Introduction
Hepatocellular carcinoma (HCC) is the most common
malignant neoplasm of the liver and the third leading cause of
cancer-related mortality worldwide (1). Although curative treatments, such as
liver transplantation and surgical resection, are available to some
patients with HCC, most patients are often diagnosed at an advanced
stage, which is not suitable for curative treatment (2). The prognosis of patients with
advanced HCC remains poor, and the therapeutic options available to
these patients are limited (3).
Sorafenib, a multikinase inhibitor, is the first
clinically approved targeted chemotherapy for HCC (4,5).
It suppresses tumor cell proliferation by targeting Raf-1
proto-oncogene, vascular endothelial growth factor receptor, and
platelet-derived growth factor receptor signaling pathways
(6). In patients with advanced
HCC, sorafenib has proven beneficial to survival; however, its main
limitation is its low objective response rates. Furthermore,
sorafenib therapy is accompanied by significant adverse effects
including dermatological, digestive, and cardiovascular toxicities
(7). The adverse effects can be
managed by sorafenib dose reduction. Therefore, it is necessary to
investigate combination treatments with sorafenib to enhance its
anticancer effects and reduce its toxicity through dose reduction.
To overcome these unmet needs of sorafenib therapy, co-treatment
with other chemotherapeutic agents has been previously evaluated
(8–10).
Bile acids synthesized from cholesterol in the liver
are necessary for the digestion and absorption of lipids; however,
elevated hydrophobic bile acid concentrations are associated with
pathological activities and promote the development of liver
cirrhosis (11). Unlike
hydrophobic bile acids, ursodeoxycholic acid (UDCA), a hydrophilic
acid, relieves cholestatic liver disease by exerting cytoprotective
activities in hepatocytes, and it has been suggested to suppress
tumorigenesis through cell cycle arrest and induction of apoptosis
(12,13). Although the antitumor role of UDCA
in HCC has been investigated, the specific molecular mechanisms are
not yet fully understood. UDCA is often administered to patients
with HCC in clinical practice and has minimal side effects. In
addition, the mechanism of the antitumor effect of UDCA differs
from that of sorafenib.
Pro-oxidant or reactive oxygen species (ROS)
activity has been demonstrated to contribute to cytotoxicity and
subsequent induction of apoptosis (14). Extracellular signal-regulated
kinase (ERK), part of the mitogen-activated protein kinase (MAPK)
superfamily, has been reported to promote apoptosis in response to
external stimuli, such as ROS (15). In addition, signal transducer and
activator of transcription 3 (STAT3) is primarily activated by
interleukin-6 (IL-6) receptor-associated Janus kinases; however,
several studies have suggested that STAT3 is influenced by
conditions such as increased pro-oxidant levels that subsequently
lead to decreased tumor growth and metastasis (16,17). STAT3 regulates the transcriptional
activation of anti-apoptotic genes, such as cyclins and
cyclin-dependent kinases (CDKs) (18). To date, investigations of STAT3
function have focused on cancer therapy.
In the present study, it was hypothesized that
co-treatment with UDCA would enhance the antitumor efficacy of
sorafenib in HCC cells. Thus, the effect of sorafenib and UDCA
co-treatment and their underlying molecular mechanisms were
investigated in HCC cells.
Materials and methods
Cell culture and drug treatment
Huh-BAT and HepG2 human HCC cells were purchased
from the Korean Cell Line Bank (Seoul, Korea). Cells were
maintained in continuous culture in Dulbecco's modified Eagle's
medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Thermo Fisher
Scientific, Inc.), penicillin (100 U/ml), and streptomycin (100
mg/ml). For the experiments, the cells were seeded in tissue
culture dishes at 37°C in a 5% CO2 humidified incubator.
They were cultured in fresh medium containing 10% FBS for 24 h and
then treated with different concentrations of UDCA (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany), sorafenib (LC Laboratories,
Woburn, MA, USA), N-acetyl cysteine (NAC; Sigma-Aldrich) or U0126
(Cell Signaling Technology, Inc., Danvers, MA, USA). Cells cultured
in medium with 10% FBS, without any additional treatments, were
used as controls.
Cell viability assay
Cells were plated in 96-well plates at a density of
5,000 cells/well and 1 day later they were incubated with sorafenib
and UDCA for 48 h. Then, the cells were incubated with 0.5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Amresco Inc., Solon, OH, USA) at 37°C for 3 h. Following removal of
the MTT solution, dimethyl sulfoxide was added to each well with
mixing for 15 min, and then the absorbance was detected at 540 nm
using a plate reader.
Annexin V/propidium iodide (PI) apoptosis
assay
Annexin V/PI staining was conducted to determine the
% of apoptotic cells in the total cell population. Following
sorafenib and UDCA treatment, cells were collected and resuspended
in binding buffer (BD Pharmingen; BD Biosciences, San Jose, CA,
USA). The cells were then incubated with 10 μg/ml PI and
Annexin V (BD Biosciences), and the fluorescence intensity was
determined using a FACSCalibur flow cytometer and BD CellQuest™ Pro
software version 5.2.1 (BD Biosciences).
Immunofluorescence assay
Cells were grown on coverslips in 12-well culture
plates and then exposed to UDCA and sorafenib for 24 h, followed by
IL-6 for 30 min. Following fixation with 4% paraformaldehyde for 20
min at room temperature, the cells were permeabilized with 100%
methanol at −20°C, blocked with 10% normal goat serum (Vector
Laboratories, Ltd., Peterborough, UK) for 1 h at room temperature
(23.8±6.3°C), incubated with phosphorylated (p)-STAT3 antibody
(1:50; cat. no. 4113; Cell Signaling Technology, Inc.) at 4°C
overnight, and then reacted with Alexa Fluor 488-conjugated
secondary antibody (1:50; cat. no. ab150113; Abcam, Cambridge, UK).
for 1 h at room temperature (23.8±6.3°C). The cell nuclei were
stained with 4′,6-diamidino-2-phenylindole, and the cells were
observed using a confocal microscope.
Measurement of ROS production
ROS levels were measured using a cellular ROS
detection assay kit (Abcam). For fluorescence microscopy, cells
were seeded in 12-well plates, exposed to UDCA or sorafenib for 24
h, and then labeled with an oxidative stress detection reagent
(green color) for 2 h, according to the manufacturer's
instructions. The fluorescence intensity was then measured using a
confocal microscope with Leica Application Suite X (Leica
Microsystems GmbH, Wetzlar, Germany). To quantify ROS levels, cells
were seeded onto 96-well black plates and then stained with ROS red
dye working solution for 1 h at 37°C in a 5% CO2
humidified incubator. Cells were treated with UDCA or sorafenib for
30 min and were then analyzed using a fluorescent microplate reader
(Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
Huh-BAT and HepG2 cells were lysed in
radioimmunoprecipitation assay buffer (Cell Signaling Technology,
Inc.). The protein concentration was determined using BCA assay
(Thermo Fisher Scientific, Inc.). The protein samples (25
μg) were separated using 8–12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and then transferred
onto polyvinylidene fluoride membranes (Millipore, Billerica, MA,
USA). After blocking with 5% skim milk in Tris-buffered saline/0.1%
Tween-20 (TBST) for 1 h, the membrane was incubated overnight at
4°C with 1:1,000 dilutions of primary antibodies against the
following proteins: β-actin (cat. no. sc-47778; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), STAT3, p-STAT3 (Tyr705; cat.
no. 4113), ERK (cat. no. 4695s), p-ERK (cat. no. 4377), caspase-9
(cat. no. 9580), cleaved caspase-9 (cat. no. 7237), caspase-3 (cat.
no. 9665), and cleaved caspase-3 (cat. no. 9664) (all from Cell
Signaling Technology, Inc.). Then, the membranes were incubated
with 1:2,500 dilutions of either goat anti-rabbit horseradish
peroxidase (HRP)-conjugated secondary antibody (cat. no. sc-2357;
Santa Cruz Biotechnology, Inc.) or goat anti-mouse HRP-conjugated
secondary antibody (cat. no. 31430; Thermo Fisher Scientific, Inc.)
in TBST for 1 h at room temperature. The blots were developed using
enhanced chemiluminescence detection reagents (Promega Corporation,
Madison WI, USA). All bands were quantified via densitometry using
ImageJ software version 1.51 (National Institutes of Health,
Bethesda, MD, USA). All western blot analyses were performed in
triplicate.
Statistical analysis
All experiments were conducted in three independent
repeats. Statistical analyses were performed using SPSS for Windows
version 22.0 (IBM Corporation, Armonk, NY, USA), and one-way
analysis of variance followed by Tukey's honest significant
difference test. P<0.05 was considered to indicate a
statistically significant difference.
Results
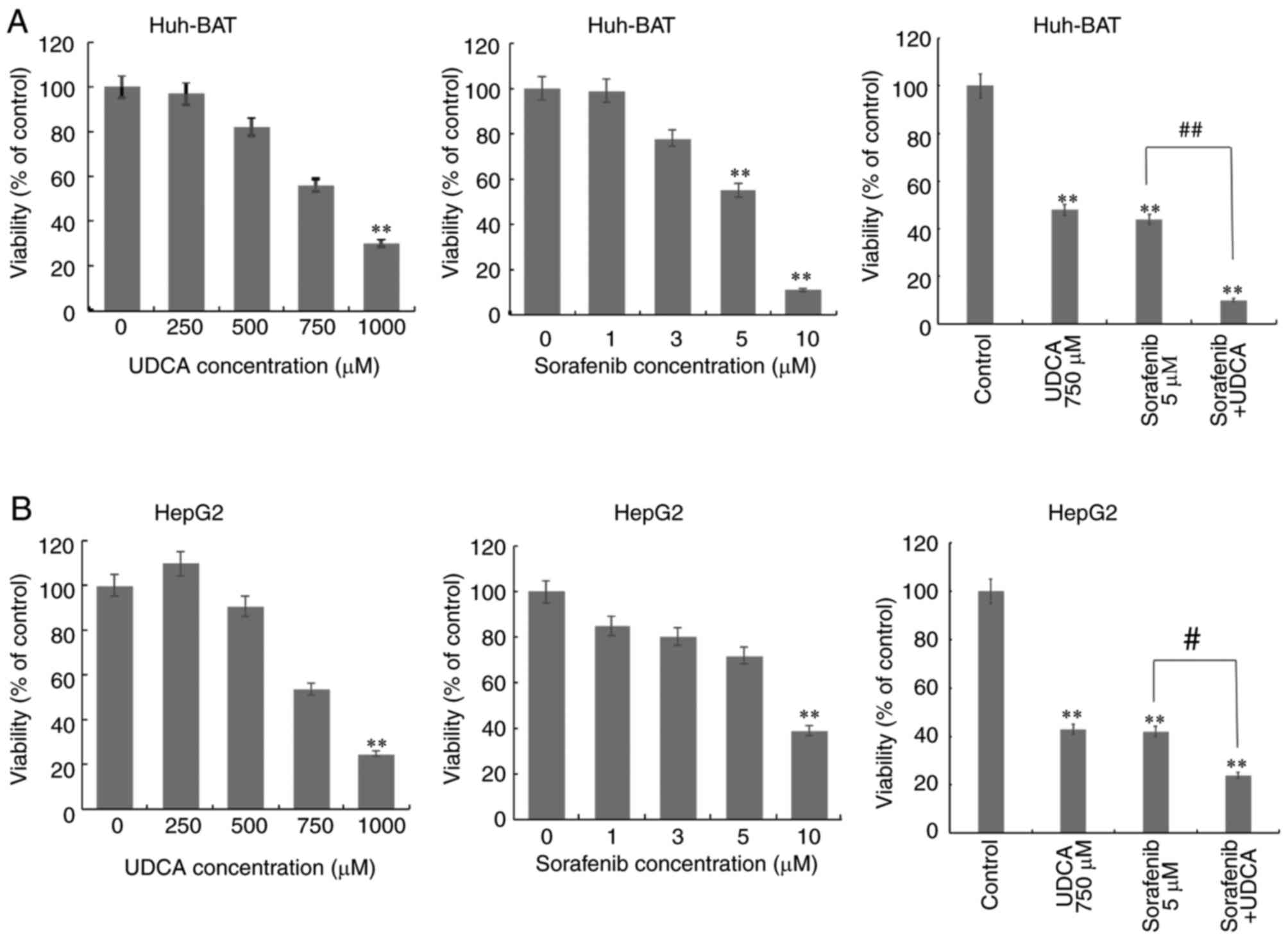
Sorafenib and UDCA inhibit HCC cell
viability
The cytotoxicity of co-treatment with sorafenib and
UDCA was evaluated in Huh-BAT and HepG2 cells. HCC cells were
treated with 0–10 μM sorafenib alone, 0–1,000 μM UDCA
alone, or 5 μM sorafenib plus 750 μM UDCA for 48 h
(Fig. 1). Treatment with
sorafenib or UDCA reduced the viability of Huh-BAT and HepG2 cells
in a concentration-dependent manner, while co-treatment with both
exhibited a significantly greater cytotoxicity compared with
sorafenib alone.
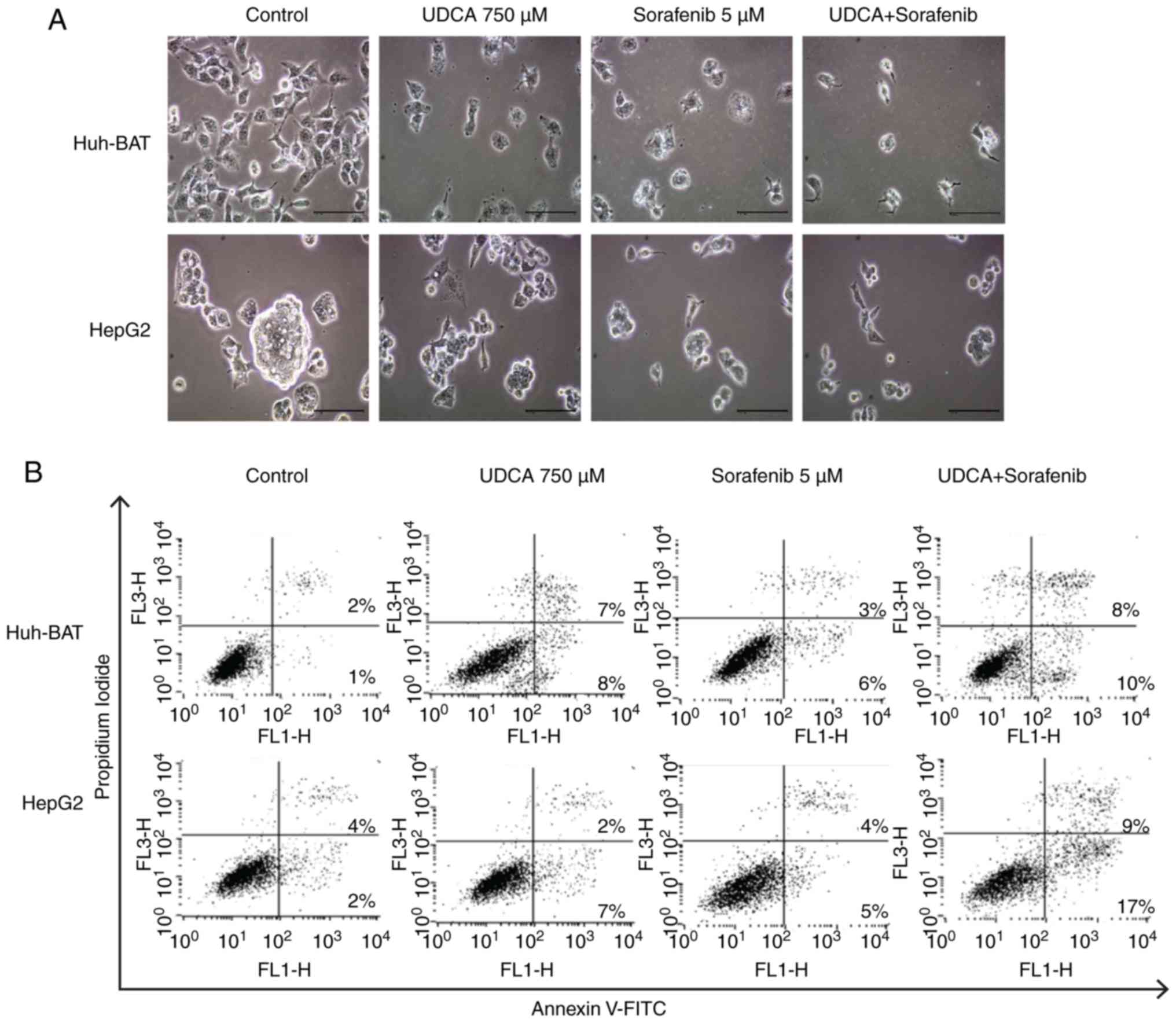
Sorafenib and UDCA induce
caspase-dependent apoptosis in HCC cells
Next, the present study evaluated whether
co-treatment affected apoptosis of Huh-BAT and HepG2 cells. As
illustrated in Fig. 2A,
microscopy image analysis of the treated cells revealed that
sorafenib, UDCA, and their co-treatment markedly altered cell
morphology. These observations were confirmed by Annexin V/PI
staining of cells treated with the indicated drugs (Fig. 2B and C). The ratio of apoptotic
cells in the untreated control Huh-BAT group was 3%, and this value
increased to 15 and 9% following treatment with UDCA and sorafenib,
respectively (Fig. 2C). Following
co-treatment with sorafenib and UDCA, the % of apoptotic cells
increased to 18% (Fig. 2C). For
HepG2 cells, the ratio of apoptotic cells in the untreated control
group was 6%, which increased to 9% following treatment with either
UDCA or sorafenib (Fig. 2C).
Apoptosis increased significantly to 26% following co-treatment
with both agents (Fig. 2C). These
findings demonstrate that cell apoptosis was induced by sorafenib
treatment, and this effect was enhanced when Huh-BAT and HepG2
cells were co-treated with sorafenib and UDCA.
To investigate the underlying mechanisms of the
apoptosis effect, caspase-3 and caspase-9 expression was evaluated
by western blotting. As illustrated in Fig. 2D, the results demonstrated that
the expression levels of both the proteins and their cleaved forms
were markedly increased in Huh-BAT and HepG2 cells co-treated with
sorafenib and UDCA, compared with the cells incubated with
sorafenib alone. These findings indicate that co-treatment induced
Huh-BAT and HepG2 cell death via caspase-dependent apoptosis.
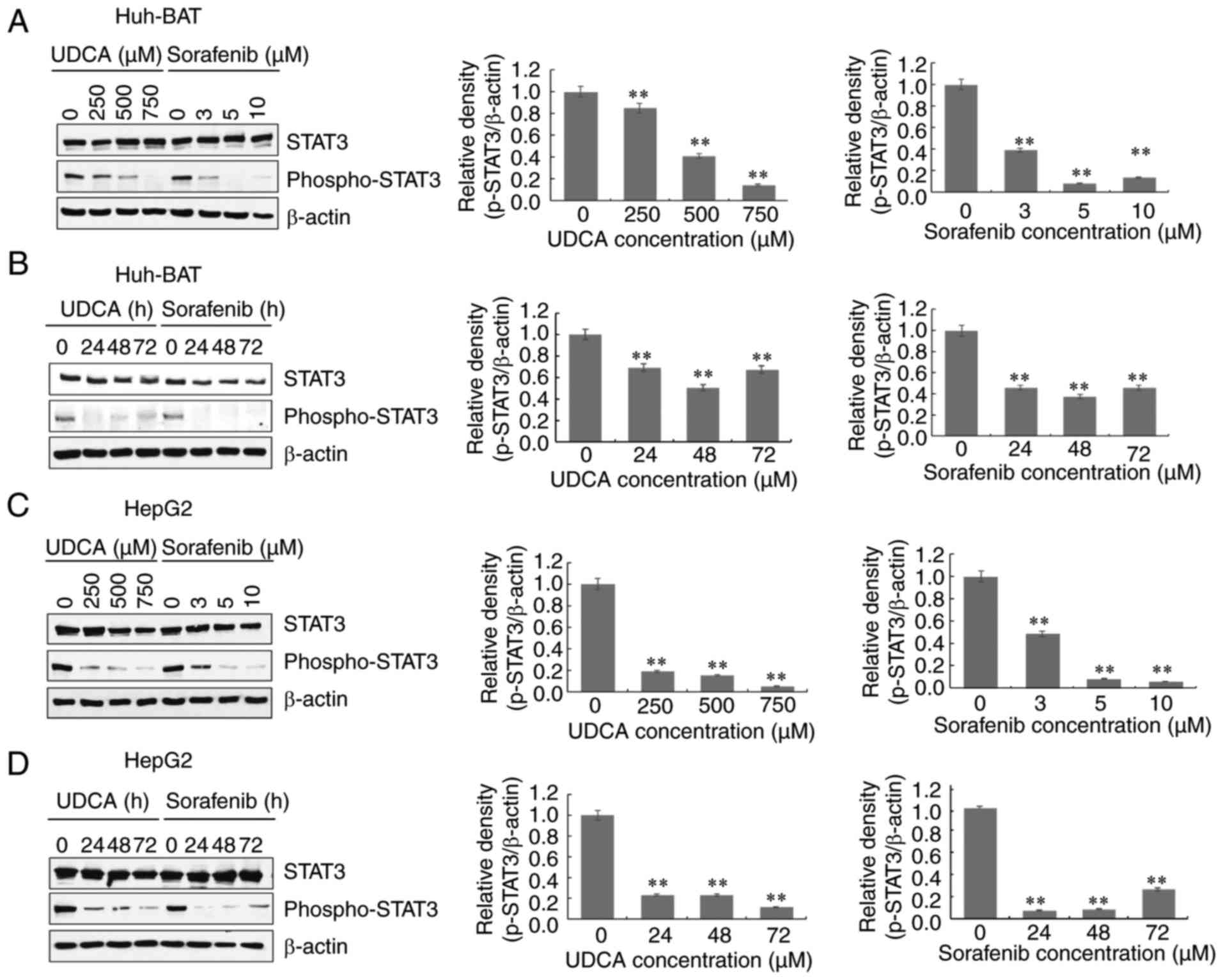
Sorafenib and UDCA suppress activation of
STAT3 in a time- and concentration-dependent manner
Next, the effects of sorafenib and UDCA co-treatment
were investigated on the phosphorylation of STAT3. Phosphorylation
of STAT3 on Try705 decreased in both Huh-BAT and HepG2 cells
treated with sorafenib or UDCA alone, both in a time- and
concentration-dependent manner (Fig.
3A-D). The levels of phosphorylated STAT3 in both cell lines
decreased following co-treatment with sorafenib and UDCA, compared
with untreated cells (Fig. 3E).
For immunofluorescence analysis, treatment with IL-6, an
inflammatory factor that induces STAT3 phosphorylation on Tyr705,
was used as a positive control. The results demonstrated that UDCA
and sorafenib suppressed STAT3 phosphorylation (Fig. 3F). These findings suggest that
co-treatment with these agents regulates phosphorylation of STAT3
on Try705 in both Huh-BAT and HepG2 cells.
Sorafenib and UDCA trigger ROS production
and ERK activation in a time- and concentration-dependent
manner
Intracellular ROS are important factors affecting
the apoptosis of cancer cells in response to DNA damage. Thus, the
generation of intercellular ROS was measured. Co-treatment with
sorafenib and UDCA induced intracellular ROS generation in Huh-BAT
and HepG2 cells, as detected by confocal microscopy (Fig. 4A and B). In the presence of
sorafenib and UDCA alone, ROS generation increased compared with
cells incubated with medium alone. The average fluorescent
intensity appeared to increase compared with the cells incubated
with sorafenib alone, although this difference was not
significant.
Next, the phosphorylation of ERK in Huh-BAT and
HepG2 cells was examined. Western blot analysis demonstrated that
ERK was phosphorylated in both those cells treated with sorafenib
or UDCA alone, in a time- and concentration-dependent manner
(Fig. 4C-F). The levels of
phosphorylated ERK in both cell lines increased following
co-treatment with sorafenib and UDCA, compared with untreated cells
(Fig. 4G). Compared with
sorafenib alone, the levels of phospho-ERK were markedly increased
following co-treatment in HepG2 cells, but this effect was not
observed in Huh-Bat cells (Fig.
4G). These observations indicate that apoptosis induced by
co-treatment with sorafenib and UDCA is associated with ROS
production and is mediated by the phosphorylation of ERK in Huh-BAT
and HepG2 cells.
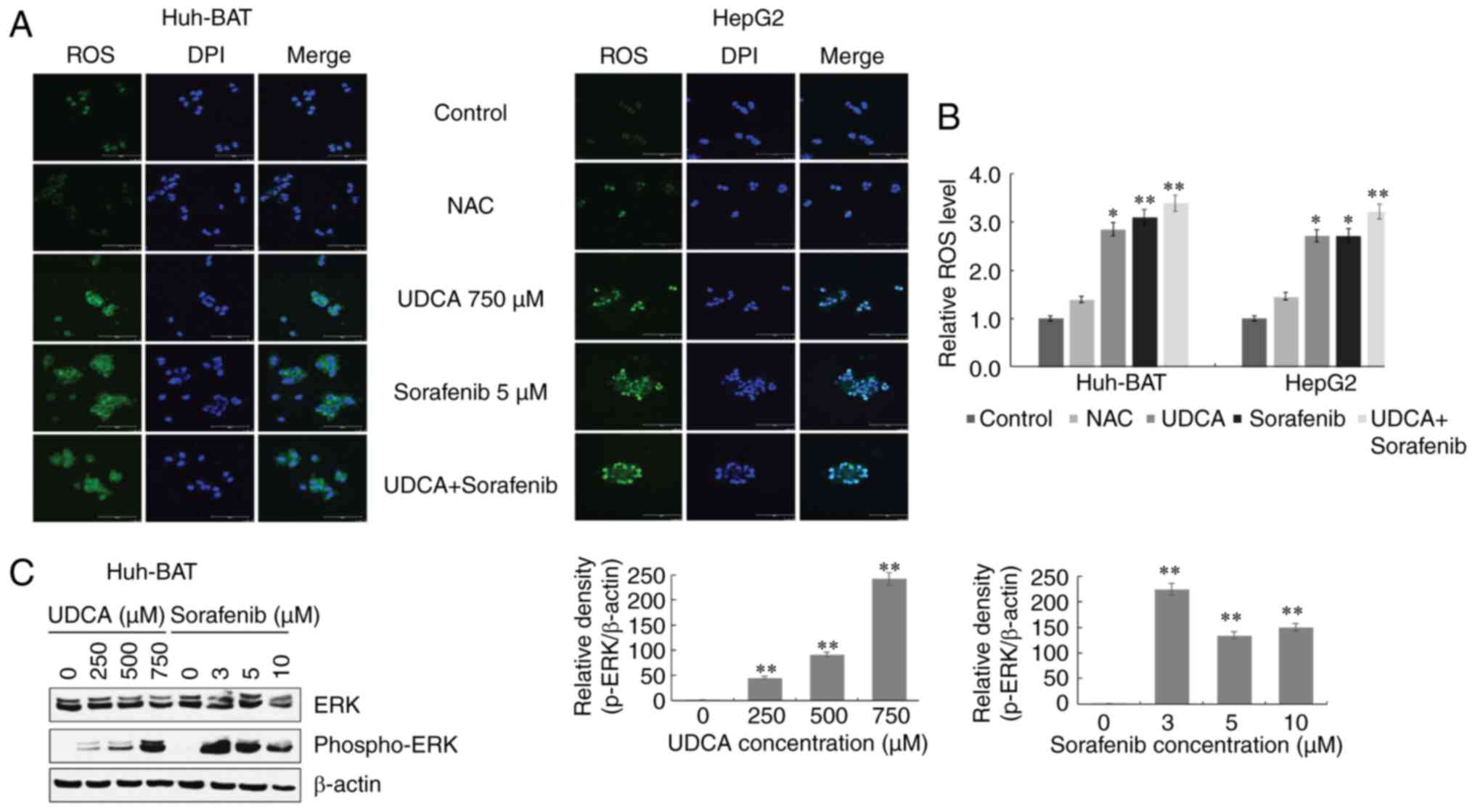
ROS production and ERK activation are
correlated with downregulation of STAT3 phosphorylation in Huh-BAT
cells co-treated with sorafenib and UDCA
To investigate the relationship among ROS
generation, ERK activation, and STAT3 dephosphorylation, Huh-BAT
cells were treated with the ROS scavenger NAC, or the ERK inhibitor
U0126 in the presence or absence of sorafenib, UDCA, or both.
Firstly, cells were pretreated with NAC to assess the role of ROS.
As presented in Fig. 5A,
pretreatment with NAC effectively inhibited sorafenib- and
UDCA-induced ROS production in Huh-BAT cells. Next, the roles of
ERK and ROS in STAT3 inactivation by sorafenib and UDCA treatment
were examined. Co-treatment with sorafenib and UDCA of Huh-BAT
cells pretreated with U0126 or NAC significantly increased
phospho-STAT3 levels and decreased those of phospho-ERK (Fig. 5B and C). These results indicate
that ROS and ERK may have an important role in the suppression of
STAT3 by sorafenib and UDCA treatment.
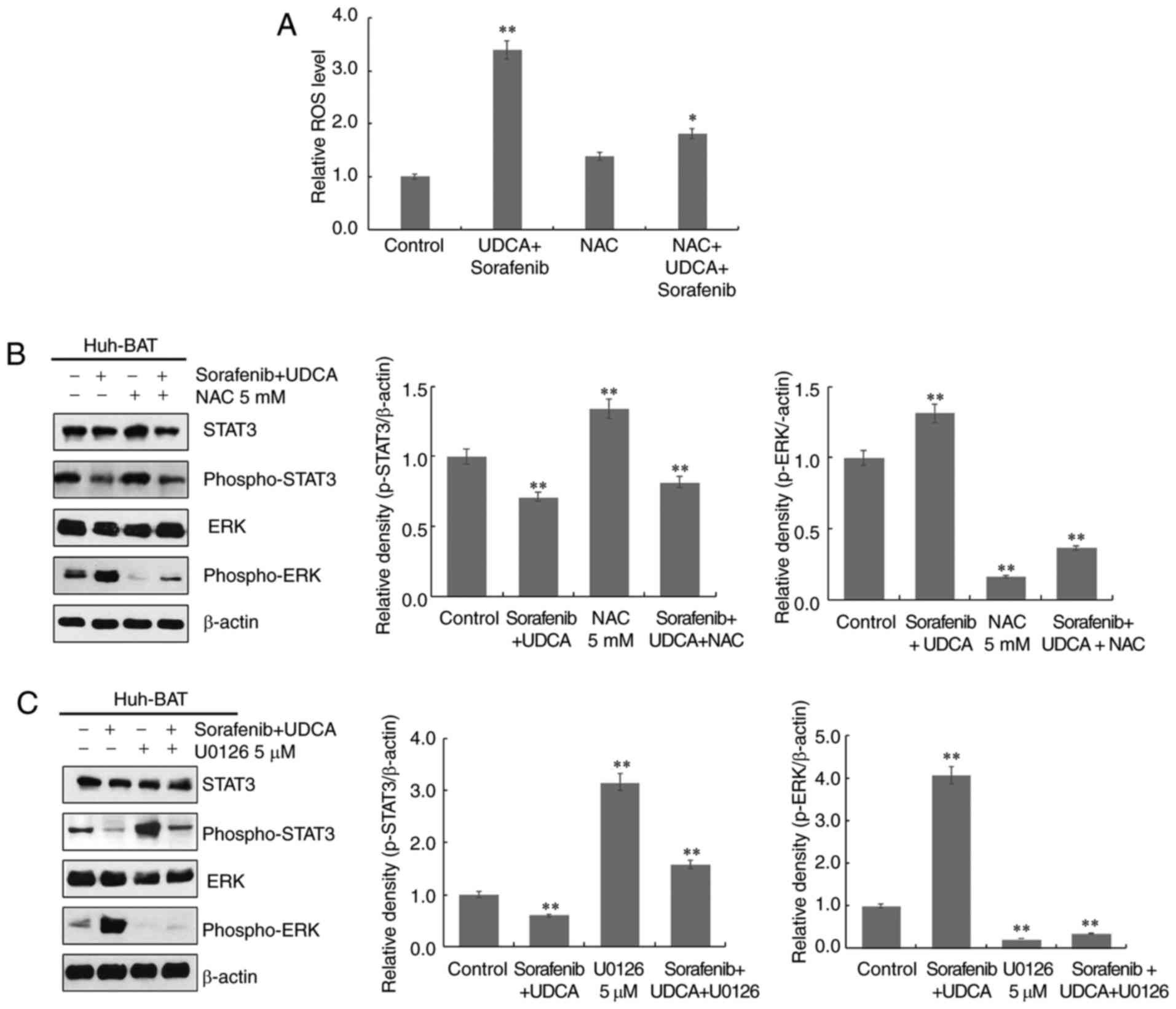 | Figure 5ROS production and ERK activation are
associated with downregulation of STAT3 phosphorylation in Huh-BAT
cells co-treated with sorafenib and UDCA. (A) Huh-BAT cells were
co-treated with 5 μM sorafenib and 750 μM UDCA for 48
h, followed by 5 mM NAC. Following treatments, ROS intensity was
measured using a fluorescence microplate reader. The relative ROS
levels were normalized to the mean intensity of ROS in the control
untreated cells (presented here as 1.0). (B) Huh-BAT cells were
pretreated with 5 mM NAC, or (C) pretreated with 5 μM U0126,
prior to 5 μM sorafenib and 750 μM UDCA treatments.
Samples were immunoblotted with primary antibodies to STAT3,
phospho-STAT3, ERK, and phospho-ERK. β-actin was used as a loading
control. *P<0.05 and **P<0.01 compared
with control untreated cells. ROS, reactive oxygen species; ERK,
extracellular signal-regulated kinase; STAT3, signal transducer and
activator of transcription 3; UDCA, ursodeoxycholic acid; NAC,
N-acetyl cysteine. |
Discussion
In the present study, co-treatment with sorafenib
and UDCA enhanced the effects of sorafenib on the inhibition of
cell viability and the induction of apoptosis, via ROS-mediated
activation of ERK and downregulation of STAT3 in HCC cells. To the
best of our knowledge, this is the first study to demonstrate the
synergistic effects of co-treatment with sorafenib and UDCA in
HCC.
Several studies have investigated the role of STAT3
signaling in sorafenib-treated HCC cells, because STAT3 regulates
the transcription of genes involved in cancer cell proliferation
and apoptosis via the phosphoinositide 3-kinase (PI3K)/AKT
signaling pathway (19).
According to Blechacz et al (20), sorafenib inhibits STAT3
phosphorylation by downregulating protein tyrosine phosphatase
non-receptor type 11 (PTPN11, also known as SHP-2) in
cholangiocarcinoma cells. In addition, Gu et al (19) suggested that inhibition of STAT3
activity reduces tumor growth and metastasis in HCC, by partially
suppressing the Raf/MAPK kinase (MEK)/ERK signaling pathway,
independent of PTPN11. The present study also demonstrated that
exposure to sorafenib suppressed STAT3 phosphorylation in HCC
cells. However, the underlying mechanism mediating the antitumor
actions of sorafenib were not consistent with those previously
reported. The present results demonstrated that the reduced
phosphorylation of STAT3 was rescued by U0126 in Huh-BAT cells,
indicating that ERK activation participated in the inhibition of
STAT3 in HCC cells. Although sorafenib is a well-known inhibitor of
the Raf/MEK/ERK signaling pathway, the mode of action of sorafenib
may vary depending on the response to stimuli under certain
conditions, such as ROS generation.
Although ROS is produced during normal aerobic
metabolism, excessive intracellular ROS generation causes oxidative
stress and leads to cell damage, ultimately inducing apoptosis
(21,22). UDCA can stimulate lipid rafts and
ROS production by acting on the cell membrane, thereby inducing
apoptosis (23). Sorafenib has
been reported to produce ROS (24). Co-treatment with these two
pro-oxidants may have synergistic effects on ROS generation.
Indeed, the present findings demonstrated that sorafenib and UDCA
co-treatment increased ROS generation and ERK activation in Huh-BAT
cells, while NAC reversed these effects.
Previous studies have demonstrated that ERK
activation induces apoptosis under specific conditions, such as
excessive ROS generation or oxidant scavenging failure (25). According to Zuo et al
(26), alternol, an antitumor
agent used in the treatment of osteosarcoma, increases ROS levels
by activating ERK and subsequently inhibiting STAT3
phosphorylation. In addition, the reduced STAT3 phosphorylation by
alternol was abrogated by pretreatment with NAC. These results are
consistent with the current findings.
The present study was limited by the small number of
cancer cell lines tested and the lack of in vivo
experiments. Further studies are needed to investigate the effects
of sorafenib and UDCA administration in vivo. In addition,
not only the present study, but other UDCA-related preclinical
studies use a relatively high concentration of UDCA as the
IC50 value (27–29). It would be more beneficial to
reduce the concentration of UDCA to protect normal cells, unless
the lower concentration reduces its antitumor efficacy. In
addition, it is necessary to identify whether reduced
concentrations of sorafenib and UDCA would have the same
cytotoxicity as the standard concentrations of sorafenib alone.
Taken together, the present results demonstrated
that sorafenib and UDCA suppressed the viability of HCC cells more
effectively compared with sorafenib alone, and promoted apoptosis.
Co-treatment with both agents inhibited STAT3 phosphorylation and
activated ERK in a ROS-dependent manner. These findings suggest
that the combination of sorafenib and UDCA may be a promising
therapeutic strategy for patients with advanced HCC.
Funding
This research was supported by the Daewoong
Pharmaceutical Company (grant nos. 800-20150450 and 800-20150451),
the Korea Health Technology R&D Project through the Korea
Health Industry Development Institute, funded by the Ministry of
Health & Welfare, Republic of Korea (grant no. HI16C1074) and
the Seoul National University Hospital Research Fund [grant no.
0420160300 (2016-1073)]
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
SL performed the experiments, analyzed the data and
wrote the manuscript. YYC critically revised the manuscript. EJC
and SJY conceived and designed the study. JL and JY analyzed the
data. YJK was involved in study design and manuscript revision and
gave final approval of the version to be published. All authors
approved the final version of the manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
CDK
|
cyclin-dependent kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FBS
|
fetal bovine serum
|
|
HCC
|
hepatocellular carcinoma
|
|
HRP
|
horseradish peroxide
|
|
KCLB
|
Korean Cell Line Bank
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
MAPK kinase
|
|
NAC
|
N-acetyl cysteine
|
|
PI
|
propidium iodide
|
|
ROS
|
reactive oxygen species
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
TBST
|
Tris-buffered saline/Tween-20
|
|
UDCA
|
ursodeoxycholic acid
|
Acknowledgments
Not applicable.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gauthier A and Ho M: Role of sorafenib in
the treatment of advanced hepatocellular carcinoma: An update.
Hepatol Res. 43:147–154. 2013. View Article : Google Scholar
|
|
4
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin
SK, Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and
safety of sorafenib in patients in the Asia-Pacific region with
advanced hepato-cellular carcinoma: A phase III randomised,
double-blind, placebo-controlled trial. Lancet Oncol. 10:25–34.
2009. View Article : Google Scholar
|
|
5
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43-9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Almhanna K and Philip PA: Safety and
efficacy of sorafenib in the treatment of hepatocellular carcinoma.
Onco Targets Ther. 2:261–267. 2009.PubMed/NCBI
|
|
8
|
Petrini I, Lencioni M, Ricasoli M,
Iannopollo M, Orlandini C, Oliveri F, Bartolozzi C and Ricci S:
Phase II trial of sorafenib in combination with 5-fluorouracil
infusion in advanced hepatocellular carcinoma. Cancer Chemother
Pharmacol. 69:773–780. 2012. View Article : Google Scholar
|
|
9
|
Wang L, Jia D, Duan F, Sun Z, Liu X, Zhou
L, Sun L, Ren S, Ruan Y and Gu J: Combined anti-tumor effects of
IFN-α and sorafenib on hepatocellular carcinoma in vitro and in
vivo. Biochem Biophys Res Commun. 422:687–692. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Y, Jiang H, Gao H, Kong J, Zhang P,
Hu S, Shi B, Zhang P, Yao M and Li Z: The monoclonal antibody CH12
enhances the sorafenib-mediated growth inhibition of hepatocellular
carcinoma xenografts expressing epidermal growth factor receptor
variant III. Neoplasia. 14:509–518. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Greim H, Trülzsch D, Roboz J, Dressler K,
Czygan P, Hutterer F, Schaffner F and Popper H: Mechanism of
cholestasis. 5. Bile acids in normal rat livers and in those after
bile duct ligation. Gastroenterology. 63:837–845. 1972.PubMed/NCBI
|
|
12
|
Alberts DS, Martinez ME, Hess LM, Einspahr
JG, Green SB, Bhattacharyya AK, Guillen J, Krutzsch M, Batta AK,
Salen G, et al: Phase III trial of ursodeoxycholic acid to prevent
colorectal adenoma recurrence. J Natl Cancer Inst. 97:846–853.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Loddenkemper C, Keller S, Hanski ML, Cao
M, Jahreis G, Stein H, Zeitz M and Hanski C: Prevention of
colitis-associated carcinogenesis in a mouse model by diet
supplementation with ursodeoxycholic acid. Int J Cancer.
118:2750–2757. 2006. View Article : Google Scholar
|
|
14
|
Sabharwal SS and Schumacker PT:
Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles'
heel? Nat Rev Cancer. 14:709–721. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan BJ and Chiu GN: Role of oxidative
stress, endoplasmic reticulum stress and ERK activation in
triptolide-induced apoptosis. Int J Oncol. 42:1605–1612. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen J, Wang J, Lin L, He L, Wu Y, Zhang
L, Yi Z, Chen Y, Pang X and Liu M: Inhibition of STAT3 signaling
pathway by nitidine chloride suppressed the angiogenesis and growth
of human gastric cancer. Mol Cancer Ther. 11:277–287. 2012.
View Article : Google Scholar
|
|
17
|
Ryu K, Susa M, Choy E, Yang C, Hornicek
FJ, Mankin HJ and Duan Z: Oleanane triterpenoid CDDO-Me induces
apoptosis in multidrug resistant osteosarcoma cells through
inhibition of Stat3 pathway. BMC Cancer. 10:1872010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wake MS and Watson CJ: STAT3 the
oncogene-still eluding therapy? FEBS J. 282:2600–2611. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gu FM, Li QL, Gao Q, Jiang JH, Huang XY,
Pan JF, Fan J and Zhou J: Sorafenib inhibits growth and metastasis
of hepatocellular carcinoma by blocking STAT3. World J
Gastroenterol. 17:3922–3932. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blechacz BR, Smoot RL, Bronk SF, Werneburg
NW, Sirica AE and Gores GJ: Sorafenib inhibits signal transducer
and activator of transcription-3 signaling in cholangiocarcinoma
cells by activating the phosphatase shatterproof 2. Hepatology.
50:1861–1870. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nordberg J and Arner ES: Reactive oxygen
species, antioxidants, and the mammalian thioredoxin system. Free
Radic Biol Med. 31:1287–1312. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lim SC, Duong HQ, Choi JE, Lee TB, Kang
JH, Oh SH and Han SI: Lipid raft-dependent death receptor 5 (DR5)
expression and activation are critical for ursodeoxycholic
acid-induced apoptosis in gastric cancer cells. Carcinogenesis.
32:723–731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Coriat R, Nicco C, Chereau C, Mir O,
Alexandre J, Ropert S, Weill B, Chaussade S, Goldwasser F and
Batteux F: Sorafenib-induced hepatocellular carcinoma cell death
depends on reactive oxygen species production in vitro and in vivo.
Mol Cancer Ther. 11:2284–2293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park BH, Lim JE, Jeon HG, Seo SI, Lee HM,
Choi HY, Jeon SS and Jeong BC: Curcumin potentiates antitumor
activity of cisplatin in bladder cancer cell lines via ROS-mediated
activation of ERK1/2. Oncotarget. 7:63870–63886. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zuo D, Zhou Z, Wang H, Zhang T, Zang J,
Yin F, Sun W, Chen J, Duan L, Xu J, et al: Alternol, a natural
compound, exerts an anti-tumour effect on osteosarcoma by
modulating of STAT3 and ROS/MAPK signalling pathways. J Cell Mol
Med. 21:208–221. 2017. View Article : Google Scholar
|
|
27
|
Pang L, Zhao X, Liu W, Deng J, Tan X and
Qiu L: Anticancer effect of ursodeoxycholic acid in human oral
squamous carcinoma HSC-3 cells through the caspases. Nutrients.
7:3200–3218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsagarakis NJ, Drygiannakis I, Batistakis
AG, Kolios G and Kouroumalis EA: A concentration-dependent effect
of ursodeoxycholate on apoptosis and caspases activities of HepG2
hepatocellular carcinoma cells. Eur J Pharmacol. 640:1–7. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Krishna-Subramanian S, Hanski ML,
Loddenkemper C, Choudhary B, Pagès G, Zeitz M and Hanski C: UDCA
slows down intestinal cell proliferation by inducing high and
sustained ERK phosphorylation. Int J Cancer. 130:2771–2782. 2012.
View Article : Google Scholar
|