Introduction
Diabetes mellitus is associated with increased risk
of heart attack, called diabetic cardiomyopathy (DCM). This
condition is one of the leading causes of mortality with common
cardiovascular complications (1).
DCM alters the diastolic and systolic cardiac functions, resulting
in myocardial ischemia and heart failure (2,3).
It is characterized by myocardial dysfunction, myocardial cell
apoptosis and myocardial fibrosis, resulting from blood glucose
metabolic abnormalities (4,5).
It is reported that high glucose (HG) can induce the expression of
inflammatory cytokines and promote myocardial cell apoptosis,
suggesting that cell apoptosis is closely associated with
cardiovascular disease (6).
Accumulating evidence has implicated oxidative stress and
inflammation induced-apoptosis as contributors to the onset and
development of DCM (7-9).
Cardiomyocyte apoptosis is a main factor in the
pathology of DCM, which causes the loss of myocardial contractile
cells, aggravates myocardial injury, accelerates the occurrence of
cardiac dysfunction and ultimately may lead to heart failure
(10,11). Another important pathological
feature of DCM is hyperglycemia-induced excess production of the
extracellular matrix (ECM), including collagen types I and III,
which are involved in cardiac remodeling and fibrosis (12). In vitro, transforming
growth factor-β (TGF-β1) plays a critical role in alteration of the
cardiac structure and function by differentiation of cardiac
fibroblasts (CFs) and ECM deposition (13). In particular, activation and
proliferation of CFs plays an important role in myocardial fibrosis
(14). Hyperglycemia induces
inflammation-associated cytokine expression, such as interleukin
(IL)-1, tumor necrosis factor-α (TNF-α) and TGF-β1, which
participate in the development and progression of myocardial
fibrosis (15,16).
Nucleotide oligomerization domain (NOD)-like
receptors are a newly discovered set of pattern-recognition
receptors. NOD2 as one of the members of the NOD-like receptor
family have been demonstrated to activate transcription factors by
mediating production of inflammatory cytokines (17,18). The association of NOD2 with the
mechanism of diabetic nephropathy, atherosclerosis, myocardial
reperfusion injury and diabetes mellitus has been revealed in
clinical and animal models (19-23). However, little is known about the
role of NOD2-mediated apoptosis and fibrosis in DCM.
The present study focused on the underlying
mechanism of NOD2 in DCM, and demonstrated the effect of NOD2
silencing on HG-induced apoptosis and fibrosis, as well as on
apoptosis and fibrosis-related proteins in a DCM model of mice. To
further investigate the association between NOD2 and apoptosis or
fibrosis-related proteins in cardiomyocytes and fibroblasts, NOD2
knockdown experiments were performed.
Materials and methods
Animal experiments and RNA
interference
8-9 week-old male C57BL/6 mice (n=40), (weighing
22-35 g) and 7-8 week-old timed pregnant C57BL/6 mice (n=10, mated
with syngeneic males) were purchased from Experimental Animal
Center of Shandong University (Jinan, China). Mice were maintained
at a temperature of 22°C and a 55% humidity controlled environment
on a 12 h light-dark cycle with free access to food and water. All
experimental protocols were approved by the Institutional Animal
Care and Use Committee of Shandong University. All surgeries were
performed under sodium pentobarbital anesthesia.
The diabetes model was established by
intraperitoneal injection of STZ (Sigma Aldrich; Merck KGaA,
Darmstadt, Germany) dissolved in 0.1 ml citrate buffer (pH 4.5)
intraperitoneally (i.p.) at 90 mg/kg per mouse for 6 consecutive
days. Control mice were injected using equal parts of citrate
buffer. After 1 week, the blood was obtained from the tail vein,
and random glucose levels were measured by glucometer (Accuchek
Advantage; Roche Diagnostics GmbH, Mannheim, Germany). Diabetes was
determined as blood glucose >16.7 mmol/l (normally 11.1 mmol/l).
Following the induction of diabetes for 12 weeks, mice were
randomly divided into 4 groups: Control (n=8); diabetes model
(n=8); NOD2 short hairpin (sh)RNA mice (n=8) injected with
1×107 UT/30 µl pLV-NOD2 shRNA (Shanghai Jikai
Biological Technology Co., Ltd., Shanghai, China) in various sites
of the left ventricle; shRNA negative control (NC) (n=8), mice were
injected with 1×107 UT/30 µl lentivehicle in
various sites of the left ventricle. 293T cells were used as an
interim cell line to generate the packaged lentivirus. After 72 h,
levels of NOD2 expression were detected in tissues to assess
efficiency of RNA interference in mice. Then, after 4 weeks,
clinical examination was performed in these groups, and mice were
sacrificed to obtain mouse blood and heart tissue for the following
experiments.
Cell culture and treatment
The primary cardiomyocytes and primary CFs were
isolated from the ventricular tissues of neonatal mice (<3 d
old) from pregnant C57BL/6 mice. Cells were seeded and incubated in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 2 mM
glutamine (Shanghai Biological Engineering Co., Ltd., Shanghai,
China) and antibiotics (100 U/ml penicillin and 100 ug/ml
streptomycin; Gibco; Thermo Fisher Scientific, Inc.), incubated at
37°C in a humidified incubator containing 5% CO2 and 95%
air. Cells were identified using α-sarcomeric actin and FSP-1 stain
to confirm that >90% of cells were cardiomyocytes and CFs,
respectively. Cultured cell populations reached 60% confluence, and
were treated in the presence or absence of HG (15-25 mM) for 48
h.
Immunofluorescence
Immunofluorescent staining was performed using
cultured primary cells. A total of 2.0×104 neonatal
cardiomyocytes and CFs were seeded on glass cover-slips (2
cm2) wells, cultured for 24 h in DMEM with 10% FBS and
washed with PBS, then fixed using 4% paraformaldehyde for 20 min at
room temperature. Cells were then washed 3 times with PBS, blocked
in 3% bovine serum albumin (BSA) for 1 h. Then cells were incubated
with anti-FSP-1 antibody (1:500; cat. no. 07-2274; EMD Millipore,
Billerica, CA, USA) and anti-α-sarcomeric actin antibody (1:200;
cat. no. A2547; Sigma-Aldrich, Merck KGaA) respectively, at 4°C
overnight. Then, cells were washed twice, and incubated with
corresponding secondary antibodies: Anti-rabbit IgG-FITC (1:200;
cat. no. F1262; Sigma-Aldrich; Merck KGaA); and IgG-CY3 antibodies
(1:400; cat. no. AP187C; Sigma-Aldrich; Merck KGaA) at 37°C for 20
min. DAPI (1:1,000; cat. no. D8417; Sigma-Aldrich; Merck KGaA) was
added to stain the nuclei. The positive cells were imaged using a
confocal laser scanning microscope (Fluoview FV1000; Olympus
Corporation, Tokyo, Japan).
Cell transfection
Small interfering RNA (siRNA) against NOD2 and NC
siRNA were purchased from Ruibo Biotechnology Co., Ltd. (Guangzhou,
China). The sequences used were as follows: Sense, 5′-AGA GAA AUU
CUU UGA ATT-3′, anti-sense 5′-UUC AAA GUU GAA UUU CUC UTG-3′ for
NOD2, and sense, 5′-UUC UCC GGA ACG UGU CAC GUT T-3′, anti-sense
5′-ACG UGA CAC GUU CGG AGA ATT-3′ for negative control sequence.
The siRNAs were diluted with OPTI-MEM medium to achieve a final
concentration of 100 nM. The cells were transfected with siRNA
using Lipofectamine® 2000 for 48 h, (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer’s protocol.
Then, cells were stimulated with 20 mM HG for 48 h to be used for
further experiments. Experiments were repeated three times.
Hemodynamic evaluation
Mice were anaesthetized with 10% sodium
pentobarbital (40 mg/kg), and the common carotid artery of mice was
separated away, then a plastic catheter containing 1% liquemine was
inserted into the left ventricles from the right common carotid
artery. The left ventricular end-diastolic pressure (LVEDP), left
ventricular systolic pressure (LVSP), maximum rates of increased
and decreased left ventricular pressure (± dP/dt max) were
determined by the multimedia biological signal acquisition and
analysis system (BD Biosciences, Franklin Lakes, NJ, USA).
Histology and immunohistochemistry
The heart tissues separated from the experimental
groups of mice were fixed with 4% paraformaldehyde for 24 h at room
temperature, embedded in paraffin using a paraffin embedding
machine and sectioned (5 µm thick). The sections were then
deparaffinized, then rehydrated with graded ethanol at room
temperature, and stained using hematoxylin for 3-5 min and eosin
for 1-2 min (H&E) and Masson’s staining (Sigma-Aldrich; Merck
KGaA) at room temperature in accordance with the manufacturer’s
protocol. For TUNEL staining of tissue, the sections were stained
with a terminal deoxynucleotidyl transferase dUTP nick end labeling
(TUNEL) kit (Boster Bioengineering Company, Wuhan, China) according
to the manufacturer’s protocol. 3.3 diaminobenzidine (DAB, Vector
Laboratories, Inc., Burlingame, CA, USA) was used as the
chromogenic substrate. Sections were then counterstained with 10%
haematoxylin for 1 min (ProSciTech, Thuringowa, Australia).
Immunostaining pictures were captured under a light microscope
(Olympus BX51; Olympus Corporation, Tokyo, Japan).
TUNEL staining
Primary cardiomyocytes were transfected with NOD2
siRNA and siRNA NC, then stimulated with 20 mM HG for 48 h. Cells
were stained using fluorescein isothiocyanate (FITC)-labeled TUNEL
for 1 h at 37°C following the protocol of the TUNEL fluorescent kit
(Beyotime Institute of Biotechnology, Haimen, China). The
TUNEL-positive cells were imaged under a laser scanning confocal
microscope (Olympus Corporation) and five fields in each image were
quantified (the number of green spots in each photograph).
ELISA
Angiontensin (Ang) II (Enzo Biochem, Inc., New York,
NY, USA), cTn I and CK-MB (R&D Systems, Minneapolis, MN, USA)
in serum of mice, or TNF-α (cat. no. MTA00B), IL-1β (cat. no.
MLB00C) and IL-6 (cat. no. DY406-05; R&D Systems, Minneapolis,
MN, USA) levels in the supernatants of cardiomyocytes were measured
by ELISA kits following the manufacturer’s protocol.
Assessment of cell proliferation
Proliferation of HG and NOD2 siRNA-treated CFs was
evaluated using the Cell Counting Kit-8 (CCK-8; MultiSciences
Biotechnology, Zhejiang, China) according to the manufacturer’s
protocol. Briefly, CFs were seeded onto 96-well plates for 12, 24,
36, 48 and 72 h, then optical density (OD) was read at a wavelength
of 450 nm with an automated microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from mouse cardiomyocytes
and CFs using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA (1 µg) was transcribed to cDNA
with PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Japan) at
37°C, 15 min and 85°C, 5 sec. The sequences for primers were as
follows: Forward, 5′-GCC TTC CTT CTA CAG CAC GT-3′; and reverse,
5′-TGG CAG GGC TCT TCT GCA AG-3 for NOD2; forward, 5′-GAA TTG CTA
TGT GTC TGG GT-3′; and reverse, 5′-CAT CTT CAA ACC TCC ATG ATG-3′
for β-actin. The expression level of genes of interest was
quantified using 20 µl reaction mixture containing 1
µg cDNA, 0.2 mmol/l of each primer and SYBR-Green (BioRad
laboratories, Inc.) in 96-well plates in a LightCycler rapid
thermal cycler system (MJ Research PTC-2000, MJ Research, Waltham,
MA, USA). RT-qPCR amplification conditions were as follows: 95°C
for 10 min, 40 cycles at 95°C for 10 sec and 60°C for 45 sec, and
relative expression of genes were analyzed by the 2−∆∆Cq
(24) calculation method and
standardized against the housekeeping gene β-actin.
Western blotting
Protein was extracted from the heart tissues and
cells using a protein extraction kit (BC3710; Beijing Solarbio
Science & Technology Co., Ltd Solarbio, Beijing, China) and
protein concentration was quantified using a BCA assay (Thermo
Fisher Scientific, Inc.) according to the manufacturer’s protocol.
Protein (20 µg) was separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto a nitrocellulose membrane (EMD Millipore).
Following blocking with 5% nonfat milk in Tris buffered saline
Tween-20, the blots were incubated with primary antibodies:
Anti-NOD2 (1:500; sc-30080; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), anti-collagen I (1:1,000; ab34710; Abcam, Cambridge, MA,
USA), anti-collagen III (1:1,000; ab7778; Abcam), anti-TGF-β
(1:1,000; ab31013; Abcam) and GAPDH (1:5,000; sc-25778; Santa Cruz
Biotechnology, Inc.) overnight at 4°C. Then, the membranes were
incubated with horseradish peroxidase-conjugated rabbit secondary
antibodies (1:2,000; K5007; Dako, Agilent Technologies, Inc., Santa
Clara, CA, USA) for 1 h at room temperature. The protein bands were
visualized using the SuperSignal chemiluminescent detection module
(34080; Pierce, Thermo Fisher Scientific, Inc.) and images were
collected using a Tanon-5200 imaging system (Tanon Science and
Technology Co., Ltd., Shanghai, China).
Statistical analysis
All statistical analysis was conducted using SPSS
software, version 18.0 (SPSS, Inc., Chicago, IL, USA). One-way
analysis of variance followed by the SNK or Dunnet’s t test was
used to evaluate the significance of differences in the data. The
experiments were repeated three times and data are presented as the
mean ± standard error of the mean. P<0.05 was considered to
indicate a statistically significant difference.
Results
NOD2 inhibition ameliorates
diabetes-induced myocardial apoptosis and fibrosis
To ascertain whether NOD2 plays a role in myocardial
apoptosis and fibrosis associated with diabetes, the present study
investigated the effect of NOD2 gene silencing in diabetic mice.
STZ induced rapid hyperglycemia in mice compared with control at 1
week following injection. After the injection of NOD2 shRNA in
diabetic mice (72 h), NOD2 shRNA treatment downregulated myocardial
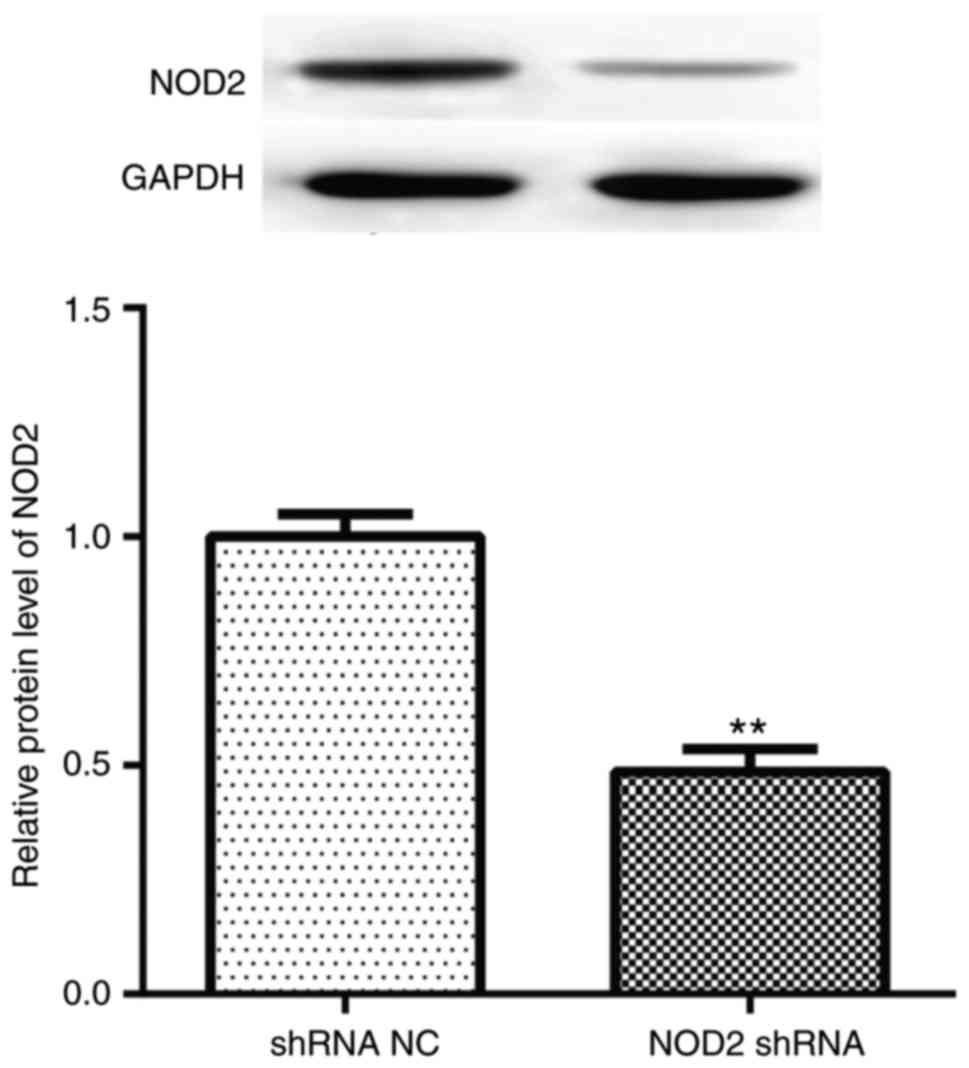
NOD2 protein levels compared with vehicle treatment (Fig. 1; P<0.01), which demonstrated
that NOD2 shRNA lentivectors were successfully introduced into the
mice. The hemodynamic indexes were first compared among the 4
groups at the end of the study. As compared with control group, the
DCM model group exhibited a significant increase in LVEDP level
(Fig. 2A; P<0.01), however the
LVSP and ± dP/dt max levels were markedly decreased (Fig. 2B-D; P<0.01). In diabetic mice,
LVEDP level was markedly reduced in NOD2 shRNA treatment group
compared with shRNA NC group (Fig.
2A; P<0.05). However, NOD2 shRNA treatment upregulated LVSP,
+dP/dt max and -dP/dt max levels compared with shRNA NC group
(Fig. 2B-D; P<0.05). In
addition, the data of the myocardial injury biomarkers revealed
that cTn I, CK-MB and Ang II in serum from DCM model were
significantly increased compared with control group. However, NOD2
gene elimination markedly reduced the levels of these factors in
diabetic mice, compared with the effect exhibited by the shRNA NC
treatment group (Fig. 2E-G;
P<0.05).
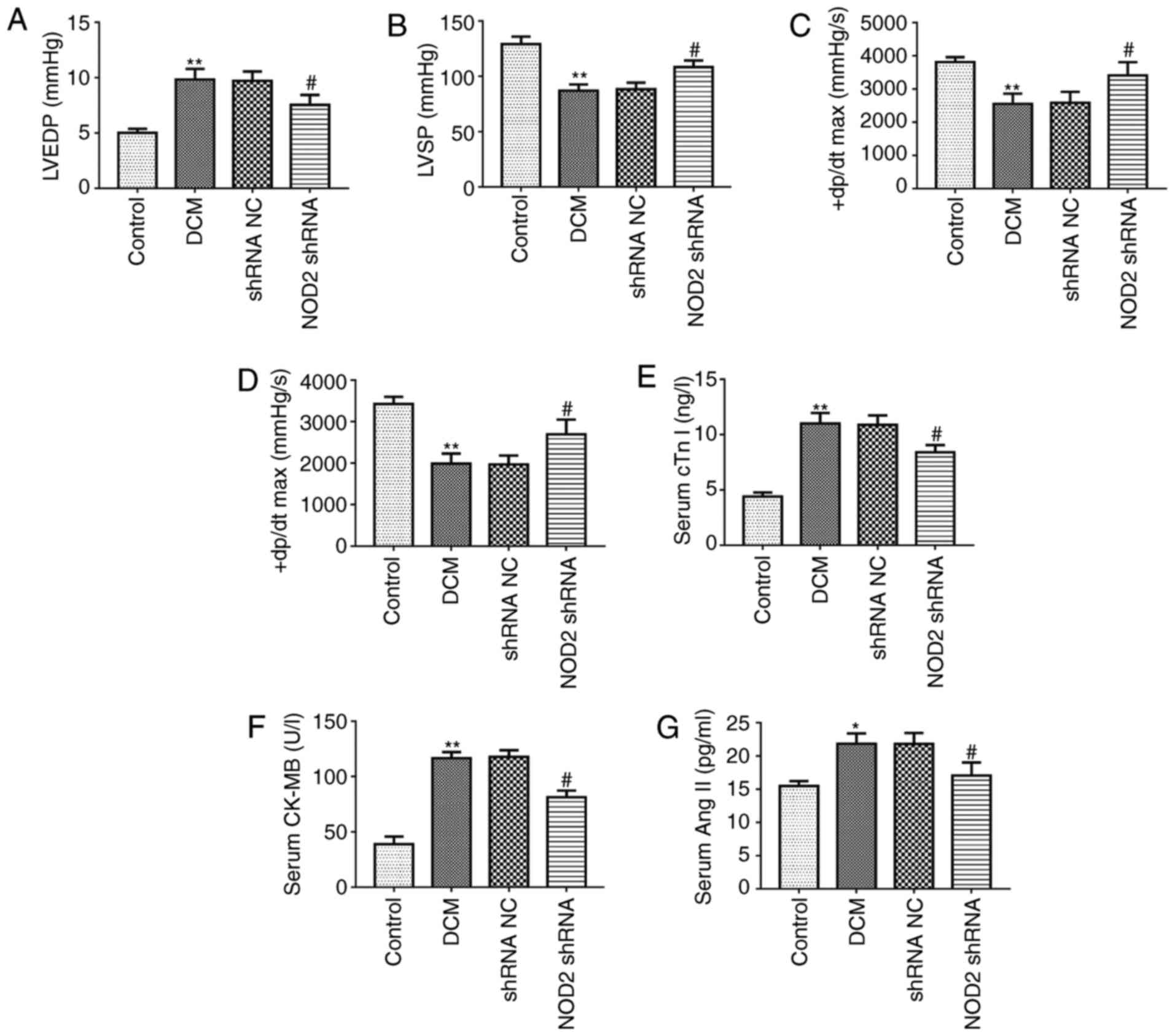 | Figure 2Pathological changes of the diabetic
mouse model. Tests of cardiac function index in each group (n=8).
Measurements of (A) LVEDP, (B) LVSP, (C) +dP/dtmax and (D)
-dP/dtmax in diabetic mice. Detection of the myocardial injury
biomarkers in mouse serum (n=8). Measurement of (E) cTn I, (F)
CK-MB, (G) Ang II. (H) Representative images of HE, Masson, TUNEL
staining. Scale bars=20 µm. Quantitative analysis of (I)
myocardial fibrosis and (J) TUNEL-positive cells.
**P<0.01 vs. control group, #P<0.05,
##P<0.01 vs. shRNA NC group. TUNEL, terminal
deoxynucleotidyl transferase dUTP nick end labeling; H&E,
hematoxylin and eosin; NOD2, nucleotide-binding oligomerization
domain 2; shRNA, short hairpin; NC, negative control; LVEDP, left
ventricular end-diastolic pressure; LVSP, left ventricular systolic
pressure; ± dP/dt max, maximum rates of increased and decreased
left ventricular pressure; DCM, diabetic cardiomyopathy. |
Heart tissues were isolated from the experimental
mice, and different staining methods were used to detect the effect
of NOD2 shRNA on pathological changes in diabetic mice. As
presented in Fig. 2H, The
pathological changes by H&E stain in control group showed that
myocardial cells were orderly arranged and the nuclei were
homogeneous. However, in the DCM group, the myocardium exhibited a
disordered arrangement, the nucleus was not uniform and part of
myocardial fiber was fractured. The NOD2 shRNA treatment lessened
injury of heart tissue in diabetic mice. Masson’s staining of heart
sections revealed more ECM in the interstitial regions of the DCM
group than myocardium of control group, and apoptosis in cardiac
tissue was assayed using TUNEL staining. Quantitative analysis of
Masson’s staining revealed a significant increase in collagen
deposition of DCM mice compared with control (Fig. 2I; P<0.01). However, NOD2 shRNA
treatment ameliorated collagen deposition (Fig. 2I; P<0.05 vs. shRNA NC).
Quantitative analysis of TUNEL stain revealed an increased number
of TUNEL-positive cells in the DCM group (Fig. 2J; P<0.01 vs. control
group). Compared to the vehicle treatment group, NOD2 silencing
weakened cell apoptosis (Fig. 2J;
P<0.01 vs. shRNA NC group).
Furthermore, in line with alteration of collagen
deposition and apoptosis detection in immunostaining, collagen I,
collagen III, TGF-β1 (Fig. 3A;
P<0.01), and apoptosis-related proteins Caspase-3 and B cell
lymphoma (Bcl)-2 associated X, apoptosis regulator (Bax) expression
levels were significantly increased in the DCM group, as
demonstrated by western blotting, but the expression of these
proteins was attenuated in diabetic mice with NOD2 shRNA treatment,
compared with vehicle-treated mice. Suppression of NOD2
significantly upregulated Bcl-2 expression, and downregulated NOD2
expression in diabetic mice (Figs.
3A and 2B; P<0.01).
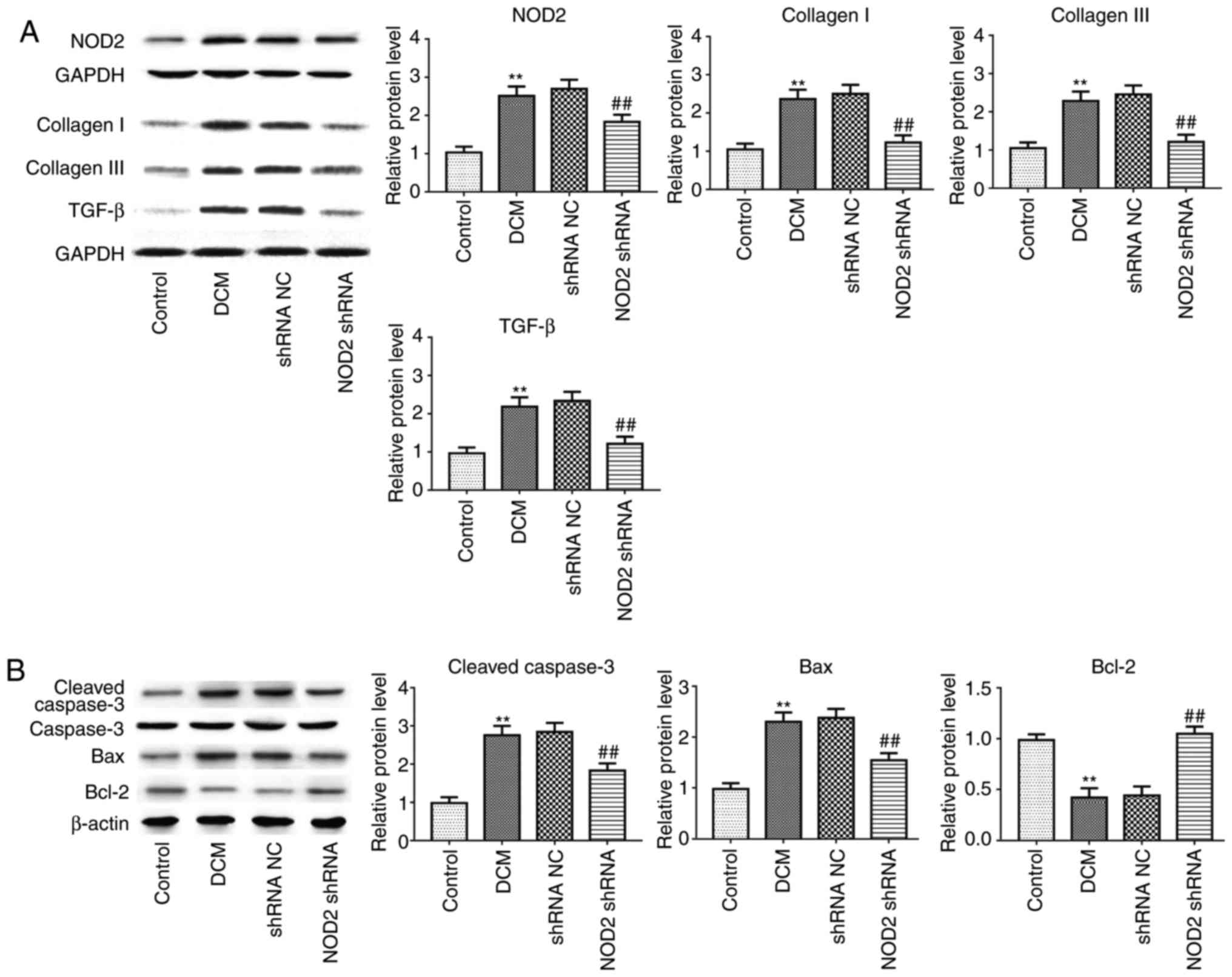 | Figure 3Suppressing NOD2 with NOD2 shRNA
attenuates collagen and apoptosis-related protein expression in
diabetic mice. (A) Western blot analysis of protein expression of
NOD2, collagen I, collagen III and TGF-β1. (B) Western blot
analysis of protein expression of Caspase-3, Bax and Bcl-2.
**P<0.01 vs. control group, ##P<0.01
vs. shRNA NC group. NOD2, nucleotide-binding oligomerization domain
2; shRNA, short hairpin; NC, negative control; DCM, diabetic
cardiomyopathy; TGF-β, transforming growth factor-β; Bcl-2, B cell
lymphoma 2; Bax, Bcl-2 associated X, apoptosis regulator. |
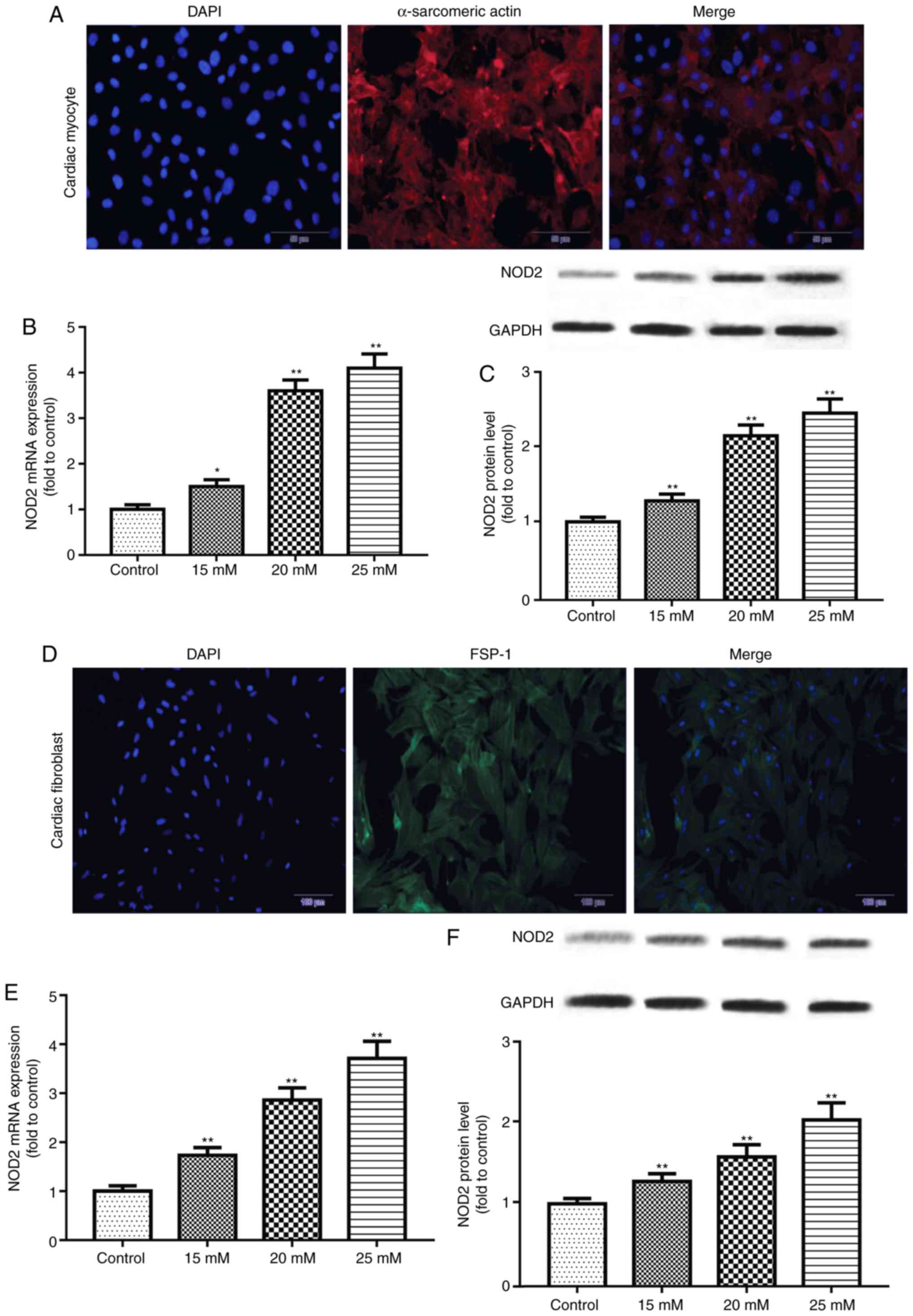
NOD2 is upregulated in HG-induced primary
cardiomyocytes and CFs
Immunofluorescence staining was performed to
identify primary cardiomyocytes and CFs. The results demonstrated
that staining of α-sarcomeric actin and FSP-1 were positive
(Fig. 4), reflecting the
isolated cells were primary cardiomyocytes and CFs. The expression
of NOD2 mRNA and protein in primary cells exposed to glucose for 48
h was further detected by RT-qPCR and western blotting. The results
revealed that NOD2 levels in primary cardiomyocytes were
significantly increased at concentrations of 15, 20 and 25 mM
glucose (Fig. 4B and C; P<0.05
vs. control group). Similarly, NOD2 level in CFs was enhanced at
concentrations of 15, 20 and 25 mM glucose treatment (Fig. 4E and F; P<0.01 vs. control
group).
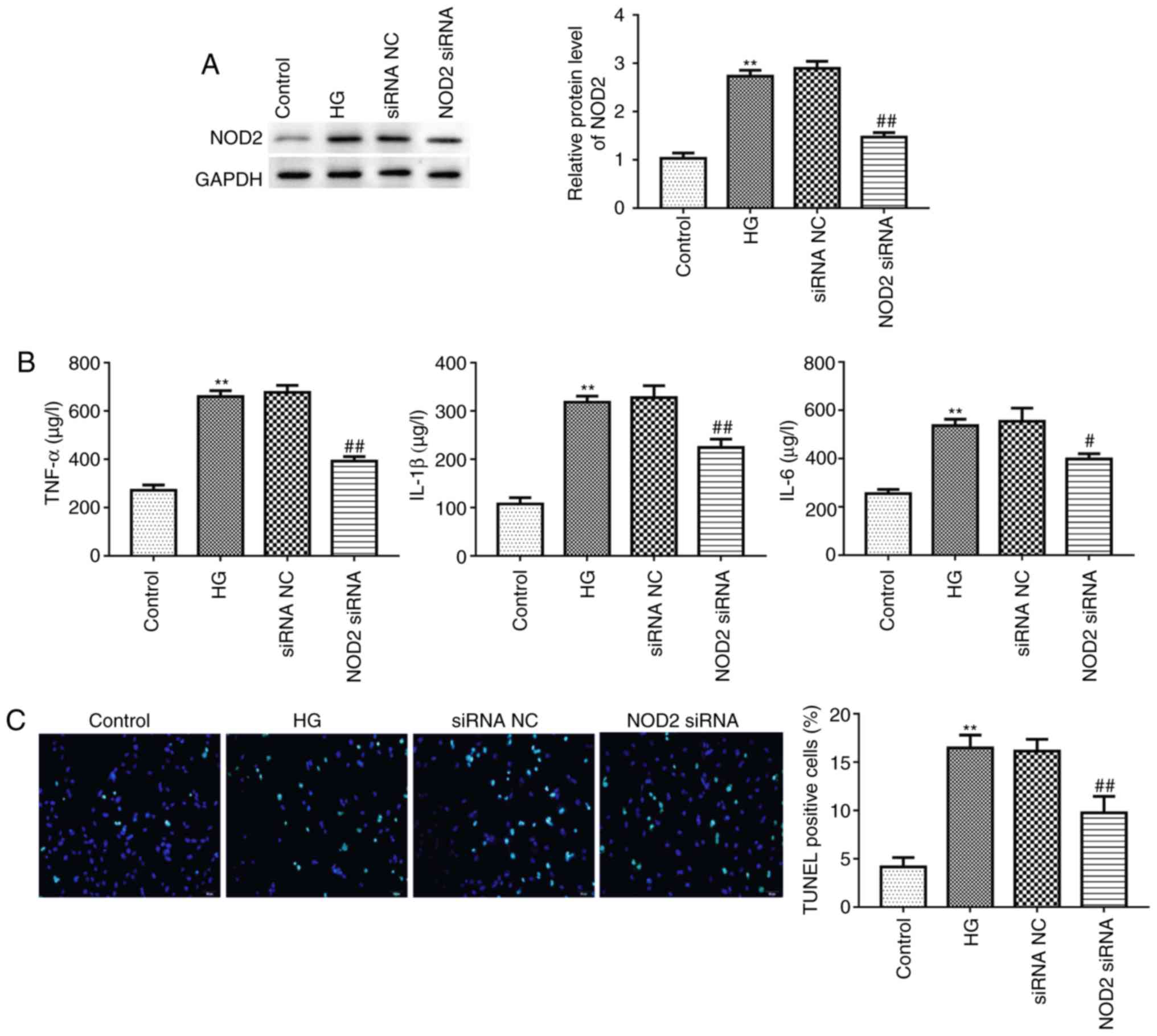
Suppressing NOD2 attenuates inflammatory
factor expression and apoptosis in cardiomyocytes
Myocardial cell apoptosis and myocardial fibrosis
are the primary pathological changes in diabetic cardiomyopathy
(5). Given this, the present
study explored the effect of NOD2 silencing on cardiomyocyte
apoptosis following 20 mM HG treatment for 48 h. The interference
efficiency of NOD2 using siRNA in primary cardiomyocytes was
evaluated by western blotting. The result revealed that treatment
with NOD2 siRNA had a marked inhibition on NOD2 protein expression
(Fig. 5A; P<0.01), reflecting
transfection efficiency of interfering RNA. It has been reported
that cardiomyocyte apoptosis is associated with inflammatory
factors (25). Next, ELISA was
used to assess the cytokine levels in different groups. As
presented in Fig. 5B, the
expression levels of TNF-α, IL-1β and IL-6 in HG-stimulated
cardiomyocytes were significantly increased compared with control
group (P<0.01). Furthermore, compared with the siRNA NC group,
NOD2 silencing significantly suppressed TNF-α, IL-1β and IL-6
expression levels (Fig. 5B;
P<0.05). In addition, the present study detected the apoptosis
of cardiomyocytes using a TUNEL assay. The results revealed that
there were more TUNEL-positive cells in HG treatment group compared
with control group (Fig. 5C;
P<0.01). The NOD2 siRNA group revealed decreased green
fluorescence compared with the siRNA NC group (Fig. 5C; P<0.01), indicating that
knockdown of NOD2 inhibited HG-induced cardiomyocyte apoptosis.
NOD2 silencing suppresses cell
proliferation and myocardial fibrosis
Next, the present study detected the effect of NOD2
silencing on fibrosis of CFs with 48 h HG incubation in
vitro. As presented in Fig.
6A, the proliferation of CFs was significantly increased in the
HG-treated group compared with control group, as detected by CCK-8
assay (P<0.01). However, compared to siRNA NC group, the cell
proliferation in NOD2 siRNA group was significantly decreased
(Fig. 6A; P<0.05). In
addition, the results of western blotting revealed that the
expression of NOD2, collagen I, collagen III, TGF-β, Caspase-3 and
Bax proteins were upregulated in HG group compared with control
group, whereas as compared with siRNA NC group, the expression
levels of these proteins in the NOD2 siRNA group were
downregulated. Conversely, Bcl-2 expression levels exhibited the
opposite trends. The afore-mentioned results suggested that NOD2
silencing suppressed collagen production in HG-induced CFs and
decreased myocardial cell apoptosis (Fig. 6B and C; P<0.01).
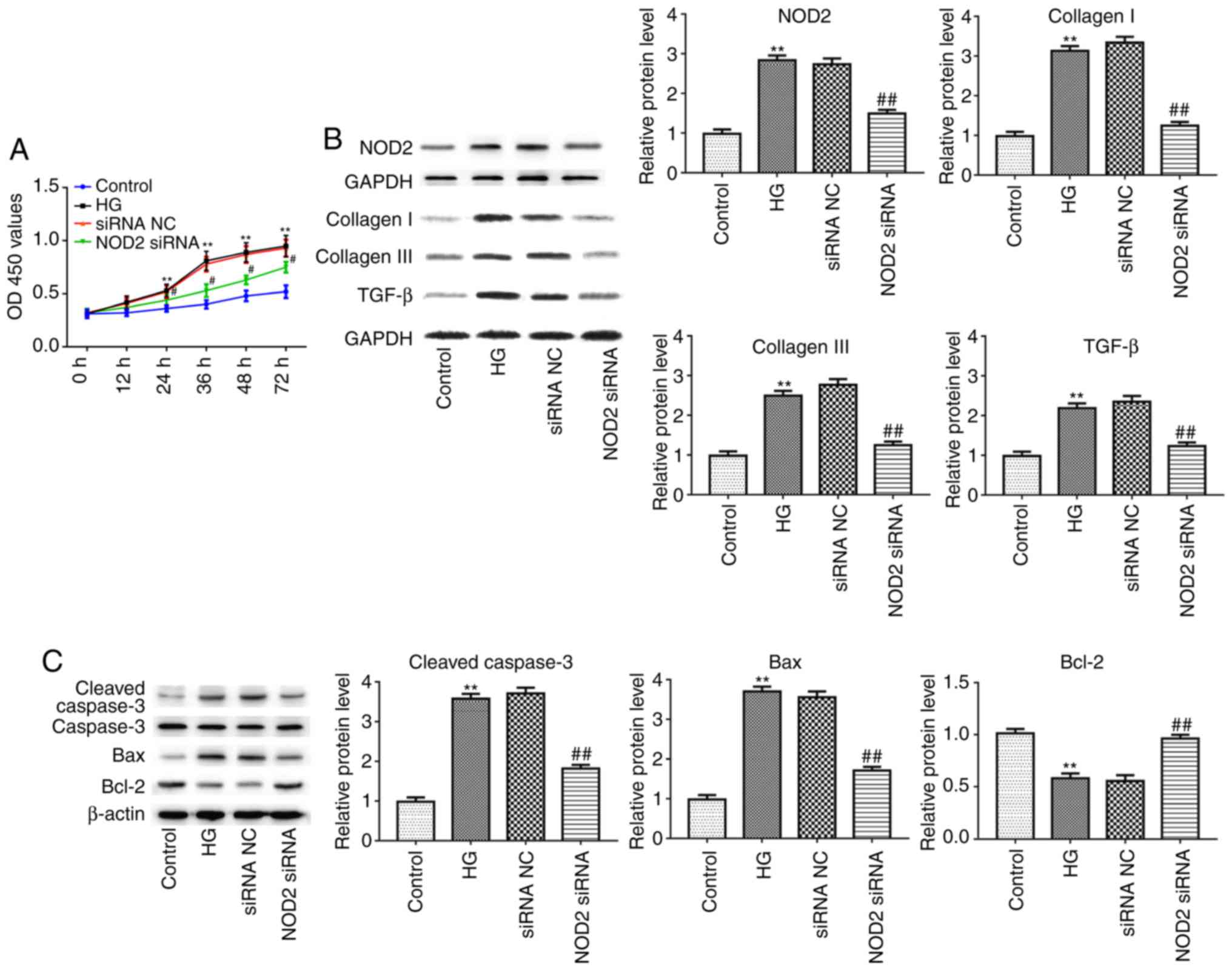 | Figure 6Effect of NOD2 silencing on cardiac
fibroblasts. (A) NOD2 silencing reduced proliferation of cardiac
fibroblasts. (B) The protein expression levels of NOD2, collagen I,
collagen III and TGF-β in cardiac fibroblasts in different groups.
(C) The protein expression levels of Caspase-3, Bax and Bcl-2 from
cardiomyocytes in different groups. **P<0.01 vs.
control group; #P<0.05, ##P<0.05 vs.
siRNA NC group. NOD2, nucleotide-binding oligomerization domain 2;
si, small interfering; NC, negative control; HG, high glucose;
TGF-β, transforming growth factor-β; Bcl-2, B cell lymphoma 2; Bax,
Bcl-2 associated X, apoptosis regulator; OD, optical density. |
Discussion
DCM is associated with diabetes-induced changes in
cardiac structure and function, and is a disorder characterized by
consistent diastolic dysfunction and increased ventricular mass
(22,26). DCM is closely associated with high
incidence and high mortality rates of heart failure in diabetic
patients (27). The possible
mechanisms for the development of DCM are myocardial metabolic
disorders and myocardial fibrosis, inflammation, myocardial
apoptosis and autophagy, resulting in left ventricular hypertrophy,
diastolic and systolic dysfunction, which ultimately form
congestive heart failure (18,28-30). DCM results from a variety of
mechanisms and signaling pathways in myocardial cells in the high
glucose state (31,32).
The present study revealed that in the DCM model,
LVEDP was significantly increased compared with control, but the
LVSP, +dP/dt max and -dP/dt max levels were markedly decreased.
However, NOD2 shRNA treatment reversed these results. In addition,
the results for pathological changes in diabetic mice revealed that
collagen deposition in DCM mice was significantly increased
compared with control. However, NOD2 silencing ameliorated collagen
deposition in the heart tissue of diabetic mice. Furthermore, TUNEL
staining indicated that the DCM group had an increased number of
TUNEL-positive cells compared with control group, and
NOD2 silencing weakened cell apoptosis. Expression
levels of collagen I, collagen III and TGF-β1 proteins were
significantly increased in the DCM group compared with control, but
the expression of these proteins in NOD2 shRNA treated mice were
lower compared with in vehicle-treated mice. These findings
suggested that NOD2 silencing ameliorated diabetes-induced
myocardial apoptosis and fibrosis.
The NOD-like receptor family is composed of more
than 20 members, each of which consist of three distinct domains.
Of these, the N-terminal effect domains include caspase recruitment
region, pyrin region or apoptotic repeat, and the most
representative is NOD2 (33,34). It has previously been demonstrated
that NOD2 is a class of endogenous substance subjected to high
blood sugar, high blood lipids, oxidants and inflammatory cell
infiltration, leading to persistent tissue injury in the
inflammatory state (35). It is
reported that NOD2 promotes the expression of nuclear factor-κB,
TNF-α, IL-1β and other inflammatory factors (20). Knockdown of NOD2 results in
reduced renal damage compared with wild type C57BL/6 mice (36). In HG-induced cardiomyocytes cells,
it was demonstrated that TNF-α, IL-1β and IL-6 levels in
HG-stimulated cardiomyocytes were significantly increased compared
with in the control group. However, NOD2 silencing suppressed
TNF-α, IL-1β and IL-6 expression and inhibited cell apoptosis.
These data indicated that knockdown of NOD2 inhibited HG-induced
inflammatory factor expression and cardiomyocyte apoptosis.
Activation of fibroblasts leads to myocardium
fibrosis (37). TGF-β, a class of
complex growth factor, has five subunits β1-β5, and TGF-β1 is a
major regulator in the mechanism of myocardial fibrosis and affects
the synthesis of collagen fibers through a series of cascade
reactions (38). In the
established DCM mouse model, NOD2, collagen I, collagen III and
TGF-β protein expression levels were significantly increased.
However, NOD2 silencing reduced NOD2, collagen I, collagen III and
TGF-β protein expression. In addition, the proliferation of CFs was
significantly increased in HG-treated cells compared with the
normal group, but compared with the siRNA NC group, the cell
proliferation in the NOD2 siRNA group was significantly decreased.
These results suggested that NOD2 silencing inhibited cell
proliferation and myocardial fibrosis.
In conclusion, the present study investigated the
role and mechanism of the NOD-like receptor NOD2 in the
pathogenesis of DCM. The results provide direct evidence that NOD2
plays a role in the process of hyperglycemia-induced cardiomyocyte
apoptosis and cardiac fibrosis. A novel effective target NOD2 for
the prevention and treatment of DCM has been screened, which has an
important theoretical significance and potential value for clinical
application.
Funding
The present study was supported by Shandong
Provincial Natural Science Foundation, China (grant no.
ZR2015HM052); key research and development plan of Shandong
Province (grant no. 2017GSF218012) and China Postdoctoral Science
Foundation (grant no. 2014M561936).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
Guarantor of integrity of the entire study: YH;
study concepts: LS, YH; study design: LS, YH, LL; definition of
intellectual content: LS, ML; analysis of pathology: ML, WY;
literature research: WW, WY; clinical studies: LS, WW, WY;
experi-mental studies: LS, YH; data acquisition: LL, WL;
statistical analysis: LS; manuscript editing: LS, LL, ML;
manuscript reviewing: LS, LL.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Animal Care and Use Committee of Shandong University
(Jinan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Hillis GS, Woodward M, Rodgers A, Chow CK,
Li Q, Zoungas S, Patel A, Webster R, Batty GD, Ninomiya T, et al:
Resting heart rate and the risk of death and cardiovascular
complications in patients with type 2 diabetes mellitus.
Diabetologia. 55:1283–1290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Adameova A and Dhalla NS: Role of
microangiopathy in diabetic cardiomyopathy. Heart Fail Rev.
19:25–33. 2014. View Article : Google Scholar
|
|
3
|
Bielecka-Dabrowa A, Wierzbicka M, Dabrowa
M and Goch A: New methods in laboratory diagnostics of dilated
cardiomy-opathy. Cardiol J. 15:388–395. 2008.
|
|
4
|
Carneiro LA, Magalhaes JG, Tattoli I,
Philpott DJ and Travassos LH: Nod-like proteins in inflammation and
disease. J Pathol. 214:136–148. 2008. View Article : Google Scholar
|
|
5
|
Zhang J, Li B, Zheng Z, Kang T, Zeng M,
Liu Y and Xia B: Protective effects of Notch1 signaling activation
against high glucose-induced myocardial cell injury: Analysis of
its mechanisms of action. Int J Mol Med. 36:897–903. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeMarco VG, Aroor AR and Sowers JR: The
pathophysiology of hypertension in patients with obesity. Nat Rev
Endocrinol. 10:364–376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lorenzo O, Picatoste B, Arescarrasco S,
Ramírez E, Egido J and Tuñón J: Potential role of nuclear factor κB
in diabetic cardiomy-opathy. Mediators Inflamm.
2011.652097:2011.
|
|
8
|
Zhang B, Qiang S, Chen Y, Pan R, Kuang S,
Liu G, Sun G and Sun X: Myricitrin alleviates oxidative
stress-induced inflammation and apoptosis and protects mice against
diabetic cardiomyopathy. Sci Rep. 7:442392017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohammadshahi M, Haidari F and Soufi FG:
Chronic resveratrol administration improves diabetic cardiomyopathy
in part by reducing oxidative stress. Cardiol J. 21:39–46. 2014.
View Article : Google Scholar
|
|
10
|
Qin WD, Liu GL, Wang J, Wang H, Zhang JN,
Zhang F, Ma Y, Ji XY, Li C and Zhang MX: Poly(ADP-ribose)
polymerase 1 inhibition protects cardiomyocytes from inflammation
and apoptosis in diabetic cardiomyopathy. Oncotarget.
7:35618–35631. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song JQ, Teng X, Cai Y, Tang CS and Qi YF:
Activation of Akt/GSK-3beta signaling pathway is involved in
inter-medin(153) protection against myocardial apoptosis induced by
ischemia/reperfusion. Apoptosis. 14:1299–1307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ares-Carrasco S, Picatoste B,
Benito-Martín A, Zubiri I, Sanz AB, Sánchez-Niño MD, Ortiz A, Egido
J, Tuñón J and Lorenzo O: Myocardial fibrosis and apoptosis, but
not inflammation, are present in long-term experimental diabetes.
Am J Physiol Heart Circ Physiol. 297:H2109–H2119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vadla GP and Vellaichamy E: Anti-fibrotic
cardio protective efficacy of aminoguanidine against streptozotocin
induced cardiac fibrosis and high glucose induced collagen up
regulation in cardiac fibroblasts. Chem Biol Interact. 197:119–128.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang HX, Zhang QF, Zeng XJ, Wang W, Tang
CS and Zhang LK: Effects of angiotensin III on protein, DNA, and
collagen synthesis of neonatal cardiomyocytes and cardiac
fibroblasts in vitro. J Cardiovasc Pharmacol Ther. 15:393–402.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang WK, Wang B, Lu QH, Zhang W, Qin WD,
Liu XJ, Liu XQ, An FS, Zhang Y and Zhang MX: Inhibition of
high-mobility group box 1 improves myocardial fibrosis and
dysfunction in diabetic cardiomyopathy. Int J Cardiol. 172:202–212.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Westermann D, Rutschow S, Jäger S,
Linderer A, Anker S, Riad A, Unger T, Schultheiss HP, Pauschinger M
and Tschöpe C: Contributions of inflammation and cardiac matrix
metalloproteinase activity to cardiac failure in diabetic
cardiomyopathy: The role of angiotensin type 1 receptor antagonism.
Diabetes. 56:641–646. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Correa RG, Milutinovic S and Reed JC:
Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and
inflammatory diseases. Biosci Rep. 32:597–608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li CJ, Lv L, Li H and Yu DM: Cardiac
fibrosis and dysfunction in experimental diabetic cardiomyopathy
are ameliorated by alpha-lipoic acid. Cardiovasc Diabetol.
11:732012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Geddes K, Magalhães JG and Girardin SE:
Unleashing the therapeutic potential of NOD-like receptors. Nat Rev
Drug Discov. 8:465–479. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu Y, Yang H, Liu LX, Yan W, Guo HJ, Li
WJ, Tian C, Li HH and Wang HX: NOD2 contributes to myocardial
ischemia/reperfusion injury by regulating cardiomyocyte apoptosis
and inflammation. Life Sci. 149:10–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perez-Chanona E, Mühlbauer M and Jobin C:
The microbiota protects against ischemia/reperfusion-induced
intestinal injury through nucleotide-binding oligomerization
domain-containing protein 2 (NOD2) signaling. Am J Pathol.
184:2965–2975. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu HQ, Zhang XY, Edfeldt K, Nijhuis MO,
Idborg H, Bäck M, Roy J, Hedin U, Jakobsson PJ, Laman JD, et al:
NOD2-mediated innate immune signaling regulates the eicosanoids in
atherosclerosis. Arterioscler Thromb Vasc Biol. 33:2193–2201. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fan Du, Han BP, Zhen H, Shang J, Wang J,
Li X, Shi X, Tang W, Bao WC, et al: NOD2 promotes renal injury by
exacerbating inflammation and podocyte insulin resistance in
diabetic nephropathy. Kidney Int. 84:265–276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408.
2001. View Article : Google Scholar
|
|
25
|
Vakhrusheva O, Smolka C, Gajawada P,
Kostin S, Boettger T, Kubin T, Braun T and Bober E: Sirt7 increases
stress resistance of cardiomyocytes and prevents apoptosis and
inflammatory cardiomyopathy in mice. Circ Res. 102:703–710. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Falcão-Pires I and Leite-Moreira AF:
Diabetic cardiomyopathy: Understanding the molecular and cellular
basis to progress in diagnosis and treatment. Heart Fail Rev.
17:325–344. 2012. View Article : Google Scholar
|
|
27
|
Li Li, Chu FX, Wang Y, Zhang X, Hu H,
Zhang Y, Wang Y, Wei Z, Jian XW, et al: NOD2 deficiency protects
against cardiac remodeling after myocardial infarction in mice.
Cell Physiol Biochem. 32:1857–1866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G,
Wang L, Peng Y, Dong X, Huang H, Si L, et al: Heme oxygenase-1
prevents cardiac dysfunction in streptozotocin-diabetic mice by
reducing inflammation, oxidative stress, apoptosis and enhancing
autophagy. PLoS One. 8:e759272013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mano Y, Anzai T, Kaneko H, Nagatomo Y,
Nagai T, Anzai A, Maekawa Y, Takahashi T, Meguro T, Yoshikawa T and
Fukuda K: Overexpression of human C-reactive protein exacerbates
left ventricular remodeling in diabetic cardiomyopathy. Circ J.
75:1717–1727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu ZW, Wang JK, Qiu C, Guan GC, Liu XH,
Li SJ and Deng ZR: Matrine pretreatment improves cardiac function
in rats with diabetic cardiomyopathy via suppressing ROS/TLR-4
signaling pathway. Acta Pharmacol Sin. 36:323–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pan Y, Wang Y, Zhao Y, Peng K, Li W, Wang
Y, Zhang J, Zhou S, Liu Q, Li X, et al: Inhibition of JNK
phosphorylation by a novel curcumin analog prevents high
glucose-induced inflammation and apoptosis in cardiomyocytes and
the development of diabetic cardiomyopathy. Diabetes. 63:3497–3511.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rubler S, Dlugash J, Yuceoglu YZ, Kumral
T, Branwood AW and Grishman A: New type of cardiomyopathy
associated with diabetic glomerulosclerosis. Am J Cardiol.
30:595–602. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Santos VN, Leite-Mór MM, Kondo M, Martins
JR, Nader H, Lanzoni VP and Parise ER: Serum laminin, type IV
collagen and hyaluronan as fibrosis markers in non-alcoholic fatty
liver disease. Braz J Med Biol Res. 38:747–753. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miki T, Yuda S, Kouzu H and Miura T:
Diabetic cardiomyopathy: Pathophysiology and clinical features.
Heart Fail Rev. 18:149–166. 2013. View Article : Google Scholar :
|
|
35
|
Teshima Y, Takahashi N, Nishio S, Saito S,
Kondo H, Fukui A, Aoki K, Yufu K, Nakagawa M and Saikawa T:
Production of reactive oxygen species in the diabetic heart. Roles
of mitochondria and NADPH oxidase. Circ J. 78:300–306. 2014.
View Article : Google Scholar
|
|
36
|
Werner S, Krieg T and Smola H:
Keratinocyte-fibroblast interactions in wound healing. J Invest
Dermatol. 127:998–1008. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang X, Ma X, Zhao M, Zhang B, Chi J, Liu
W, Chen W, Fu Y, Liu Y and Yin X: H2 and H3 relaxin inhibit high
glucose-induced apoptosis in neonatal rat ventricular myocytes.
Biochimie. 108:59–67. 2015. View Article : Google Scholar
|
|
38
|
Zhang YC, Mou YL and Xie YY: Research
progress in relations between renin angiotensin system and diabetic
cardiomyopathy. Sheng Li Ke Xue Jin Zhan. 42:269–275. 2011.In
Chinese. PubMed/NCBI
|