Introduction
Myocardial ischemia-reperfusion injury is considered
a more severe form of myocardial injury than ischemia alone, due to
aggravated oxidative stress (1).
Oxidative stress refers to an imbalance between pro-oxidants and
antioxidants (2), and is
associated with the pathogenesis of myocardial ischemia-reperfusion
injury.
To control the cellular redox environment, cells
contain two primary redox regulatory systems: The glutathione
(GSH)/GSH reductase (GSR)/GSH peroxidase (GPx)/glutaredoxin (Grx)
pathway, and the thioredoxin (Trx)/peroxiredoxin (Prx) pathway
(3). These two major
thiol-dependent antioxidant systems serve key roles in the defense
against oxidative stress (4,5).
Trx, Grx and Prx have been characterized as regulators of the
intracellular redox state, and are increasingly being recognized
for their specific roles in redox signaling. It has previously been
reported that these antioxidants regulate cell survival and death
through redox-dependent signal transduction pathways during
oxidative stress (2). Therefore,
the redox states of these antioxidants are critical for the fate of
cells under oxidative conditions.
Maintenance of the GSH redox couple, GSH/GSH
disulfide (GSSG), is achieved by recycling via the pentose
phosphate pathway and GSH biosynthesis. N-acetylcysteine (NAC),
which is a precursor of GSH, is widely used and has attracted great
interest as a thiol-containing antioxidant and modulator of the
intracellular redox state (6). In
addition, it has been demonstrated that repletion of GSH levels
through NAC protects against oxidative stress-induced cell death
though scavenging of free radicals (7). Furthermore, NAC can inhibit
oxidative stress-induced apoptosis of cardiomyo-cytes by decreasing
caspase-3 activity (8,9). Wang et al (10), revealed that NAC and allopurinol
reduce myocardial ischemia-reperfusion injury in diabetes by
activating the phosphoinositide 3-kinase (PI3K)/protein kinase B
(Akt) and Janus kinase 2/signal transducer and activator of
transcription 3 pathways. Kumar and Sitasawad demonstrated that NAC
prevents glucose/glucose oxidase-induced oxidative stress by
normalizing mitochondrial membrane potential, inhibiting cytochrome
c release, increasing B-cell lymphoma 2 (Bcl-2) expression,
decreasing Bcl-2-associated X protein expression and activating
procaspase-9 in H9c2 cells (11).
The present study investigated whether NAC protects
cardiomyocytes from oxidative damage by regulating the redox status
of intracellular antioxidant proteins. The results revealed that
NAC pretreatment alleviated the oxidation of intracellular
antioxidants, such as Trx1, Prx1 and GSR, in order to protect
cardiomyocytes from oxidative stress-induced apoptosis.
Materials and methods
Reagents and antibodies
NAC (cat. no. A9165), N-ethylmaleimide (NEM; cat.
no. E3876) and trichlo-roacetic acid (cat. no. T0699) were obtained
from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany, China).
Chloromethyl-dichlorofluorescein diacetate (CM-H2DCFDA; cat. no.
C6827) was purchased from Invitrogen; Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). Anti-Prx1 (cat. no. 8499), anti-Trx1 (cat.
no. 2429), anti-PTEN (cat. no. 9552), anti-green fluorescent
protein (GFP; cat. no. 2956S), anti-apoptosis signal-regulating
kinase 1 (ASK1; cat. no. 8662S), anti-Akt (cat. no. 9272S) and
anti-phosphorylated (p-ASK1; Thr845) (cat. no. 3765) and p-Akt
(Thr308) (cat. no. 9275) antibodies were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA), and anti-GSR (cat.
no. sc-133245) was obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Anti-GAPDH (cat. no. BM3874) was purchased from
Wuhan Boster Biological Technology Ltd. (Wuhan, Hubei, China).
Caspase-3 (cat. no. K106), caspase-8 (cat. no. K113) and caspase-9
(cat. no. K119) assay kits were purchased from BioVision, Inc.
(Milpitas, CA, USA).
Cell culture
H9c2 cells (cat. no. CRL-1446; American Type Culture
Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified
Eagle’s medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal calf serum (Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin and 100 µg/ml streptomycin in a
humidified atmosphere containing 5% CO2 at 37°C.
Subsequently, the cells were seeded at varying densities in 6-well
plates or 6-cm dishes, incubated for 24 h, and then treated with
NAC or H2O2 for the desired time periods.
MTT assay for cell viability
The MTT assay is a standard method used to assess
cell viability. Briefly, H9c2 cells were washed twice with PBS,
detached with 0.25% trypsin and made into a single cell suspension.
Cells were seeded into 96-well microtiter plates at a density of
3-5×103 cells/well in 200 µl culture medium.
Following incubation with 4 mM NAC for 1 h at 37°C, the cells were
treated with 0.75 mM H2O2 and were incubated
for various durations. Subsequently, 100 µl MTT solution
(0.5 mg/ml) was added to each well and the cells were incubated for
4 h at 37°C, after which, 150 µl dimethyl sulfoxide was
added to each well. The absorbance was measured at 570 nm, and the
values were used to calculate the relative ratio of viable cells to
total cells.
Apoptosis assay
H9c2 cells were pretreated with 4 mM NAC for 1 h, ad
were then treated with 0.75 mM H2O2 for 0, 12
and 24 h. A total of 2-3×104 cells were collected in a
tube and were washed twice with PBS. Subsequently, the cells were
centrifuged at 10,000 × g for 10 min at 4°C; the supernatant was
removed and the cell pellets were resus-pended in 500 µl
binding buffer containing 5 µl Annexin V-fluorescein
isothiocyanate and 5 µl propidium iodide (cat. no. 556547;
BD Pharmingen; BD Biosciences, Franklin Lakes, NJ, USA). Following
incubation at room temperature in the dark for 15 min, cell
apoptosis was assessed by fluorescence-activated cell sorting
(Beckman Coulter, Inc., Brea, CA, USA).
GSH and GSSG measurements
The levels of GSH and GSSG were measured using a GSH
assay kit (cat. no. 703002; Cayman Chemical Company, Ann Arbor, MI,
USA), which employs a spectrophotometric GSR recycling assay. H9c2
cells were pretreated with 4 mM NAC for 1 h, and were then treated
with 0.75 mM H2O2 for 0, 15 and 30 min. A
total of 3-5×106 cells were washed twice with chilled
PBS, scraped into cold buffer containing 0.2 M 2-(N-morpholino)
ethanesulfonic acid, 50 mM phosphate and 1 mM EDTA (pH 6.0),
sonicated (20-25 kHz) on ice twice for 20 sec, and centrifuged at
10,000 × g for 15 min at 4°C. The supernatant was removed and
deproteinated for analysis of total GSH and GSSG, according to the
manufacturer’s protocol. Reduced GSH levels were calculated from
the total GSH and GSSG levels. All determinations were normalized
to protein content, as determined using a bicinchoninic acid (BCA)
protein assay kit (cat. no. 23227; Thermo Fisher Scientific, Inc.).
The absorbance was measured at 405 nm using a plate reader at 5-min
intervals for 30 min.
ROS measurement
ROS production was measured using the cell-permeable
probe, CM-H2DCFDA. Cells were plated 24 h prior to analysis in
6-well plates. Cells were incubated with CM-H2DCFDA at a
concentration of 10 µM for 30 min at 37°C. Subsequently, the
cells were washed twice with PBS and incubated with 4 mM NAC for 1
h, followed by treatment with 0.75 mM H2O2
for 15 and 30 min. Fluorescence was quantified by flow cytometry
(Gallios; Beckman Coulter, Inc.) with excitation and emission
wavelengths of 485 and 530 nm, respectively.
Caspase-3, -8 and -9 activity assay
Caspase-3, -8 and -9 activities were measured using
caspase colorimetric assay kits (BioVision, Inc.). Briefly, total
cellular protein levels were quantified using the bicinchoninic
acid protein assay kit (cat. no. 23227; Thermo Fisher Scientific,
Inc.) and reacted with the corresponding substrates: DEVD-pNA,
IETD-pNA and LEHD-pNA. Caspase-3, -8 and -9 activities were
subsequently measured as the optical density of the cleaved
substrate, ρNA, at 405 nm using a microplate reader (ELX-800;
Bio-Tek Instruments, Inc., Winooski, VT, USA).
NEM-alkylated redox western blotting
For mitochondrial reduction-oxidation-sensitive
green fluorescent protein (mito-roGFP) analysis, 3-5×106
H9c2 cells were transiently transfected with mito-roGFP (provided
by Dr S. James Remington, University of Oregon, Eugene, OR, USA)
using Lipofectamine® 2000 (cat. no. 11668019;
Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h at 37°C.
H9c2 cells (3-5×106) were treated with
0.75 mM H2O2 for 0, 0.25, 0.5, 1, 2 and 3 h
and NEM-alkylated redox western blotting was performed to detect
the redox states of GSR and PTEN. Alternatively, following
incubation with 4 mM NAC for 1 h at 37°C, the cells
(3-5×106) were treated with 0.75 mM
H2O2 for 0, 15 and 30 min, and NEM-alkylated
redox western blotting was performed to detect the redox state of
mito-GFP, Prx1, Trx1, GSR and PTEN. The present study analyzed the
redox states of mito-GFP, Prx1, Trx1, GSR and PTEN using methods
reported by Poynton and Hampton (12). Cells were washed twice with
ice-cold PBS immediately after treatment. Subsequently, cells were
precipitated with chilled trichloroacetic acid (10%) for 30 min at
4°C, centrifuged at 12,000 × g for 10 min and washed twice with
100% acetone. The protein pellets were dissolved in nonreducing
buffer (100 mM Tris-HCl, pH 6.8; 2% SDS; and either 40 mM NEM for
Trx, GSR, GFP and PTEN or 100 mM NEM for Prx1). Trx1 redox forms
were separated by 17% non-reducing SDS-PAGE; Prx1, GSR and PTEN
redox forms were separated by 15% non-reducing SDS-PAGE. For
reducing SDS-PAGE, 5% dithiothreitol was added to the samples (30
or 50 µg protein was loaded onto gels) and the proteins were
transferred onto polyvinylidene fluoride membranes. Subsequently,
membranes were blocked with 5% dry milk in TBS-0.1% Tween-20 (TBST)
for 1 h at room temperature, and were incubated with the GFP
(1:1,000), GSR (1:1,000), Prx1 (1:1,000), Trx1 (1:1,000) and PTEN
(1:1,000) antibodies diluted in TBST and 0.2% bovine serum albumin
(cat. no. TS-38839; Thermo Fisher Scientific, Inc.) overnight at
4°C. After washing three times with PBS (15 min/wash), the
membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit secondary antibodies (1:5,000; cat no. BS13278;
Bioworld Technology, Inc., St. Louis Park, MN, USA) for 1 h at room
temperature. Bands were semi-quantified using ImageJ (1.48v)
software (National Institutes of Health, Bethesda, MD, USA).
SDS-PAGE and immunoblotting
To detect ASK, p-ASK, Akt and p-Akt expression, cell
lysates were extracted using 1X SDS buffer (0.5 M Tris-HCl, pH 6.8;
20% SDS; 10% glycerol). The concentration of the samples was
determined using a BCA protein assay kit (cat. no. 23227; Thermo
Fisher Scientific, Inc.). Subsequently, 30 or 50 µg protein
was separated by 12% glycine SDS-PAGE and transferred onto
polyvinylidene fluoride membranes. Subsequently, membranes were
blocked with 5% dry milk in TBST for 1 h at room temperature, and
were incubated with ASK (1:1,000), p-ASK (1:1,000), Akt (1:1,000),
p-Akt (1:1,000), GAPDH (1:1,000) and GFP (1:1,000) antibodies
diluted in TBST and 0.2% bovine serum albumin overnight at 4°C. The
subsequent immunoblotting steps were conducted as
aforementioned.
Statistical analysis
Each experiment was repeated three times. For
western blotting, one representative image is shown in the figures.
Results are presented as the means ± standard deviation.
Statistical significance was assessed by one-way analysis of
variance followed by Tukey’s multiple comparisons test using
GraphPad Prism Version 7.0 (GraphPad Software, Inc., La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
NAC pretreatment attenuates
H2O2-induced cytotoxicity, and inhibits
H2O2-induced activation of caspase-3, -8 and
-9 in H9c2 cells
To investigate the effects of NAC pretreatment on
H2O2-induced H9c2 cell death, cell viability
was measured based on the uptake and reduction of MTT to an
insoluble formazan dye. Compared with in the control group, the
viability of H9c2 cells was significantly decreased by
H2O2 in a dose- and time-dependent manner
(Fig. 1A and B). Based on these
results, 0.75 mM H2O2 was used in subsequent
experiments. As shown in Fig. 1C,
4 mM NAC pretreatment for 1 h
markedly enhanced cell viability during
H2O2-induced oxidative injury. The dose of
NAC was chosen based on previous in vitro studies (11,13).
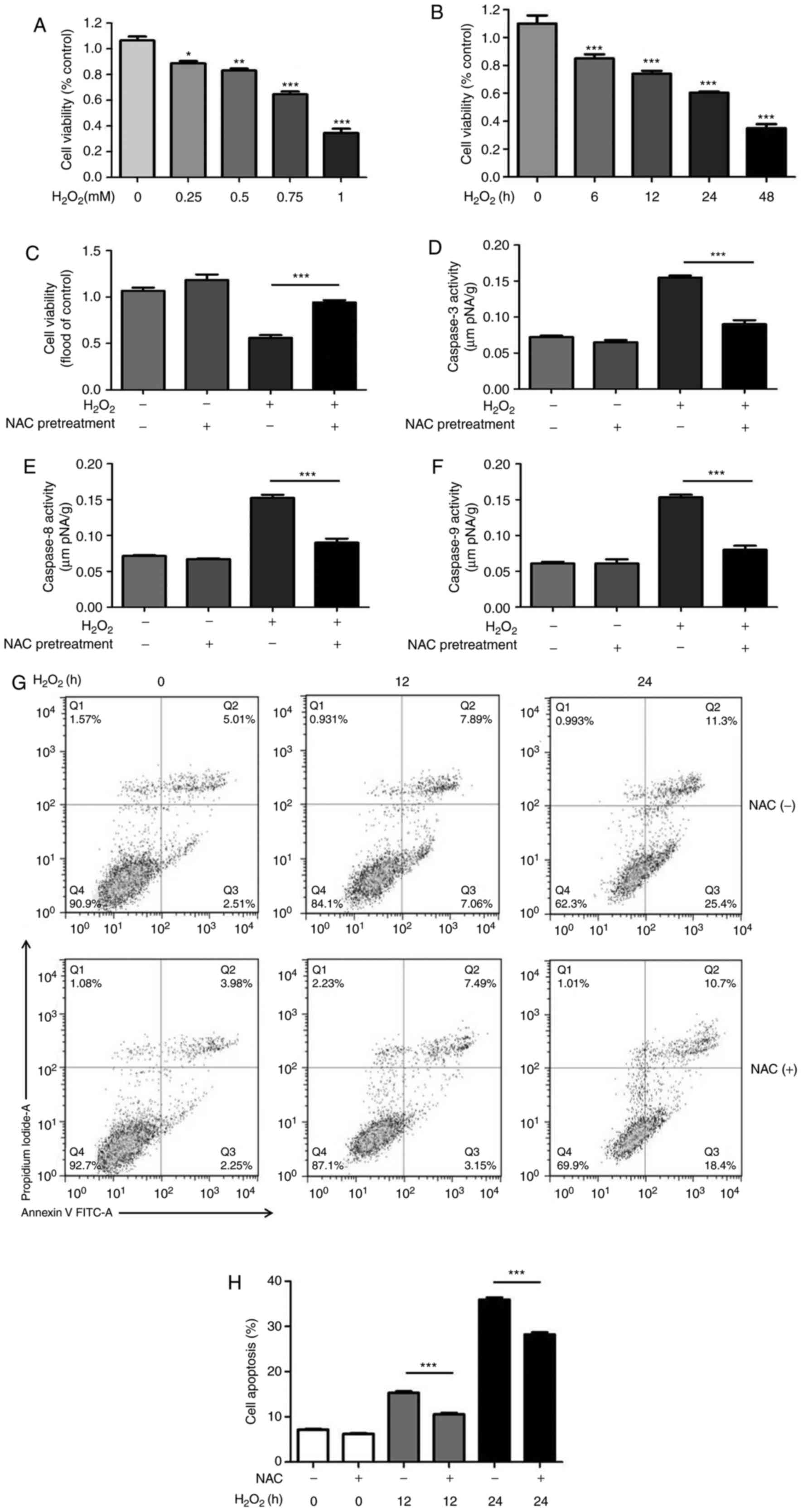 | Figure 1NAC pretreatment attenuates
H2O2-induced cytotoxicity. (A) H9c2 cells
were treated with various concentrations of
H2O2 (0.25, 0.5, 0.75 or 1.0 mM) for 24 h.
(B) H9c2 cells were treated with 0.75 mM H2O2
for 0, 6, 12, 24 and 48 h. MTT assay was used to measure cell
viability. Data are presented as the means ± standard deviation
(n=8 per group). (C) H9c2 cells were pretreated with 4 mM NAC for 1
h and were then incubated with 0.75 mM H2O2
for 24 h. MTT was used to measure cell viability. Data are
presented as the means ± standard deviation (n=8). Enzymatic
activities of (D) caspase-3, (E) caspase-8 and (F) caspase-9 were
measured using a colorimetric assay. (G and H) H9c2 cells were
pretreated with 4 mM NAC for 1 h and were then incubated with 0.75
mM H2O2 for the indicated times (0, 12 and 24
h), and flow cytometric analysis of cell apoptosis was conducted.
(G) Representative plots of flow cytometric analysis are presented.
(H) Results of flow cytometry indicated that NAC significantly
reduced the percentage of apoptotic H9c2 cells. Data are presented
as the means ± standard deviation (n=6). *P<0.05,
**P<0.01, ***P<0.001. FITC, fluorescein
isothiocyanate; H2O2, hydrogen peroxide; NAC,
N-acetylcysteine. |
To determine whether NAC pretreatment suppresses
H2O2-induced apoptosis, the activities of
initiator (caspase-8 and -9) and effector caspases (caspase-3) were
measured using colorimetric assays. The results revealed that NAC
pretreatment decreased the activation of caspase-3, -8 and -9
induced by H2O2 (Fig. 1D-F). To further confirm whether
NAC alleviates H2O2-induced H9c2 cell
apoptosis, flow cytometric analysis was conducted. The results
indicated that NAC significantly reduced the percentage of
apoptotic H9c2 cells (Fig. 1G and
H), thus suggesting that NAC pretreatment may protect H9c2 cell
from H2O2-induced cell death.
NAC regulates the redox state of the
cytoplasm and mitochondria in H9c2 cells
Increasing evidence has demonstrated that repletion
of GSH levels through NAC protects against oxidative stress-induced
cell death though the scavenging of free radicals. ROS levels were
measured by flow cytometry using the CM-H2DCFDA dye. As shown in
Fig. 2A, ROS levels in the
control and NAC groups were low. Conversely, increased ROS levels
were observed in the H2O2-stimulated cells,
thus indicating that oxidative stress was induced, whereas these
levels were significantly attenuated by NAC pretreatment. NAC is a
precursor of GSH; in order to determine the effects of NAC
pretreatment on intracellular redox state, total and reduced GSH
levels were determined using a spectrophotometric GSR recycling
assay. The results revealed that NAC pretreatment markedly
increased the levels of total and reduced GSH (Fig. 2B and C).
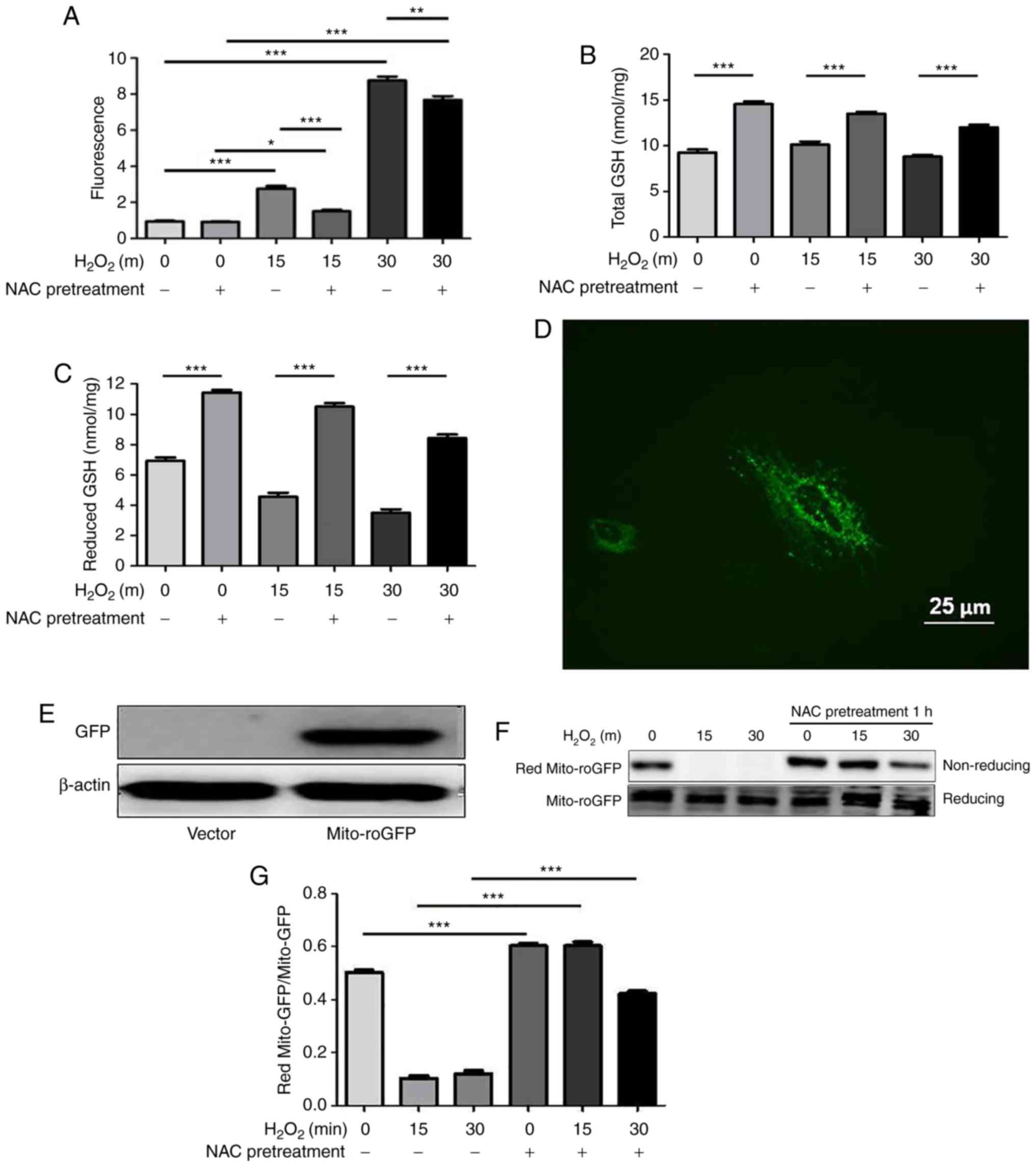 | Figure 2NAC pretreatment decreases levels of
reactive oxygen species and increases GSH under oxidative
conditions in H9c2 cells. (A) H9c2 cells were incubated with
CM-H2DCFDA dye at a concentration of 10 µM for 30 min at
37°C. Cells were pretreated with 4 mM NAC for 1 h and were then
treated with 0.75 mM H2O2 for the indicated
durations (15 and 30 min). Fluorescence was quantified using flow
cytometry with excitation and emission wavelengths of 485 and 530
nm, respectively. Values are presented as the means ± standard
deviation (n=6). Intracellular (B) total and (C) reduced GSH levels
were determined using a GSH reductase recycling assay. (D)
Immunofluorescence of mito-roGFP (green) in H9c2 cells
(magnification, x400). Scale bar, 25 µm. (E) Western
blotting revealed the expression levels of mito-roGFP and β-actin.
(F and G) H9c2 cells were transfected with mito-roGFP or vector for
48 h and were then treated with 0.75 mM H2O2
for the indicated times (15 and 30 min). Non-reducing western
blotting was performed to detect the redox state of mito-roGFP.
Data are presented as the means ± standard deviation (n=6).
*P<0.05, **P<0.01,
***P<0.001. GFP, green fluorescent protein; GSH,
glutathione; H2O2, hydrogen peroxide;
mito-roGFP, mitochondrial redox-sensitive GFP; NAC,
N-acetylcysteine. |
To directly assess the redox state in mitochondria,
mito-roGFP was used, as described in our previous study (13), whose fluorescent properties at two
excitation wavelengths change in response to formation of an
engineered disulfide bond. This method allows for dynamic
monitoring of the subcellular redox status of GSH/GSSG within
living cells. The present study transiently transfected
mitochondrial mito-roGFP into H9c2 cells. As shown in Fig. 2D-G, redox western blotting
revealed that NAC significantly increased the reduced forms of
mito-roGFP in response to oxidative stress. These results suggested
that NAC pretreatment may modify cytoplasmic and mitochondrial
redox states.
NAC pretreatment attenuates
H2O2-induced oxidation of Trx1, Prx1 and
GSR
Trx and Prx are important thiol peroxidases that
regulate intracellular ROS levels. Under oxidative stress, Prx or
Trx oxidation products accumulate in cells. The accumulation of
oxidized Trx/Prx may disrupt cellular redox homeostasis, promote
apoptotic signaling pathways and determine the fate of cells
(2). To determine the effects of
NAC pretreatment on the redox state of antioxidant proteins in H9c2
cells under oxidative conditions, the redox states of Trx1 and Prx1
were measured via non-reducing SDS-PAGE. As reported in our
previous study (13), NEM
derivatization can be used to identify reduced Trx1 since it
migrates faster than the oxidized form (Fig. 3A, upper panel). Following
exogenous application of H2O2, the reduced
form of Trx1 was decreased, thus suggesting an expected shift to
the oxidized state, which could not be detected under this
condition (Fig. 3A, upper panel).
Conversely, there was no difference in total Trx1 levels following
H2O2 treatment under reducing conditions
(Fig. 3A, lower panel). As shown
in Fig. 3A and B, reduced Trx1
was markedly increased by NAC pretreatment under oxidative
conditions. Similar to previous study (13), the oxidized form of Prx1 was
detected following H2O2 stimulation using the
NEM derivatization method (Fig.
3C, upper panel), which migrated slower than the reduced form
and disappeared under reducing conditions (Fig. 3C, lower panel). NAC pretreatment
significantly attenuated oxidation of Prx1 by
H2O2, as shown by a decrease in the oxidized
form of Prx1 (Fig. 3C, upper
panel and Fig. 3D).
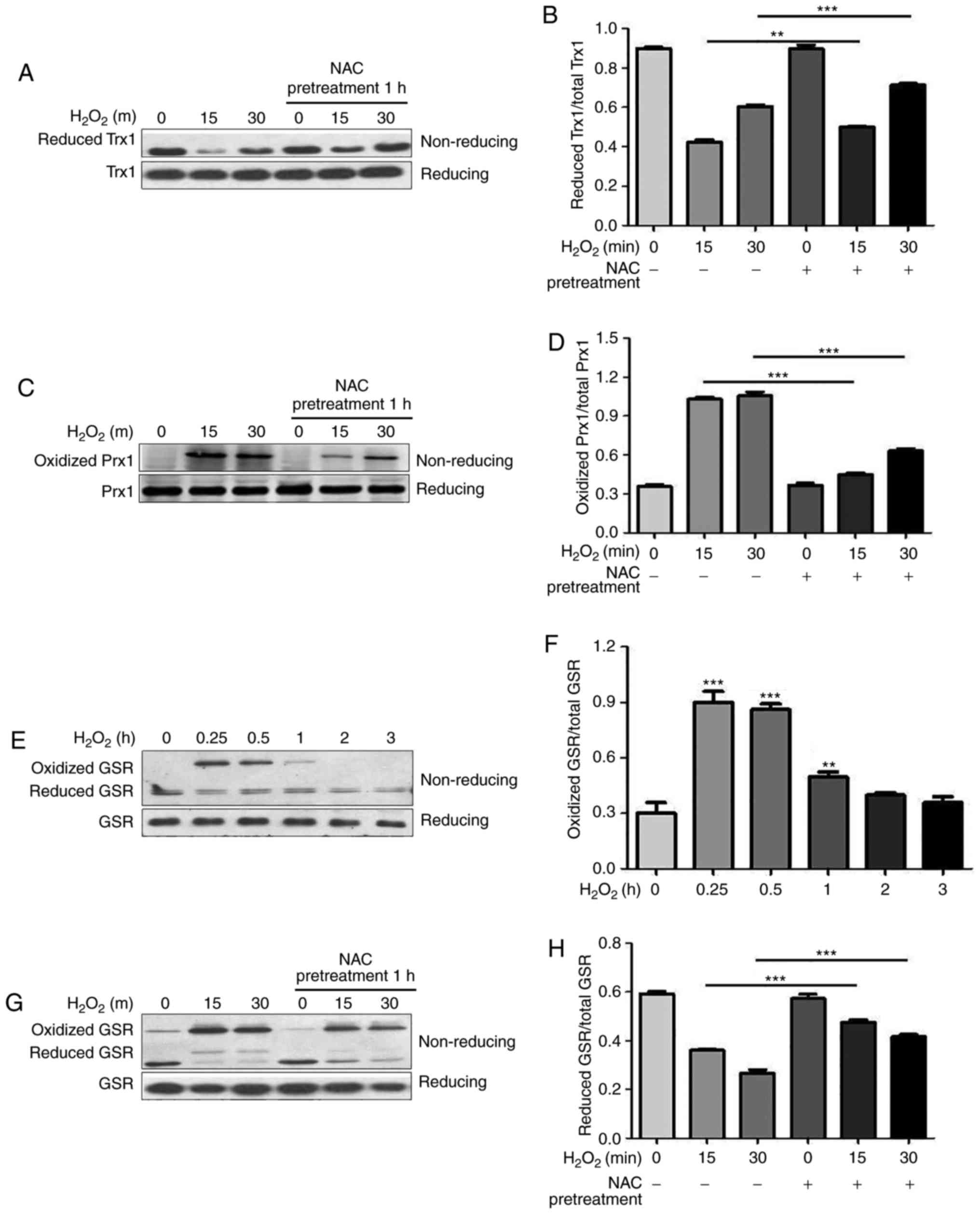 | Figure 3NAC pretreatment attenuates
H2O2-induced oxidation of Trx1, Prx1 and GSR,
and decreases phosphorylation of ASK1. N-ethylmaleimide-alkylated
redox western blotting was performed to detect the redox states of
(A and B) Trx1, (C and D) Prx1 and (E-H) GSR. For reducing
SDS-PAGE, 5% dithiothreitol was added to the samples. (B, D, F and
H) Reduced and oxidized forms of Trx1, Prx1 and GSR were
semi-quantified using ImageJ software. Data are presented as the
means ± standard deviation (n=3). (I) Western blotting was
performed to detect total and p-ASK1. (J) p-ASK1 was
semi-quantified using ImageJ software. Data are presented as the
means ± standard deviation (n=3). **P<0.01,
***P<0.001 as indicated, or vs. the 0 h
H2O2 group. ASK1, apoptosis signal-regulating
kinase 1; GSR, glutathione reductase; H2O2,
hydrogen peroxide; NAC, N-acetylcysteine; p, phosphorylated; Prx1,
peroxiredoxin 1; Trx1, thioredoxin 1. |
GSR is an enzyme that catalyzes the reduction of
GSSG to GSH, which is a critical molecule in resisting oxidative
stress and maintaining the reducing environment of the cell. The
present study demonstrated that GSR immediately underwent oxidation
in response to H2O2 and returned to its
reduced form 1 h following H2O2 treatment
(Fig. 3E and F). Redox
western blotting revealed that GSR displayed a high molecular
weight band (~150 kDa) following stimulation with
H2O2 using the NEM derivatization method
(Fig. 3G, upper panel), which
disappeared under reducing conditions (Fig. 3E, lower panel). NAC pretreatment
significantly shifted GSR levels from the oxidized to the reduced
form (Fig. 3G, upper panel and
Fig. 3H). The redox states of
Trx, Prx and GSR were modified by NAC pretreatment during oxidative
stress, thus indicating that the cytoprotective function of NAC is
associated with regulation of the redox state of intracellular
antioxidants.
NAC pretreatment decreases
H2O2-induced phosphorylation of ASK1
ASK1, which is a serine/threonine protein kinase, is
a ROS-sensitive mitogen-activated protein kinase (MAPK) kinase
kinase and a key mediator in cell death. The active form of ASK1
(p-ASK1) functions as a signaling node for several MAPK kinases,
resulting in activation of p38MAPK and c-Jun N-terminal kinase
(JNK), and regulation of cell survival and death (14). Trx1 has been reported to directly
bind to the N-terminal regulatory domain of ASK1, resulting in
inhibition of apoptosis. The interaction between Trx1 and ASK1 is
highly dependent on the redox status of Trx. Reduced Trx1 interacts
with and inhibits ASK1 activity, whereas oxidized Trx1 has
decreased affinity for ASK1 (15). The present study revealed that NAC
pretreatment increased the levels of reduced Trx1 under oxidative
conditions; therefore, this study investigated whether NAC
pretreatment influences H2O2-induced
phosphorylation of ASK1 in H9c2 cells. As shown in Fig. 3I, phosphorylation of ASK1 was
induced following treatment with H2O2 for 15
min. Conversely, NAC pretreatment alleviated activation of ASK1
(Fig. 3J), which is consistent
with the increased levels of reduced Trx1 (Fig. 3A).
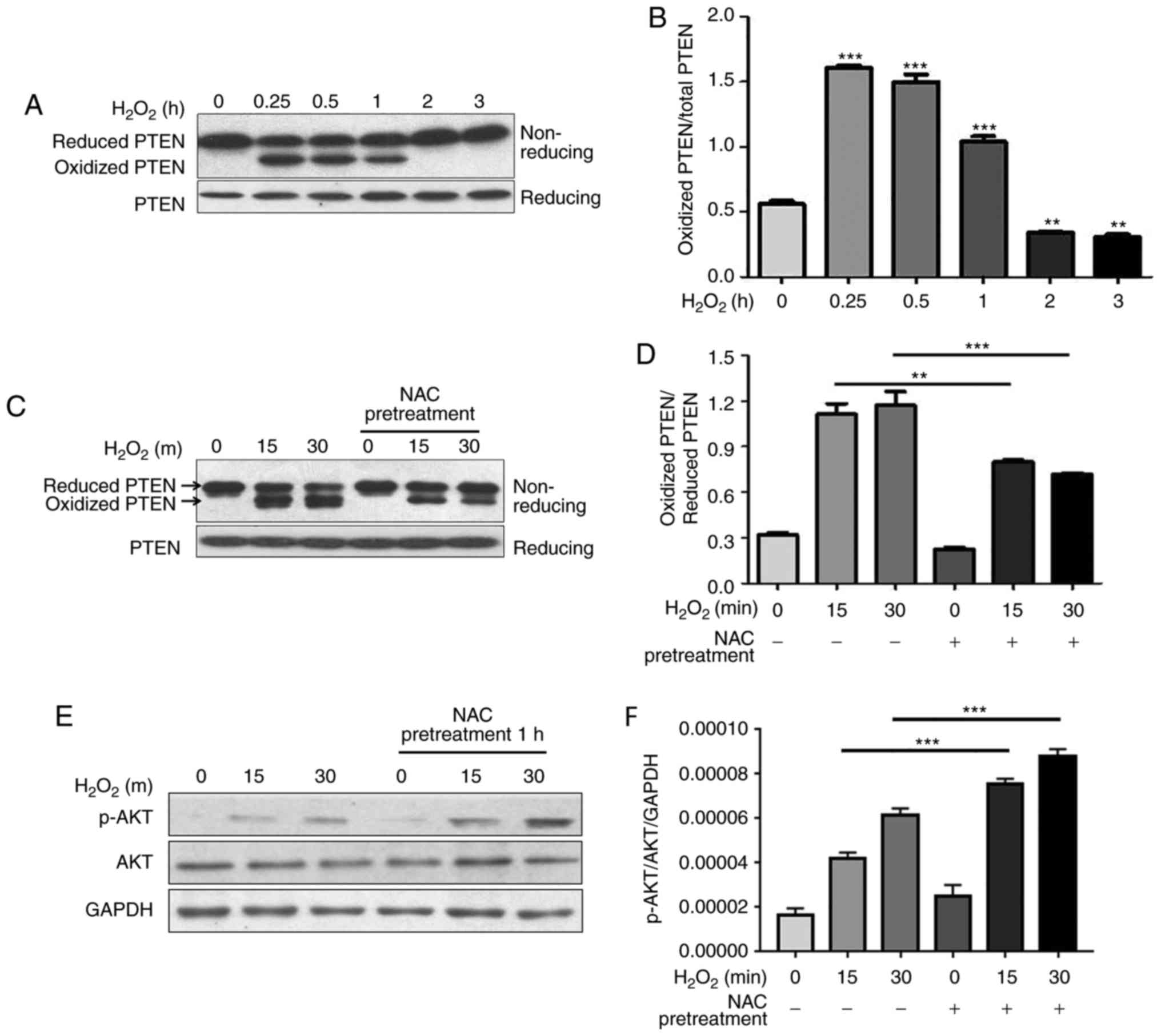
NAC pretreatment decreases
H2O2-induced oxidation of PTEN and increases
H2O2-induced phosphorylation of AKT
The tumor suppressor PTEN is a phosphatase that
dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate, which is the lipid
product of the class I PI3Ks, and suppresses growth and
proliferation of several cell types (16). The phosphatase activity of PTEN is
redox-dependent and is inhibited by
H2O2-induced transient oxidation of PTEN,
which subsequently activates PI3K-dependent signaling, including
AKT phosphorylation, and promotes cell survival (17). Cao et al and Schwertassek
et al reported that Trx1 and Prx1 increase the reduced form
of PTEN by protecting it from oxidation-induced inactivation
(18,19). The present study demonstrated that
NAC pretreatment protected Trx1 and Prx1 from
H2O2-induced oxidation. In the present study,
the redox state of PTEN and phosphorylation of AKT were
investigated. The present results demonstrated that PTEN underwent
oxidation following H2O2 stimulation in H9c2
cells, and the reduced form was recovered ~2 h after
H2O2 treatment (Fig. 4A and B). Notably, the levels of
H2O2-induced oxidized PTEN were decreased by
NAC pretreatment, which corresponded with an increase in the
reduced form (Fig. 4C and D).
Unexpectedly, phosphorylation of AKT was significantly increased by
NAC pretreatment (Fig. 4E and
F).
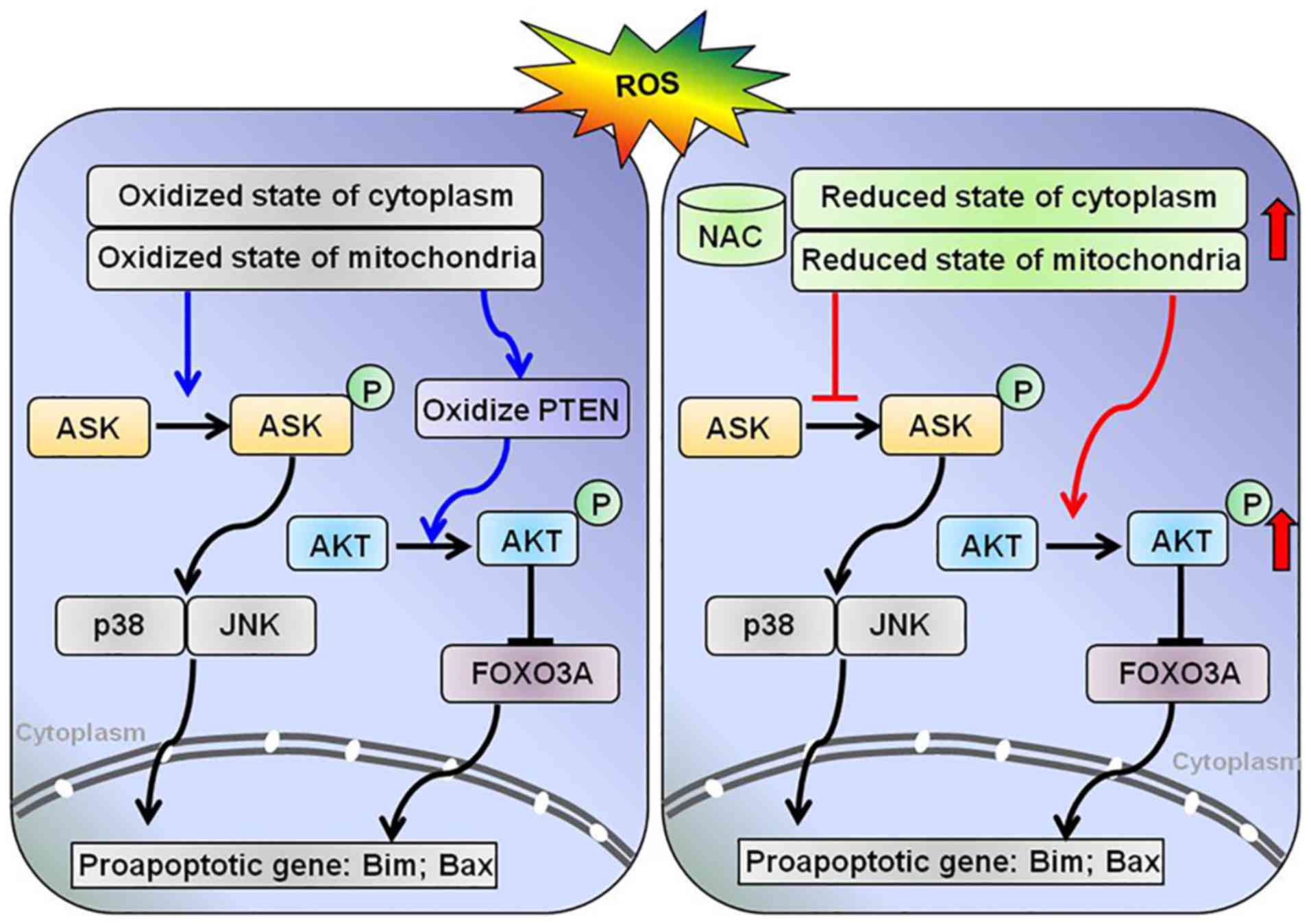
Discussion
The antioxidant activity of NAC results from its
free radical-scavenging properties, which occur either directly by
modulating the redox potentials of thiols, or by increasing
cellular levels of GSH. The present study revealed that NAC
pretreatment protected H9c2 cells from
H2O2-induced apoptosis by modifying the
intracellular redox state and modulating downstream redox signaling
pathways, including the proapoptotic kinase ASK1 and the
prosurvival kinase AKT (Fig.
5).
Oxidative stress is caused by an imbalance between
reduced and oxidized biomolecules within cells, in favor of the
latter, thus resulting in alterations that are deleterious to vital
cell functions (20). As a
consequence, numerous cellular compounds undergo redox
modifications, some of which function in signaling transduction.
Numerous key regulators of redox signaling are members of the Trx
family, including Trx1 and Prx1. Trx1 and Prx1 are characterized by
their active site motifs that contain one or two cysteine residues
(2). These thiol groups are
essential for the reduction of protein disulfide bonds,
deglutathionylation and denitrosylation. During these processes,
the active sites of Trx form a protein disulfide bond. Usually,
oxidation of Trx is reduced by Trx reductase, which uses NADPH as a
cofactor (21). The present study
demonstrated that repletion of GSH by NAC attenuated the oxidation
of Trx1 under oxidative conditions in H9c2 cells. Szadkowski and
Myers also reported that pretreatment of endothelial cells with NAC
resulted in partial protection of Trx1 from oxidation by acrolein,
a reactive aldehyde (22). The
underlying mechanism is possibly due to decreased formation of
protein disulfides in the active sites of Trx1 or increased
reduction of oxidized Trx1 by Trx reductase.
ASK1 is activated by ROS via Trx1 and/or Grx1.
Reduced Trx1 and Grx1 bind to the N- and C-terminal domains of
ASK1, respectively, thereby inhibiting its activation.
Stress-induced oxidation of Trx1 or Grx1 leads to their
dissociation from the complex and subsequent activation of the
kinase (15,23,24). In addition, binding of Trx1 to
ASK1 targets the kinase for degradation via the ubiquitin pathway
(25). Notably, this study
demonstrated that NAC pretreatment attenuated
H2O2-induced oxidation of Trx1 and Prx1, and
decreased phosphorylation of ASK1, which may subsequently result in
a reduction in caspase-3, -8 and -9 activities. This finding may be
associated with inhibition of the p38/JNK signaling pathway.
The GSH/GPx/Grx signaling cascade is another
important antioxidant pathway (26). GPx and Grx use reduced GSH to
reduce hydrogen/lipid peroxide or oxidation products; during these
processes, reduced GSH is oxidized to GSSG; GSSG can be reduced
back to GSH by GSR and NADPH. Oxidation of GSR may contribute to
the increased levels of GSSG and the decreased GSH/GSSG ratio
induced by H2O2. The present study revealed
that the oxidized form of GSR was decreased in the NAC pretreatment
group, which indicated that redox regulation of GSR may be
important to intracellular redox status.
The oxidation of PTEN induced by
H2O2 activates the AKT signaling pathway and
protects cells from oxidative stress-induced cell death. In the
present study, pretreatment with NAC markedly attenuated PTEN
oxidation induced by H2O2, which was expected
to decrease the phosphorylation of AKT. However, the expression
levels of p-AKT were increased by NAC pretreatment. The
phosphorylation of AKT is necessary for its anti-apoptotic function
and cell survival signal transduction in response to oxidative
stress. These data indicated that the phosphorylation of AKT under
oxidative conditions is regulated by both oxidation of PTEN and
other factors. Murata H et al reported that, under oxidative
stress, AKT is transiently phosphorylated and undergoes disulfide
bond formation between Cys-297 and Cys-311; dephosphorylation
corresponds with an increased association with protein phosphatase
2A in H9c2 cells. In addition, Grx may protect AKT from
H2O2-induced oxidation and maintain
phosphorylation of AKT, thus inhibiting apoptosis (27). The redox status of Grx was
difficult to detect in the present study, due to the poor quality
of the Grx antibody and the lack of an appropriate redox blot
protocol for Grx. The AKT signaling pathway serves a pivotal role
in inhibiting apoptosis through activation of downstream effector
molecules. Transcription factor forkhead box O3 (FoxO3a) is one of
the most important downstream targets of AKT signaling and a
crucial regulator of pro-apoptotic genes, such as Bcl-2-like
protein 11 (28,29). It has been reported that
inhibition of AKT promotes FOXO3a-dependent apoptosis in several
cell types (30). Based on these
findings, it may be hypothesized that NAC inhibits
H2O2-induced apoptosis via the AKT/FOXO3a
signaling pathway (Fig. 5).
Physiological, homeostatic and intracellular ROS are
maintained at low levels by various enzyme systems. ROS are
becoming increasingly appreciated as second messengers and
signaling molecules that regulate various physiological responses
(31). At excessive
concentrations, H2O2 can cause biological
damage. Conversely, it also promotes oxidation-induced inactivation
of phosphatases, such as PTEN, thus mediating cell survival signal
transduction. Oxidative stress is originally defined as an
imbalance between the production of oxidants or ROS, and their
elimination by antioxidants. The redefinition of ‘oxidative stress’
is based on alterations in observable post-translational protein
thiol modifications that are central to redox regulation and
control (26). Redox western blot
analysis and redox-sensitive GFPs provide means to quantify
thiol/disulfide redox alterations in specific subcellular
compartments (26). It is
important to analyze the redox modification of protein thiols to
understand the role of redox modifications on protein function.
In conclusion, to the best of our knowledge, the
present study is the first to reveal that NAC pretreatment
alleviates oxidation of intracellular antioxidant proteins, in
order to inhibit oxidative stress-induced cardiomyocyte apoptosis.
These findings may broaden the clinical applications of NAC in
ROS-asssociated diseases, such as myocardial infarction and
Alzheimer’s disease.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81270279 and
81471897) and the Hunan Natural Science Foundation (grant no.
2013JJ1009).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
HZ conceived the study and designed the experiments.
XL, LW and JC performed the experiments. KL, ML and HW collected
and analyzed the experimental results. XL drafted and revised the
article. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanschmann EM, Godoy JR, Berndt C,
Hudemann C and Lillig CH: Thioredoxins, glutaredoxins, and
peroxire-doxins-molecular mechanisms and health significance: From
cofactors to antioxidants to redox signaling. Antioxid Redox
Signal. 19:1539–1605. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holmgren A, Johansson C, Berndt C, Lönn
ME, Hudemann C and Lillig CH: Thiol redox control via thioredoxin
and glutare-doxin systems. Biochem Soc Trans. 33:1375–1377. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lillig CH, Berndt C and Holmgren A:
Glutaredoxin systems. Biochim Biophys Acta. 1780:1304–1317. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu J and Holmgren A: The thioredoxin
antioxidant system. Free Radic Biol Med. 66:75–87. 2014. View Article : Google Scholar
|
|
6
|
Samuni Y, Goldstein S, Dean OM and Berk M:
The chemistry and biological activities of N-acetylcysteine.
Biochim Biophys Acta. 1830.4117–4129. 2013.
|
|
7
|
Mayer M and Noble M: N-acetyl-L-cysteine
is a pluripotent protector against cell death and enhancer of
trophic factor-mediated cell survival in vitro. Proc Natl Acad Sci
USA. 91:7496–7500. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peng YW, Buller CL and Charpie JR: Impact
of N-acetylcysteine on neonatal cardiomyocyte ischemia-reperfusion
injury. Pediatr Res. 70:61–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duan JL, Wang JW, Guan Y, Yin Y, Wei G,
Cui J, Zhou D, Zhu YR, Quan W, Xi MM and Wen AD: Safflor yellow A
protects neonatal rat cardiomyocytes against anoxia/reoxygenation
injury in vitro. Acta Pharmacol Sin. 34:487–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang T, Mao X, Li H, Qiao S, Xu A, Wang J,
Lei S, Liu Z, Ng KF, Wong GT, et al: N-Acetylcysteine and
allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via
adiponectin and attenuated myocardial postischemic injury in
diabetes. Free Radic Biol Med. 63:291–303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar S and Sitasawad SL: N-acetylcysteine
prevents glucose/glucose oxidase-induced oxidative stress,
mitochondrial damage and apoptosis in H9c2 cells. Life Sci.
84:328–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poynton RA and Hampton MB: Peroxiredoxins
as biomarkers of oxidative stress. Biochim Biophys Acta.
1840.906–912. 2014.
|
|
13
|
Zhang H, Limphong P, Pieper J, Liu Q,
Rodesch CK, Christians E and Benjamin IJ: Glutathione-dependent
reductive stress triggers mitochondrial oxidation and cytotoxicity.
FASEB J. 26:1442–1451. 2012. View Article : Google Scholar :
|
|
14
|
Nishida K and Otsu K: The role of
apoptosis signal-regulating kinase 1 in cardiomyocyte apoptosis.
Antioxid Redox Signal. 8:1729–1736. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saitoh M, Nishitoh H, Fujii M, Takeda K,
Tobiume K, Sawada Y, Kawabata M, Miyazono K and Ichijo H: Mammalian
thioredoxin is a direct inhibitor of apoptosis signal-regulating
kinase (ASK)1. EMBO J. 17:2596–2606. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leslie NR, Kriplani N, Hermida MA,
Alvarez-Garcia V and Wise HM: The PTEN protein: Cellular
localization and post-translational regulation. Biochem Soc Trans.
44:273–278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leslie NR, Bennett D, Lindsay YE, Stewart
H, Gray A and Downes CP: Redox regulation of PI 3-kinase signalling
via inactivation of PTEN. EMBO J. 22:5501–5510. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao J, Schulte J, Knight A, Leslie NR,
Zagozdzon A, Bronson R, Manevich Y, Beeson C and Neumann CA: Prdx1
inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J.
28:1505–1517. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schwertassek U, Haque A, Krishnan N,
Greiner R, Weingarten L, Dick TP and Tonks NK: Reactivation of
oxidized PTP1B and PTEN by thioredoxin 1. FEBS J. 281:3545–3558.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Harris C and Hansen JM: Oxidative stress,
thiols, and redox profiles. Methods Mol Biol. 889:325–346. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Whayne TF Jr, Parinandi N and Maulik N:
Thioredoxins in cardiovascular disease. Can J Physiol Pharmacol.
93:903–911. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szadkowski A and Myers CR: Acrolein
oxidizes the cytosolic and mitochondrial thioredoxins in human
endothelial cells. Toxicol. 243:164–176. 2008. View Article : Google Scholar
|
|
23
|
Song JJ and Lee YJ: Differential role of
glutaredoxin and thioredoxin in metabolic oxidative stress-induced
activation of apoptosis signal-regulating kinase 1. Biochem J.
373:845–853. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song JJ, Rhee JG, Suntharalingam M, Walsh
SA, Spitz DR and Lee YJ: Role of glutaredoxin in metabolic
oxidative stress. Glutaredoxin as a sensor of oxidative stress
mediated by H2O2. J Biol Chem.
277:46566–46575. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y and Min W: Thioredoxin promotes ASK1
ubiquitination and degradation to inhibit ASK1-mediated apoptosis
in a redox activity-independent manner. Circ Res. 90:1259–1266.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hansen JM, Go YM and Jones DP: Nuclear and
mitochondrial compartmentation of oxidative stress and redox
signaling. Annu Rev Pharmacol Toxicol. 46:215–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Murata H, Ihara Y, Nakamura H, Yodoi J,
Sumikawa K and Kondo T: Glutaredoxin exerts an antiapoptotic effect
by regulating the redox state of Akt. J Biol Chem. 278:50226–50233.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu D, Liang M, Dang H, Fang F, Xu F and
Liu C: Hydrogen protects against hyperoxia-induced apoptosis in
type II alveolar epithelial cells via activation of PI3K/Akt/Foxo3a
signaling pathway. Biochem Biophys Res Commun. 495:1620–1627. 2018.
View Article : Google Scholar
|
|
29
|
Wang YQ, Cao Q, Wang F, Huang LY, Sang TT,
Liu F and Chen SY: SIRT1 protects against oxidative stress induced
endothelial progenitor cells apoptosis by inhibiting FOXO3a via
FOXO3a ubiquitination and degradation. J Cell Physiol.
230:2098–2107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Das TP, Suman S, Alatassi H, Ankem MK and
Damodaran C: Inhibition of AKT promotes FOXO3a-dependent apoptosis
in prostate cancer. Cell Death Dis. 7:e21112016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rhee SG, Bae YS, Lee SR and Kwon J:
Hydrogen peroxide: A key messenger that modulates protein
phosphorylation through cysteine oxidation. Sci STKE.
2000:pe12000.
|