Introduction
As a neurodegenerative disease, Alzheimer’s disease
(AD) accounts for 50-70% of all dementia cases. With the aging of
the population, it is expected that the worldwide prevalence of AD
will quadruple by 2050 as compared with the reported rate of 26.6
million cases in 2006, and ~43% of AD patients require a high level
of care (1). The main symptoms of
AD patients include cognitive decline, accompanied by psychological
and behavioral abnormalities, decline in the ability to perform
daily activities, depression and sleep disorder. As one of the
common symptoms, sleep disorder severely affects the quality of
life of patients (2).
AD-associated sleep disorder is increasingly
prominent, and studies on mechanism of AD-associated sleep disorder
and new therapies are of great value. However, the molecular
mechanism of AD-associated sleep disorder remain unclear.
Kondratova and Kondratov (3) have
suggested that the sleep disorder in patients with AD may be
associated with the degeneration of the suprachiasmatic nucleus,
pineal gland, hypothalamus and brain nuclei. Furthermore, it has
recently been reported that sleep disorder may accelerate the AD
neurodegeneration and cognitive decline (4,5). A
previous study has suggested that insufficient sleep facilitates
the accumulation of amyloid β (Aβ), which has been confirmed as one
of the main pathological characteristics of AD (6). Neurofibrillary tangles composed of
an excessive amount of phosphorylated tau (p-tau) protein have been
confirmed as the second pathology of AD. Therefore, Aβ and abnormal
tau protein have a central role in the neuropathology of AD
(7,8). A previous study has also confirmed
that a long tau was associated with the delayed sleep phase
disorder, which may be the reflection of an abnormal circadian
timing system (9). The interplay
and synergy between Aβ and tau induce sleep disorder and accelerate
the pathogenesis of AD (10,11).
Sleep is produced when the internal inhibition
process spreads to the cerebral cortex and subcortical structure.
Sleep serves an essential role in the regulation of synaptic weight
in the brain; for instance, it reduces the slow wave activity of
the brain caused by the accumulation of synaptic enhancement in
wakefulness (12). The
neuropeptides orexin A (also known as hypocretin-1) and orexin
neurons express A1 adenosine receptor (A1R) secreted from lateral
hypothalamus (LH) neurons are known to be critical modulators of
the sleep/wakefulness system (13,14). In addition, it has been reported
that orexinergic system disorder may alter the sleep-wake rhythms
and influence AD pathology (15).
It has also been reported that blockade of adenosine A1R in the
orexinergic LH induced a significant increase in wakefulness;
however, adenosine A1R antagonists are able to reverse this effect
and may serve as potential preventive or therapeutic strategies in
AD patients (16,17).
Activation of adenosine A1R can accelerate sleep,
whereas adenosine A1R blockade can promote awakening and
compensatory sleep (17,18). A previous study has also reported
that orexin A, which may increase arousal levels, promoted Aβ
production and was associated with the regulation of the sleep-wake
cycle. Furthermore, recent studies have focused on the associations
of orexin A with p-tau (19).
However, whether the changes in Aβ and p-tau levels are associated
with orexin A secretion in the sleep-wake cycle remains unknown.
Based on this preliminary evidence, the aim of the present study
was to investigate the regulatory effect of Aβ and p-tau on sleep
disorder in AD patients.
Materials and methods
Establishment of an AD animal model
Experiments were performed on male 5xFAD mice (15-25
g), which overexpressed human APP and PS1 with 5 familial AD
mutations. 50 mice (1-year-old) were purchased from Kay Biological
Technology (Shanghai) Co. Ltd (Shanghai, China) All the mice were
free access to food and water and were kept in an undisturbed and
clean environment with 12 h/12 h dark-light cycle, 90% relative
humidity and 25°C. The animals were acclimatized to the laboratory
environment for 1 week before the experiments. The AD mouse model
was constructed as previously described in which 1-year old 5xFAD
mice spontaneously exhibit AD-like pathological changes (20-22), and mice received cerebral
injection of Aβ 25-35 (cat. no. ym-Y-0044, Yuanmu, China) 3 weeks
prior to tests. Mice anesthetized with 400 mg/kg chloral hydrate
(i.p.) (23) received an
injection stereotactically, directly into the lateral ventricles,
of aggregated Aβ25-35 peptide (5 µg/mouse i.c.v.) through a
Hamilton syringe (VWR International, Fontenay-sous-Bois, France).
Before use, Aβ 25-35 was diluted in sterile saline to a
concentration of 0.5 mM and was maintained at 37°C for 7 days to
pre-age the peptide (24). A
water maze experiment was used to detect the behavioral changes of
the mice. All animals were treated in accordance with the
guidelines of Shandong University (Jinan, China), and experiments
were approved by the Animal Research Committee of Shandong
University.
Morris water maze
The Morris water maze test was conducted in a
circular pool with a diameter of 120 cm, a height of 50 cm and a
depth of 30 cm. The pool was divided into 4 quadrants, and a
platform with a diameter of 10 cm and height of 28 cm was placed in
the middle of one of the quadrants. The back of the head of the
mouse was marked with picric acid for tracking. A marker that hung
over the wall of the pool and a number of constant reference
objects around the pool were used as visual cues. The water
temperature was maintained at 25°C, and the hidden platform
experiment was conducted 4 times per day for 5 successive days.
Mice were dropped from 4 different locations and the escape latency
time spent to find the hidden platform was monitored by a video
tracking system. If the mouse did not find the hidden platform
within 2 min, the researcher guided the mouse to the platform and
permitted to stay on the platform for 10 sec, after which the mouse
was returned to the cage, and the incubation period was recorded as
2 min. The experiment consisted of two parts, in part 1, the mice
were dropped into the 4 different quadrants, termed west (w), east
(e), north (n) and south (s) respectively, each day for 5 days in
the following order: Day 1, w-s-e-n; day 2, s-e-n-w; day 3:
e-n-w-s; day 4, n-w-s-e; and day 5, w-s-e-n. In part 2, on day 6,
the hidden platform was removed and the mice were put into the
water from a quadrant that did not contain the platform. The number
of times that the mice crossed the original location of the
platform within 2 min was recorded. During part 1 and part 2, the
total distance that the mice swam, and the time the mice spent in
the platform quadrant were recorded.
Sleep analysis
The sleep disturbances and sleep-wake state of AD
mice were recorded and analyzed with polysomnography, in which
electroencephalogram (EEG) and electromyography (EMG) results were
recorded and used to analyzed the non-rapid eye-movement sleep and
rapid eye-movement sleep. In order to obtain the EEG and EMG scan,
EEG and EMG electrodes were implanted into the skull simultaneously
(NeuroLogger, TSE Systems GmbH, Bad Homburg, Germany). For EEG
recording, two stainless steel screws attached to wire electrodes
were placed over the right frontal and parietal bones. For EMG
recording, two wire electrodes were directly inserted into the neck
musculature.
During the study, age matched mice were used as a
control. Briefly, after 2 days of habituation to the device, 5xFAD
mice and age matched controls underwent EEG recordings, which
commenced on day 3. Body movements, and brain EEG activity was
recorded and stored at 200 samples per second with a high-pass
filter of 0.25 Hz and low-pass filter of 70 Hz. Digitized data were
downloaded offline to a computer. EEG and EMG signals were recorded
in a computer and then assessed with sleep-scoring software
(SIRENIA® SEIZURE PRO).
Cell culture
The human SH-SY5Y cell line was purchased from the
American Type Culture Collection (ATCC® CRL-2266™;
Manassas, VA, USA) and cultured in Eagle’s minimum essential medium
(cat. no. M2279; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
supplemented with 10% fetal bovine serum (cat. no. F4135;
Sigma-Aldrich; Merck KGaA), 100 IU/ml penicillin (cat. no.
10378016; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
100 mg/ml streptomycin (cat. no. 15140-122; Gibco; Thermo Fisher
Scientific, Inc.), 1% non-essential amino acids (cat. no.
11140-050; Invitrogen; Thermo Fisher Scientific, Inc.) and 1%
glutamine (cat. no. 11090-081; Gibco; Thermo Fisher Scientific,
Inc.) at 37°C with 5% CO2. SH-SY5Y cells were treated
with or without the tau inhibitor TRx 0237. SH-SY5Y cells were
grouped into negative control (SH-SY5Y) and Aβ 25-35 (SH-SY5Y+Aβ
25-35) or TRx 0237 (100 nM; cat. no. T7003; TargetMol, Boston, MA,
USA) (SH-SY5Y+Aβ 25-35+TRx 0237). SH-SY5Y cells (1 ×105 cells/well)
were seeded in 6-well plates and transfected with vehicle, Aβ 25-35
or TRx 0237 for 48 h. Before use, Aβ 25-35 was diluted in sterile
saline to a concentration of 0.5 mM and was maintained at 37°C for
7 days to pre-age the peptide (24). The aged Aβ solution was diluted to
40 µM for use.
Oligonucleotide transfection
Negative control (NC) and small interfering RNAs
(siRNAs) targeting adenosine A1R (si-A1R) or orexin A (si-orexin A)
were purchased from Shanghai GeneChem Co., Ltd. (Shanghai, China).
SH-SY5Y cells (1×105 cells/well) were seeded in 6-well
plates and transfected with NC, si-A1R or si-orexin A for 48 h
using Lipofectamine 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer’s protocol.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Mice anesthetized with 400 mg/kg chloral hydrate
(i.p.) (23) were decapitated on
the day after all the behavioral and neurocognitive tests finished,
and the whole brains of which olfactory bundle, optic nerve,
cerebellum and medulla were removed were collected and frizzed at
-80°C for PCR and western blotting assays.Total RNA from the
brain tissues or cells was extracted using TRIzol reagent (cat. no.
15596-018; Invitrogen; Thermo Fisher Scientific, Inc.), cDNA was
synthesized using 1 µg total RNA and then reverse
transcribed to cDNA using an RT assay (DBI Bioscience, Newark, DE,
USA). Subsequently, the relative expression of different target
genes and controls was analyzed using a SYBR-Green PCR Master Mix
kit (cat. no. RR420A; Takara Bio, Inc., Otsu, Japan), and the qPCR
reactions were performed on an ABI 7500 Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The primer
sequences for the genes were as follows: Tau, 5′-GTG GCC AGG TGG
AAG TAA AA-3′ (forward) and 5′-TGG AAG ACA CAT TGC TGA GG-3′
(reverse); orexin A, 5′-GCA TAT CGG CCG CTT TAA TA-3′ (forward) and
5′-GGG TCC TCG AGT CTC TTT CC-3′ (reverse); adenosine A1R, 5′-TGT
AGG TGC CTT GGT CAT CC-3′ (forward) and 5′-ATC CCT GCT CTT CTT GCT
GT-3′ (reverse); GAPDH, 5′-GCC ATC ACA GCA ACA CAG AA-3′ (forward)
and 5′-GCC ATA CCA GTA AGC TTG CC-3′ (reverse). Cycling conditions
included denaturation at 95°C for 2 min followed by annealing at
94°C for 20 sec for 40 cycles, and extension at 58°C for 20 sec. On
the basis of exponential amplification of the target gene as well
as a calibrator, the quantity of amplified molecules at the
quantification cycle was given by 2−ΔΔCq. The data was
assayed with the comparative 2−ΔΔCq method (25) to determine the expression levels
of target genes.
Western blot analysis
Total proteins from the tissues or cells were lysed
in radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Haimen, China) containing protease inhibitors
(BIOSS, Beijing, China), and the protein concentration was
determined using a BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.). Equal amount of total proteins was then separated by 10%
SDS-PAGE and transferred onto a polyvinylidene difluoride membrane
(cat. no. PK-NEF1002; PerkinElmer, Inc., Boston, MA, USA). Next,
fat-free milk (5%) was used to block the membranes for 2 h at room
temperature. Following blocking, the membranes were incubated with
primary antibodies overnight at 4°C. The blots were subsequently
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:4,000) at room temperature for 1 h (Wuhan Boster
Biological Technology, Ltd., Wuhan, China; cat. no. BA1054) and
then developed using an enhanced chemiluminescence detection kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer’s
protocol. The primary antibodies used included anti-GAPDH
(dilution, 1:2,000; ab8245), anti-tau (dilution, 1:1,000; ab10439),
anti-p-tau (dilution, 1:1,000; ab4841), anti-A1R (dilution,
1:1,500; ab82477) and anti-orexin (dilution 1:1,500; ab77370; all
purchased from Abcam, Cambridge, MA, USA) antibodies.
Statistical analysis
The data were analyzed by the Student’s t-test and
analysis of variance (ANOVA) using IBM SPSS software (version 19.0;
IBM Corp., Armonk, NY, USA). The Student-Newman-Keuls post hoc test
was used to calculate the P-value for pairwise comparisons
following ANOVA. Paired t-test was used to analyze comparisons
between the groups and paired data. Unpaired t-test was used to
analyze comparisons between 2 groups. Each experiment was repeated
at least 3 times. Data are presented as the means ± standard
deviation. A statistically significant difference was considered to
be denoted by P<0.05.
Results
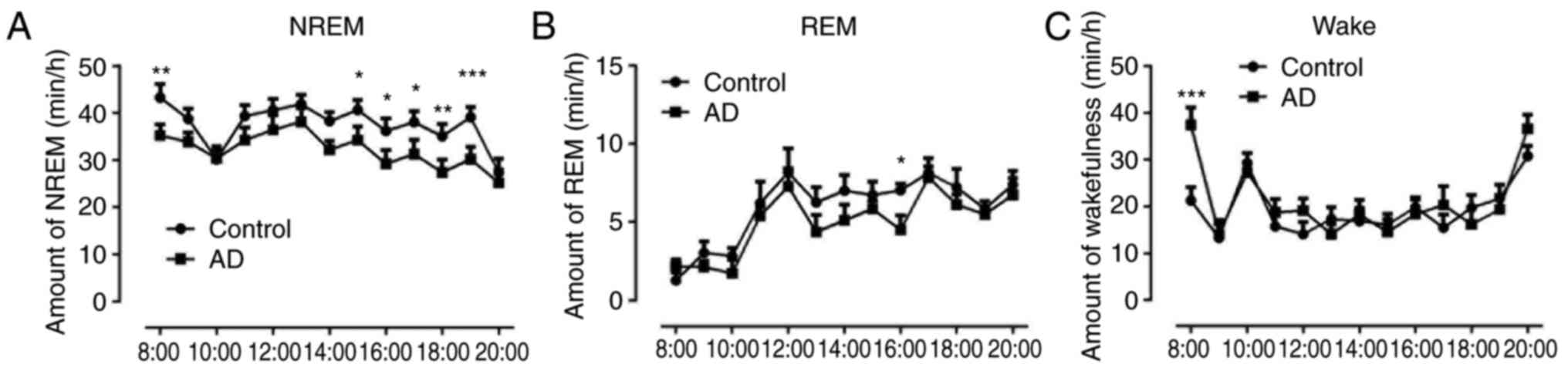
Decreased non-rapid eye movement (NREM)
sleep and increased wakefulness in AD mice
The sleep patterns in the control and AD groups were
compared, and as mice are nocturnal animals, the sleep cycle was
from 8 am to 8 pm. The results indicated that the NREM sleep in AD
mice was significantly decreased compared with that in the control
group (P<0.05, P<0.01 and P<0.001; Fig. 1A). The results also demonstrated
that non-rapid eye movement (NREM) sleep in AD mice was similar to
the normal amount observed in control animals at other time points,
except for at 16:00 (P<0.05) (Fig.
1B). The wakefulness in AD mice at 8:00 was markedly increased
compared with that of the control group (P<0.001), however, it
was equal to the normal amount of wakefulness observed in control
animals at other hourly time points (Fig. 1C).
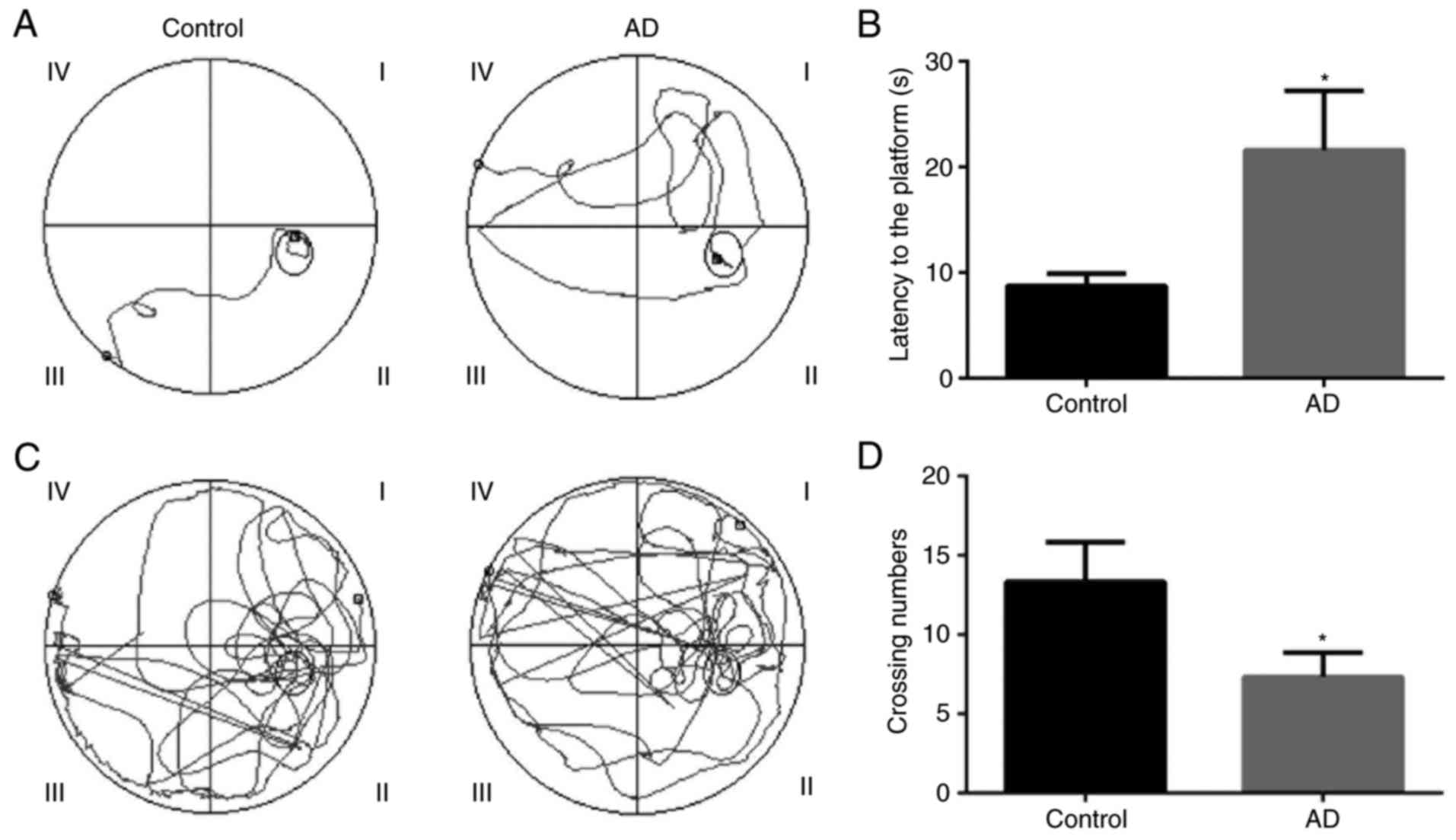
Behavioral changes in AD model mice
The Morris water maze test was applied to confirm
the behavioral changes of AD mice. The tracing of the movement of
mice during the hidden platform test and the probe trial test for
the control and AD model mice are presented in Fig. 2. The results revealed that the
escape latency of the hidden platform test was significantly longer
in the AD model mice as compared with that in the control mice
(P<0.05; Fig. 2A and B).
Therefore, it is suggested that the AD model was successfully
established in the mice. In addition, the results indicated that
the number of platform-crossings for the AD model mice was
significantly lower compared with that of the control mice
(P<0.05; Fig. 2C and D). It
is, thus, suggested that the learning ability of AD model mice was
worse in comparison with that of control group mice.
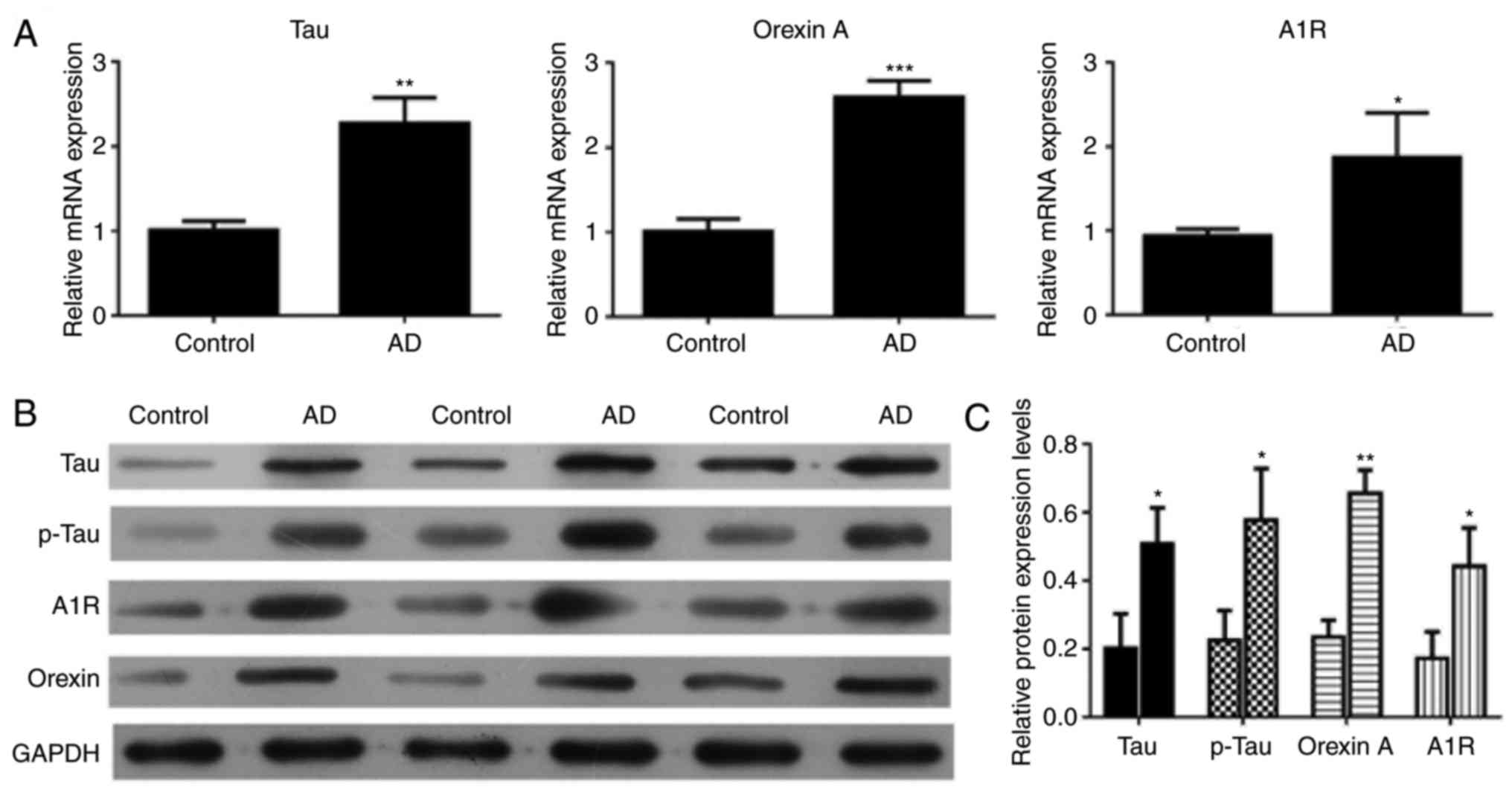
Upregulation of tau, p-tau, orexin A and
adenosine A1R in the brain tissue of 5xFAD mice
RT-qPCR and western blot analysis were conducted to
validate the expression levels of different genes in the control
and AD mice. The RT-qPCR results demonstrated that the relative
expression levels of tau, orexin A and adenosine A1R were markedly
upregulated in AD mice compared with the control mice (P<0.01,
P<0.001 and P<0.05, respectively; Fig. 3A). The western blot results were
consistent with the RT-qPCR results (Fig. 3B). The tau, p-tau, orexin A and
adenosine A1R expression levels were higher in AD mice compared
with those in control mice (P<0.05; Fig. 3C).
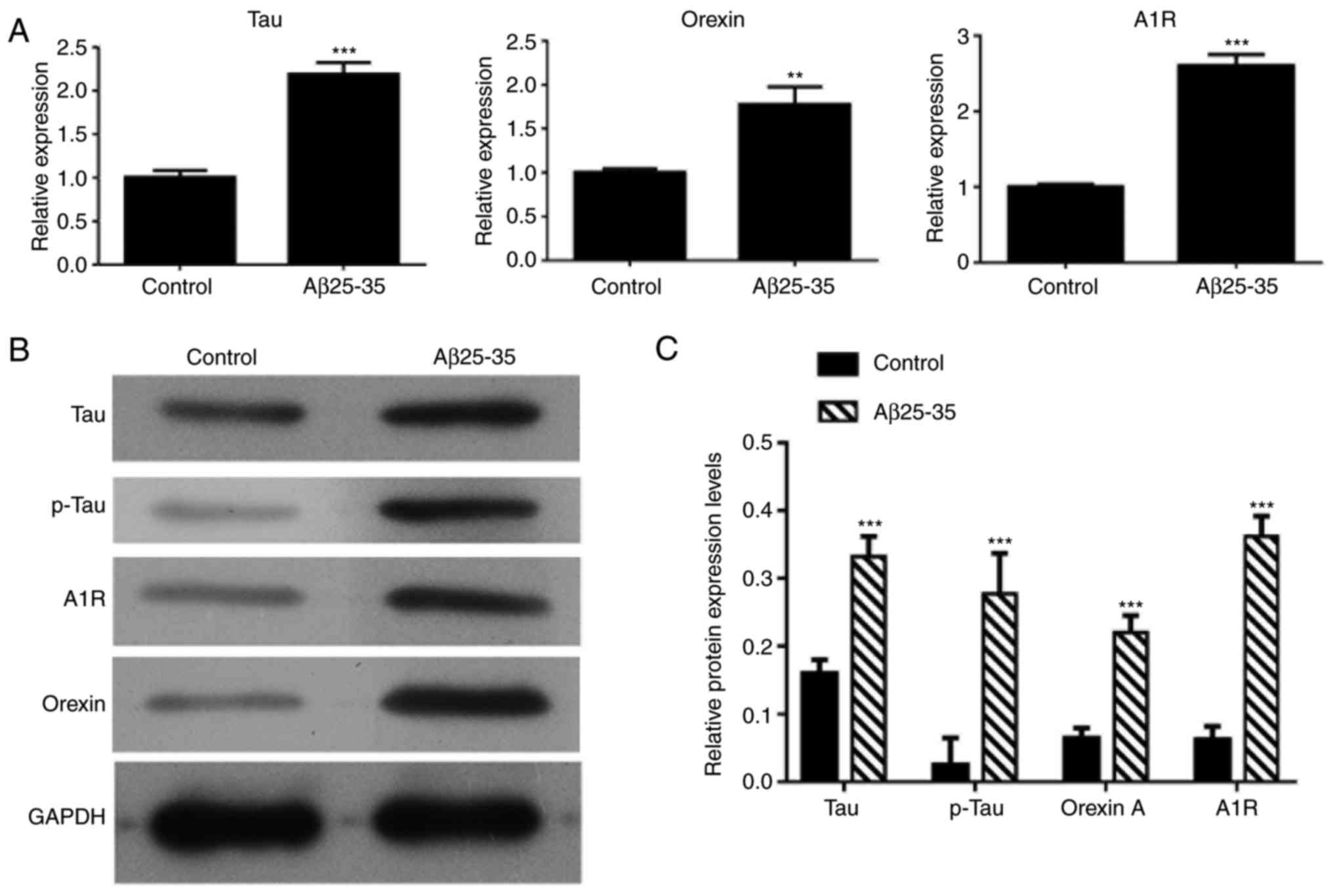
Treatment with Aβ25-35 upregulates the
expression levels of tau, p-tau, orexin A and adenosine A1R in
SH-SY5Y cells
RT-qPCR and western blot analysis were also used to
detect the mRNA and protein expression levels, respectively, in
SH-SY5Y cells treated with Aβ25-35. As shown in Fig. 4, the mRNA expression levels of
tau, orexin A and adenosine A1R were significantly increased in
Aβ25-35-treated SH-SY5Y cells as compared with the control group
cells (P<0.01), while significant increase was also observed in
the protein levels of tau, p-tau, orexin A and adenosine A1R.
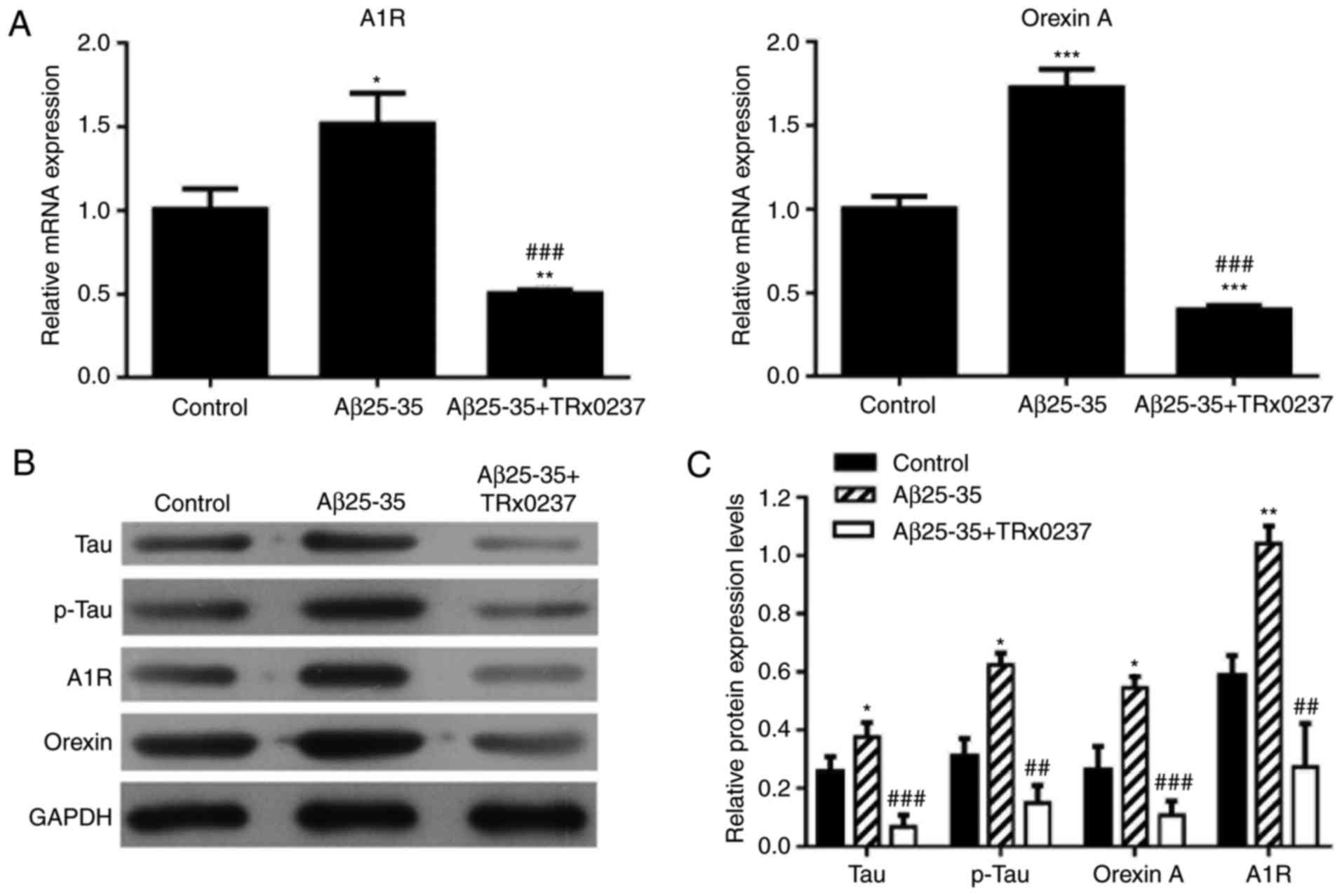
Changes in mRNA and protein expression
levels following treatment with Aβ25-35, or a combination of
Aβ25-35 and TRx 0237
There was a significant increase in the mRNA
relative expression levels of adenosine A1R and orexin A following
Aβ25-35 treatment; however, the high expression levels of adenosine
A1R and orexin A were significantly reduced following Aβ25-35 and
TRx 0237 co-treatment (P<0.05; Fig. 5A). Next, the protein expression
levels of tau, p-tau, orexin A and adenosine A1R were determined by
western blot analysis. Overexpression of tau, p-tau, orexin A and
adenosine A1R was observed in the Aβ25-35-treated group compared
with the control group. However, TRx 0237 co-treatment
significantly reversed the promoting effects of Aβ25-35 on tau,
p-tau, orexin A and adenosine A1R expression (Fig. 5B and C).
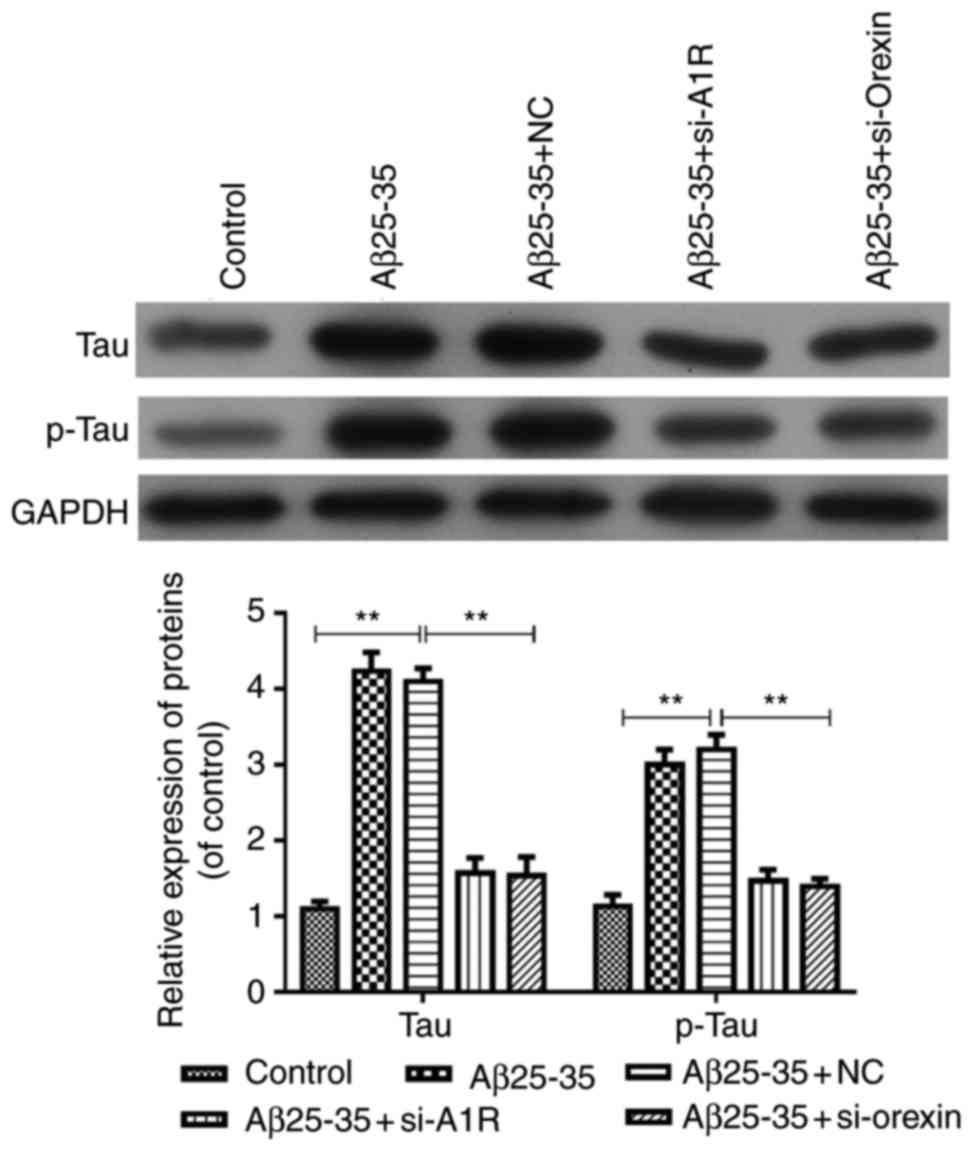
Silencing of adenosine A1R and orexin by
siRNA transfection inhibits tau and p-tau expression mediated by
Aβ25-35 in SH-SY5Y cells
The current study further explored the effects of
adenosine A1R and orexin A knockdown on tau and p-tau expression
induced by Aβ25-35 in the AD cell model. As shown in Fig. 6, the results indicated that tau
and p-tau group compared with the control. However, these levels
were significantly downregulated in the Aβ25-35 + si-A1R Aβ25-35 +
si-orexin A groups compared with the Aβ25-35 and Aβ25-35 + NC
groups (P<0.05; Fig. 6).
Therefore, these data suggested that adenosine A1R or orexin
knockdown inhibited the Aβ25-35-mediated expression levels of tau
and p-tau in the AD cell model.
Discussion
AD is the most common type of neurodegenerative
disease that affects the cognitive functions of the elderly
(26). Currently, no therapies
are available to halt or reverse the progression of this disease.
In the present study, it was observed that NREM sleep was decreased
and wakefulness was increased in AD mice. In the Morris water maze
assay, a shorter escape latency time indicates better memory, and
increased number of platform crossings indicates better learning
ability (27,28). The present study in 5xFAD mice
revealed a longer escape latency and lower number of platform
crossings compared with control group, which indicated a
deteriorated memory and learning ability in 5xFAD mice.
Aβ serves a pivotal and potentially causative role
in the pathogenesis of AD, and has thus become a major therapeutic
target (29). At present, studies
have confirmed that AD neurodegeneration started with the
aggregation of non-soluble monomeric Aβ peptides, and that the
deposition of Aβ in plaques is the main driver of AD pathogenesis.
Epidemiological studies have reported that patients with AD
suffered from sleep dysfunction (2,30,31). Sleep disorder has been confirmed
to decrease clearance of Aβ in the brain and accelerate AD
pathology. In the present study, the interaction between sleep
disorder and AD was further explored.
In recent years, it has been documented that the
aggregation of Aβ can induce abnormal phosphorylation of tau
protein and lead to nerve fiber formation (32). In addition, a higher level of tau
protein has been confirmed as a marker of rapid cognitive decline,
while the abundance of neurofibrillary tangles was correlated with
the severity of dementia (33,34). Previous studies (20-22) investigated neurocognitive function
of the 5xFAD mice, using an AD animal model with a Morris water
maze assay. In addition, in a study performed by Kang et al
(10) suggested that the levels
of soluble Aβ are positively associated with the time awake and
negatively associated with the time asleep. In the present study,
the 5xFAD mice AD model was used, and the data indicated a
significantly decreased NREM sleep and increased wakefulness in the
mice.
The orexinergic system is a key regulator of sleep
onset. The neuropeptide orexin A (also known as hypocretin-1)
produced by LH neurons has a significant impact on sleep-wake cycle
regulation (13,35). It has been suggested that orexin
regulated both the diurnal wake and nocturnal sleep periods through
reducing REM sleep and slow-wave sleep, and increasing wakefulness
(36,37). Additionally, adenosine inhibits
excitatory synaptic transmission of orexin neurons via the receptor
adenosine A1R. Blockade of adenosine A1R in the orexinergic LH
induced a significant increase in wakefulness, while it decreased
sleep in the sleep-wake cycle (16,17). Consequently, it can be predicted
that the sleep disorder may be associated with the abnormal
regulation of Aβ and tau on the adenosine A1R axis. In the present
study, the expression levels of orexin A and adenosine A1R were
found to be markedly upregulated in the brain tissue of AD mice,
and similar results were observed in SH-SY5Y cells treated with
Aβ25-35. In addition, treatment with the tau inhibitor TRx 0237 not
only decreased the tau and p-tau expression levels, but also
reversed the promoting effects of Aβ25-35 on orexin A and adenosine
A1R expression levels. Adenosine A1R or orexin knockdown also
inhibited tau and p-tau expression mediated by Aβ25-35 in the AD
cell model. In fact, a previous publication indicated that
increased orexin A promoted wakefulness and sleep fragmentation,
consequently promoting p-tau accumulation (38). In the current study, it was
confirmed that orexin A expression was positively correlated with
the wakefulness time and tau expression induced by Aβ accumulation,
which provides further evidence on the regulatory network of sleep
disorder in AD pathology.
In conclusion, the present study highlighted that
sleep disorder in 5xFAD mice with AD-like neurocognitive changes is
positively associated with the accumulation of Aβ and tau, which
induced the overactivation of the orexinergic system. Therefore,
these findings suggest that Aβ and tau can be considered as novel
biomarkers of sleep disorder in AD pathology. Furthermore, Aβ and
tau appear to serve key roles in sleep regulation by regulating the
expression levels of orexin A and adenosine A1R. However, further
studies are required to validate how silencing A1R and orexin by
siRNA transfection affects the sleep-wake cycle changes. It is also
necessary to investigate the deeper mechanisms underlying the
actions of adenosine A1R and orexin in sleep disorder in AD.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The data used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors’ contributions
ZL, FW and MT conceived and designed the experiment.
ZL, FW, MT, YZ and XW carried out the experiments. ZL, FW, and MT
acquired the reagents and materials. ZL and FW analyzed the data.
ZL FW and MT wrote the manuscript. All authors approved the final
version of the manuscript.
Ethics approval and consent to
participate
All animals were treated in accordance with the
guidelines of Shandong University (Jinan, China), and experiments
were approved by the Animal Research Committee of Shandong
University.
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brookmeyer R, Johnson E, Ziegler-Graham K
and Arrighi HM: Forecasting the global burden of Alzheimer’s
disease. Alzheimers Dement. 3:186–191. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moran M, Lynch CA, Walsh C, Coen R,
Coakley D and Lawlor BA: Sleep disturbance in mild to moderate
Alzheimer’s disease. Sleep Med. 6:347–352. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kondratova AA and Kondratov RV: The
circadian clock and pathology of the ageing brain. Nat Rev
Neurosci. 13:325–335. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ooms S, Overeem S, Besse K, Rikkert MO,
Verbeek M and Claassen JA: Effect of 1 night of total sleep
deprivation on cerebrospinal fluid β-amyloid 42 in healthy
middle-aged men: A randomized clinical trial. JAMA Neurol.
71:971–977. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peter-Derex L, Magnin M and Bastuji H:
Heterogeneity of arousals in human sleep: A
stereo-electroencephalographic study. Neuroimage. 123:229–244.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mander BA, Winer JR, Jagust WJ and Walker
MP: Sleep: A novel mechanistic pathway, biomarker, and treatment
target in the pathology of Alzheimer’s disease? Trends Neurosci.
39:552–566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hyman BT: New neuropathological criteria
for Alzheimer disease. Arch Neurol. 55:1174–1176. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tanaka T, Mayuyama D and Takeda M:
Alzheimer disease and tau protein. Rinsho Shinkeigaku.
52:1171–1173. 2012.In Japanese. View Article : Google Scholar
|
|
9
|
Campbell SS and Murphy PJ: Delayed sleep
phase disorder in temporal isolation. Sleep. 30:1225–1228. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth
LP, Cirrito JR, Fujiki N, Nishino S and Holtzman DM: Amyloid-beta
dynamics are regulated by orexin and the sleep-wake cycle. Science.
326:1005–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morishima-Kawashima M and Ihara Y:
Alzheimer’s disease: Beta-amyloid protein and tau. J Neurosci Res.
70:392–401. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vyazovskiy VV, Olcese U, Lazimy YM,
Faraguna U, Esser SK, Williams JC, Cirelli C and Tononi G: Cortical
firing and sleep homeostasis. Neuron. 63:865–878. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Lecea L, Kilduff TS, Peyron C, Gao X,
Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT,
Bartlett FS II, et al: The hypocretins: Hypothalamus-specific
peptides with neuroexcitatory activity. Proc Natl Acad Sci USA.
95:322–327. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sakurai T, Amemiya A, Ishii M, Matsuzaki
I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP,
Wilson S, et al: Orexins and orexin receptors: A family of
hypothalamic neuropeptides and G protein-coupled receptors that
regulate feeding behavior. Cell. 92:573–585. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liguori C: Orexin And Alzheimer’s disease.
Curr Top Behav Neurosci. 33:305–322. 2017. View Article : Google Scholar
|
|
16
|
Thakkar MM, Winston S and McCarley RW:
Orexin neurons of the hypothalamus express adenosine A1 receptors.
Brain Res. 944:190–194. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thakkar MM, Engemann SC, Walsh KM and
Sahota PK: Adenosine and the homeostatic control of sleep: Effects
of A1 receptor blockade in the perifornical lateral hypothalamus on
sleep-wakefulness. Neuroscience. 153:875–880. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alam MN, Kumar S, Rai S, Methippara M,
Szymusiak R and McGinty D: Role of adenosine A1 receptor
in the perifornical-lateral hypothalamic area in sleep-wake
regulation in rats. Brain Res. 1304:96–104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cedernaes J, Osorio RS, Varga AW, Kam K,
Schiöth HB and Benedict C: Candidate mechanisms underlying the
association between sleep-wake disruptions and Alzheimer’s disease.
Sleep Med Rev. 31:102–111. 2017. View Article : Google Scholar
|
|
20
|
Gu L, Wu D, Tang X, Qi X, Li X, Bai F,
Chen X, Ren Q and Zhang Z: Myelin changes at the early stage of
5XFAD mice. Brain Res Bull. 137:285–293. 2018. View Article : Google Scholar
|
|
21
|
Aytan N, Choi JK, Carreras I, Crabtree L,
Nguyen B, Lehar M, Blusztajn JK, Jenkins BG and Dedeoglu A:
Protective effects of 7,8-dihydroxyflavone on neuropathological and
neurochemical changes in a mouse model of Alzheimer’s disease. Eur
J Pharmacol. 828:9–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maiti P, Paladugu L and Dunbar GL: Solid
lipid curcumin particles provide greater anti-amyloid,
anti-inflammatory and neuroprotective effects than curcumin in the
5xFAD mouse model of Alzheimer’s disease. BMC Neurosci. 19:72018.
View Article : Google Scholar
|
|
23
|
Shao J, Lin M, Li Y, Li X, Liu J, Liang J
and Yao H: In vivo blood glucose quantification using Raman
spectroscopy. PLoS One. 7:e481272012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Harkany T, Abrahám I, Kónya C, Nyakas C,
Zarándi M, Penke B and Luiten PG: Mechanisms of beta-amyloid
neurotoxicity: Perspectives of pharmacotherapy. Rev Neurosci.
11:329–382. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar
|
|
26
|
Kantarci K: Molecular imaging of Alzheimer
disease pathology. AJNR Am J Neuroradiol. 35(Suppl 6): S12–S17.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo K, Yin G, Zi XH, Zhu HX and Pan Q:
Effect of selective serotonin reuptake inhibitors on expression of
5-HT1AR and neurotransmitters in rats with vascular dementia. Genet
Mol Res. 15: View Article : Google Scholar : 2016.PubMed/NCBI
|
|
28
|
Liu ZJ, Li ZH, Liu L, Tang WX, Wang Y,
Dong MR and Xiao C: Curcumin attenuates beta-amyloid-induced
neuroinlammation via activation of peroxisome
proliferator-activated receptor-gamma function in a rat model of
Alzheimer’s disease. Front Pharmacol. 7:2612016. View Article : Google Scholar
|
|
29
|
Helal M, Hingant E, Pujo-Menjouet L and
Webb GF: Alzheimer’s disease: Analysis of a mathematical model
incorporating the role of prions. J Math Biol. 69:1207–1235. 2014.
View Article : Google Scholar
|
|
30
|
McCurry SM, Logsdon RG, Vitiello MV and
Teri L: Treatment of sleep and nighttime disturbances in
Alzheimer’s disease: A behavior management approach. Sleep Med.
5:373–377. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vitiello MV and Prinz PN: Alzheimer’s
disease. Sleep and sleep/wake patterns. Clin Geriatr Med.
5:289–299. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rosenwasser AM: Functional neuroanatomy of
sleep and circadian rhythms. Brain Res Rev. 61:281–306. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arriagada PV, Growdon JH, Hedley-Whyte ET
and Hyman BT: Neurofibrillary tangles but not senile plaques
parallel duration and severity of Alzheimer’s disease. Neurology.
42:631–639. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Braak H and Braak E: Neuropathological
stageing of Alzheimer- related changes. Acta Neuropathol.
82:239–259. 1991. View Article : Google Scholar
|
|
35
|
Saper CB, Scammell TE and Lu J:
Hypothalamic regulation of sleep and circadian rhythms. Nature.
437:1257–1263. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Arrigoni E, Mochizuki T and Scammell TE:
Activation of the basal forebrain by the orexin/hypocretin
neurones. Acta Physiol. 198:223–235. 2010. View Article : Google Scholar
|
|
37
|
Lee MG, Hassani OK and Jones BE: Discharge
of identified orexin/hypocretin neurons across the sleep-waking
cycle. J Neurosci. 25:6716–6720. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Di Meco A, Lauretti E, Vagnozzi AN and
Praticò D: Zileuton restores memory impairments and reverses
amyloid and tau pathology in aged Alzheimer’s disease mice.
Neurobiol Aging. 35:2458–2464. 2014. View Article : Google Scholar : PubMed/NCBI
|