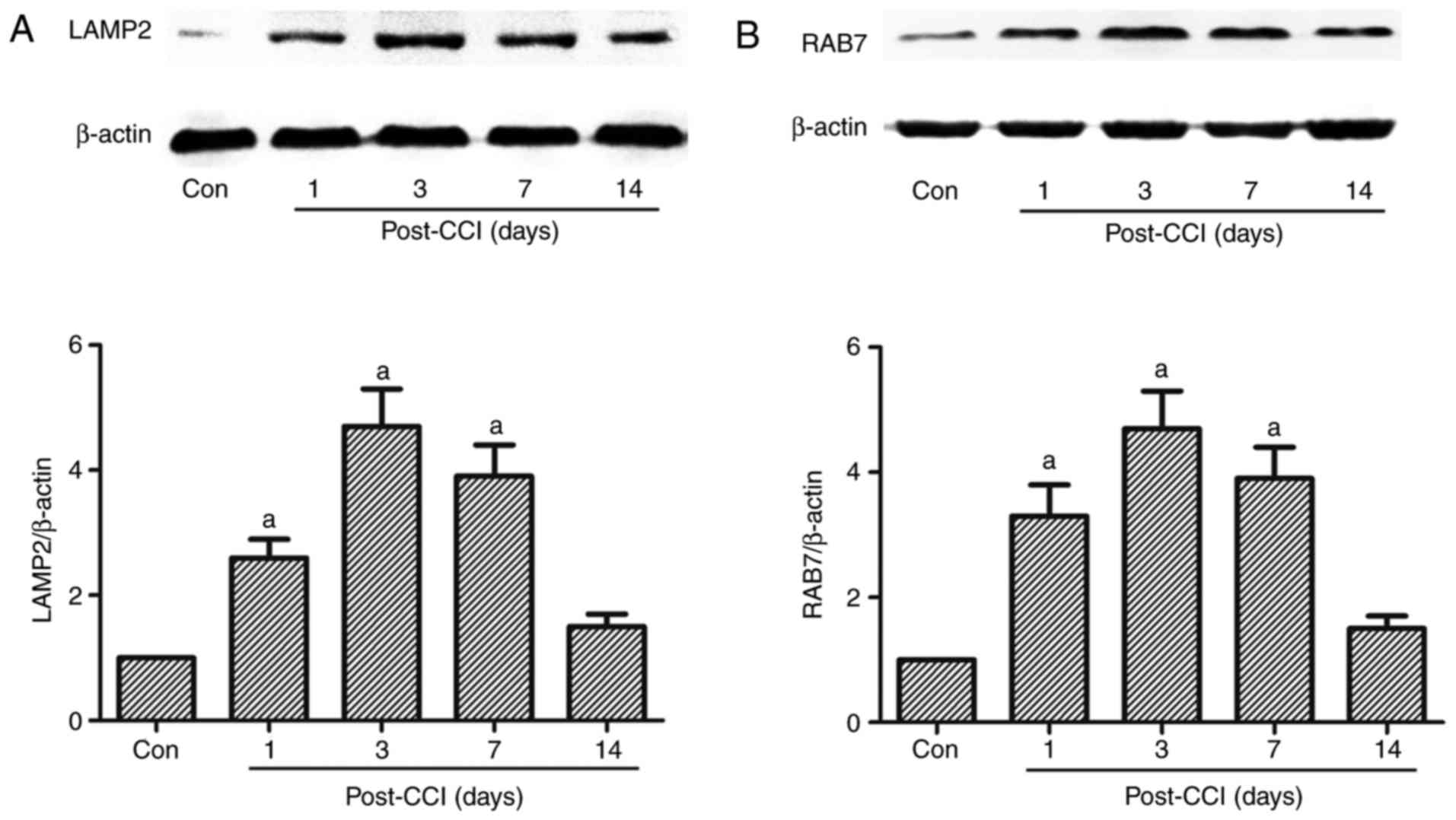
Expression of the lysosome-related
proteins, LAMP2 and RAB7, in a rat model of neuropathic pain
induced by CCI of the sciatic nerve
Autophagy is the dynamic process of sequestering
cytoplasmic proteins or organelles into the lytic component; this
process requires the fusion of autophagosomes and lysosomes
(14,16,17). If the fusion of autophagosomes and
lysosomes is blocked, damaged or malfunctioning proteins or
organelles cannot be recycled. LAMP2 and RAB7 are required for the
fusion of autophagosomes with lysosomes (14). LAMP2, a heavily glycosylated
protein, constitutes the majority of all membrane proteins in the
lysosome. RAB7 is a member of the Rab family involved in transport
to late endosomes and in the biogenesis of the perinuclear lysosome
compartment (14). In the present
study, the expression of LAMP2 and RAB7 increased from day 1 to 14
after CCI, and this increase was significant on days 1, 3 and 7
compared with the Con group, peaking at 3 days after CCI; however,
no significant increase was observed 14 days after CCI (Fig. 5).
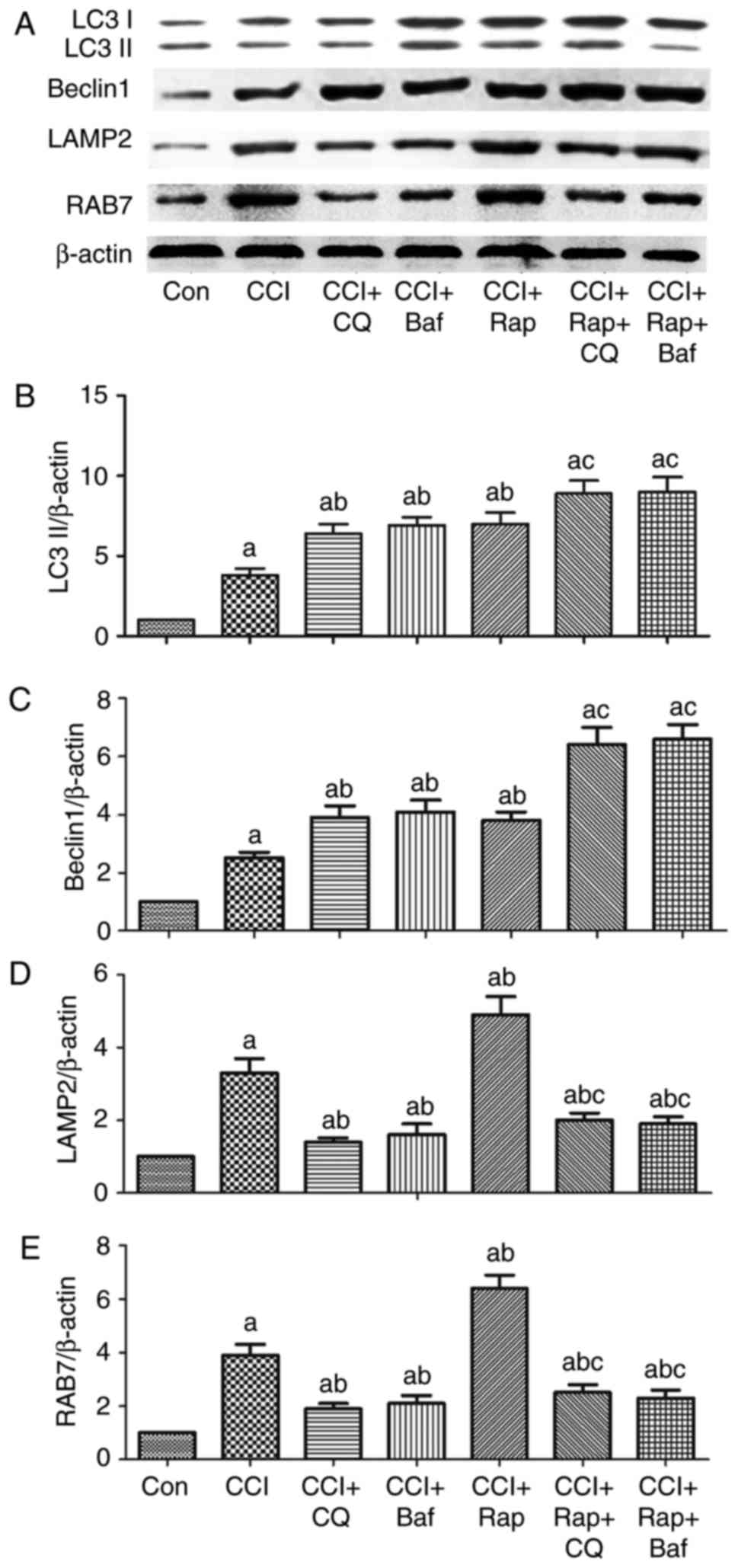
Effect of lysosomal inhibitors, Baf and
CQ, on the expression of the autophagy-related proteins, LC3II and
Beclin 1, and that of the lysosome-related proteins, LAMP2 and
RAB7, in a rat model of CCI
Autophagic activation does not only depend on the
increased synthesis or lipidation of LC3 and Beclin 1, but also on
a series of regulatory proteins orchestrating different autophagic
steps, from autophagosome formation to fusion with the lysosome and
subsequent release of the breakdown products (35). On the one hand, the increase in
the levels of the autophagy markers, LC3 or Beclin 1, was
attributed to the potent autophagic activation; on the other hand,
the increase was due to the inhibition of the fusion of
autophagosomes and lysosomes when autophagic activity was not
profound (35). To better
elucidate the mechanisms responsible for the increase in the levels
of the autophagy-related proteins, LC3 and Beclin 1, in neuropathic
pain, the lysosomal inhibitors, Baf A1 and CQ, were used to block
the fusion process of autophagosomes and lysosomes.
Compared with the Con group, CCI induced an increase
in the expression levels of LC3II and Beclin 1 (Figs. 1 and 3). The autophagy agonist, Rap, and the
lysosomal inhibitors, Baf A1 and CQ, markedly increased the
expression levels of LC3II and Beclin 1 compared with the CCI group
(Fig. 6A–C). LC3II and Beclin 1
expression further increased by treatment with Rap and CQ, or Rap
and Baf A1 compared with the CCI + CQ, CCI + Baf, or CCI + Rap
groups (Fig. 6A–C).
Compared with the Con group, CCI induced an increase
in LAMP2 and RAB7 expression (Figs.
5, and 6A, D and E). Compared
with the CCI group, the autophagy inducer, Rap, increased the
expression of LAMP2 and RAB7 (Fig.
6A, D and E), whereas the lysosomal inhibitors, Baf A1 and CQ,
markedly inhibited LAMP2 and RAB7 expression. Therefore, the
expression of LAMP2 and RAB7 decreased significantly following
treatment with Rap and CQ, or Rap and Baf A1 compared with the CCI
+ Rap group (Fig. 6A, D and
E).
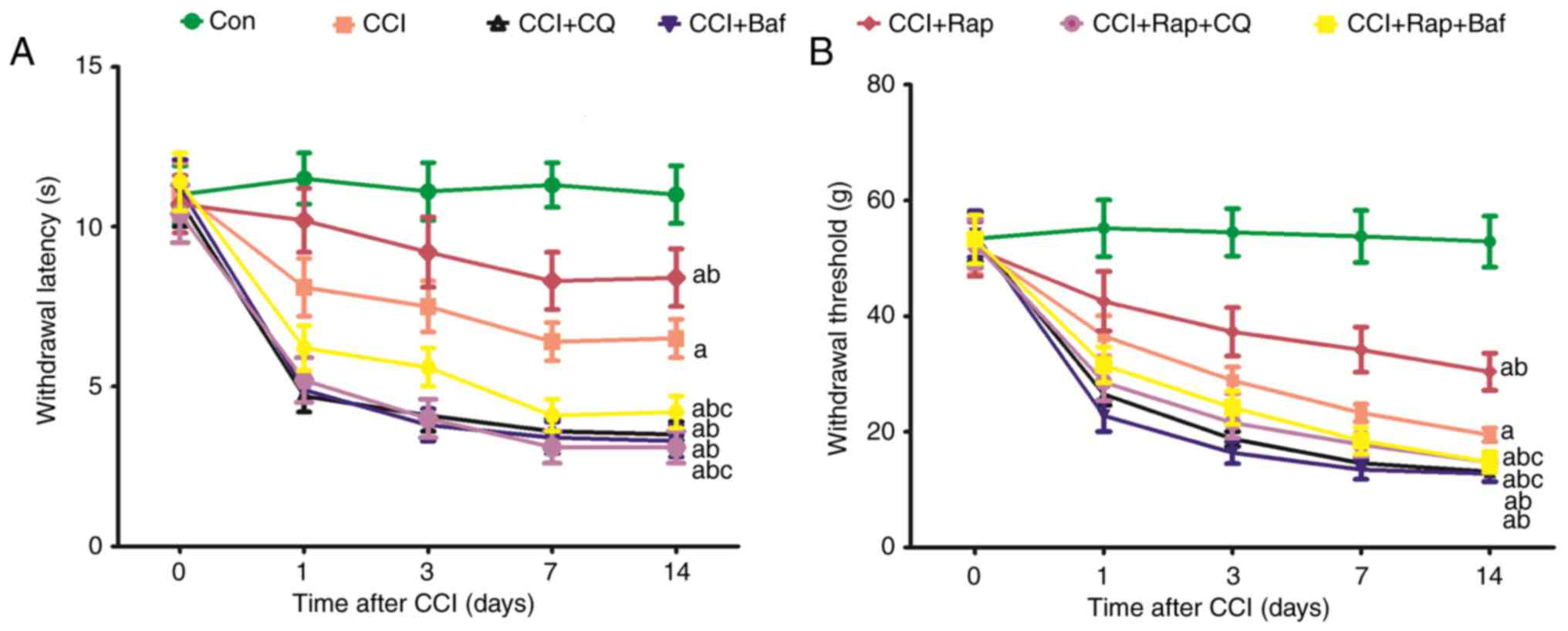
Effect of the lysosomal inhibitors, Baf
A1 and CQ, on allodynia and hyperalgesia in a rat model of CCI
CCI induced a rapid decrease in the threshold of
mechanical allodynia and thermal hyperalgesia compared with the Con
group (Figs. 4A and B; and
7A and B). The threshold of
mechanical allodynia and thermal hyperalgesia markedly decreased at
1, 3, 7 and 14 days after CCI (Figs.
4A and B; and 7). The
lysosomal inhibitors, Baf A1 CCI group (Fig. 7). These results indicated that
inhibiting the fusion of autophagosomes and lysosomes aggravates
the decline of the threshold of mechanical and thermal
hyperalgesia.
Discussion
Neuropathic pain is a chronic adynamic condition,
which is caused by a primary injury or dysfunction in the nervous
system and is primarily characterized by abnormal sensory
perception of pain persistent pain, such as allodynia and
hyperalgesia (1). Neuropathic
pain caused by peripheral nerve injury is associated with
pathological changes in the damaged peripheral nerves, dorsal root
ganglion (DRG) and the activation of astrocytes, which play a
critical role in the maintenance of a persistent pain state
following peripheral nerve injury. The findings of the present
study were as follows: i) Neuropathic pain induced an increase in
the levels of the autophagy-related proteins, LC3II and Beclin 1,
and the lysosome-related proteins, LAMP2 and RAB7, at different
time points in the rats with CCI-induced neuropathic pain; ii) the
effect of the autophagy inducer, Rap, and inhibitor, 3-MA,
alleviated and exacerbated allodynia, hyperalgesia and GFAP
expression in rat model of neuropathic pain, respectively; iii) the
autophagy inducer, Rap, increased the expression of LC3II, Beclin
1, LAMP2 and RAB7, whereas the lysosomal inhibitors, Baf and CQ,
inhibited the fusion of autophagosomes and lysosomes, increased the
expression of LC3II and Beclin 1, and attenuated the expression of
LAMP2 and RAB7 in rats with neuropathic pain; iv) the lysosomal
inhibitors, Baf and CQ, also aggravated allodynia and hyperalgesia
in rats with neuropathic pain. On the whole, these results
demonstrate that peripheral nerve injury activates autophagy, which
is involved in the renewal and regeneration of the injured
peripheral nerves.
Peripheral nerve injury may lead to a state of
chronic neuropathic pain, characterized by dysesthesia,
hyperalgesia and allodynia (36).
As the etiology and mechanisms underlying neuropathic pain remain
unclear, the currently available treatments, including
anticonvulsant agents, local anesthetics and opioids, are often
unsatisfactory. Therefore, it is imperative to elucidate the
mechanisms associated with the occurrence of neuropathic pain in
order to develop more effective therapies. CCI has been frequently
used in a rat model to induce signs of neuropathic pain (37,38), which manifests with decreased
thermal and mechanical nociceptive thresholds following CCI of the
sciatic nerve. Of the several experimental animal models available,
signs of mechanical allodynia and thermal hyperalgesia are most
evident in the neuropathic pain of the nerve ligation model. In the
present study, we also examined the changes in mechanical allodynia
and thermal hyperalgesia following CCI of the sciatic nerve.
Consistent with previous findings (37,38), neuropathic pain induced by CCI
generated allodynia and hyperalgesia, manifesting as a markedly
decreased threshold of mechanical allodynia and thermal
hyperalgesia 1 day after CCI, remaining constant for at least 14
days.
Astrocytes are abundant, constituting 40–50% of all
glial cells (39). Under
physiological conditions, astrocytes are relatively static
(40); however, following injury
or under disease conditions, they may become activated and
participate in the pathogenesis of neurological disorders (41). Astrocyte activation is responsible
for the maintenance of chronic pain, which has important
implications for the development of therapeutics. Astrocytes are
closely associated with neurons and blood vessels, and are crucial
for the nutrition, support and protection of neurons. It is
estimated that a single astrocyte enwraps 4–6 neuronal somata and
is in contact with 300–600 neuronal dendrites (41). The intermediate filament protein,
GFAP, is not only a marker of astrocytes in the CNS, but also
facilitates the maintenance of pain. Intrathecal GFAP antisense
oligonucleotide treatment has been reported to alleviate
neuropathic pain behaviors in animals with nerve injury (9). The present study also focused on
astrocyte activation and GFAP expression in neuropathic pain. It
was demonstrated that neuropathic pain induced by CCI could
activate astrocytes, which exhibited a higher GFAP expression until
day 7 after CCI. Astrocyte activation participated in maintaining
the pain state.
Cell death is a complex and well controlled process,
including apoptosis and autophagy. Apoptosis, the main mechanism
and pathogenesis of programmed cell death (type I cell death), has
been extensively investigated and well verified. However,
autophagy, type II cell death, participates in the initial and
developmental process of disease, which is evoked in response to
various stresses that finally lead to apoptosis (43); however, its pathogenesis remains
obscure. As is known, autophagy is an evolutionarily conserved
regulated process and gate-keeping mechanism through which damaged
cytoplasmic macromolecules, organelles and redundant proteins are
degraded via lysosomes to stabilize intracellular homeostasis and
metabolism and to recycle cellular nutrients (44,45). Autophagic stress is responsible
for Parkinson's (46,47), Huntington's (48) and Alzheimer's disease (49,50), stroke (51), and other neuropathies (52). In addition, changes in autophagy
regulation have been observed in neuropathic pain (35,53) and traumatic brain injury (54). LC3 and Beclin 1 are two crucial
markers of autophagy, which are closely associated with the
autophagic process, particularly in its early stages (55). LC3 is associated with the
formation of autophagosomes. After becoming conjugated to the lipid
phosphatidylethanolamine, LC3I is cleaved to form LC3II and
localizes to the autophagosome membrane. Beclin 1, a key autophagic
protein localized to the PAS, forms a complex by interacting with
PI3KC3/Vps34 to control autophagic nucleation and promote autophagy
in mammals; the suppression of Beclin 1 expression impairs
autophagy (31,55,56). p62, which is used as an autophagy
marker, is the first protein reported to have such an adaptor
function in autophagy, and is degraded by autophagy activation.
Therefore, there is a reported association between the inhibition
of autophagy and increased levels of p62 (32,57,58).
Our team always focuses on the autophagy in
different model and focus on the different mechanism in neuropathic
pain (59-62). In the present study, neuropathic
pain activated autophagy, manifesting as an increase in LC3II and
Beclin 1 expression from day 1 to 3 after CCI by sciatic nerve
ligation, and then gradually declined by day 14 of the experiment,
although it remained higher compared with that in the Con group.
The accumulation of p62 or increased p62 levels are frequently used
as signs of autophagy impairment (57,63). p62 is a major LC3-interacting
protein, which possesses a C-terminal ubiquitin-binding domain and
a short LC3-interacting region sequence (63,64). The present study also examined the
changes in the levels of p62. p62 expression was decreased in rats
with neuropathic pain, with the lowest expression being observed 3
days after CCI, and increasing again until day 14 of the
experiment. Treatment with Rap, a well-known inducer of autophagy,
further activated autophagy by increasing autophagosome formation
and autophagosome-lysosome fusion. 3-MA is generally accepted as a
specific inhibitor of autophagy. In the present study, Rap and 3-MA
were used to induce and inhibit autophagy, respectively. Treatment
with 3-MA and Rap notably inhibited and induced the expression of
LC3II and Beclin 1, respectively, and resulted in p62 accumulation
and degradation, respectively, in rats subjected to CCI. 3-MA and
Rap were not only used to examine the expression of
autophagy-related proteins, but also to investigate the effects of
autophagy on withdrawal threshold, withdrawal latency and astrocyte
activation. Neuropathic pain led to a decrease in the withdrawal
threshold, withdrawal latency and astrocyte activation, and
increased GFAP expression; 3-MA caused a further decrease in the
withdrawal threshold, withdrawal latency and astrocyte activation,
whereas Rap significantly reversed the effects of the autophagy
inhibitors on withdrawal threshold, withdrawal latency and
astrocyte activation. These results indicate that there is a
negative association between LC3II, Beclin 1 and p62, and after the
nerve is injured by ligation, tissues and cells react to stress and
activate autophagy-mediated physiological and pathological changes
in the rat body. However, after the nerve damage reaches a certain
extent, autophagy impairment leads to tissue and cell
dysfunction.
The autophagosome formation pathway consists of
several stages, including initiation (formation of a PAS, leading
to an isolation membrane, or phagophore), vesicle elongation,
autophagosome maturation and cargo sequestration, and
autophagosome-lysosome fusion. In the final phase, autophagosomal
contents are degraded by lysosomal acid hydrolases and released for
metabolic recycling (14). An
increase in autophagy can promote LC3II and Beclin 1 protein
accumulation; however, whether LC3II and Beclin 1 accumulation
fully explain the results of autophagy increase or
autophagosome-lysosome fusion malfunction remains unclear. In
lysosomal storage diseases, defects in specific lysosomal
hydrolases have been shown to result in lysosomal dysfunction and
autophagy impairment (65,66).
It has been reported that lysosomal dysfunction contributes to the
pathological accumulation of autophagosomes, neuronal dysfunction
and death (67). Therefore, the
entire process of autophagy, including autophagosome formation and
the fusion of autophagosomes and lysosomes, should be taken into
consideration to accurately evaluate autophagy. In an attempt to
elucidate this matter, it was found that the lysosomal inhibitors,
Baf and CQ, inhibited the late-stage fusion of autophagosomes and
lysosomes. LAMP2A, apart from acting as a receptor of autophagy, is
also indispensable for efficient fusion of autophagosomes and
lysosomes (68,69). RAB7 is necessary for the
maturation of late autophagosomes and the fusion of autophagosomes
with lysosomes (70). In the
present study, we found that the expression of LAMP2 and RAB7
increased from day 1 to 14 after CCI; these expression leveks
peaked at day 3 and gradually declined thereafter. The lysosomal
inhibitors, Baf and CQ, markedly increased the accumulation of
LC3II and Beclin 1 and inhibited the expression of LAMP2 and RAB7.
Rap increased the expression of LAMP2 and RAB7 in rats with
neuropathic pain when compared with the CCI group. LC3 accumulation
persisted due to the increased autophagy following the fusion of
autophagosomes with lysosomes (15,71). Once fusion is inhibited, LC3II
accumulates, even if autophagy is no longer occurring. In the
present study, compared with the CCI group, Rap + CQ or Rap + Baf
treatment led to the excessive accumulation of LC3II and Beclin 1
and a decrease in LAMP2 and RAB7 levels. It was also observed that
the lysosomal inhibitors, Baf and CQ, exacerbated mechanical and
thermal hyperalgesia via a reduction in the withdrawal threshold
and withdrawal latency. These results indicated that neuropathic
pain induced autophagy, thereby increasing LC3II, Beclin 1, LAMP2
and RAB7 levels; the autophagy inducer, Rap, further increased the
expression of LC3II, Beclin 1, LAMP2 and RAB7. The inhibition of
the late stage of autophagy (the fusion stage of autophagosomes
with lysosomes) increase LC3II and Beclin 1 levels, and decreased
those of the lysosome proteins, LAMP2 and RAB7.
In conclusion, this study demonstrates that
neuropathic pain induces the expression of the autophagy-related
proteins, LC3II and Beclin 1, and that of the lysosome-related
proteins, LAMP2 and RAB7. Increased autophagy alleviates mechanical
and thermal hyperalgesia and astrocyte activation. The
autophagy-lysosomal pathway was found to be responsible for the
neuropathic pain induced by CCI of the sciatic nerve.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YY conceived and designed the study. HC, YH, KX, YC
and HW conducted the experimental protocols in the present study.
HC, YC and HW acquired analytical reagents and tools. YH, YB, YW
and AD performed data analysis. YY and HC prepared the manuscript.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Institutional Animal Care and Use Committee of Tianjin Medical
University and were performed in accordance with the National
Institutes of Health Guide for Care and Use of Laboratory Animals.
All efforts were made to minimize animal suffering and the number
of animals used.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sandkühler J: Models and mechanisms of
hyperalgesia and allodynia. Physiol Rev. 89:707–758. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ji RR, Kohno T, Moore KA and Woolf CJ:
Central sensitization and LTP: Do pain and memory share similar
mechanisms? Trends Neurosci. 26:696–705. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jensen TS and Finnerup NB: Allodynia and
hyperalgesia in neuropathic pain: Clinical manifestations and
mechanisms. Lancet Neurol. 13:924–935. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang W, Wang W, Wang Y, Huang J, Wu S and
Li YQ: Temporal changes of astrocyte activation and glutamate
transporter-1 expression in the spinal cord after spinal nerve
ligation-induced neuropathic pain. Anat Rec (Hoboken). 291:513–318.
2008. View
Article : Google Scholar
|
|
5
|
Watkins LR, Milligan ED and Maier SF:
Spinal cord glia: New players in pain. Pain. 93:201–205. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhuang ZY, Wen YR, Zhang DR, Borsello T,
Bonny C, Strichartz GR, Decosterd I and Ji RR: A peptide c-Jun
N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after
spinal nerve ligation: Respective roles of JNK activation in
primary sensory neurons and spinal astrocytes for neuropathic pain
development and maintenance. J Neurosci. 26:3551–3560. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhuang ZY, Gerner P, Woolf CJ and Ji RR:
ERK is sequentially activated in neurons, microglia, and astrocytes
by spinal nerve ligation and contributes to mechanical allodynia in
this neuropathic pain model. Pain. 114:149–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eliasson C, Sahlgren C, Berthold CH,
Stakeberg J, Celis JE, Betsholtz C, Eriksson JE and Pekny M:
Intermediate filament protein partnership in astrocytes. J Biol
Chem. 274:23996–24006. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim DS, Figueroa KW, Li KW, Boroujerdi A,
Yolo T and Luo ZD: Profiling of dynamically changed gene expression
in dorsal root ganglia post peripheral nerve injury and a critical
role of injury-induced glial fibrillary acidic protein in
maintenance of pain behaviors (corrected). Pain. 143:114–122. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eng LF, Ghirnikar RS and Lee YL: Glial
fibrillary acidic protein: GFAP-thirty-one years (1969–2000).
Neurochem Res. 25:1439–1451. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mccall MA, Gregg RG, Behringer RR, Brenner
M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY and
Messing A: Targeted deletion in astrocyte intermediate filament
(Gfap) alters neuronal physiology. Proc Natl Acad Sci USA.
93:6361–6366. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shibuki K, Gomi H, Chen L, Bao S, Kim JJ,
Wakatsuki H, Fujisaki T, Fujimoto K, Katoh A, Ikeda T, et al:
Deficient cerebellar long-term depression, impaired eyeblink
conditioning, and normal motor coordination in GFAP mutant mice.
Neuron. 16:587–599. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tanaka H, Katoh A, Oguro K, Shimazaki K,
Gomi H, Itohara S, Masuzawa T and Kawai N: Disturbance of
hippocampal long-term potentiation after transient ischemia in GFAP
deficient mice. J Neurosci Res. 67:11–20. 2002. View Article : Google Scholar
|
|
14
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hsieh Ch, Pai PY, Hsueh HW, Yuan SS and
Hsieh YC: Complete induction of autophagy is essential for
cardioprotection in sepsis. Ann Surg. 253:1190–1200. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jaeger PA and Wyss-Coray T:
All-you-can-eat: Autophagy in neurodegeneration and
neuroprotection. Mol Neurodegener. 4:162009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu YD, Wang ZB, Han G and Zhao P:
Hyperbaric oxygen treatment attenuates neuropathic pain by
elevating autophagy flux via inhibiting mTOR pathway. Am J Transl
Res. 9:2629–2638. 2017.PubMed/NCBI
|
|
19
|
Guo JS, Jing PB, Wang JA, Zhang R, Jiang
BC, Gao YJ and Zhang ZJ: Increased autophagic activity in dorsal
root ganglion attenuates neuropathic pain following peripheral
nerve injury. Neurosci Lett. 599:158–163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tateda S, Kanno H, Ozawa H, Sekiguchi A,
Yahata K, Yamaya S and Itoi E: Rapamycin suppresses microglial
activation and reduces the development of neuropathic pain after
spinal cord injury. J Orthop Res. 35:93–103. 2017. View Article : Google Scholar
|
|
21
|
Piao Y, Gwon DH, Kang DW, Hwang TW, Shin
N, Kwon HH, Shin HJ, Yin Y, Kim JJ, Hong J, et al: TLR4-mediated
autophagic impairment contributes to neuropathic pain in chronic
constriction injury mice. Mol Brain. 11:112018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Erlich S, Alexandrovich A, Shohami E and
Pinkas-Kramarski R: Rapamycin is a neuroprotective treatment for
traumatic brain injury. Neurobiol Dis. 26:86–93. 2017. View Article : Google Scholar
|
|
23
|
Carloni S, Buonocore G and Balduini W:
Protective role of autophagy in neonatal hypoxia-ischemia induced
brain injury. Neurobiol Dis. 32:329–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang G, Yue Z, Talloczy Z, Hagemann T, Cho
W, Messing A, Sulzer DL and Goldman JE: Autophagy induced by
Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK
and mTOR signaling pathways. Hum Mol Genet. 17:1540–1555. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qin AP, Liu CF, Qin YY, Hong LZ, Xu M,
Yang L, Liu J, Qin ZH and Zhang HL: Autophagy was activated in
injured astrocytes and mildly decreased cell survival following
glucose and oxygen deprivation and focal cerebral ischemia.
Autophagy. 6:738–753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bennett GJ and Xie YK: A peripheral
mononeuropathy in rat that produces disorders of pain sensation
like those seen in man. Pain. 33:87–107. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF,
Hsueh HW, Lee PH and Hsieh YC: Lc3 over-expression improves
survival and attenuates lung injury through increasing
autophagosomal clearance in septic mice. Ann Surg. 257:352–363.
2013. View Article : Google Scholar
|
|
28
|
Takahashi W, Watanabe E, Fujimura L,
Watanabe-Takano H, Yoshidome H, Swanson PE, Tokuhisa T, Oda S and
Hatano M: Kinetics and protective role of autophagy in a mouse
cecal ligation and puncture-induced sepsis. Crit Care. 17:R1602013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yalcin I, Choucair-Jaafar N, Benbouzid M,
Tessier LH, Muller A, Hein L, Freund-Mercier MJ and Barrot M:
beta(2)-adrenoceptors are critical for antidepressant treatment of
neuropathic pain. Ann Neurol. 65:218–225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bianchi M, Sacerdote P,
Ricciardi-Castagnoli P, Mantegazza P and Panerai AE: Central
effects of tumor necrosis factor alpha and interleukin-1 alpha on
nociceptive thresholds and spontaneous locomotor activity. Neurosci
Lett. 148:76–80. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mizushima N and Hara T: Intracellular
quality control by autophagy: How does autophagy prevent
neurodegeneration? Autophagy. 2:302–304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Svensson CI and Brodin E: Spinal
astrocytes in pain processing: Non-neuronal cells as therapeutic
targets. Mol Interv. 10:25–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao YJ and Ji RR: Targeting astrocyte
signaling for chronic pain. Neurotherapeutics. 7:482–493. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Berliocchi L, Maiarù M, Varano GP, Russo
R, Corasaniti MT, Bagetta G and Tassorelli C: Spinal autophagy is
differently modulated in distinct mouse models of neuropathic pain.
Mol Pain. 11:32015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Woolf CJ and Mannion RJ: Neuropathic pain:
Aetiology, symptoms, mechanisms, and management. Lancet.
353:1959–1964. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hamidi GA, Jafari-Sabet M, Abed A,
Mesdaghinia A, Mahlooji M and Banafshe HR: Gabapentin enhances
anti-nociceptive effects of morphine on heat, cold, and mechanical
hyperalgesia in a rat model of neuropathic pain. Iran J Basic Med
Sci. 17:753–759. 2014.
|
|
38
|
Lattanzi R, Maftei D, Marconi V,
Florenzano F, Franchi S, Borsani E, Rodella LF, Balboni G,
Salvadori S, Sacerdote P and Negri L: Prokineticin 2 upregulation
in the peripheral nervous system has a major role in triggering and
maintaining neuropathic pain in the chronic constriction injury
model. Biomed Res Int. 2015:3012922015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aldskogius H and Kozlova EN: Central
neuron-glial and glial-glial interactions following axon injury.
Prog Neurobiol. 55:1–26. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nimmerjahn A, Kirchhoff F and Helmchen F:
Resting microglial cells are highly dynamic surveillants of brain
parenchyma in vivo. Science. 308:1314–1318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rossi DJ, Brady JD and Mohr C: Astrocyte
metabolism and signaling during brain ischemia. Nat Neurosci.
10:1377–1386. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Halassa MM, Fellin T, Takano H, Dong JH
and Haydon PG: Synaptic islands defined by the territory of a
single astrocyte. J Neurosci. 27:6473–6477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Booth LA, Tavallai S, Hamed HA,
Cruickshanks N and Dent P: The role of cell signalling in the
crosstalk between autophagy and apoptosis. Cell Signal. 26:549–555.
2014. View Article : Google Scholar :
|
|
44
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Anglade P, Vyas S, Javoy-Agid F, Herrero
MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC and
Agid Y: Apoptosis and autophagy in nigral neurons of patients with
Parkinson's disease. Histol Histopathol. 12:25–31. 1997.PubMed/NCBI
|
|
47
|
Nixon RA: The role of autophagy in
neurodegenerative disease. Nat Med. 19:983–997. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kegel KB, Kim M, Sapp E, McIntyre C,
Castaño JG, Aronin N and DiFiglia M: Huntingtin expression
stimulates endosomal-lysosomal activity, endosome tubulation, and
autophagy. J Neurosci. 20:7268–7278. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cataldo AM, Hamilton DJ, Barnett JL,
Paskevich PA and Nixon RA: Properties of the endosomal-lysosomal
system in the human central nervous system: Disturbances mark most
neurons in populations at risk to degenerate in Alzheimer's
disease. J Neurosci. 16:186–199. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rubinsztein DC: The roles of intracellular
protein-degradation pathways in neurodegeneration. Nature.
443:780–786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Den YH, He HY, Yang LQ and Zhang PY:
Dynamic changes in neuronal autophagy and apoptosis in the ischemic
penumbra following permanent ischemic stroke. Neural Regen Res.
11:1108–1114. 2016. View Article : Google Scholar
|
|
52
|
Boellaard JW, Kao M, Schlote W and
Diringer H: Neuronal autophagy in experimental scrapie. Acta
Neuropathol. 82:225–228. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Berliocchi L, Russo R, Maiarù M, Levato A,
Bagetta G and Corasaniti MT: Autophagy impairment in a mouse model
of neuropathic pain. Mol Pain. 7:832011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sarkar C, Zhao Z, Aungst S, Sabirzhanov B,
Faden AI and Lipinski MM: Impaired autophagy flux is associated
with neuronal cell death after traumatic brain injury. Autophagy.
10:2208–2222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li JP, Yang YX, Liu QL, Zhou ZW, Pan ST,
He ZX, Zhang X, Yang T, Pan SY, Duan W, et al: The pan-inhibitor of
Aurora kinases danusertib induces apoptosis and autophagy and
suppresses epithelial-to-mesenchymal transition in human breast
cancer cells. Drug Des Devel Ther. 9:1027–1062. 2015.PubMed/NCBI
|
|
56
|
Wang W, Fan H, Zhou Y, Duan P, Zhao G and
Wu G: Knockdown of autophagy-related gene BECLIN1 promotes cell
growth and inhibits apoptosis in the A549 human lung cancer cell
line. Mol Med Rep. 7:1501–1505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang QJ, Ding Y, Kohtz DS, Mizushima N,
Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N and Yue Z:
Induction of autophagy in axonal dystrophy and degeneration. J
Neurosci. 26:8057–8068. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ling H, Chen H, Wei M, Meng X, Yu Y and
Xie K: The effect of autophagy on inflammation cytokines in renal
ischemia/reperfusion injury. Infammation. 39:347–356. 2016.
View Article : Google Scholar
|
|
60
|
Dong AL, Chen HG, Xie KL and Yu YH: Role
of autophagy in lung injury in septic mice. Chin J Anesthesiol.
35:1124–1127. 2015.
|
|
61
|
Wang HX, Huo XD, Chen HG, Li B, Liu J, Ma
WT, Wang XJ, Xie KL, Yu YH and Shi KM: Hydrogen-rich saline
activated autophagy via HIF-1α pathways in neuropathic pain model.
BioMed Res Int. 2018:Article ID 4670834. 2018.
|
|
62
|
Dong A, Wang L, Wang YY, Bian YX, Yu YH
and Xie KL: Role of autophagy in hydrogen-induced reduction of lung
injury in septic mice. Chin J Anesthesiol. 37:632–634. 2017.In
Chinese.
|
|
63
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ciani B, Layfield R, Cavey JR, Sheppard PW
and Searle MS: Structure of the ubiquitin-associated domain of p62
(SQSTM1) and implications for mutations that cause Paget's disease
of bone. J Biol Chem. 278:37409–37412. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Settembre C, Fraldi A, Jahreiss L,
Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C,
Rubinsztein DC and Ballabio A: A block of autophagy in lysosomal
storage disorders. Hum Mol Genet. 17:119–129. 2008. View Article : Google Scholar
|
|
66
|
Settembre C, Fraldi A, Rubinsztein DC and
Ballabio A: Lysosomal storage diseases as disorders of autophagy.
Autophagy. 4:113–114. 2008. View Article : Google Scholar
|
|
67
|
Dehay B, Martinez-Vicente M, Caldwell GA,
Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M and Bezard E:
Lysosomal impairment in Parkinson's disease. Mov Disord.
28:725–732. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Saftig P and Eskelinen EL: Live longer
with LAMP-2. Nat Med. 14:909–910. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T,
Xiao GD, Yang YP and Liu CF: The autophagy-lysosome pathway: A
novel mechanism involved in the processing of oxidized LDL in human
vascular endothelial cells. Biochem Biophys Res Commun.
394:377–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gutierrez MG, Munafó DB, Berón W and
Colombo MI: Rab7 is required for the normal progression of the
autophagic pathway in mammalian cells. J Cell Sci. 117:2687–2697.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hsieh YC, Athar M and Chaudry IH: When
apoptosis meets autophagy: Deciding cell fate after trauma and
sepsis. Trends Mol Med. 15:129–138. 2009. View Article : Google Scholar : PubMed/NCBI
|