Introduction
Foxhead box L2 (FOXL2), a single-exon gene located
at 3q23, belongs to the forkhead family of transcription factors,
and is a regulatory mediator in organogenesis (1). It was the earliest known marker of
sexual dimorphism in ovarian differentiation in mammals and may
have multiple roles in ovarian somatic cell differentiation and
follicle development and/or maintenance (2). In addition, sequence comparison of
FOXL2 from several vertebrate species has shown that it is a
conserved early regulator of vertebrate ovarian development and
that early defects in ovarian formation are likely to contribute to
the blepharophimosis ptosis epicanthus-inversus syndrome (BPES)
phenotype (3).
FOXL2 is required for commitment to ovary
differentiation (4). Several
novel insights into ovarian follicle development have arisen from
the investigation of relevant knockout mouse models (5), and somatic sexual reprogramming of
adult ovaries to testes by FOXL2 ablation has been demonstrated in
mice (6). The absence of
secondary follicles and oocyte atresia in Foxl2lacZ
homozygous mutant ovary granulosa cells shows that the Foxl2 gene
is essential for granulosa cell differentiation and ovary
maintenance (7). In
Foxl2lacZ homozygous mutant mice, the transition from
pseudosquamous to cuboidal granulosa cells in the primary follicle
cannot occur, indicating FOXL2 may be a regulator of the granulosa
cell tumor differentiation process (8). Therefore, fully understanding the
function and regulation of FOXL2 may reveal novel avenues for the
treatment of ovarian granulosa cell tumors and female infertility
in the future. However, the detailed molecular mechanisms involved
remain to be fully elucidated. Therefore, investigation into the
expression of FOXL2 in human cells is likely to provide useful
information.
FOXL2 has several potential gene targets, including
gonadotropin releasing hormone receptor (GnRHR), glycoprotein
hormone α subunit, follistatin (FST), follicle-stimulating hormone
(FSH)-β and cytochrome P450 family (CYP)17A1 (9). In addition, steroidogenic acute
regulatory protein (StAR) and CYP19A1, two transcriptional targets
relevant to granulosa cell biology, have been reported previously
in relation to the role of FOXL2 in a granulosa tumor cell line
(10). StAR is a late
differentiation marker in granulosa cells in pre-ovulatory
follicles and CYP19A1 is the gene that encodes aromatase. StAR
catalyzes the translocation of cholesterol from the outer to the
inner mitochondrial membrane, allowing it to be processed into
steroid hormones, whereas aromatase is an enzyme that is also
involved in steroidogenesis and the conversion of androgens to
estrogens in granulosa cells (10).
Clustered regularly interspaced short palindromic
repeats (CRISPR) together with CRISPR-associated (Cas) proteins are
an elegant adaptive defense mechanism in bacteria and archaea,
which provide acquired resistance to invading viruses and plasmids
(11-13). Studies have shown that type II
CRISPR systems can create a synthetic single-guide RNA (sgRNA)
consisting of a fusion of CRISPR RNA (crRNA) and trans-activating
crRNA (tracrRNA), which can direct site-specific DNA cleavage by
the Cas9 nuclease (14). CRISPR
and Cas proteins recognize site-specific nucleases directly through
gRNA to generate a DNA double-strand break (DSB) at a specified
locus. DSBs can then be repaired by homologous recombination or
error-prone non-homologous end joining, resulting in a mutant
carrying deletions or insertions at the cut site, leading to an
open reading frame shift (15).
Therefore, this system can efficiently induce targeted genetic
modifications, and use short RNA to direct the degradation of
foreign nucleic acids (15). The
CRISPR/Cas system has been adapted as an efficient gene-targeting
technology with the potential for multiplexed genome editing
(16). Reports have demonstrated
that CRISPR/Cas-mediated gene editing allows the simultaneous
targeting of multiple genes and has been utilized not only in
cultured human cells (17-19)
but also in animal model establishment (20,21) and cancer therapy (22-24).
It was hypothesized that FOXL2 can be effectively
disrupted using CRISPR, therefore, the aim of the present study was
to create a stable FOXL2-knockout cell line in KGN human ovarian
granulosa cells. This cell line may then assist with future
investigations of FOXL2 and its mechanism of action.
Materials and methods
Cell culture
The KGN human ovarian granulosa cells were supplied
by Professor Yiming Mu of the Chinese PLA General Hospital
(Beijing, China) and human embryonic kidney (293T and 293A) cells
were obtained from the American Type Culture Collection (Manassas,
VA, USA). The KGN cells were incubated in Dulbecco’s modified
Eagle’s medium: Nutrient Mixture F-12 (DMEM/F-12; Hyclone; GE
Healthcare Life Sciences, Logan, UT, USA), and the 293T and 293A
cells were incubated in DMEM (Hyclone; GE Healthcare Life Sciences)
supplemented with 10% fetal bovine serum (FBS; Hyclone; GE
Healthcare Life Sciences) and antibiotics at 37°C in a humidified
atmosphere (Thermo Fisher Scientific, Inc.; Waltham, MA, USA) with
5% CO2.
Plasmid construction
Three G (N19) NGG sequences, where N stands for any
A, T, G, or C nucleotide, downstream of the FOXL2 translation start
site (ATG) were selected as potential target sites with the
assistance of the Optimized CRISPR Design website (http://crispr.mit.edu/). The resulting three target
sequences were as follows: F404: GCG CGC CGG CTT TGT CAT GAT GG;
F425: GGC CAG CTA CCC CGA GCC CGA GG; and F446: GGA CGC GGC GGG GGC
CCT GCT GG.
The following primers were then synthesized: F404F,
5′-GAA AGG ACG AAA CAC CGC GCG CCG GCT TTG TCA TG AGTT TTA GAG CTA
GAA AT-3′ and F404R, 5′-ATT TCT AGC TCT AAA ACT CAT GAC AAA GCC GGC
GCG CGG TGT TTC GTC CTT TC-3′; F425F, 5′-GAA AGG ACG AAA CAC CG G
CCA GCT ACC CCG AGC CCG GTT TTA GAG CTA GAA AT-3′ and F425R, 5′-ATT
TCT AGC TCT AAA ACC GGG CTC GGG GTA GCT GGC CGG TGT TTC GTC CTT
TC-3′; F446F, 5′- GAA AGG ACG AAA C ACC GGA CGC GGC GGG GGC CCTGCG
TT T TAG AGC TAG AAA T-3′ and F446R, 5′-ATT TCT AGC TC TAAA ACG CAG
GGC CCC CGC CGC GTC CGG TGT TTC GT C CTT TC-3′; CRIF, 5′-GTA TTT
CGA TTT CTT GGC TTT ATA TAT CTT GTG GAA AGG ACG AAA CAC CG-3′ and
CRIR, 5′-GTT GAT AAC GGA CTA GCC TTA TTT TAA CTT GCT ATT TCT AGC
TCT AAA AC-3′.
To construct the F404 gRNA lentivirus plasmid, F404F
and F404R were denatured and annealed to act as the template for
CRIF and CRIR polymerase chain reaction (PCR) amplification. PCR
was conducted using the following parameters: An initial
denaturation at 94°C for 3 min, followed by 20 cycles of
amplification (94°C for 20 sec, 53°C for 20 sec, and 72°C for 20
sec) with a final extension step at 72°C for 10 min. The PCR
products were then inserted into the pLKO.1 plasmid using Gibson
master mix according to the manufacturer’s protocol (New England
Biolabs, Beverly, MA, USA). The F425 and F446 gRNA plasmids were
also constructed in a similar manner. A flag-tagged Cas9 gene was
cloned into the pShuttle-CMV plasmid using the BglII and
XhoI restriction sites. Following linearizing with
PmeI, the pShuttle-CMV-Cas9 was co-transformed with the
pAdEasy-1 plasmid into Escherichia coli strain
BJ5183.(Stratagene, La Jolla, CA, USA) by electroporation using a
Bio-Rad Gene Pulser electroporator (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Lentivirus and adenovirus packaging
To package the gRNA lentivirus, the gRNA plasmid and
the packaging plasmids pMD and SPA were co-transfected into 293T
cells. At 48 h post-transfection, the medium was harvested and used
to infect KGN cells. The recombinant Ad plasmid was linearized with
PacI and transfected into AD-293 cells (Stratagene) to
produce the adenovirus.
Cell genomic DNA extraction and target
sequence amplification
The genomic DNA of KGN cells derived from single
colony was extracted using a kit (Tiangen Biotech Co., Ltd.,
Beijing, China) following the manufacturer’s protocol. The sequence
including the target sites was amplified with F296F and F818R
primers and purified with a kit (Tiangen Biotech Co., Ltd.). The
resulting PCR product was 523 bp. The sequences of the primers were
as follows: F296F: 5′-CAT CCG CAG TCT CCA GAA GTT TGA-3′; F818R:
5′-AGG CCG GGT CCA GCG TCC AGT AGT-3′.
T7 Endonuclease I (T7 EI) digestion
Prior to T7 EI digestion, the PCR product was
denatured, by placing the reaction tube in a 500-ml beaker of 94°C
water, and annealed, by allowing it to cool to room temperature.
Following cooling, the PCR product was digested with T7 EI at 37°C
for 2 h and then assayed with agarose gel electrophoresis.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted and purified with the Takara
MiniBEST Universal RNA Extraction kit (Takara Bio, Inc., Tokyo,
Japan) according to the manufacturer’s protocol. For cDNA
synthesis, 2 μg of RNA was reverse transcribed with
PrimeScript RT reagent kit (Takara Bio, Inc.) according to the
manufacturer’s protocol. The real-time PCR was standardised using
SYBR FAST qPCR mix (Takara Bio, Inc.). The primers were as follows:
β-actin, forward 5′-GTC CCT CAC CCT CCC AAA AG-3′ and reverse
5′-GCT GCC TCA ACA CCT CAA CCC-3′; StAR, forward 5′-TGG GCA TCC TTA
GCA ACC A-3′ and reverse 5′-GGG ACC ACT TTA CTC ATC ACT TTG T-3′;
and CYP19A, forward 5′-TGA GGA TCC CTT TGG ACG AA-3′ and reverse
5′-AAT AAC CTT GGA TTT TAA CCA CGA TAG-3′. qPCR was conducted using
the following parameters: An initial denaturation at 95°C for 5
min, followed by 25 cycles of amplification (95°C for 30 sec, 60°C
for 40 sec, and 72°C for 1 min) with a final extension step at 72°C
for 10 min. The PCR results were quantified using the Eppendorf
realplex 4S RTqPCR system. The qPCR data were analyzed with the
comparative 2-ΔΔCq method (25) using β-actin to normalize the
expression data. Values are presented as changes relative to the
KGN wt values, which were set to 1.
Western blot analysis
The cells were collected and lysed with buffer [50
mM Tris-HCl, 150 mM NaCl, 1% NP40, 0.1% SDS, 1 mM DTT (pH 7.5)] and
protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany)
and sonicated briefly. Following centrifugation (12,000 × g) at 4°C
for 15 min, the supernatant was collected, and protein
concentrations were determined using the BCA protein quantification
assay kit (Thermo Fisher Scientific, Inc.). The supernatant was
boiled in Laemmli buffer and analyzed by western blot analysis; 40
μg of protein homogenates were loaded onto a denaturing 10%
sodium dodecyl sulfate-polyacrylamide gel using a Mini Protean
apparatus (Bio-Rad Laboratories, Inc.) and transferred onto
polyvinylidene difluoride membranes (Merck KGaA, Darmstadt,
Germany). The membrane was blocked with Tris-buffered saline
containing 5% non-fat dry-milk at room temperature for 2 h, then
exposed to primary antibodies overnight at 4°C. Based on the
described reactivity, anti-FOXL2 antibody (1:1,000; cat. no.
ab188584, Abcam, Cambridge, MA, USA) and anti-flag antibody
(1:2,000; cat. no. F2555, Sigma-Aldrich; Merck KGaA) were used for
detection of the transfection effectiveness, and anti-StAR antibody
(1:1,000; cat. no. sc-166821, Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), and anti-CYP19A antibody (1:1,000; cat. no.
sc-374176, Santa Cruz Biotechnology, Inc.) were used to evaluate
the disruption of FOXL2. Anti-cyclin D1 antibody (1:1,000; cat. no.
ab134175, Abcam), anti-cyclin-dependent kinase (CDK)4 antibody
(1:1,000; cat. no. ab108357, Abcam), and anti-CDK6 antibody
(1:1,000; cat. no. ab124821, Abcam) were used to detect knockout of
the cell cycle. Anti-β-actin antibody (1:5,000; cat. no. ab8227,
Abcam) was used as a protein loading control. The secondary
antibody (anti-rabbit or anti-mouse peroxidase-conjugated IgG,
1:5,000; cat. nos. ab6721 and ab6789, Abcam) was incubated with the
membrane for 1 h at room temperature. The reaction products were
revealed by enhanced chemiluminescence (ECL Plus; Thermo Fisher
Scientific, Inc.). The intensities of the digitally detected bands
were evaluated by densitometry using Image-Pro Plus software
(Version X; Media Cybernetics, Inc., Silver Springs, MD, USA).
Flow cytometric analysis of cell cycle
and apoptosis
The cells (1×106) were collected
following trypsinization and washed twice with PBS. The cells were
then stained using the cycle staining kit and Annexin V-FITC/PI
apoptosis kit (Multi Sciences Biotech Co., Ltd., Hangzhou, China)
according to the manufacturer’s protocol, respectively. In the cell
cycle assay, 2×105 cells for each sample were measured
with a Becton Dickinson FACSCalibur flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA) and analyzed with ModFit software version
3.0 (Verity Software House; Topsham, ME, USA). Cell cycle status
was analyzed using propidium iodide (PI) as a specific fluorescent
dye probe. For the apoptosis assay, 1×105 cells were
measured and analyzed with CellQuest software version 2.0 (BD
Biosciences). Annexin V(+)/PI(-) and Annexin V(+)/PI(+) represent
the cells in early apoptosis and late apoptosis/necrosis
respectively and the proportion of these cells out of the total
number of cells analyzed were determined.
xCELLigence real-time cell proliferation
analysis
Cell proliferation experiments were performed using
the xCELLigence Real Time Cell Analyzer (RTCA) DP instrument (ACEA
Biosciences, San Diego, CA, USA). The cells were seeded in 16-well
plates (E-plate 16 ACEA Biosciences, Inc.) at a density
2×103 and 4×103 cells in 150 μl medium
per well, and impedance measurements were monitored at regular time
intervals. The cell growth curves were automatically recorded with
the xCELLigence system in real time. The area of growth covered in
an E-plate due to cell adhesion was recorded as the cell index
(CI). A high CI indicates more cell adhesion and vice versa. The
experiment was allowed to run for 3 days and each experiment was
performed in triplicate. Data were analyzed using RTCA software
version 1.2.
Cell viability assay by BrdU labelling
and Cell Counting Kit-8 (CCK-8) assay
The cellular viabilities of the FOXL2-knockout and
wt KGN cells were determined using two methods: BrdU labelling and
a CCK-8 assay. The BrdU labelling assays were performed using the
APC-BrdU Flow kit (BD Biosciences, cat. no. 552598) according to
the manufacturer’s protocol. Briefly, during the final 1 h of
culture, the cells were pulsed with 10 μM of BrdU APC and
7-AAD. The stained cells were analyzed on a flow cytometer using a
low flow rate. For the CCK-8 assay, the cells were seeded in
96-well plates at a density of 1×104 cells per well and
incubated for 24 h. The cells were then treated with different
concentrations of FBS (0, 2, 5 and 10%) for 24 h. Following
treatment, CCK-8 (10 μl/well) was added to the wells and the
cells were incubated at 37°C for 1 h. Subsequently, the OD value
was we detected at 450 nm (A450) using a microplate
spectrophotometer.
Statistical analysis
The experimental data are expressed as the mean ±
standard deviation. For statistical analysis, Student’s t-test for
quantitative data was processed with Excel version 2007 (Microsoft
Corporation, Redmond, WA, USA). P<0.05 was considered to
indicate a statistically significant difference. Error bars
represent the standard deviation of at least three independent
experiments.
Results
Knockout of FOXL2 in KGN cells using
CRISPR/Cas9
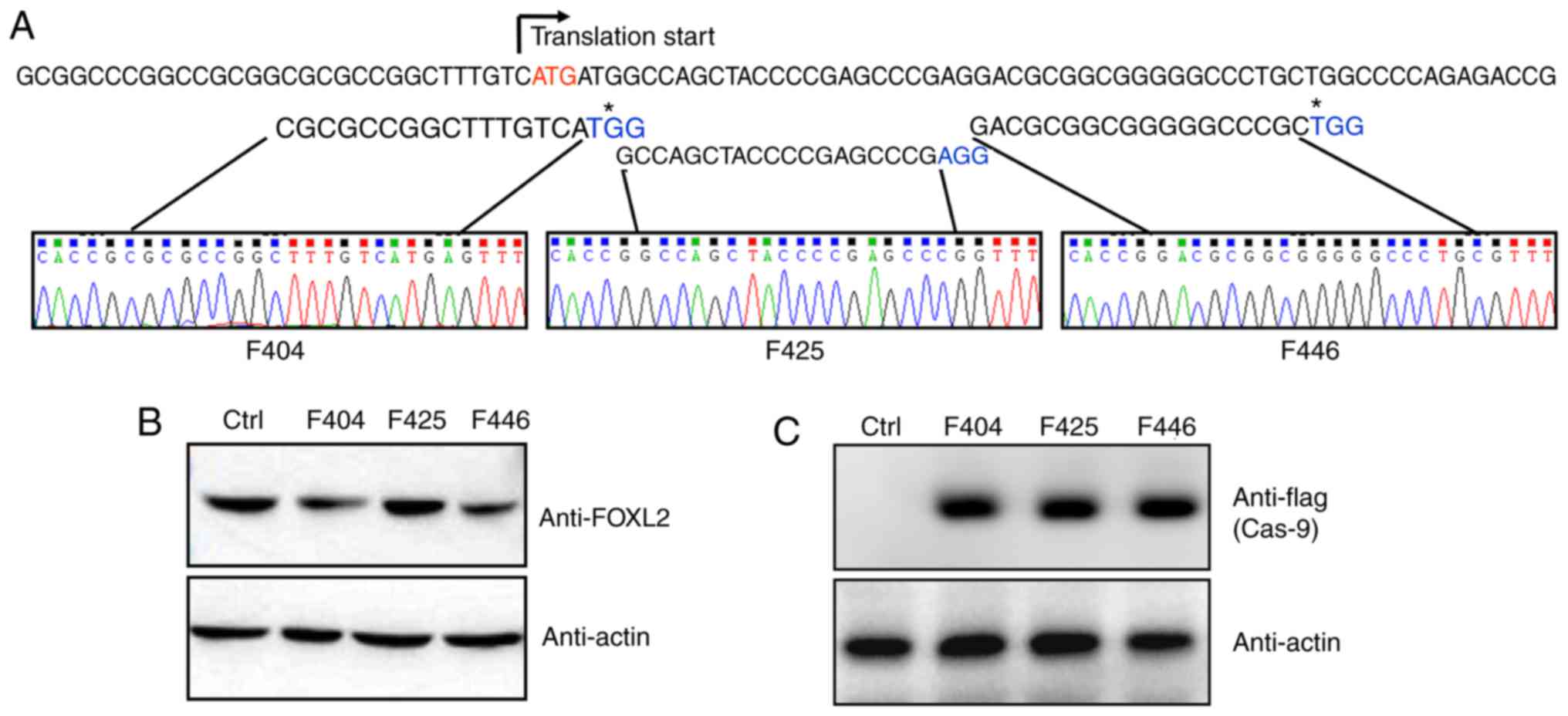
Three potential G (N19) NGG target sites were
screened just downstream of the translation start site. All three
gRNA sequences were cloned into the pLKO.1 lentivirus vector, and
were confirmed by sequencing (Fig.
1A). In addition, as shown in Fig. 1B, gRNA lentivirus infection did
not alter the expression of FOXL2. The gRNA lentivirus-infected KGN
cells were further infected with flag-Cas9 adenovirus and the
expression of Cas9 was monitored by anti-flag western blot analysis
(Fig. 1C).
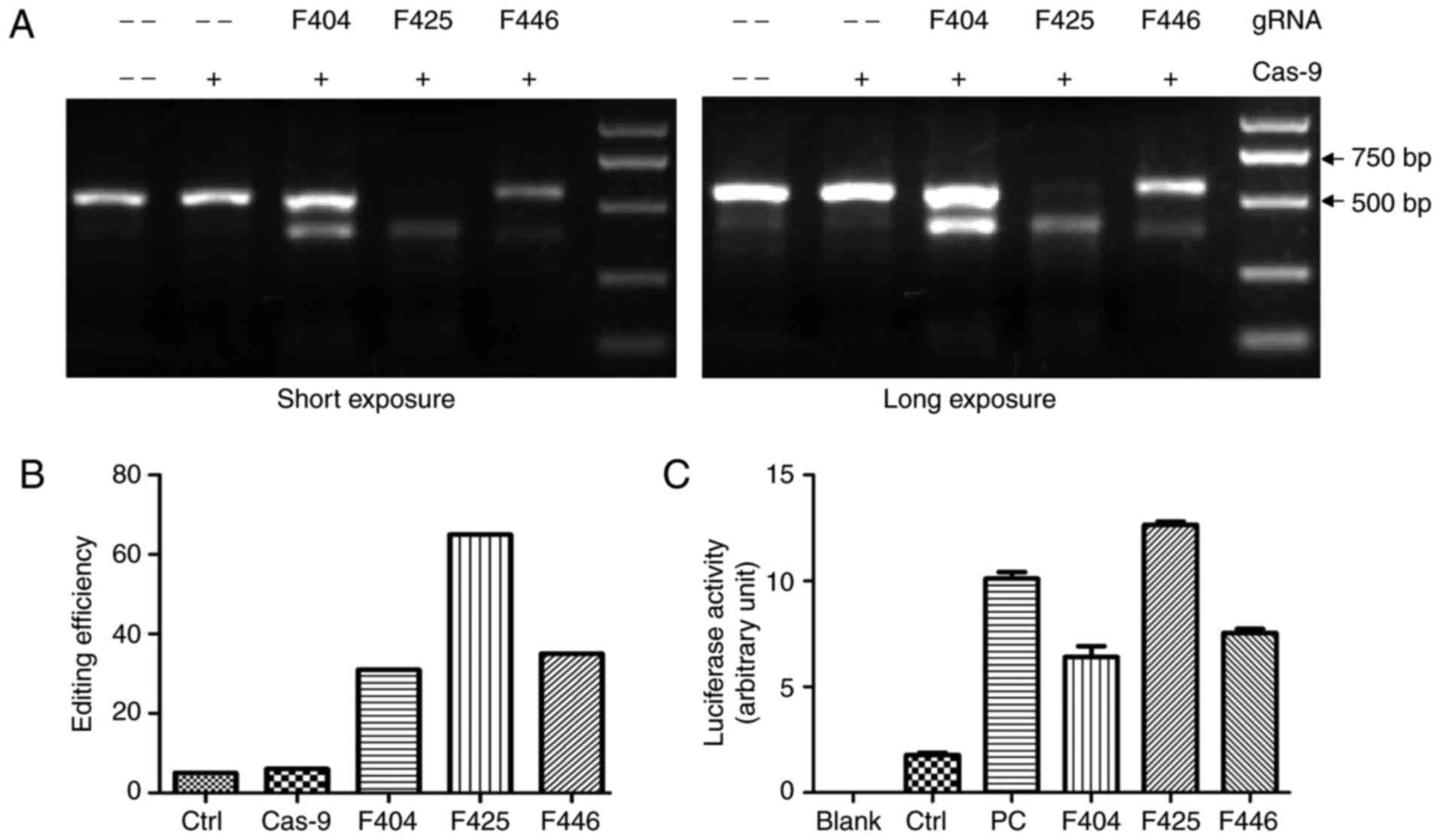
Validation of CRISPR/Cas9-mediated
FOXL2-knockout
To assess the efficacy of Cas9-mediated genome
editing, the T7 EI digestion of PCR products from the genomic DNA
of control and infected KGN cells was analyzed by agarose gel
electrophoresis. As shown in Fig.
2A, the PCR products derived from the double infected cells
were cut to produce bands of the predicted size, whereas the PCR
products of control cells remained almost intact. Quantitative
analysis revealed that the cells infected with F425 gRNA were most
efficiently edited by Cas9 (Fig.
2B). The luciferase reporter assay results also confirmed that
F425 had the highest efficiency of the three targets (Fig. 2C).
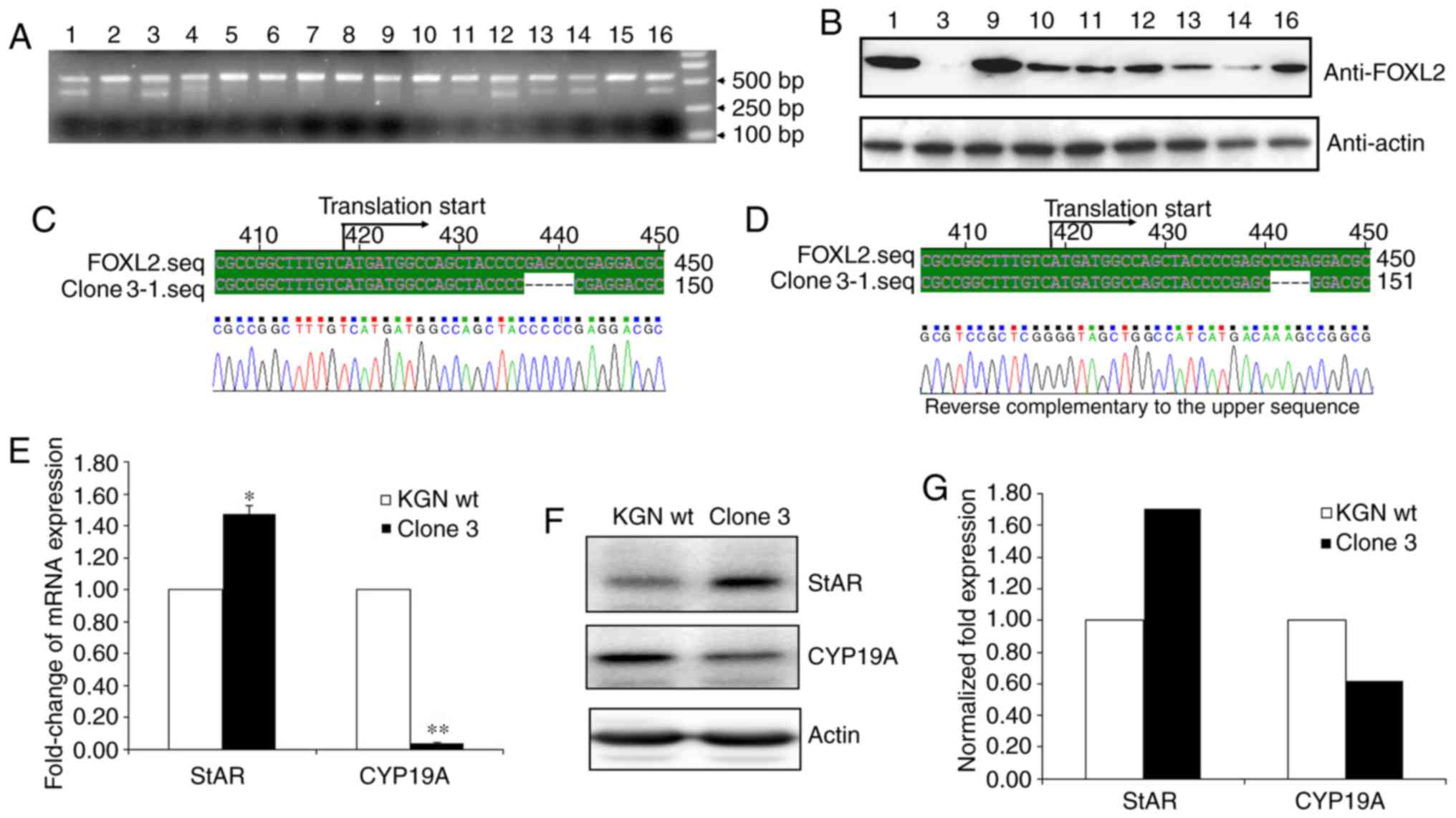
A total of 16 F425 single cell clones were then
screened and the genomic DNA of the cells was extracted to perform
T7 EI digestion following PCR amplification. As shown in Fig. 3A, the majority of the PCR products
from different clones were digested by T7 EI, which included clones
1, 3, 9, 10, 11, 12, 13, 14, and 16. The results of the western
blot analysis further indicated that clone 3 had been fully
disrupted at the FOXL2 locus (Fig.
3B). To further investigate whether FOXL2 has been completely
disrupted in clone 3, the sequence that included the target sites
was cloned into the T vector and 50 positive clones were sequenced.
The sequencing results showed only two genotypes of the target
sites, one of which lacked five base pairs and the other lacked
four base pairs (Fig. 3C and
D).
To further rule out the potential disruption of
FOXL2, StAR and CYP-19A, two major reported downstream modulators
of FOXL2, were measured. Previous reports have identified that
FOXL2 positively regulates StAR and negatively regulates CYP19A
(10). As expected, the results
showed that StAR was markedly elevated and CYP19A was markedly
decreased in clone 3 at the mRNA (Fig. 3E) and protein (Fig. 3F and G) levels. Therefore, the
results confirmed that FOXL2 had been successfully knocked out in
clone 3 using the CRISPR/Cas9 method.
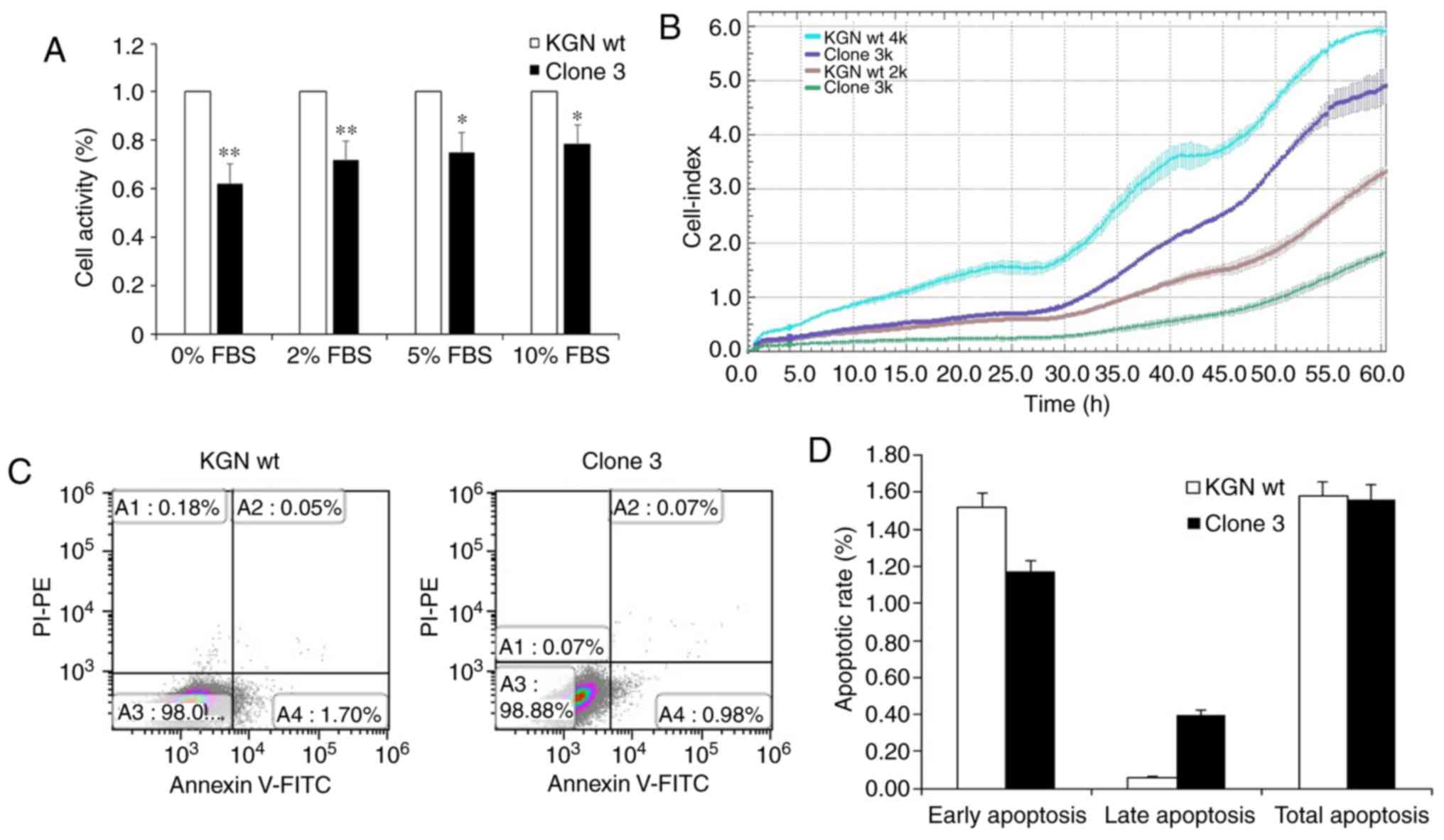
FOXL2 knockout results in decreased
proliferation of KGN cells without affecting apoptosis
To further investigate whether the physiological
activities of KGN cells were influenced by FOXL2 knockout,
proliferation, cell cycle and apoptosis assays were been performed.
The viability assay results using CCK-8 detection showed that, in
the presence of different concentrations of FBS (0, 2, 5 and 10%),
the proliferation rate of clone 3 was significantly inhibited,
compared with that in the wt cells (Fig. 4A). In addition, the proliferation
rate inhibition in clone 3 was confirmed by the xCELLigence RTCA
assay (Fig. 4B). However, no
significant changes were observed in total apoptosis following the
knockout of FOXL2 (Fig. 4C and
D). Therefore, the results indicated that FOXL2 knockout mainly
attenuated the proliferation of KCN cells.
FOXL2 knockout restricts cell cycle
progression at the G0/G1 phase
The present study then measured the effects of FOXL2
knockout on cell cycle. The results showed that the percentage of
cells in the G0/G1 phases in clone 3 cells, which lacked FOXL2, was
significantly enhanced, whereas the percentage in the S phase was
decreased; this suggested that proliferation was inhibited through
cell cycle arrest at G0/G1 following FOXL2 knockout (Fig. 5A and B). The BrdU labelling assays
with flow cytometry also confirmed that the clone 3 cells had
enhanced G0/G1 phases (Fig. 5C and
D). In addition, the results of the western blot analysis
demonstrated that the expression of cyclin D1 and CDK4 were
upregulated whereas that of CDK6 was downregulated (Fig. 5E and F). This suggested that the
knockout of FOXL2 arrested cell cycle progression at the G0/G1
phase. Therefore, the results indicated that knocking out FOXL2 in
KGN cells inhibited the proliferation of cells and promoted cell
cycle arrest at the G0/G1 phase.
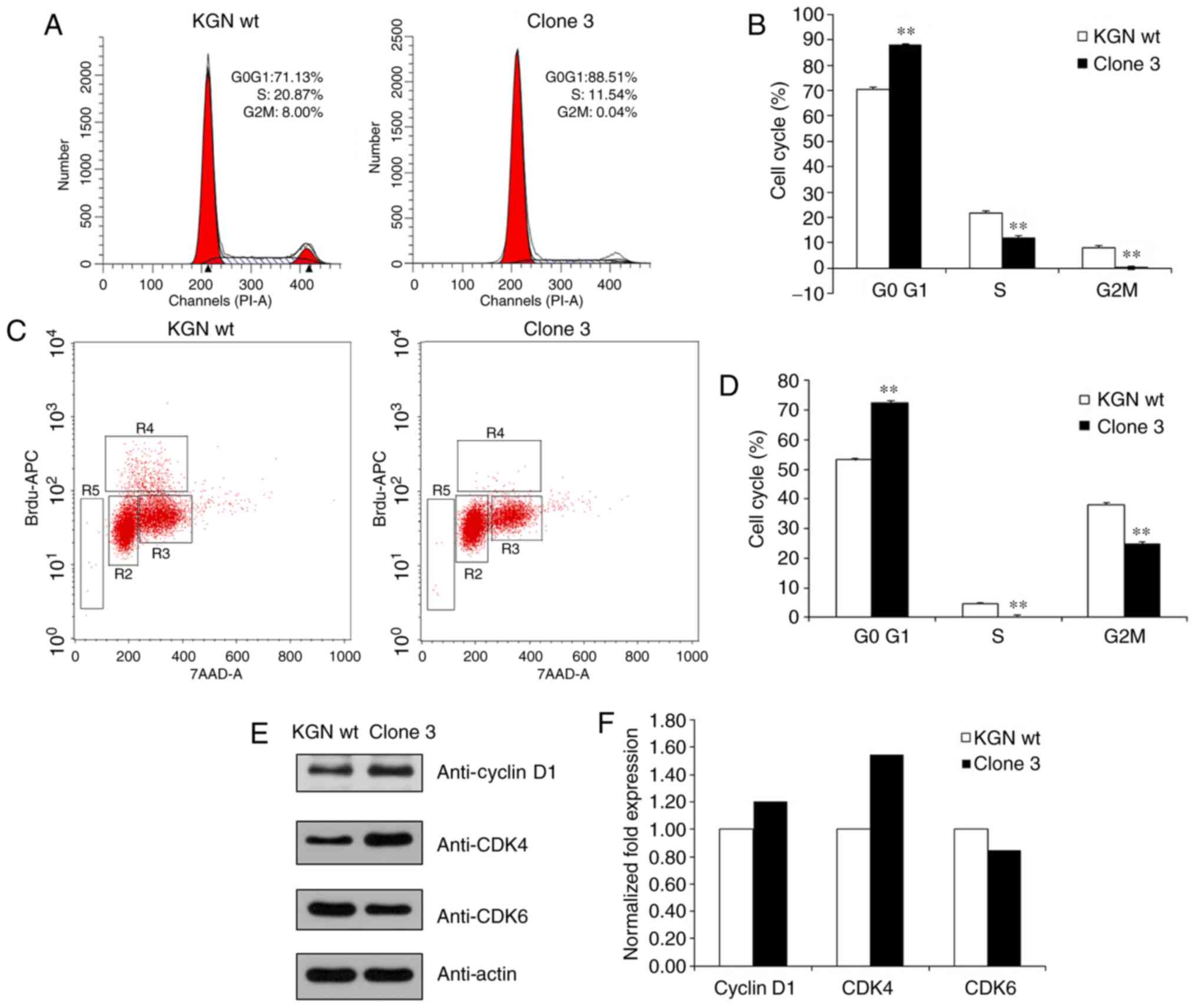 | Figure 5Knockout of FOXL2 attenuates the cell
cycle. (A) Analysis of cell cycle distribution of KGN cells with
FOXL2 knocked out. (B) Analysis of the cell cycle phases; the bar
graph showing the G0/G1, S, and G2/M phases. **P<0.01
vs. KGN wt. (C) Measurement of cell-incorporated BrdU (with APC
anti-BrdU) and total DNA content (with 7-AAD). The cells were
cultured with 10 μM of BrdU for 1 h. As shown by the regions
applied to the 7-AAD, vs. BrdU dot plot, flow cytometric analysis
of cells stained with the reagents provided in the BrdU Flow kit
allowed for the discrimination of cell subsets that were apoptotic
(R5, 0.17 or 0.05% of cells) or resided in the G0/G1 (R2, 82.79 or
85.73%), S (R4, 5.17 or 3.50%), or G2+M (R3, 10.77 or 9.38%) phases
of the cell cycle and had recently synthesized DNA. (D) Analysis of
the cell cycle phases; the bar graph shows the percentage of cells
in the G0/G1, S, and G2/M phases. **P<0.01 vs. KGN
wt. (E) Protein levels of cyclin D1, CDK4 and CDK6 were determined
by western blot analysis. (F) Levels of cyclin D1, CDK4, and CDK6
were quantified using the Quantity One software. Quantitative
analysis has been performed once. FOXL2, Forkhead box L2; wt,
wild-type; CDK, cyclin-dependent kinase. |
Discussion
In the present study, FOXL2 was disrupted using the
CRISPR method, and stable knockout in the KGN human ovarian
granulosa cell line was screened. During the investigations, three
sites (F404, F425 and F446) around the ATG start codon on FOXL2 DNA
sequence were constructed in gRNA lentivirus. The results showed
that F425-targeted gene editing had the highest efficiency among
the three candidates. Subsequently, KGN cells infected with F425
gRNA and Cas9 were screened for total FOXL2 knockout. The results
of western blot analysis and DNA sequencing identified that one of
the screened clones had both FOXL2 alleles fully disrupted. Further
investigation indicated that the knockout of FOXL2 inhibited
proliferation and restricted cell cycle progression at the G0/G1
phase. Furthermore, the expression levels of cell cycle mediators
cyclin D1 and CDK4 were reduced, which confirmed the disruption of
FOXL2 in this KGN cell clone, which may be associated with cell
cycle attenuation.
The stable cell line with knockout of FOXL2
demonstrated in the present study is likely to provide a useful
tool for further investigations into the mechanism of action of
FOXL2. FOXL2 is a single-exon gene which belongs to the forkhead
transcription factor family (1).
The protein sequence of FOXL2 contains a characteristic forkhead
box DNA-binding domain and a polyalanine tract of 14 residues of
unknown function, which is strictly conserved among eutherian
mammals (26). Germline mutations
of FOXL2 are responsible for BPES (MIM 110100). Previous studies
have also produced models in which the expression of FOXL2 is
knocked out. Using a Cre/lox approach, Tran et al (27) selectively ablated FOXL2 in murine
anterior pituitary gonadotrope cells and demonstrated that
conditional knockout mice developed overtly normally but were
subfertile in adulthood. Shi et al (28) disrupted the expression of FOXL2 in
mice using piggyBac insertion and successfully mimicked BPES
syndrome. Previous investigations in FOXL2-/- knockout
mice revealed that FOXL2 is critical for ovarian determination and
the proper differentiation of granulosa cells. However, the
detailed mechanisms by which FOXL2 is able to mediate cellular
processes remain to be fully elucidated.
Since the first mammalian applicable CRISPR/Cas9
study was published in 2013 (16), this technique has been
successfully used to disrupt several genes in a wide variety of
developing organisms (14,29-32).
In the present study, FOXL2 was disrupted with CRISPR and a stable
knockout cell line was screened in KGN human ovarian granulosa
cells. The overall GC content of FOXL2 cDNA is ~64% and, in its
coding region, this is >72%, which limits the gRNA selection due
to its high GC content around the ATG site. Fortunately, the free
online bioinformatical gRNA design tool (http://crispr.mit.edu/) developed by Hsu et
al(33) enabled the selection
of three potentially useful gRNA sites in the present study.
F425-targeted gene editing exhibited the highest efficiency among
the three candidates, and both FOXL2 alleles were fully disrupted
in one of the cell clones, as shown in the through western blot
analysis and DNA sequencing results. In this clone, the expression
of StAR was increased and that of CYP19A was decreased, as expected
when FOXL2 is disrupted (10).
Therefore, the results reported herein may provide a useful tool to
delineate the function of FOXL2 and its mutants in human
granulose-like cell lines.
In the present study, it was also found that the
expression levels of cell cycle mediators cyclin D1 and CDK4 were
reduced and, unexpectedly, CDK6 was increased. Cyclin D1 is
reported to be a key element in cell proliferation, whereas CDK4
and CDK6 preferably associate with D-type cyclins during the G1
phase (34). The flow cytometry
results indicated that the knockout of FOXL2 restricted cell cycle
progression at the G0/G1 phase but did not influence total cell
apoptosis. Therefore, these results suggest that the knockout of
FOXL2 induced cell cycle arrest. FOXL2 is able to form functional
heterodimers with the TGF-â effector transcription factor mothers
against decapentaplegic (Smad)3 (35), and an increase in the expression
of Smad3 alone can induce the accumulation of cells at G0/G1 arrest
through upregulation of the cell cycle progression inhibitors
p15/INK4b, p16/INK4a, p19/ARF and p21/CIP1 (36). Therefore, the activity of Smad3
relative protein may require further investigation in
FOXL2-knockout cells.
In conclusion, the present study established a
stable cell line with disrupted FOXL2 using the CRISPR method in
KGN human ovarian granulosa cells. The knockout of FOXL2 restricted
cell cycle progression at the G0/G1 phase, but did not influence
total cell apoptosis. The expression levels of cell cycle mediators
cyclin D1 and CDK4 were reduced, which confirmed that the
disruption of FOXL2 may be associated with cell cycle arrest.
Acknowledgments
The authors would like to thank Professor Yiming Mu
(Chinese PLA General Hospital, Beijing, China) for providing the
KGN human ovarian granulosa-like cells.
Funding
This study was funded by the National Natural
Science Foundation of China (grant no. 81471880).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors’ contributions
BT and YZhang performed the experiments,
participated in collecting data, and drafted the manuscript. WZ
performed the statistical analysis and participated in its design.
YZhu and SY contributed to study design, supervised the work,
analyzed data and assisted in drafting the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Crisponi L, Deiana M, Loi A, Chiappe F,
Uda M, Amati P, Bisceglia L, Zelante L, Nagaraja R, Porcu S, et al:
The putative forkhead transcription factor FOXL2 is mutated in
blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet.
27:159–166. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cocquet J, Pailhoux E, Jaubert F, Servel
N, Xia X, Pannetier M, De Baere E, Messiaen L, Cotinot C, Fellous M
and Veitia RA: Evolution and expression of FOXL2. J Med Genet.
39:916–921. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Loffler KA, Zarkower D and Koopman P:
Etiology of ovarian failure in blepharophimosis ptosis epicanthus
inversus syndrome: FOXL2 is a conserved, early-acting gene in
vertebrate ovarian development. Endocrinology. 144:3237–3243. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ottolenghi C, Omari S, Garcia-Ortiz JE,
Uda M, Crisponi L, Forabosco A, Pilia G and Schlessinger D: Foxl2
is required for commitment to ovary differentiation. Hum Mol Genet.
14:2053–2062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burns KH, Owens GE, Fernandez JM, Nilson
JH and Matzuk MM: Characterization of integrin expression in the
mouse ovary. Biol Reprod. 67:743–751. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Uhlenhaut NH, Jakob S, Anlag K,
Eisenberger T, Sekido R, Kress J, Treier AC, Klugmann C, Klasen C,
Holter NI, et al: Somatic sex reprogramming of adult ovaries to
testes by FOXL2 ablation. Cell. 139:1130–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schmidt D, Ovitt CE, Anlag K, Fehsenfeld
S, Gredsted L, Treier AC and Treier M: The murine winged-helix
transcription factor Foxl2 is required for granulosa cell
differentiation and ovary maintenance. Development. 131:933–942.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalfa N, Philibert P, Patte C, Ecochard A,
Duvillard P, Baldet P, Jaubert F, Fellous M and Sultan C:
Extinction of FOXL2 expression in aggressive ovarian granulosa cell
tumors in children. Fertil Steril. 87:896–901. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moumne L, Batista F, Benayoun BA,
Nallathambi J, Fellous M, Sundaresan P and Veitia RA: The mutations
and potential targets of the forkhead transcription factor FOXL2.
Mol Cell Endocrinol. 282:2–11. 2008. View Article : Google Scholar
|
|
10
|
Rosario R, Araki H, Print CG and Shelling
AN: The transcriptional targets of mutant FOXL2 in granulosa cell
tumours. PLoS One. 7:e462702012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Horvath P and Barrangou R: CRISPR/Cas, the
immune system of bacteria and archaea. Science. 327:167–170. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Terns MP and Terns RM: CRISPR-based
adaptive immune systems. Curr Opin Microbiol. 14:321–327. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wiedenheft B, Sternberg SH and Doudna JA:
RNA-guided genetic silencing systems in bacteria and archaea.
Nature. 482:331–338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai
SQ, Sander JD, Peterson RT, Yeh JR and Joung JK: Efficient genome
editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol.
31:227–229. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cong L, Ran FA, Cox D, Lin S, Barretto R,
Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA and Zhang F:
Multiplex genome engineering using CRISPR/Cas systems. Science.
339:819–823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Yang H, Shivalila CS, Dawlaty MM,
Cheng AW, Zhang F and Jaenisch R: One-step generation of mice
carrying mutations in multiple genes by CRISPR/Cas-mediated genome
engineering. Cell. 153:910–918. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang T, Wei JJ, Sabatini DM and Lander ES:
Genetic screens in human cells using the CRISPR-Cas9 system.
Science. 343:80–84. 2014. View Article : Google Scholar
|
|
18
|
Shalem O, Sanjana NE, Hartenian E, Shi X,
Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG and
Zhang F: Genome-scale CRISPR-Cas9 knockout screening in human
cells. Science. 343:84–87. 2014. View Article : Google Scholar :
|
|
19
|
Lee PC, Truong B, Vega-Crespo A, Gilmore
WB, Hermann K, Angarita SA, Tang JK, Chang KM, Wininger AE, Lam AK,
et al: Restoring ureagenesis in hepatocytes by CRISPR/Cas9-mediated
genomic addition to arginase-deficient induced pluripotent stem
cells. Mol Ther Nucleic Acids. 5:e3942016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tu Z, Yang W, Yan S, Guo X and Li XJ:
CRISPR/Cas9: A powerful genetic engineering tool for establishing
large animal models of neurodegenerative diseases. Mol
Neurodegener. 10:352015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X,
Xiong JW and Xi JJ: Genome editing with RNA-guided Cas9 nuclease in
zebrafish embryos. Cell Res. 23:465–472. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao S, He Z and Chen C:
CRISPR/Cas9-mediated genome editing of epigenetic factors for
cancer therapy. Hum Gene Ther. 26:463–471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gebler C, Lohoff T, Paszkowski-Rogacz M,
Mircetic J, Chakraborty D, Camgoz A, Hamann MV, Theis M, Thiede C
and Buchholz F: Inactivation of cancer mutations utilizing
CRISPR/Cas9. J Natl Cancer Inst. 109:pii: djw1832016. View Article : Google Scholar
|
|
24
|
Kawamura N, Nimura K, Nagano H, Yamaguchi
S, Nonomura N and Kaneda Y: CRISPR/Cas9-mediated gene knockout of
NANOG and NANOGP8 decreases the malignant potential of prostate
cancer cells. Oncotarget. 6:22361–22374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Verdin H and De Baere E: FOXL2 impairment
in human disease. Horm Res Paediatr. 77:2–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tran S, Zhou X, Lafleur C, Calderon MJ,
Ellsworth BS, Kimmins S, Boehm U, Treier M, Boerboom D and Bernard
DJ: Impaired fertility and FSH synthesis in gonadotrope-specific
Foxl2 knockout mice. Mol Endocrinol. 27:407–421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi F, Ding S, Zhao S, Han M, Zhuang Y, Xu
T and Wu X: A piggyBac insertion disrupts Foxl2 expression that
mimics BPES syndrome in mice. Hum Mol Genet. 23:3792–3800. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Doudna JA and Charpentier E: Genome
editing. The new frontier of genome engineering with CRISPR-Cas9.
Science. 346:12580962014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hsu PD, Lander ES and Zhang F: Development
and applications of CRISPR-Cas9 for genome engineering. Cell.
157:1262–1278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harrison MM, Jenkins BV, O’Connor-Giles KM
and Wildonger J: A CRISPR view of development. Genes Dev.
28:1859–1872. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sander JD and Joung JK: CRISPR-Cas systems
for editing, regulating and targeting genomes. Nat Biotechnol.
32:347–355. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hsu PD, Scott DA, Weinstein JA, Ran FA,
Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al: DNA
targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol.
31:827–832. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Blount AL, Schmidt K, Justice NJ, Vale WW,
Fischer WH and Bilezikjian LM: FoxL2 and Smad3 coordinately
regulate follistatin gene transcription. J Biol Chem.
284:7631–7645. 2009. View Article : Google Scholar :
|
|
36
|
Huang SM, Lu KT and Wang YC: ATM/ATR and
SMAD3 pathways contribute to 3-indole-induced G1 arrest
in cancer cells and xenograft models. Anticancer Res. 31:203–208.
2011.PubMed/NCBI
|