Introduction
Pulmonary fibrosis is a common occurrence in the
final stages of various lung diseases, including acute lung injury,
drug reactions, sarcoidosis and autoimmune disease. Due to the lack
of timely and effective intervention, the majority of patients who
succumb to the disease exhibit respiratory failure 3-5 years
following diagnosis (1).
Therefore, identifying appropriate anti-fibrotic therapy is of
vital importance (2-4).
Although the underlying mechanisms of pulmonary
fibrosis are complex, previous studies have demonstrated that lung
fibrosis develops from the maladaptive regulation of repair
processes following lung injury and inflammation, where various
profibrotic factors precipitate the formation of α smooth muscle
actin (α-SMA)-expressing myofibroblasts, which in turn synthetize
and secrete immoderate extracellular matrix (ECM) components,
replacing normal lung tissue and driving lung fibrosis (5-7).
It has been established that myofibroblasts originate from resident
differentiated lung fibroblasts (8). The most important hallmark of
fibroblast activation and differentiation is the expression of
α-SMA (9). Transforming growth
factor β1 (TGF-β1), a multifunctional molecule, is considered to be
the most potent inducing mediator of fibroblast activation
(10). TGF-β1 promotes fibroblast
migration, proliferation and differentiation and promotes the
production of ECM (11). The
disruption of TGF-β1 mediated signaling inhibits fibroblast
activation, thus preventing or improving pulmonary fibrotic
response in vivo and in vitro (12).
Previous studies have revealed a strong association
between inflammation, fibroblast activation and lung fibrosis
(5-7). Infiltrating inflammatory cells
within injured lung tissue release a high number of
profibrogenicmediators, which activate fibroblasts and induce lung
fibrosis (13). Anti-inflammatory
therapeutics are effective in extenuating pulmonary fibrosis
(14). Lipoxins (LXs) are
endogenous eicosanoids, which are generated either via 5- and
15-lipoxygenases, or via 5- and 12-lipoxygenases. They serve as the
‘stop signal’ for inflammation and exert potent anti-inflammatory
and pro-resolution properties (15). LXA4, as a principle LX,
has been demonstrated to exert protective effects in various
inflammation-associated diseases, including paracetamol-induced
acute hepatic liver injury and lipopolysaccharide (LPS)-induced
acute lung injury (16,17). In addition, LXA4 serves
primary roles in the regulation of tissue repair following
inflammation, particularly in renal, skin and pulmonary fibrosis
(18-20). In the dermal fibrosis model,
LXA4 is important for the inhibition of fibroblast
proliferation and activation (19). LXA4 acts through a
specific G protein-coupled-receptor termed ALX to exert its
multicellular effects. BML-111 is a lipoxinA4 receptor
(ALX) agonist and exerts its biological activity by binding to ALX
(21). Previous studies have
identified two different ALXs (ALX1/FPR-rs1 and ALX2/FPR2) in mice
(22-25). BML-111 was initially considered to
exert inhibitory effects similar to that of LXA4 by
inhibiting LTB4-induced neutrophil migration (26). Previous studies have demonstrated
that BML-111 exhibits anti-inflammatory and pro-resolving effects
in haemorrhagic shock-induced lung injury and ventilator-induced
lung injury (27-29). Furthermore, a previous study
revealed that BML-111 exerts protective effects on carbon
tetrachloride (CCl4)-induced hepatic fibrosis in rats
(30). However, whether BML-111
affects fibroblast activation and lung fibrosis remains
unknown.
In the present study, it was demonstrated that
BML-111 reduces the expression of α-SMA, fibronectin and total
collagen induced by TGF-β1 in NIH3T3 cells, and that it interferes
with TGF-β1 associated signaling pathways. The results of the
current study indicated that BML-111 inhibits the activation of
fibroblasts and exerts direct anti-fibrotic affects. In addition,
BML-111 treatment markedly improved murine survival rates in the
BLM intratracheal mouse model, while BOC-2
(N-tertbutyloxy-carbonyl-phenyalanine-le-ucyl-
phenyalanine-leucyl-phenyalanine) partially weakened the effects of
BML-111. Furthermore, it was concluded that BML-111 alleviates
BLM-induced pulmonary fibrosis by binding to ALX, and that these
mechanisms may be involved in the anti-inflammatory response and in
the inhibition of fibroblast activation.
Materials and methods
Cell culture
NIH3T3 cells were obtained from China Center for
Type Culture Collection (Wuhan, China) and were cultured in
Dulbecco’s Modified Eagle’s medium (DMEM; HyClone; GE Healthcare
Life Sciences, Logan, UT, USA) to 75% confluence. The cells were
then serum-starved for 12 h prior to each experiment. To select an
optimal concentration of BML-111 (Cayman Chemical, Ann Arbor, MI,
USA), cells were treated with varying concentrations (1, 10, 100,
200 and 500 nM) of BML-111 or vehicle (0.035% methanol) for 30 min
at 37°C prior to the addition of 5 ng/ml TGF-β1 (PeproTech Inc.,
Rocky Hill, NJ, USA) for 24 h at 37°C. Although BML-111 at
concentrations of 1 and 10 nM did not appear to affect a-SMA
protein levels, the other concentrations of BML-111 substantially
suppressed TGF-β1-induced a-SMA expression, with 200 and 500 nM
concentrations producing the most notable effects. Notably, there
was no substantial difference between these two concentrations.
Therefore 200 nM BML-111 was selected for subsequent experiments.
To assess whether the action of BML-111 is associated with ALX, 10
µM BOC-2 (Phoenix Pharmaceuticals, Inc., Burlingame, CA,
USA) was supplemented to cells prior to BML-111 treatment for 30
min.
RNA isolation and reverse-transcriptase
(RT) polymerase chain reaction (PCR)
Total RNA was isolated from cultured cells using the
TRIzol reagent (Invitrogen; Thermo Fisher Scientific Inc., Waltham,
MA, USA). RNA reverse transcription was performed using an ReverTra
Ace kit (Toyobo Life Science, Osaka, Japan). Briefly, the reaction
was incubated in steps of 65°C for 5 min, 37°C for 15 min, 95°C for
5 min and held at −20°C. The amplified products of PCR were
resolved using 2% agarose gel electrophoresis. The primers utilized
were as follows: 5′-GGC AAC TCT GTT GAG GAA AG-3′ and
5′-GGCTCTCGGTAGACGAGA-3′ for ALX homeobox 1 (ALX1)/formyl peptide
receptor related sequence 1 (FPR-rs1); and 5′-GTC AA-G ATC AAC AGA
AGA AAC C-3′ and 5′-GGG CTC TCT CAA GAC TAT AAG G-3′ for ALX
homeobox 2 (ALX2)/formyl peptide receptor 2 (FP-R2); and 5′-CTG AGA
GGG AAA TCG TGC GT-3′ and 5′-CCA CAG GAT TCC ATA CCC AAG A-3′ for
actin (25).
Immunofluorescence
For the detection of the expression of FPR2, NIH3T3
cells were cultured in DMEM at 37°C for 24 h on sterile glass cover
slips in 6-well plates and treated as aforementioned. Cells were
then fixed with 4% paraformaldehyde. Following permeabilization,
washing and blocking, the cells were incubated with rabbit
anti-FPR2 anti-bodies (1:100; cat. no. AFR-002; Alomone Labs,
Jerusalem, Israel) at 4°C overnight and then washed and incubated
with a fluorescent-labeled secondary antibody (fluorescein
isothiocyanate-labelled goat anti-rabbit immunoglobulin G; 1:50;
cat. no. AS1110; Aspen Biological, Wuhan, China) for 45 min at
37°C. After washing again, over slips were mounted with anti-fade
mounting medium (Beyotime Institute of Biotechnology, Haimen,
China) on slides, and observed using a confocal microscope (Olympus
FluoviewFV500).
Western blotting
Total protein was extracted using a Total Protein
Extraction kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China).
The protein concentration was determined using a BCA Protein Assay
kit (cat. no. KGP902; KeyGen Biotech Co., Ltd). A total of 30
µg protein from lung tissue or cells were separated on 8 or
10% SDS-PAGE, respectively and transferred onto a polyvinylidene
difluoride membrane (Merck KGaA, Darmstadt, Germany). Membranes
were blocked using 5% skimmed milk for 1 h at 4°C. After incubating
with primary antibodies overnight at 4°C, the membrane was
incubated with secondary antibodies [horseradish peroxidase (HRP)
conjugated-Goat anti-Rabbit immunoglobulin G (IgG); 1:5,000; cat.
no. ANT020 or HRP conjugated-Goat anti-Mouse IgG; 1:5,000; cat. no.
ANT019] at room temperature for 1 h. Immunoreactive bands were
detected using the Supersignal West Pico chemiluminescent substrate
system (Pierce; Thermo Fisher Scientific Inc.) and analyzed using
Quantity One Version 4.6.3 Image software (Bio-Rad Laboratories
Inc., Hercules, CA, USA). The primary antibodies used were as
follows: anti-fibronectin (1:1,000; cat. no. 1574-1), anti-α-SMA
(1:1,000; cat. no. 1184-1), anti-pAkt (1:500; cat. no. 3188-1),
anti-Akt (1:1,000; cat. no. 1085-1; each Epitomics; Abcam,
Cambridge, UK); anti-mothers against decapentaplegic homolog
(Smad)2/3 (1:500; cat. no. 5678), anti-phosphorylated (p) Smad2
(1:500; cat. no. 3108), anti-pSmad3 (1:500; cat. no. 9520), anti-p
extracellular signal-regulated kinase (ERK; 1:500; cat. no. 4370),
anti-ERK (1:1,000; cat. no. 4695; each Cell Signaling Technology
Inc., Danvers, MA, USA) and anti-GAPDH (1:4,000; cat. no.
LF-MA20175; Young In Frontier Co., Ltd., Seoul, Korea).
Collagen content determination
A total of 100 µl lung homogenates were
extracted using 0.5 M acetic acid containing 0.6% pepsin and 200
µl NIH3T3 culture medium were mixed with 1 ml of Sircol dye
reagent for 30 min at room temperature. Total collagen content was
determined using the Sircol collagen assay kit (Biocolor Ltd.,
County Antrim, UK) according to manufacturer’s protocol.
Cell viability assay
A total of 100 µl cells were seeded in
96-well plates (104 cells/ml), following serum starvation for 12 h.
Cells were then pre-treated with or without BML-111 and BOC-2 for
30 min at 37°C and cultured with or without TGF-β1 (5 ng/ml) for 24
h at 37°C. Subsequently, MTT was added to the culture medium and
cells were incubated for a further 4 h at 37°C. The medium was then
removed and 100 µl of dimethyl sulfoxide was added to each
well and mixed for a further 10 min. The absorbance at 570 nm was
determined using Sunrise™ (Tecan, Groedig, Austria).
Mice and grouping
C57BL/6 male mice (age, 6-8 weeks; weight, 20-25 g;
n=76) were purchased from Beijing HFK Bioscience Co., Ltd.
(Beijing, China) and housed in a specific pathogen-free animal
facility. The mice were maintained under pathogen-free conditions,
constant temperature, 22±2°C; humidity, 40-50%; 12 h light/dark
cycle and were given food and water ad libitum. The
BLM-induced pulmonary fibrosis mouse model was established and
validated according to a previously described method (31). Mice were randomly divided into 4
groups: A saline-injected group (sham group; n=10), a BLM-injected
group treated with saline (untreated group; n=22), a BLM
intratracheal injection group treated with BML-111 (BML-111 group;
n=22), and a BOC-2 group (n=22). BOC-2 (Phoenix Pharmaceuticals,
Inc., Burlingame, CA, USA) was injected to the BLM-injected mice
treated with BML-111 before giving BML-111 30 min. Mortality was
assessed daily until day 21 following BLM instillation. All
surviving mice were then euthanized on the 21st day using an
intraperitoneal injection of 200 mg/kg ketamine with 10 mg/kg
xyalzine, followed by a thoracotomy (32-34). Lungs were removed during this
procedure and at −80°C for further analysis. Mice were sacrificed
if the following humane endpoints were observed within the 21-day
period: Body weight loss of >20%, impaired mobility, inability
to retrieve food or water, labored breathing (increased respiratory
rate and effort) and no response to external stimuli.
The use of mice within the present study was
reviewed and approved by the Institutional Animal Care and Use
Committee of Tongji Medical College, Huazhong University of Science
and Technology (Huazhong, China). All animal studies (including the
mice euthanasia procedure) were completed in compliance with the
regulations and guidelines of Huazhong University institutional
animal care and conducted according to the AAALAC and the IACUC
guidelines.
Histological analysis
Lung samples were fixed in 10% formalin for 24 h at
room temperature, embedded in paraffin and sectioned onto slides at
a thickness of 4-5 µm. Sections were stained with Masson’s
trichrome for detection of collagen deposition. For Masson’s
trichrome staining, tissue sections were stained with Weigert’s
hematoxylin stain for 10 min at room temperature and rinsed in
lukewarm water for 5 min, immersed in acid ponceau/solferino for 15
min at room temperature, phosphomolybdic acid for 10 min at room
temperature, and finally incubated with 2% aniline blue for 15 min
at room temperature. The samples were also stained with hematoxylin
and eosin (H&E) for 7 min and 15 sec respectively, at room
temperature, for lung injury evaluation. Fibrosis scoring was
evaluated according to Masson’s trichrome and calculated as
previously described by Ashcroft et al (35). The grade of lung fibrosis was
scored as follows: 0, no pulmonary fibrosis; 1, mild pulmonary
fibrosis, the affected area was <20%; 2, moderately pulmonary
fibrosis, involvement of area of 20-50%; 3, severe pulmonary
fibrosis, the affected area was >50%.
Hydroxyproline assay
Lung tissues were minced and hydrolyzed in 0.5 ml of
6 mol/l HCl for 6 h at 100°C. After adjusting the pH to 6.0-6.8,
activated carbon was added to hydrolyzation products (25 mg
activated carbon with 4 ml diluted hydrolysate) diluted with
distilled water. Samples were centrifuged at 1,445.5 × g for 10 min
at room temperature and the supernatant was used to measure the
hydroxyproline content with a Hydroxyproline assay kit (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China; cat. no.
A030-3) according to the manufacturer’s protocol.
ELISA for TGF-β1, IL-1β and TNF-α in
bronchoalveolar lavage fluid (BALF)
BALF was acquired according to a previously
described method (36) and
measured using ELISA kits (Wuhan Boster Biological Technology.,
Ltd., Wuhan, China) according the manufacturer’s protocol.
Statistical analysis
Survival rates were evaluated using the log-rank
(Mantel-Cox) test. Results were expressed as mean ± standard
deviation and analyzed using one-way analysis of variance analysis
followed by a Bonferoni post hoc test. Prism 5.0 (GraphPad
Software, Inc., La Jolla, CA, USA) was used to perform statistical
analysis. P<0.05 was considered to indicate a statistically
significant result.
Results
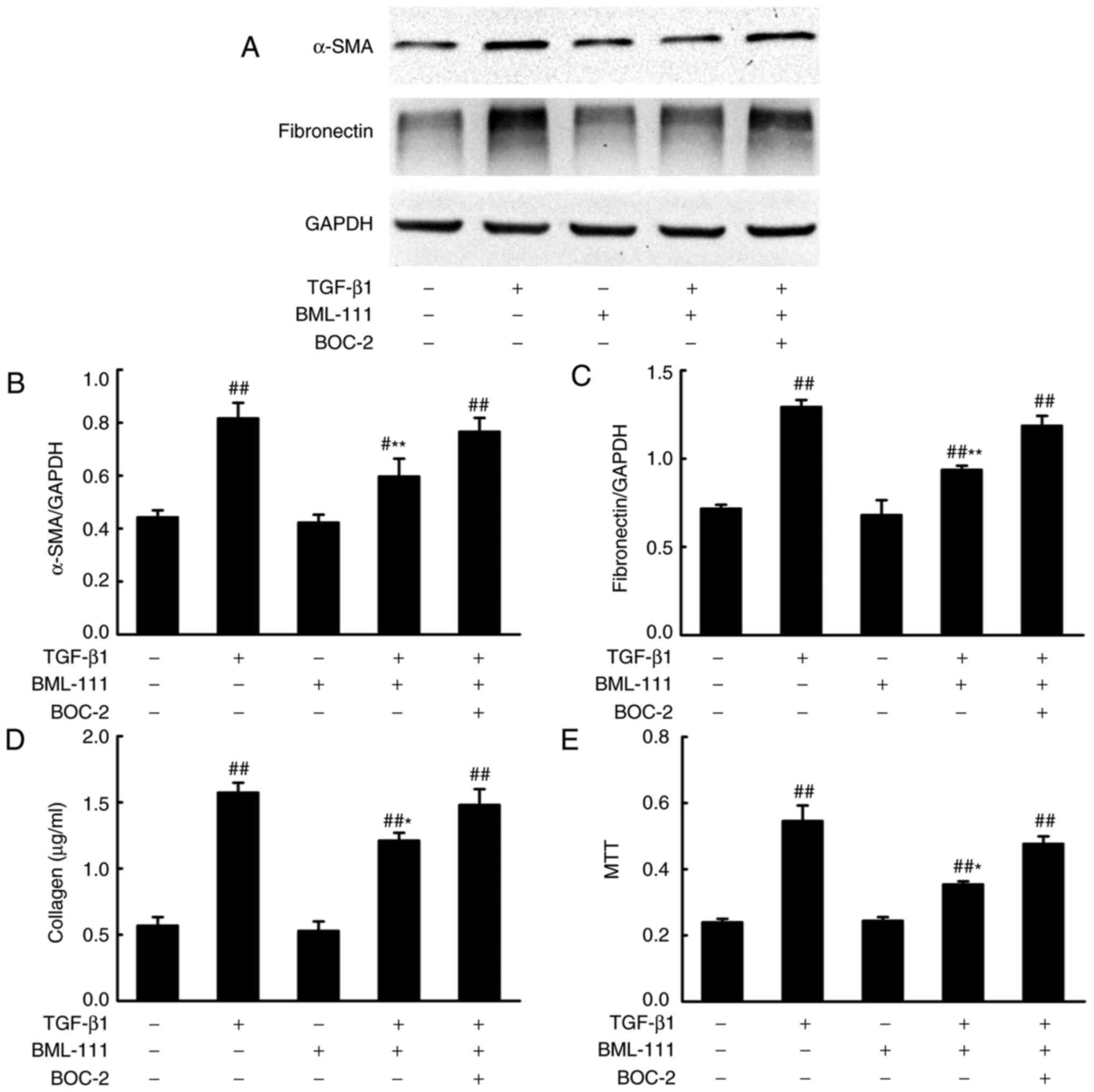
BML-111 inhibits TGF-β1 induced NIH3T3
cell activation in vitro
BML-111 functions by binding to its corresponding
receptor. As detected by PCR, NIH3T3 cells expressed rs1 and FPR2
(Fig. 1A and B). Activated lung
fibroblasts, which express α-SMA and synthetize elevated ECM,
serves a primary role in fibrogenesis. To determine whether BML-111
inhibits fibro-blast activation, the effect of BML-111 on
TGF-β1-induced NIH3T3 viability, α-SMA expression and the
expression of various ECM components including the total collagen
protein and fibronectin, was assessed. The results demonstrated
that the stimulation of NIH3T3 cells with TGF-β1 significantly
increases cell viability and the production of α-SMA, total
collagen protein and fibronectin. NIH3T3 cell pretreatment with
BML-111 markedly inhibits TGF-β1-induced NIH3T3 proliferation and
the expression of α-SMA, total collagen protein and fibronectin. To
further assess the role of the ALX in BML-111 activity, BOC-2 was
added to the cells prior to treatment with BML-111. The results
indicated that BOC-2 pretreatment inhibits the effect of BML-111
(Fig. 2).
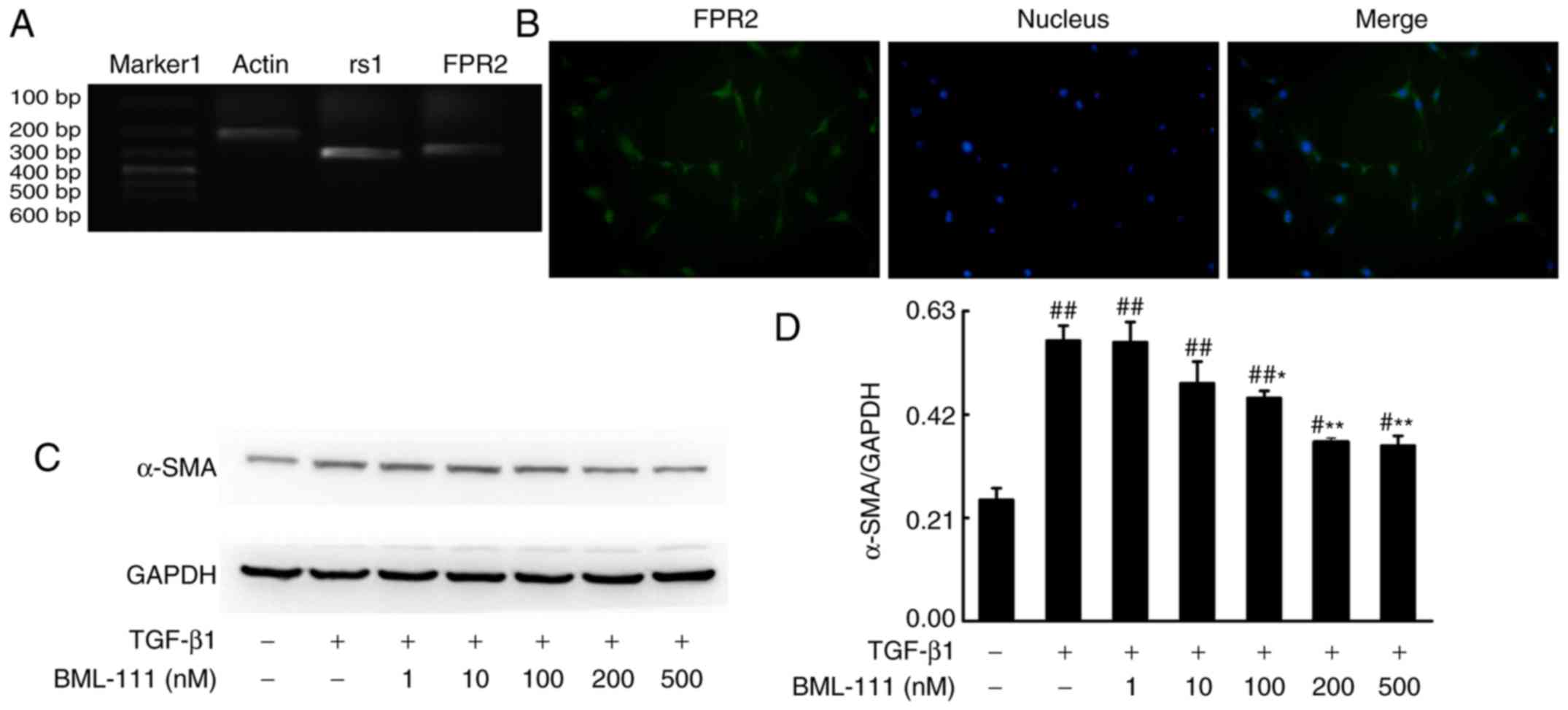 | Figure 1BML-111 decreased TGF-β1-induced
NIH3T3 cell α-SMA expression in a dose-dependent manner. (A) NIH3T3
cells express rs1 and (B) FPR2. Cells were pretreated with a
vehicle (0.035% ethanol) or BML-111 (1, 10, 100, 200 and 500 nM)
for 30 min and then treated with TGF-β1 (5 ng/ml) for 24 h. (C) The
expression of α-SMA was assessed using western blotting and (D)
quantified. Similar results were obtained from at least 3 sections.
Data are expressed as the mean ± standard deviation.
#P<0.05 and ##P<0.01 vs. the vehicle
group. *P<0.05 and **P<0.01 vs. the
TGF-β1 group in the absence of BML-111. Magnification, ×200.
TGF-β1, Transforming growth factor-β1; α-SMA, smooth muscle α
actin; rs1, related sequence 1; FPR2, formyl peptide receptor;
marker 1, Trans DNA ladder (Tiangen Biotech, Co., Ltd., Beijing,
China). |
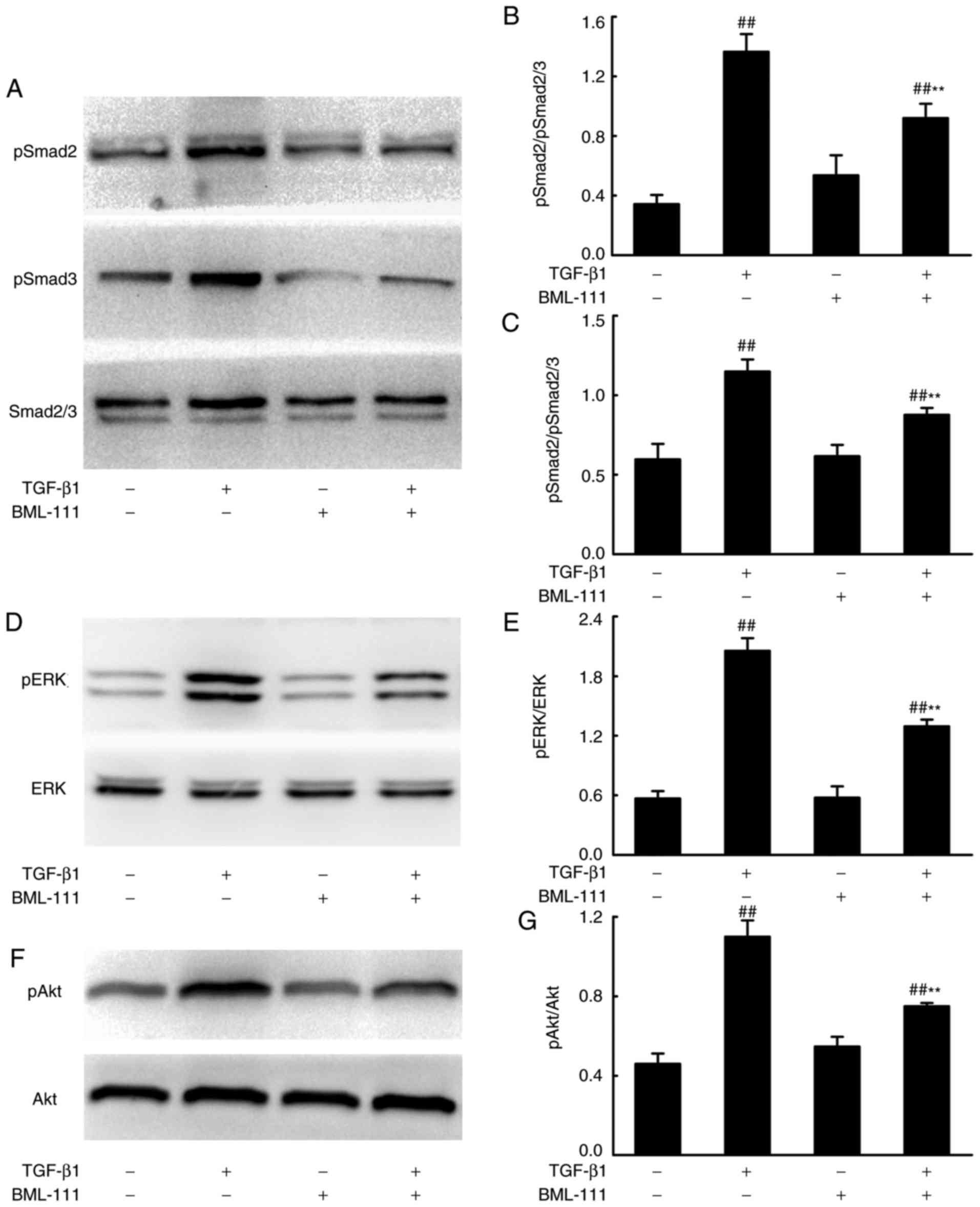
BML-111 suppresses Smad-dependent and
Smad-independent signaling pathways in TGF-β1-induced NIH3T3
cells
Smad-dependent and -independent signaling pathways
mediate the pro-fibrotic effects of TGF-β1. To assess whether
BML-111 mediated fibrosis is regulated by these pathways, the
effect of BML-111 on TGF-β1-induced Smad2, Smad3, ERK and Akt
phosphorylation in NIH3T3 cells were analyzed using western
blotting. The results demonstrated that BML-111 significantly
reduces pSmad2, pSmad3, pERK and pAkt levels in cells stimulated by
TGF-β1 (Fig. 3).
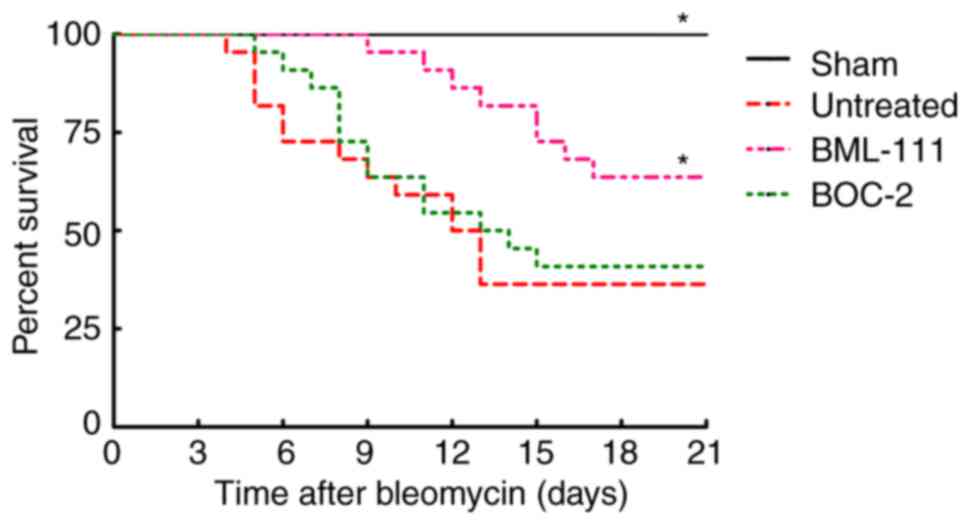
BML-111 improves mice survival rate
following BLM instillation
The survival rate of mice was monitored for 21 days
following BLM injection. As presented in Fig. 4, no mice succumbed in the Sham
group. BLM instillation without treatment led to a greater
increased murine survival rate than the sham group, which primarily
occurred between days 7 and 14. However, the majority of mice in
the BML-111 group succumbed between day 11 and 18, pretreatment
with BML-111 delayed and decreased mortality in mice with pulmonary
fibrosis, and BOC-2 counteracted this effect of BML-111.
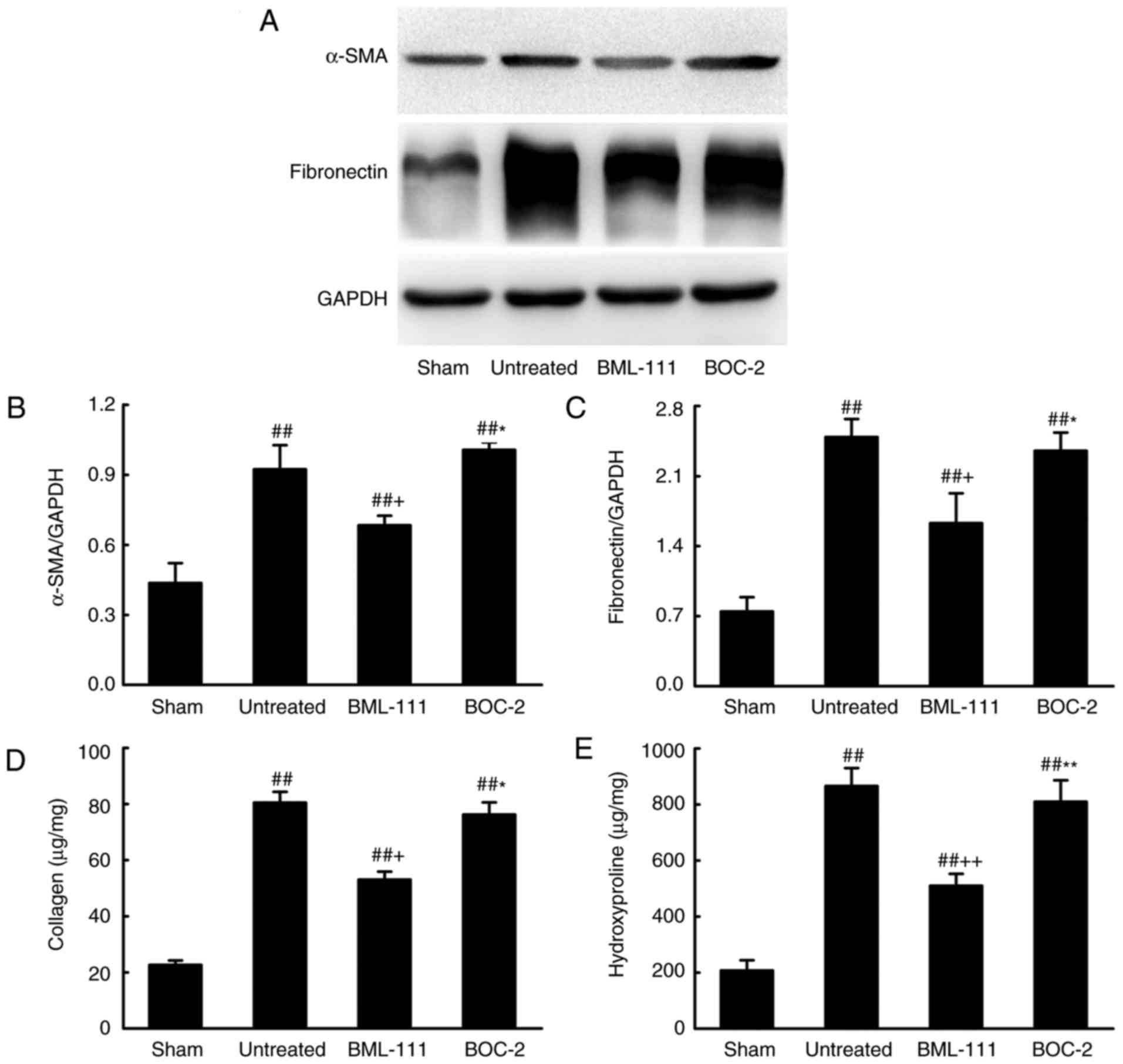
BML-111 decreases BLM-induced pulmonary
fibrosis
BML-111 demonstrated a high efficiency in protecting
lungs from fibrosis. As observed on H&E and Massion-stained
slides (Fig. 5A-C), no
pathological changes were observed in the lung tissue of the Sham
group. Untreated, BOC-2 and BML-111 groups exhibited inflammatory
cell infiltration, alveolar space collapse, alveolar wall
thickening and extracellular collagen deposition. However the
observed changes were less severe in the BML-111 group. The lung
tissues of BLM-treated mice exhibited significantly upregulated
levels of ECM and α-SMA, which is typical of fibrosis. However,
these levels were significantly suppressed following BML-111
treatment (Fig. 6). In addition,
the BLM injection administered to the untreated group significantly
upregulated the production of TGF-β1, TNF-α and IL-1β in BALF
compared with the Sham group. The BML-111 group exhibited a
reduction in these cytokines compared with the BLM and BOC-2 group,
and BOC-2 counteracted the effect of BML-111 (Fig. 5D-F).
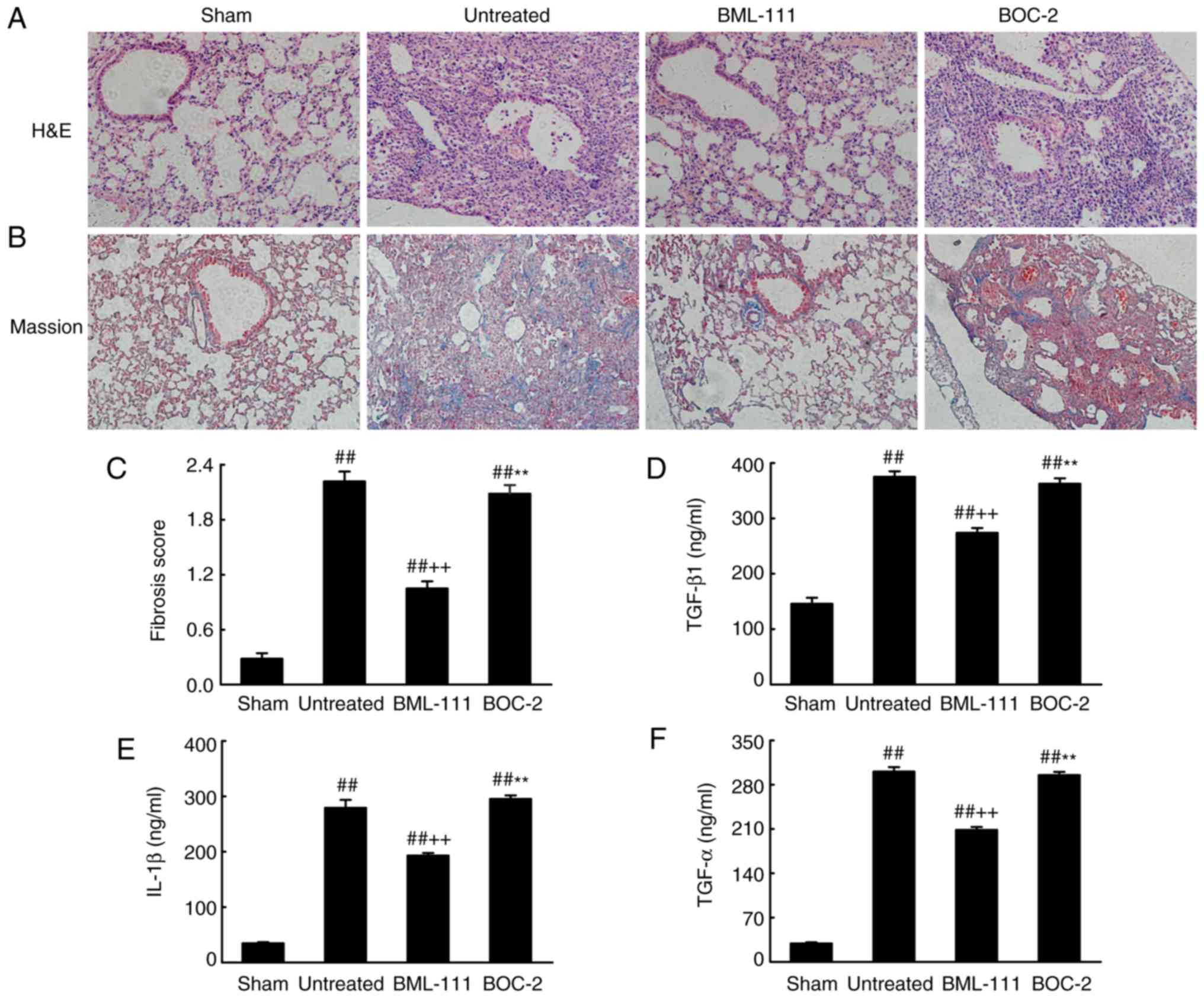 | Figure 5BML-111 treatment mitigated the
destruction of lung architecture and production of TGF-β1, TNF-α
and IL-1β in BALF following BLM iniection. Mice were treated with
50 µl saline (Sham group) or 2 mg/kg BLM (untreated group,
BML-111 group and BOC-2 group) at day 0 were intraperitoneally
administered with 1 ml of saline (Sham group and untreated group)
or 1 mg/kg of BML-111 (BML-111 group and BOC-2 group) in the
presence or absence of 50 µg/kg BOC-2 (BOC-2 group) prior to
the administration of BML-111. Mice were then sacrificed on day 21
and the extent of pulmonary injury and fibrosis were assessed using
(A) H&E and (B) Masson’s trichrome staining (magnification,
×100). (C) Fibrotic score was measured using the Ashcroft method.
Levels of (D) TGF-β1, (E) TNF-α and (F) IL-1β in BALF were
determined using ELISA. Data are expressed as mean ± standard
deviation (n=8). ##P<0.01 vs. the Sham group.
++P<0.01 vs. the untreated group.
**P<0.01 vs. the BML-111 group. TGF-β1, Transforming
growth factor-β1; TNF-α, tumor necrosis factor α; IL-1β,
interleukin 1β; BALF, bronchoalveolar lavage fluid; BLM, bleomycin;
BOC-2,
N-tert-butyloxy-carbonyl-phenyalanine-le-ucyl-phenyalanine-leucyl-phenyalanine. |
Discussion
Lung fibrosis can be divided into two stages: The
inflammatory and fibrotic stage. During the inflammatory stage,
inflammatory cells infiltrate into the area of injury, attempt to
clear tissue debris, and replace damaged cells (7). In the fibrotic phase, activated
cytokines, including TGF-β1, induce the formation of
myofibroblasts, which synthesize excessive ECM components and thus
precipitate tissue remodeling (11). Previously, it has been
demonstrated that BML-111 effectively mitigates the inflammatory
response following lung injury (21). However, studies have assessed its
direct anti-fibrotic actions. The present study demonstrated that
BML-111 treatment inhibited TGF-β1 mediated NIH3T3 proliferation
and activation as well as the synthesis and expression of ECM
components. Furthermore, it was also revealed that BML-111
attenuated fibrotic changes following BLM instillation in mice and
that thisprotective effect was partially counteracted following
BOC-2 pretreatment.
BML-111 is a commercially stable ALX agonist, which
possesses excellent anti-inflammatory and pro-resolving action,
similar to that of LXA4. BML-111 suppresses pulmonary
inflammatory reactions in ventilator/hemorrhagic shock/LPS-induced
lung injury (27,29,37). Previously, it has been
demonstrated that human lung fibroblasts in fibrotic lung tissue
express ALX and that LXA4 decreases TGF-β1 and
connective tissue growth factor dependent profibrotic activity in
human lung myofibroblasts (38,39). Although it has been revealed that
BML-111 inhibitsCCl4-induced hepatic fibrosis in
vivo, no assessment has been made on its effect on fibro-blast
activation in vitro (30).
The present study detected the expression of ALX in NIH3T3 cells
and demonstrated that BML-111 treatment inhibited TGF-β1 triggered
increases of α-SMA in a dose-dependent manner, with a maximum
effect at 200 nM. Furthermore, 200 nM BML-111 treatment mitigated
TGF-β1-induced cell proliferation and the production of ECM,
including total collagen and fibronectin. This indicated that
BML-111 treatment inhibits fibroblast activation and exerts a
direct anti-fibrosis effect in vitro. However, the activity
of BML-111 is abrogated by BOC-2, which indicates that this effect
is receptor dependent.
TGF-β1 mediated fibroblast activation occurs
primarily via Smad-dependent and independent pathways. To further
investigate the mechanisms by which BML-111 inhibits
TGF-β1-mediated NIH3T3 cell activation, the downstream components
of TGF-β1 signaling were determined. The results indicated that
BML-111 inhibits TGF-β1-induced phosphorylation of Smad2 and Smad3.
Previous studies have also demonstrated that phosphorylated Smad2
and Smad3 integrate with Smad4 and translocate into the nucleus
from the cytoplasm, where they ultimately activate the
transcription of pro-fibrotic genes, including collagen I,
fibronectin and α-SMA through reaction cascades (40,41). The current study demonstrated that
BML-111 treatment inhibits the phosphorylation of
non-Smad-dependent pathways including ERK and Akt. Previous studies
have revealed that phosphorylated ERK and Akt are involved in
TGF-β1 induced ECM increase, while ERK is involved in fibroblast
proliferation (42-44). Thus, these results indicate that
BML-111 acts as an anti-fibrotic agent by inhibiting the TGF-β1
associated signaling pathway.
BLM is widely used to induce pulmonary fibrosis in
mice. Following BLM instillation, mice are infected with acute
alveolitis within 2-3 days, where upon profibrosis media is
released, initiating ECM synthesis and the progression to fibrosis
(45). The anti-fibrotic effects
of BML-111 were examined using this model in the current study
(45). The results indicated that
BML-111 treatment markedly reduced the destruction of lung tissue
and structure. In addition, BML-111 inhibited BLM-induced
expression of α-SMA and ECM accumulation. These results were
consistent with in vitro results. Furthermore, it was
revealed that IL-1β, TNF-α and TGF-β1 in BALF was also decreased
following BML-111 administration. IL-1β and TNF-α are vital
pro-inflammatory cytokines that cause the further release of
fibrosis media and perpetuate the fibrotic cascade (13). Additionally, TGF-β1 also
counteracts fibroblast apoptosis and attenuates ECM degradation
(46-48). The results of the present study
also demonstrated that BML-111 administration delayed and decreased
the mortality of mice. This confirmed that BML-111 exerts a
protective effect on lung fibrosis, which may be attributed to
anti-inflammatory action, the downregulation of TGF-β1 expression
and the inhibition of fibroblasts activation. Similarly, this
effect is receptor dependent.
To conclude, the present study demonstrated that
BML-111 treatment suppresses TGF-β1-induced fibroblast α-SMA
protein synthesis and total collagen and fibronectin expression, by
suppressing Smad-dependent and Smad-independent signaling pathways.
Furthermore, BML-111 inhibited TGF-β1 levels and the synthesis of
inflammatory mediators in BLM-induced pulmonary fibrosis. These
results indicate that BML-111 may be used as a potential agent for
the treatment of pulmonary fibrosis.
Funding
The present study was supported by grants obtained
from the National Natural Science Foundation of China (grant nos.
30930089, 81372036, 81671890, 81500064, 81601669 and 81500436) and
the Key Clinical Project of Ministry of Health of China (grant. No.
2010-47).
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors’ contributions
YDJ, SLY and YS produced substantial contributions
to the conception and design of the present study. YDJ, ZLL, CXC,
BL and JG performed the experiments. ZLL, YXW and LC analyzed the
data. YDJ and ZLL drafted the paper. YS edited and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The use of mice within the present study was
reviewed and approved by the Institutional Animal Care and Use
Committee of Tongji Medical College, Huazhong University of Science
and Technology (Huazhong, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
King TJ Jr, Tooze JA, Schwarz MI, Brown KR
and Cherniack RM: Predicting survival in idiopathic pulmonary
fibrosis: Scoring system and survival model. Am J Respir Crit Care
Med. 164:1171–1181. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dempsey OJ: Clinical review: Idiopathic
pulmonary fibrosis-past, present and future. Respir Med.
100:1871–1885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Noble PW, Barkauskas CE and Jiang D:
Pulmonary fibrosis: Patterns and perpetrators. J Clin Invest.
122:2756–2762. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
du Bois RM: Strategies for treating
idiopathic pulmonary fibrosis. Nat Rev Drug Discov. 9:129–140.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wynn TA: Cellular and molecular mechanisms
of fibrosis. J Pathol. 214:199–210. 2008. View Article : Google Scholar
|
|
6
|
Wynn TA: Integrating mechanisms of
pulmonary fibrosis. J Exp Med. 208:1339–1350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Todd NW, Luzina IG and Atamas SP:
Molecular and cellular mechanisms of pulmonary fibrosis.
Fibrogenesis Tissue Repair. 5:112012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Phan SH: Biology of fibroblasts and
myofibroblasts. Proc Am Thorac Soc. 5:334–337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cutroneo KR, White SL, Phan SH and Ehrlich
HP: Therapies for bleomycin induced lung fibrosis through
regulation of TGF-beta1 induced collagen gene expression. J Cell
Physiol. 211:585–589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sivakumar P, Ntolios P, Jenkins G and
Laurent G: Into the matrix: Targeting fibroblasts in pulmonary
fibrosis. Curr Opin Pulm Med. 18:462–469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee CG, Cho S, Homer RJ and Elias JA:
Genetic control of transforming growth factor-beta1-induced
emphysema and fibrosis in the murine lung. Proc Am Thorac Soc.
3:476–477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Caraci F, Gili E, Calafiore M, Failla M,
La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A and Vancheri
C: TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK
activation in the transition of human lung fibroblasts into
myofibroblasts. Pharmacol Res. 57:274–282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wynn TA and Ramalingam TR: Mechanisms of
fibrosis: Therapeutic translation for fibrotic disease. Nat Med.
18:1028–1040. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pourgholamhossein F, Rasooli R,
Pournamdari M, Pourgholi L, Samareh-Fekri M, Ghazi-Khansari M,
Iranpour M, Poursalehi HR, Heidari MR and Mandegary A: Pirfenidone
protects against paraquat-induced lung injury and fibrosis in mice
by modulation of inflammation, oxidative stress, and gene
expression. Food Chem Toxicol. 112:39–46. 2018. View Article : Google Scholar
|
|
15
|
Romano M, Cianci E, Simiele F and
Recchiuti A: Lipoxins and aspirin-triggered lipoxins in resolution
of inflammation. Eur J Pharmacol. 760:49–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xia J, Zhou XL, Zhao Y, Zhu YQ, Jiang S
and Ni SZ: Roles of lipoxin A4 in preventing paracetamol-induced
acute hepatic injury in a rabbit model. Inflammation. 36:1431–1439.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang X, Li Z, Jiang S, Tong X, Zou X,
Wang W, Zhang Z, Wu L and Tian D: Lipoxin A4 exerts protective
effects against experimental acute liver failure by inhibiting the
NF-κB pathway. Int J Mol Med. 37:773–780. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Börgeson E, Docherty NG, Murphy M, Rodgers
K, Ryan A, O’Sullivan TP, Guiry PJ, Goldschmeding R, Higgins DF and
Godson C: Lipoxin A4 and benzolipoxin A4
attenuate experimental renal fibrosis. FASEB J. 25:2967–2979. 2011.
View Article : Google Scholar
|
|
19
|
Krönke G, Reich N, Scholtysek C,
Akhmetshina A, Uderhardt S, Zerr P, Palumbo K, Lang V, Dees C,
Distler O, et al: The 12/15-lipoxygenase pathway counteracts
fibroblast activation and experimental fibrosis. Ann Rheum Dis.
71:1081–1087. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martins V, Valença SS, Farias-Filho FA,
Molinaro R, Simões RL, Ferreira TPE, Silva PM, Hogaboam CM, Kunkel
SL, Fierro IM, et al: ATLa, an aspirin-triggered lipoxin A4
synthetic analog, prevents the inflammatory and fibrotic effects of
bleomycin-induced pulmonary fibrosis. J Immunol. 182:5374–5381.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gong J, Guo S, Li HB, Yuan SY, Shang Y and
Yao SL: BML-111, a lipoxin receptor agonist, protects haemorrhagic
shock-induced acute lung injury in rats. Resuscitation. 83:907–912.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fiore S, Maddox JF, Perez HD and Serhan
CN: Identification of a human cDNA encoding a functional high
affinity lipoxin A4 receptor. J Exp Med. 180:253–260. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takano T, Fiore S, Maddox JF, Brady HR,
Petasis NA and Serhan CN: Aspirin-triggered 15-epilipoxin A4 (LXA4)
and LXA4 stable analogues are potent inhibitors of acute
inflammation: Evidence for anti-inflammatory receptors. J Exp Med.
185:1693–1704. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao JL, Chen H, Filie JD, Kozak CA and
Murphy PM: Differential expansion of the N-formylpeptide receptor
gene cluster in human and mouse. Genomics. 51:270–276. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang YP, Wu Y, Li LY, Zheng J, Liu RG,
Zhou JP, Yuan SY, Shang Y and Yao SL: Aspirin-triggered lipoxin A4
attenuates LPS-induced pro-inflammatory responses by inhibiting
activation of NF-κB and MAPKs in BV-2 microglial cells. J
Neuroinflammation. 8:952011. View Article : Google Scholar
|
|
26
|
Lee TH, Lympany P, Crea AE and Spur BW:
Inhibition of leukotriene B4-induced neutrophil migration by
lipoxin A4: structure-function relationships. Biochem Biophys Res
Commun. 180:1416–1421. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li HB, Wang GZ, Gong J, Wu ZY, Guo S, Li
B, Liu M, Ji YD, Tang M, Yuan SY, et al: BML-111 attenuates
hemorrhagic shock-induced acute lung injury through inhibiting
activation of mitogen-activated protein kinase pathway in rats. J
Surg Res. 183:710–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gong J, Li HB, Guo S, Shang Y and Yao SL:
Lipoxin receptor agonist, may be a potential treatment for
hemorrhagic shock-induced acute lung injury. Med Hypotheses.
79:92–94. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li H, Wu Z, Feng D, Gong J, Yao C, Wang Y,
Yuan S, Yao S and Shang Y: BML-111, a lipoxin receptor agonist,
attenuates ventilator-induced lung injury in rats. Shock.
41:311–316. 2014. View Article : Google Scholar
|
|
30
|
Zhou XY, Yu ZJ, Yan D, Wang HM, Huang YH,
Sha J, Xu FY, Cai ZY and Min WP: BML-11, a lipoxin receptor
agonist, protected carbon tetrachloride-induced hepatic fibrosis in
rats. Inflammation. 36:1101–1106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Izumo T, Kondo M and Nagai A: Effects of a
leukotriene B4 receptor antagonist on bleomycin-induced pulmonary
fibrosis. Eur Respir J. 34:1444–1451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kelly MN, Zheng M, Ruan S, Kolls J,
D’Souza A and Shellito JE: Memory CD4+ T cells are
required for optimal NK cell effector functions against the
opportunistic fungal pathogen Pneumocystis murina. J Immunol.
190:285–295. 2013. View Article : Google Scholar
|
|
33
|
Yang J, Nan C, Ripps H and Shen W:
Destructive changes in the neuronal structure of the FVB/N mouse
retina. PLoS One. 10:e1297192015.
|
|
34
|
AVMA Guodelines for the Euthanasia of
Animals. 2013.48
|
|
35
|
Ashcroft T, Simpson JM and Timbrell V:
Simple method of estimating severity of pulmonary fibrosis on a
numerical scale. J Clin Pathol. 41:467–470. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu J, Wei Y, Luo Q, Xu F, Zhao Z, Zhang
H, Lu L, Sun J, Liu F, Du X, et al: Baicalin attenuates
inflammation in mice with OVA-induced asthma by inhibiting NF-κB
and suppressing CCR7/CCL19/CCL21. Int J Mol Med. 38:1541–1548.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang M, Chen L, Li B, Wang Y, Li S, Wen A,
Yao S and Shang Y: BML-111 attenuates acute lung injury in
endotoxemic mice. J Surg Res. 200:619–630. 2016. View Article : Google Scholar
|
|
38
|
Roach KM, Feghali-Bostwick CA, Amrani Y
and Bradding P: Lipoxin A4 Attenuates constitutive and
TGF-β1-dependent profibrotic activity in human lung myofibroblasts.
J Immunol. 195:2852–2860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu SH, Wu XH, Lu C, Dong L and Chen ZQ:
Lipoxin A4 inhibits proliferation of human lung fibroblasts induced
by connective tissue growth factor. Am J Respir Cell Mol Biol.
34:65–72. 2006. View Article : Google Scholar
|
|
40
|
Mendoza JA, Jacob Y, Cassonnet P and Favre
M: Human papillomavirus type 5 E6 oncoprotein represses the
transforming growth factor beta signaling pathway by binding to
SMAD3. J Virol. 80:12420–12424. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hasegawa M, Matsushita Y, Horikawa M,
Higashi K, Tomigahara Y, Kaneko H, Shirasaki F, Fujimoto M,
Takehara K and Sato S: A novel inhibitor of Smad-dependent
transcriptional activation suppresses tissue fibrosis in mouse
models of systemic sclerosis. Arthritis Rheum. 60:3465–3475. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Runyan CE, Schnaper HW and Poncelet AC:
The phosphatidylinositol 3-kinase/Akt pathway enhances
Smad3-stimulated mesangial cell collagen I expression in response
to transforming growth factor-beta1. J Biol Chem. 279:2632–2639.
2004. View Article : Google Scholar
|
|
43
|
Xiao L, Du Y, Shen Y, He Y, Zhao H and Li
Z: TGF-beta 1 induced fibroblast proliferation is mediated by the
FGF-2/ERK pathway. Front Biosci (Landmark Ed). 17:2667–2674. 2012.
View Article : Google Scholar
|
|
44
|
Hinz B: Myofibroblasts. Exp Eye Res.
142:56–70. 2016. View Article : Google Scholar
|
|
45
|
Mouratis MA and Aidinis V: Modeling
pulmonary fibrosis with bleomycin. Curr Opin Pulm Med. 17:355–361.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kolb M, Margetts PJ, Anthony DC, Pitossi F
and Gauldie J: Transient expression of IL-1beta induces acute lung
injury and chronic repair leading to pulmonary fibrosis. J Clin
Invest. 107:1529–1536. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sime PJ, Marr RA, Gauldie D, Xing Z,
Hewlett BR, Graham FL and Gauldie J: Transfer of tumor necrosis
factor-alpha to rat lung induces severe pulmonary inflammation and
patchy interstitial fibrogenesis with induction of transforming
growth factor-beta1 and myofibroblasts. Am J Pathol. 153:825–832.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sheppard D: Transforming growth factor
beta: A central modulator of pulmonary and airway inflammation and
fibrosis. Proc Am Thorac Soc. 3:413–417. 2006. View Article : Google Scholar : PubMed/NCBI
|