Introduction
Ischemic cerebrovascular disease poses an increasing
threat to human health due to its high rate of incidence and
resulting disability (1). Rate of
recurrence and mortality associated with ischemic cerebrovascular
disease in Beijing, China was estimated as 27% (1). Cerebral ischemia/reperfusion injury
is the leading cause of poor prognosis and high rate of severe
disability observed in the clinical practice (2-4).
The pathophysiology of this disease involves a variety of immune
cells of the central nervous system, mainly microglia, which
excessively activate and release a large quantity of oxygen free
radicals, inflammatory cytokines and other pro-inflammatory
compounds. The inflammatory injury then leads to neuronal death and
increases the damage resulting from the stroke (5-7).
Therefore, the focus of stroke research has become the
identification of an effective drug that can suppress inflammation
and improve patient prognosis.
Propofol is widely used in clinical practice as an
intravenous anesthetic due to its rapid induction and recovery
times (8). Recent research has
identified that the mechanism of action of propofol in sedation may
be associated with adenosine receptors (9,10).
Propofol inhibits adenosine reabsorption and increases the
concentrations of extracellular adenosine (11). Adenosine receptors A1 and A2 are
widely present in brain tissues. Subsequent to activation by
adenosine, A1 and A2 receptors exert neuroprotective effects by
increasing the intracellular cyclic adenosine monophosphate level,
promoting glycogen decomposition, inhibiting the activation of
central nervous system immune cells and improving the utilization
rate of metabolic substrates (12-14).
Therefore, in the present study, it was hypothesized
that propofol may activate the A2b receptor and inhibit microglial
activation, thereby reducing inflammatory injury following cerebral
infarction. Through a number of in vitro and in
vivoexperiments, alterations in the microglia activation
conditions, the levels of cytotoxic molecules and the expression of
inflammatory factors were detected following treatment with
propofol.
Materials and methods
Microglia isolation and culture
All procedures involving animals were reviewed and
approved by the Institutional Clinical Experiments Committee and
Institutional Review Board of the Hainan Medical University
(Haikou, China). A total of 90 male Sprague-Dawley (SD) rats
weighing 250-300 g were sacrificed by overdose of anesthetic at 3
days after birth. The microglia isolation procedure was performed
as previously described (15,16). Briefly, brains were removed and
washed with sterile phosphate-buffered saline (PBS). Meninges and
brain blood vessels were stripped, and the bilateral cerebral
cortex was cut into sections and digested for 10 min in trypsin.
Next, the cells were filtered (port size, 70 µm), seeded on
poly-L-lysine-coated flasks at a density of 4×105/ml and
cultured in Dulbecco’s modified Eagle’s medium (DMEM)-F12 medium
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% fetal bovine serum (FBS; Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C. After the cells had grown to cover the
bottom of the culture flasks, the flasks were shaken using a
rotatory shaker (Cellnest Shaker; Sino-Biotop, Shanghai, China) at
a speed of 200 rpm for 2 h at 37°C. Subsequently, cells
(1×106/ml) were collected and seeded into 250 ml flasks
and 6-well culture plates. Non-adherent cells were washed away
after 1 h, and fresh medium was added into all the culture flasks
and plates.
For in vitro experiments, five groups of
microglia were stimulated for 24-48 h respectively with 1
µg/ml lipopoly-saccharide (LPS; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany; n=8), with LPS and 1, 3 or 5 µg/ml
propofol (AstraZeneca plc, Cambridge, UK; n=8), or with LPS, 5
µg/ml propofol and 100 µM MRS agar (Sigma-Aldrich;
Merck KGaA) (n=8). The control group was only treated with DMEM/F12
medium (n=8).
Reactive oxygen species (ROS) and nitric
oxide (NO) detection
For ROS detection, pretreated microglia in each
group were incubated with the molecular probe
2′,7′-dichlorodihydro-fluorescein diacetate (DCFH-DA; 10 µM;
Sigma-Aldrich; Merck KGaA) in serum-free medium at 37°C for 1 h in
the dark and then washed twice with PBS (17,18). ROS levels were measured at 492/520
nm using a microplate reader (Synergy HT; BioTek Instruments, Inc.,
Winooski, VT, USA).
The total NO levels in microglia were measured using
the Griess reagent kit (Invitrogen; Thermo Fisher Scientific,
Inc.). Pretreated microglia were incubated with Griess reagent in
serum-free medium at 37°C for 20 min in the dark and then washed
twice with PBS (19). The control
group only contained complete medium and Griess reagent. NO levels
were measured at 540 nm using the microplate reader.
Immunofluorescence staining of
F-actin
Microglia from each test group were respectively
harvested, seeded (1×106/ml) and stimulated on glass
cover slips in 12-well poly-L-lysine-coated culture plates at 37°C.
Following fixation and permeabilization, 3% goat serum (R&D
Systems, Inc., Minneapolis, MN, USA) was used to block non-specific
microglial proteins for 0.5 h at room temperature. Subsequently,
cells were stained with 5 µg/ml FITC-phalloidin (Invitrogen;
Thermo Fisher Scientific, Inc.) at 24°C for 1 h and washed with PBS
(20-22). Next, 100 ng/ml DAPI (Invitrogen;
Thermo Fisher Scientific, Inc.) was added into the culture plates,
and the nuclei were stained for 15 min. Finally, cells were imaged
using a confocal imaging system (Leica TCS SPE; Leica Microsystems
GmbH, Wetzlar, Germany).
MTT assay
Microglia from each test group were seeded on glass
coverslips in 96-well culture plates (1×104 cells per
well) at 37°C. Next, 5% MTT solution (0.2 mg/ml; Sigma-Aldrich;
Merck KGaA) was added into each well (20 µl per well) on the
following day. After 4 h, dimethyl sulfoxide (D4540; Sigma-Aldrich;
Merck KGaA) was added to each well (200 µl per well).
Finally, a microplate reader was used to measure the absorbance of
each well at 490 nm.
In vitro migration assay
Microglia were harvested and seeded into the upper
chamber of a 12-well Transwell plate (0.65 µm; Corning,
Inc., Corning, NY, USA). The lower chamber contained 1 µg/ml
LPS, LPS and 5 µg/ml propofol, or LPS with 5 µg/ml
propofol and 100 µM MRS agar. The control group only
contained culture medium. After 12-h incubation at 37°C, cells on
the lower chamber were fixed and stained with crystal violet. The
number of migrating microglia was observed under a light microscope
(Leica DVM6; Leica Microsystems GmbH).
In vitro scratch wound assay
Microglia (1×106/ml) were plated onto a
12-well tissue culture plate and allowed to reach near-confluence
overnight at 4°C. Mitomycin (5 µg/ml; Bio-Rad Laboratories,
Inc., Hercules, CA, USA) was added to the culture medium to inhibit
cell proliferation at 2 h before the scratch wound was applied.
Each well was scratched across the center using a sterile P-200
pipet tip to create an artificial in vitro wound. Cells in
each group were treated as described earlier. After 12-h incubation
at 37°C, the cells were washed with PBS and stained with 0.5%
crystal violet. Imaging as performed by light microscopy and
analyzed with ImageJ software (version 1.48; National Institutes of
Health, Bethesda, MD, USA).
Animal transient middle cerebral artery
occlusion (tMCAO) model
A total of 40 male SD rats (age, 8-12 weeks; weight,
220-250 g) were selected and purchased from the Experimental Animal
Center of Hainan Medical University 5 days before the experiments.
All procedures performed with animals were approved by the
Institutional Clinical Experiments Committee of the Second
Affiliated Hospital of Hainan Medical University (Haikou, China).
Rats were housed at 21-26°C with a humidity of 65±5%, 0.03%
CO2 and 12 h light/dark cycle with free access to water
and food. They were fasted for 6 h before tMCAO. Briefly, 2.5%
sodium pentobarbital (36 mg/kg; Sigma-Aldrich; Merck KGaA) was
injected into the abdominal cavity of rats. Next, the common,
internal and external carotid arteries of the neck were exposed by
blunt dissection. A 1.8 cm-long nylon filament (diameter, 0.24-0.28
mm; Biospes Co., Ltd., Chongqing, China) was inserted via a cut of
the internal carotid artery, and the middle cerebral artery was
occluded by inserting the nylon filament (23,24). Rats were kept at 37°C during the
entire surgical procedure. The nylon filament was untied 2 h after
tMCAO. Finally, the rats in the experimental group were treated
with propofol (100 mg/kg; Sigma-Aldrich; Merck KGaA), while the
rats without any surgery in the sham group and rats subjected to
tMCAO in control group were only treated with saline (100 mg/kg;
Sigma-Aldrich; Merck KGaA) (25,26). A total of 10 animals were used for
each group.
BrdU immunofluorescence
At 2 days after tMCAO challenge, SD rats were
intravenously treated with BrdU (50 mg/kg; Sigma-Aldrich; Merck
KGaA) two times at 12-h intervals. Then, rats were euthanized using
2.5% sodium pentobarbital. Cryostat brain sections (20 µm)
from the frontal to the occipital poles were cut using a
cryomicrotome (HM525 NX; Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The sections were then incubated with HCl (2N) at 37°C
for 30 min, and borate buffer was used to neutralize the HCl
through three washes. Next, the sections were blocked with 1% FBS
in 0.3% Triton X-100 at 37°C for 10 min and stained using a BrdU
cell proliferation assay kit (Cell Signaling Technology, Inc.,
Danvers, MA, USA) following the manufacturer’s protocol. Microglia
were labeled with a primary antibody against ionized calcium
binding adaptor molecule 1 (Iba1; cat. no. 019-19741; 1:500
dilution; Wako Pure Chemical Industries, Ltd., Osaka, Japan) at 4°C
overnight. In addition, 702- Rat IgG1 kappa antibody (cat. no.
67-4301-80; 1:500; Thermo Fisher Scientific, Inc.) was used as the
control antibody. Subsequently, the cells were stained with an
Alexa488-conjugated secondary antibody (cat. no. S11223; 1:1,000
dilution; Thermo Fisher Scientific, Inc.) after the unbound primary
antibody was washed three times using PBS. Finally, cells were
imaged using laser scanning confocal microscopy (TCS SP8 DLS; Leica
Microsystems GmbH). The number of double immune positive microglia
in 10 different fields-of-view was analyzed using ImageJ
software.
ELISA
At 12 h after tMCAO, the cerebrospinal fluid (CSF)
of SD rats was extracted following a previously described method
(27). Subsequently, the levels
of interleukin (IL)-1β, IL-6 and tumor necrosis factor-α (TNF-α) in
the CSF were measured using ELISA kits (cat. nos. EK0393, EK0412
and EK0526; Boster Biological Technology, Ltd., Wuhan, China)
following the manufacturer’s protocol.
Statistical analysis
Experimental values are reported as the mean ±
standard deviation, and the one-way analysis of variance followed
by Student-Newman-Keuls post hoc test was used to evaluate
statistically significant differences between groups. Differences
were considered to be statistically significant when the P-value
was <0.05. Statistical evaluations were performed with the SPSS
version 13 software package (SPSS, Inc., Chicago, IL, USA).
Results
Propofol inhibits NO and ROS
overexpression induced by LPS in microglia through the A2b
receptor
ROS and NO are the major cytotoxic factors secreted
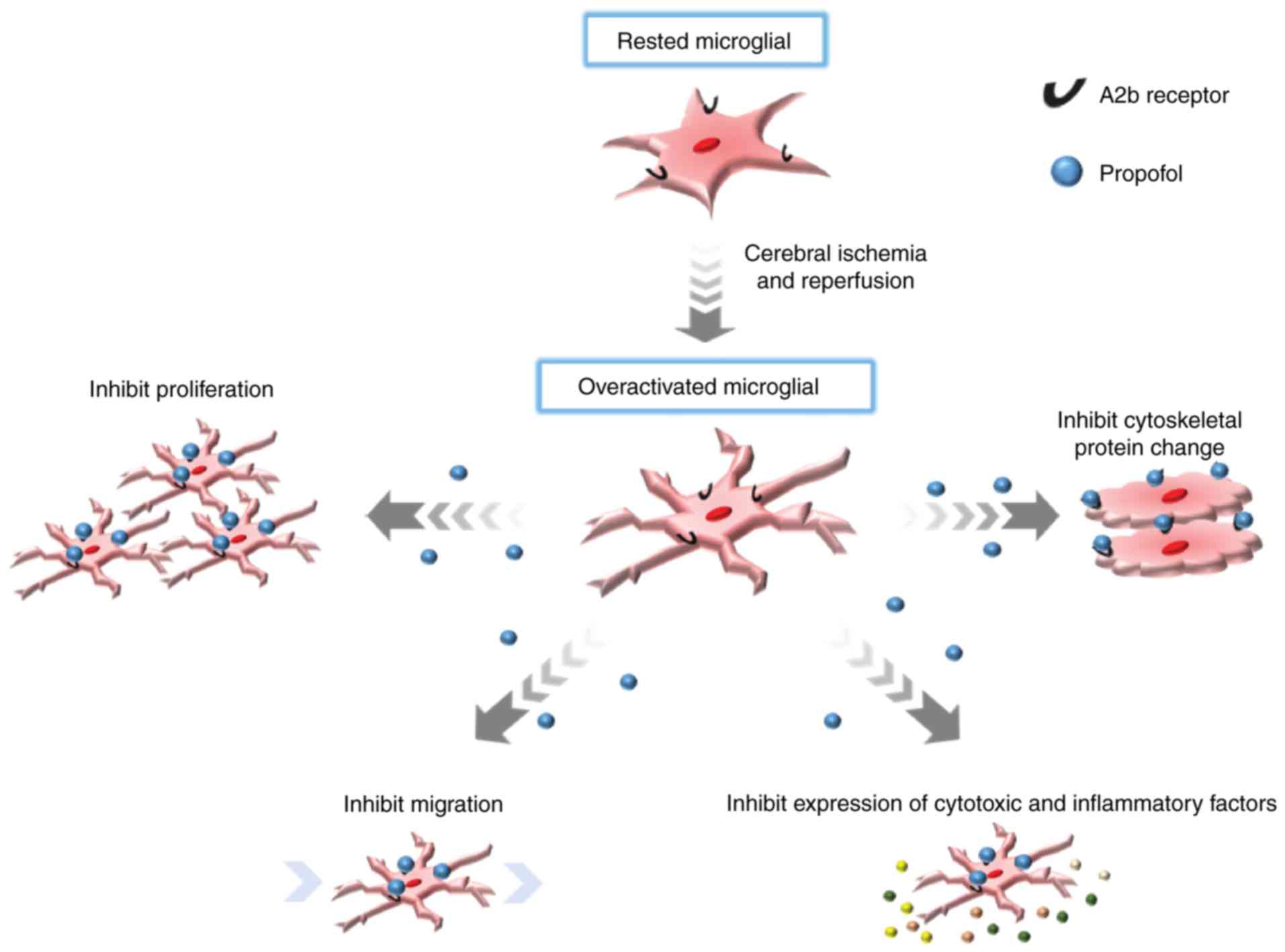
by overactive microglia (Fig. 1).
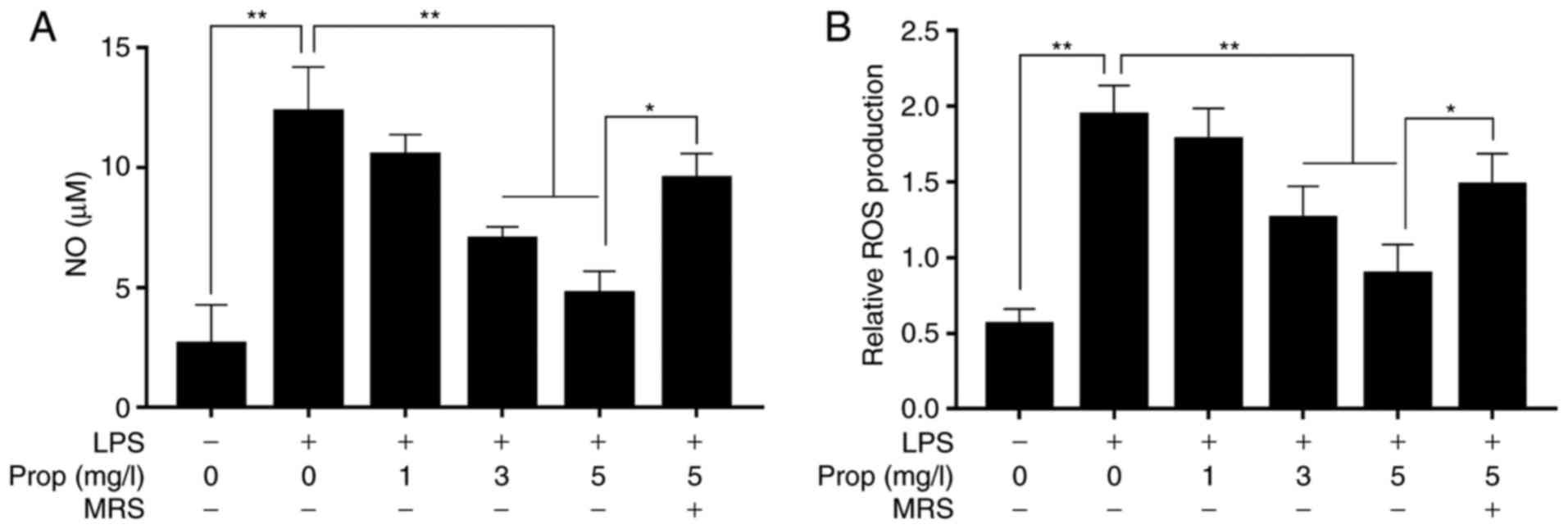
The present study results revealed that the expression levels of
ROS and NO in microglia were 0.58 and 2.52 µM in untreated
cells (Fig. 2). Following
stimulation with LPS (1 µg/ml), the levels of ROS and NO
increased by 379.3 and 491.6% (both P<0.01), respectively.
However, the ROS and NO levels decreased by 8.3 and 12.2% as
compared with the LPS alone group when LPS-treated microglia were
stimulated with propofol (1 mg/l) (P<0.01). When the
concentration of propofol was increased to 3 mg/l, the ROS and NO
levels in LPS-treated cells decreased by 31.6 and 39.5%,
respectively, and further decreased by 52.3 and 49.8% following
treatment with 5 mg/l propofol (P<0.01). However, the effect of
propofol (5 mg/l) on microglia was inhibited by 100 µM MRS
agar, an A2b receptor antagonist, and the levels of ROS and NO were
increased by 169.5 and 172.4% (P<0.05), respectively, as
compared with those in the LPS and propofol (5 mg/l) group
(Fig. 2A and B).
Propofol inhibits structural alterations
in cytoskeletal protein F-actin in microglia through A2b
receptors
The actin cytoskeleton serves an important role in
the morphological alteration of cells, and structural change in
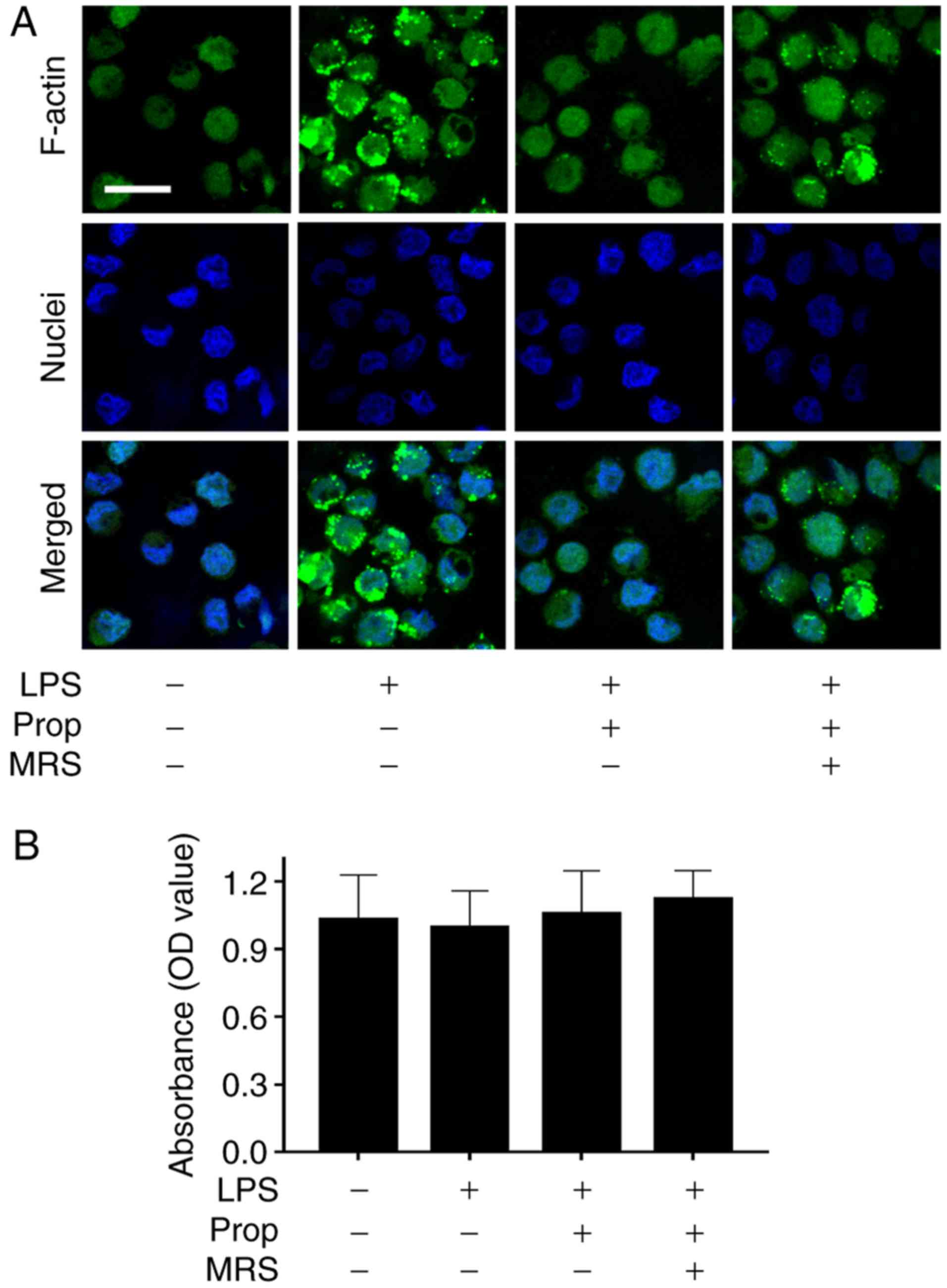
F-actin is one of the results of microglial activation (Fig. 1) (28). In the present study, the changes
in F-actin were detected by fluorescence staining (Fig. 3A). The results revealed that the
fluorescence intensity of F-actin in microglia was low in untreated
control cells, whereas it significantly increased following the
addition of LPS. The fluorescence intensity in microglia strongly
decreased subsequent to treatment with propofol, which demonstrated
that propofol inhibited the structural alterations in the
cytoskeletal protein in microglia. However, the effect of propofol
on microglia was suppressed by the addition of 100 µM MRS
agar (Fig. 3A). MTT results
demonstrated that microglial survival rates were similar among all
four experimental groups, as the OD value was ~1.0 and there was no
significant difference between any of the groups (Fig. 3B).
Propofol inhibits LPS-induced abnormal
migration of microglia via the A2b receptor
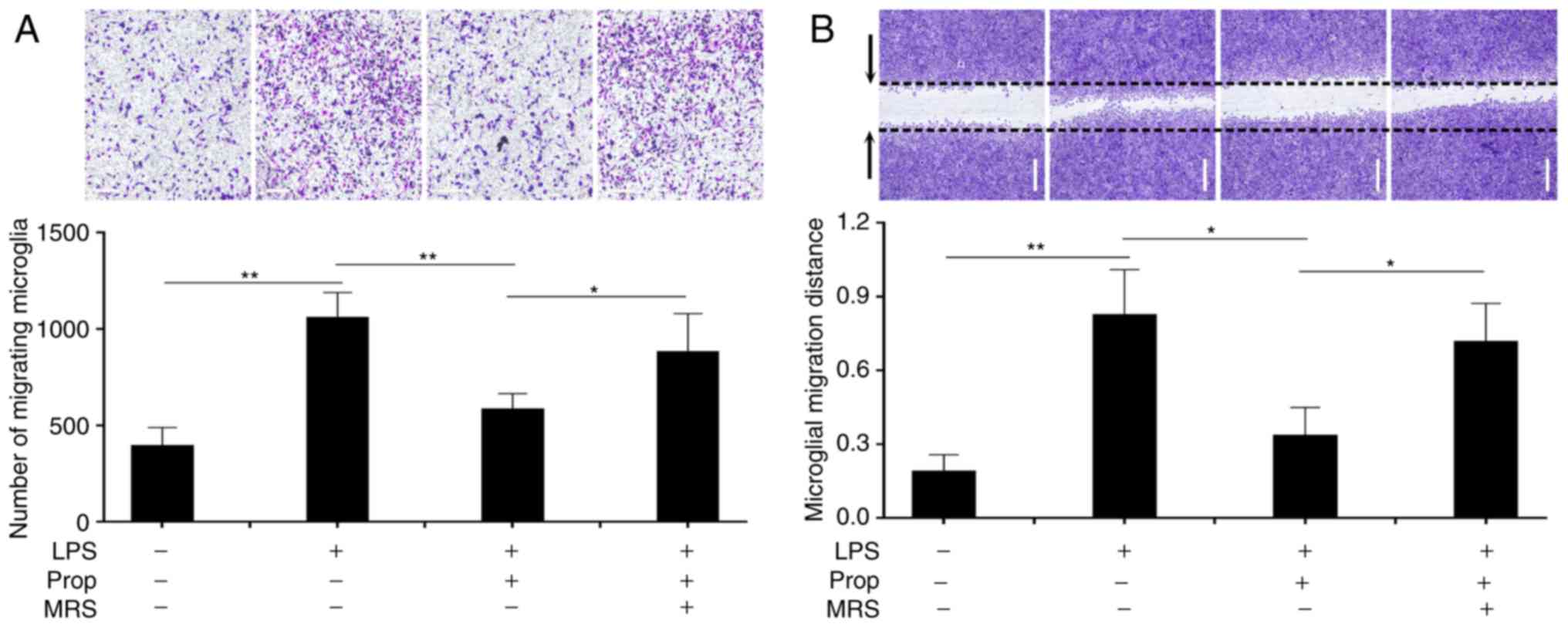
Microglia migration increased abnormally following
cerebral infarction (Figs. 1 and
4) (29). In untreated control cells, the
number of migrating cells was 473 and the migration distance was
0.195 mm (Fig. 4). Subsequent to
treatment with LPS, microglial migration was significantly
increased, as the number of migrating cells and the migration
distance had increased to 236.7 and 412.5% (P<0.01),
respectively. However, microglial migration decreased following
treatment with propofol (5 mg/ml), as the number of migrating cells
and the migration distance markedly decreased by 46.4 and 54.1%
(P<0.01), respectively. These results demonstrated that propofol
inhibited microglial migration following cerebral ischemia. Similar
to the aforementioned results of other experiments, the effect of
propofol was inhibited by MRS agar treatment, as the number of
migrating cells and the migration distance in the MRS agar-treated
group increased to 137.6 and 196.4% (P<0.05), respectively,
compared with that of the LPS and propofol (5 mg/l) group (Fig. 4A and B).
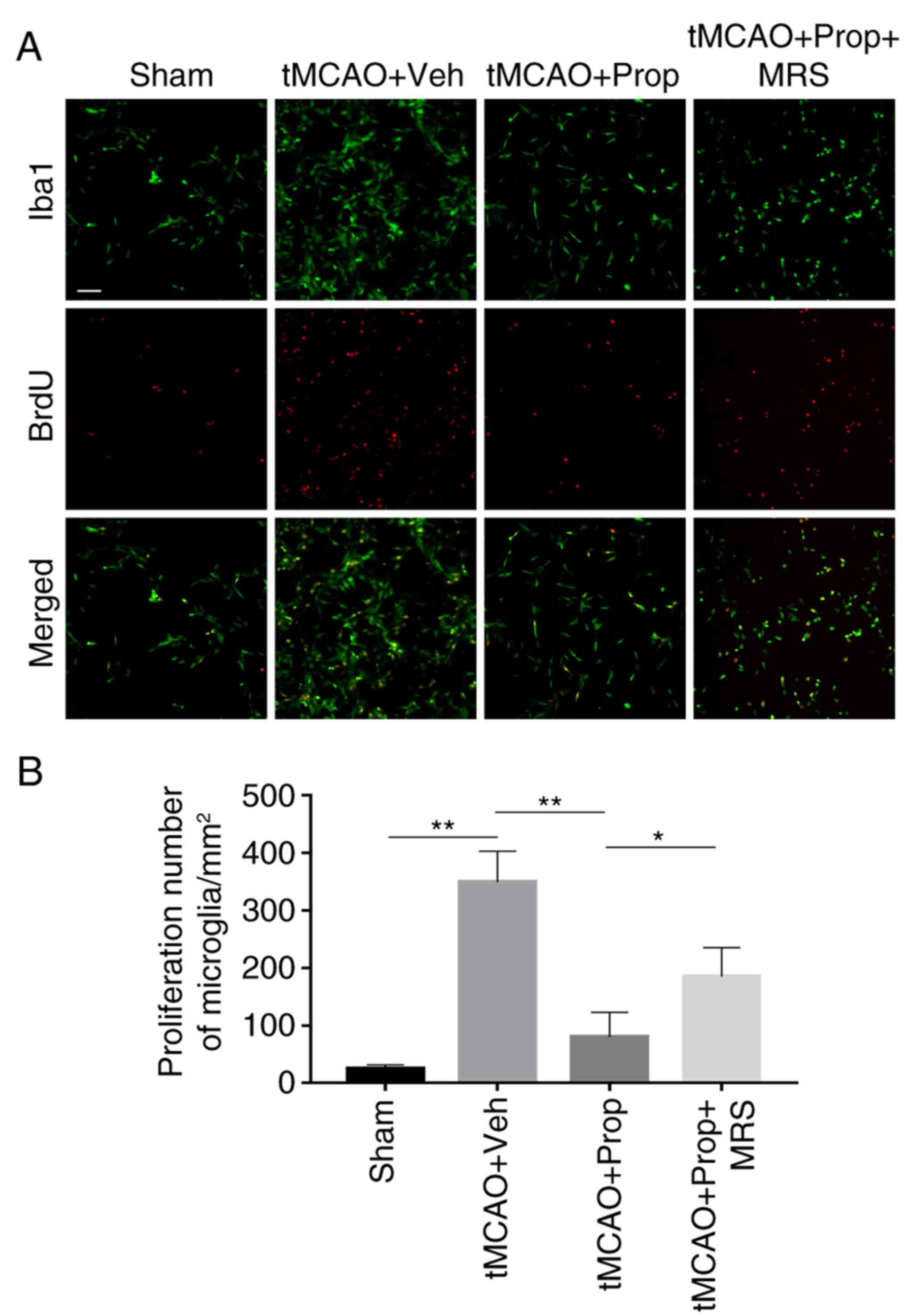
Propofol inhibits abnormal proliferation
of microglia in the tMCAO model through the A2b receptor
Abnormal proliferative potential of microglia is
associated with brain ischemia and reperfusion (30). In order to assess the effect of
propofol on microglial proliferation, brain sections were processed
with double-immunofluorescence with BrdU and an antibody against
Iba1, a microglia-specific marker. In the control group, microglia
exhibited a low proliferative potential, the number of
double-positive cells was 28.6/mm2. Microglial
proliferative potential increased significantly subsequent tMCAO,
and the number of double-positive cells increased to
367.2/mm2 at 3 days following tMCAO (P<0.01). Upon
treatment with propofol (5 mg/l), the number of double-positive
cells in the tMCAO model rats decreased to 92/mm2
(P<0.01). However, the effect of propofol was significantly
inhibited by the addition of MRS agar, as the number of microglia
increased to 196/mm2 in the MRS agar-treated group
(P<0.05) (Fig. 5A and B).
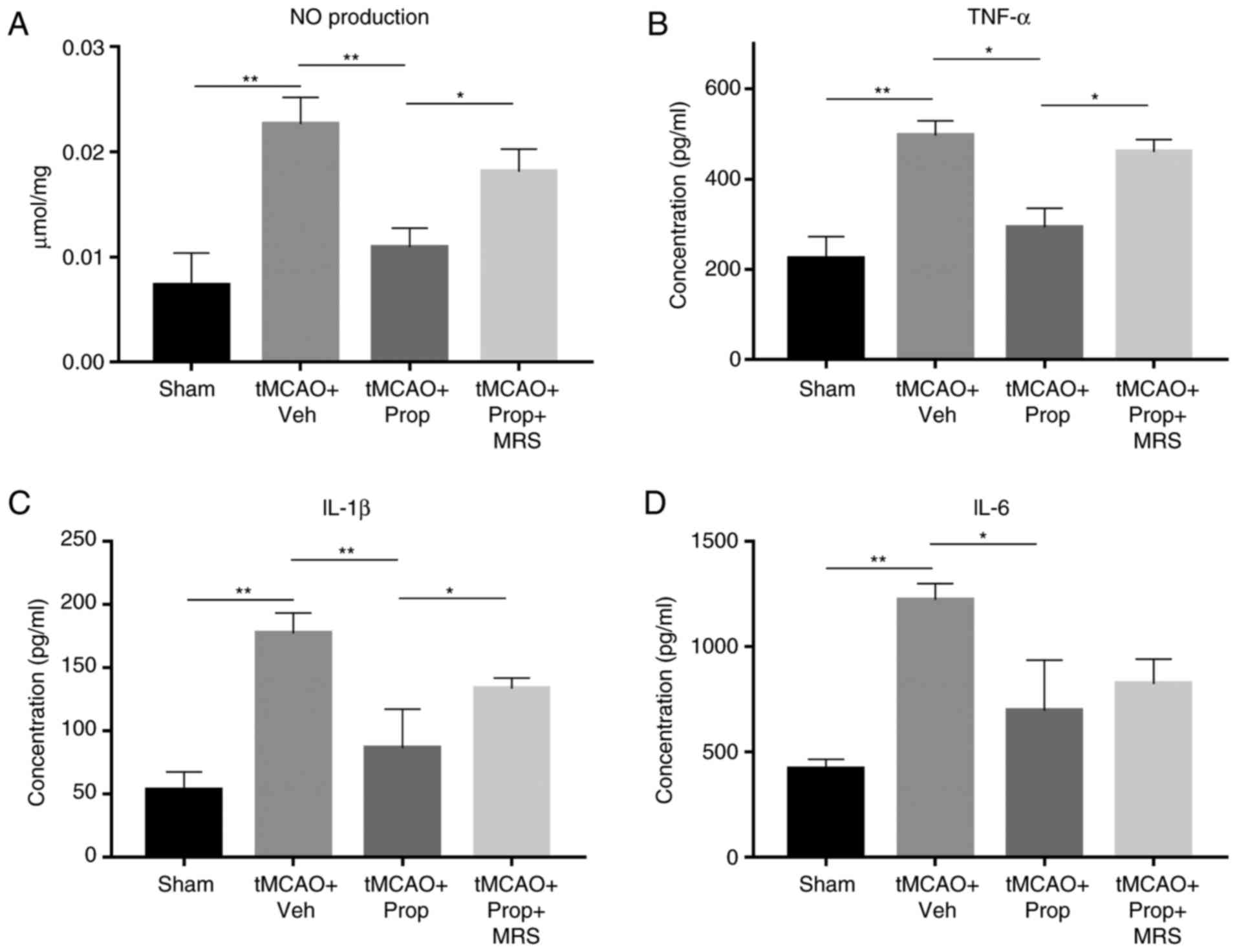
Propofol inhibits the overexpression of
microglial inflammatory factors in the tMCAO model via the A2b
receptor
Following cerebral ischemia and reperfusion,
microglia and other associated inflammatory cells release a large
quantity of pro-inflammatory and cytotoxic factors, including IL-6,
IL-1β, TNF-α and NO. In the control group, the inflammatory
response remained at a low level, and the measured levels of IL-6,
IL-1β and TNF-α in the CSF were 461, 53 and 226 pg/ml,
respectively, while the level of NO was 0.0088 µmol/mg. The
inflammatory response was significantly enhanced at 3 days after
tMCAO challenge, and the levels of IL-6, IL-1β, TNF-α and NO
increased to 294.1, 312.4, 255.7 and 283.4%, respectively (all
P<0.01), compared with those observed in the control group.
Following treatment with propofol (5 mg/ml), the inflammatory
response decreased, and the production of IL-6, IL-1β, TNF-α and NO
decreased by 61 (P<0.05), 49 (P<0.01), 63 (P<0.05) and 47%
(P<0.01), respectively. However, the effect of propofol on
microglia and other inflammatory cells was significantly inhibited
by MRS agar treatment, as the production of IL-1β, TNF-α and NO in
the CSF increased by 1.33-, 1.41- and 1.39-fold (P<0.05),
respectively, when compared with the LPS and propofol-treated alone
group. Notably, the expression of IL-6 was less affected by MRS
agar treatment, and the IL-6 production only increased to 1.15-fold
that of the propofol-treated group in the MRS-treated group, with
no significant difference observed (Fig. 6).
Discussion
Excessive activation of microglia is considered to
be the main cause of cerebral infarction injury in the central
nervous system (31-32). Secreted cytotoxic and inflammatory
cytokines can lead to neuritis, immune response and damage of the
nervous system, and may be the main pathological basis of several
neurodegenerative diseases (33).
A number of previous studies had found that propofol has
neuroprotective effects in animal models (34-36). However, there is a lack of studies
on the mechanism of action of propofol regarding its
neuroprotective role (37). In
the present study, the effects of propofol on suppressing
inflammation were examined, and it was attempted to identify a
suitable dose for clinical use.
LPS can induce microglia to release abundant
quantities of NO and ROS. These factors are major cytotoxic
substances released following ischemic stroke (38,39). Comparing microglia from each
experimental group, the current study observed that propofol was
able to inhibit the overexpression of NO and ROS, and these results
are in agreement with recent evidence (40). In addition, the experimental
results of the present study revealed that there was a negative
correlation between the expression of cytotoxic factors and the
propofol dose within a certain concentration range. Simultaneously,
the effect of propofol on microglial activityand survival was also
investigated by MTT assay, and the results indicated that propofol
treatment has no significant effect on microglial activity. These
findings are important for future clinical applications of
propofol.
Following ischemic stroke, activated microglia
migrates rapidly to the site of injury and mediates the
inflammatory reaction (28).
Comparing the number of migrated cells in the Trans well experiment
and scratch injury assays conducted in the present study, it was
observed that propofol inhibited the abnormal proliferation of
overactive microglia. These in vitro experimental results
preliminarily demonstrate that propofol is able to inhibit the
aberrant migration of overactive microglia and the overexpression
of cytotoxic factors.
It is known that cytoskeletal proteins serve an
important role in maintaining cellular morphology and function
(41). In the present study,
fluorescence staining of microglial F-actin demonstrated that
propofol treatment inhibited the structural alterations induced by
LPS in the microglial cytoskeleton induced.
A tMCAO rat model was also employed in the current
study to assess microglial proliferation and inflammatory factor
secretion in vivo. It was observed that propofol inhibited
the proliferation and activation of microglia. In addition, the
levels of IL-1β, IL-6, TNF-α and NO were evidently low in the
propofol-treated group, which indicated that propofol attenuated
inflammation in tMACO. Although it remains uncertain whether all
these inflammatory factors are secreted only by microglia, it is
likely that propofol inhibits the overall levels of inflammatory
factors in the brain subsequent to ischemic stroke.
In the aforementioned experiments, an MRS
agartreatment group was used to determine the mechanism of action
of propofol on cerebral inflammation. The results suggested that
MRS agar, an A2b receptor antagonist, effectively blocked nearly
all the effects of propofol on microglia. This supports the
hypothesis that the A2b receptor serves a significant role in the
inflammation-attenuating effects of propofol. However, blocking the
A2b receptor did not significantly affect the level of IL-6,
suggesting that propofol may also fulfill its anti-inflammatory
function via another route.
In conclusion, propofol was demonstrated to inhibit
excessive microglial activation, abnormal proliferation and
migration, as well as the release of cytotoxic and inflammatory
factors, and its effects were likely mediated by the A2b receptor.
Therefore, the present study determined a feasible drug and an
appropriate clinical dosage for the purpose of reducing
inflammation and improving the prognosis in cerebral infarction. In
addition, A2b receptor may be not the only receptor through which
propofol acts on microglia, and other pathways remain to be
explored (42). Determining the
ideal reaction time, propofol dosage and possible side effects will
be the direction of future research.
Acknowledgments
Not applicable.
Funding
This study was supported by the 2016 Cultivation
Fund of Hainan Medical University (grant no. HY2016-07) and the
Industry Research Project of Hainan Health and Family Planning
Commission (grant no. 1601032054A2001).
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
HY took part in all experiments and was the major
contributor in writing the manuscript. XW analyzed and interpreted
the data regarding cell experiments. FK and ZC established the
tMCAO model and conducted other experiments in vivo. YM
analyzed and interpreted data regarding experiments in vivo.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures involving animals were reviewed and
approved by the Institutional Clinical Experiments Committee and
Institutional Review Board of the Hainan Medical University
(Haikou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu J, Zhao D, Wang W, Sun JY, Li Y and
Jia YN: Trends regarding the incidence of recurrent stroke events
in Beijing. Zhonghua Liu Xing Bing Xue Za Zhi. 28:437–440. 2007.In
Chinese. PubMed/NCBI
|
|
2
|
Labeyrie C, Cauquil C, Sarov M, Adams D
and Denier C: Cerebral infarction following subcutaneous
immunoglobulin therapy for chronic inflammatory demyelinating
polyradiculo-neuropathy. Muscle Nerve. 54:166–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Numata K, Suzuki M, Mashiko R and Tokuda
Y: Lethal bilateral cerebral infarction caused by moyamoya disease.
OJM. 109:5012016.
|
|
4
|
Zhan R, Xu K, Pan J, Xu Q, Xu S and Shen
J: Long noncoding RNA MEG3 mediated angiogenesis after cerebral
infarction through regulating p53/X4 axis. Biochem Biophys Res
Commun. 490:700–706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kwon SK, Ahn M, Song HJ, Kang SK, Jung SB,
Harsha N, Jee S, Moon JY, Suh KS, Lee SD, et al: Nafamostat
mesilate attenuates transient focal ischemia/reperfusion-induced
brain injury via the inhibition of endoplasmic reticulum stress.
Brain Res. 1627:12–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang S, Gao L, Lu F, Wang B, Gao F, Zhu G,
Cai Z, Lai J and Yang Q: Transcription factor myocyte enhancer
factor 2D regulates interleukin-10 production in microglia to
protect neuronal cells from inflammation-induced death. J
Neuroinflammation. 12:332015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brown A: Understanding the MIND phenotype:
Macrophage/ microglia inflammation in neurocognitive disorders
related to human immunodeficiency virus infection. Clin Transl Med.
4:72015. View Article : Google Scholar
|
|
8
|
Pessach I and Paret G: PICU propofol use,
where do we go from here? Pediatr Crit Care Med. 17:273–275. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi SS, Yang WZ, Chen Y, Chen JP and Tu
XK: Profol reduces inflammatory reaction and ischemic brain damage
in cerebral ischemia in rats. Neurochem Res. 39:793–799. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Woldegerima N, Rosenblatt K and Mintz CD:
Neurotoxic properties of propofol sedation following traumatic
brain injury. Crit Care Med. 44:455–456. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu J, Gao XF, Ni W and Li JB: Effects of
propofol on P2X7 receptors and the secretion of tumor necrosis
factor-α in cultured astrocytes. Clin Exp Med. 12:31–37. 2012.
View Article : Google Scholar
|
|
12
|
Perígolo-Vicente R, Ritt K,
Gonçalves-de-Albuquerque CF, Castro-Faria-Neto HC, Paes-de-Carvalho
R and Giestal-de-Araujo E: IL-6, A1 and A2aR: A crosstalk that
modulates BDNF and induces neuroprotection. Biochem Biophys Res
Commun. 449:477–482. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zywert A, Szkudelska K and Szkudelski T:
Effects of adenosine A1 receptor antagonism on insulin
secretion from rat pancreatic islets. Physiol Res. 60:905–911.
2011.
|
|
14
|
Ohnishi M, Urasaki T, Ochiai H, Matsuoka
K, Takeo S, Harada T, Ohsugi Y and Inoue A: Selective enhancement
of wnt4 expression by cyclic AMP-associated cooperation between rat
central astrocytes and microglia. Biochem Biophys Res Commun.
467:367–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JB, Yu YM, Kim SW and Lee JK:
Anti-inflammatory mechanism is involved in ethyl pyruvate-mediated
efficacious neuroprotection in the postischemic brain. Brain Res.
1060:188–192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang AL, Yu AC, He QH, Zhu X and Tso MO:
AGEs mediated expression and secretion of TNFα in rat retinal
microglia. Exp Eye Res. 84:905–913. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo H, Hu LM, Wang SX, Wang YL, Shi F, Li
H, Liu Y, Kang LY and Gao XM: Neuroprotective effects of
scutellarin against hypoxic-ischemic-induced cerebral injury via
augmentation of antioxidant defense capacity. Chin J Physiol.
54:399–405. 2011.
|
|
18
|
Hands S, Sajjad MU, Newton MJ and
Wyttenbach A: In vitro and in vivo aggregation of a fragment of
huntingtin protein directly causes free radical production. J Biol
Chem. 286:44512–44520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee JK, Chung J, McAlpine FE and Tansey
MG: Regulator of G-protein signaling-10 negatively regulates NF-κB
in microglia and neuroprotects dopaminergic neurons in
hemiparkinsonian rats. J Neurosci. 31:11879–11888. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shen K, Tolbert CE, Guilluy C, Swaminathan
VS, Berginski ME, Burridge K, Superfine R and Campbell SL: The
vinculin C-terminal hairpin mediates F-actin bundle formation,
focal adhesion, and cell mechanical properties. J Biol Chem.
286:45103–45115. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gorbatyuk V, Nguyen K, Podolnikova NP,
Deshmukh L, Lin X, Ugarova TP and Vinogradova O: Skelemin
association with αIIbβ3 integrin: A
structural mode. Biochemistry. 53:6766–6775. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Liu Y and Haas TA: Skelemin in
integrin αIIbβ3 mediated cell spreading.
Biochemistry. 52:681–689. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zuhayra M, Zhao Y, von Forstner C, Henze
E, Gohlke P, Culman J and Lützen U: Activation of cerebral
peroxisome proliferator-activated receptors γ (PPARγ) reduces
neuronal damage in the substantia nigra after transient focal
cerebral ischaemia in the rat. Neuropathol Appl Neurobiol.
37:738–752. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao LJ, Wang J, Hao PP, Sun CL and Chen
YG: Effects of ulinastatin, a urinary trypsin inhibitor, on
synaptic plasticity and spatial memory in a rat model of cerebral
ischemia/reperfusion injury. Chin J Physiol. 54:435–442. 2011.
|
|
25
|
Stekiel TA, Conteny SJ, Roman RJ, Weber
CA, Stadnicka A, Bosnjak ZJ, Greene AS and Moreno C:
Pharmacogenomic strain differences in cardiovascular sensitivity to
propofol. Anesthesiology. 115:1192–1200. 2011.PubMed/NCBI
|
|
26
|
Tanaka K, Tsutsumi YM, Kinoshita M, Kakuta
N, Hirose K, Kimura M and Oshita S: Differential effects of
propofol and isoflurane on glucose utilization and insulin
secretion. Life Sci. 88:96–103. 2011. View Article : Google Scholar
|
|
27
|
Mariappan TT, Kurawattimath V, Gautam SS,
Kulkarni CP, Kallem R, Taskar KS, Marathe PH and Mandlekar S:
Estimation of the unbound brain concentration of P-glycoprotein
substrates or nonsubstrates by a serial cerebrospinal fluid
sampling technique in rats. Mol Pharm. 11:477–485. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wessler S, Gimona M and Rieder G:
Regulation of the actin cytoskeleton in Helicobacter pylori-induced
migration and invasive growth of gastric epithelial cells. Cell
Commun Signal. 9:272011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Davalos D, Grutzendler J, Yang G, Kim JV,
Zuo Y, Jung S, Littman DR, Dustin ML and Gan WB: ATP mediates rapid
microglial response to local brain injury in vivo. Nat Neurosci.
8:752–758. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Campanella M, Sciorati C, Tarozzo G and
Beltramo M: Flow cytometric analysis of inflammatory cells in
ischemic rat brain. Stroke. 33:586–592. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jin YL, Luo HL, Liu L, Cheng CF and Ho LC:
Inhibitory effects of different concentrations of curcumin on
excessive activation of microglia cultured in vitro. J Clin Rehabil
Tissue Eng Res. 15:6951–6955. 2011.
|
|
32
|
Stertz L, Magalhães PV and Kapczinski F:
Is bipolar disorder an inflammatory condition? The relevance of
microglia activation. Curr Opin Psychiatry. 26:19–26. 2013.
View Article : Google Scholar
|
|
33
|
Kokubu Y, Yamaguchi T and Kawabata K: In
vitro model of cerebral ischemia by using brain microvascular
endothelial cells derived from human induced pluripotent stem
cells. Biochem Biophys Res Commun. 486:577–583. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou R, Yang Z, Tang X, Tan Y, Wu X and
Liu F: Propofol protects against focal cerebral ischemia via
inhibition of microglia-mediated proinflammatory cytokines in a rat
model of experimental stroke. Plos One. 8:e827292013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li J, Han B, Ma X and Qi S: The effects of
propofol on hippocampal caspase-3 and Bcl-2 expression following
forebrain ischemia-reperfusion in rats. Brain Res. 1356:11–23.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harman F, Hasturk AE, Yaman M, Arca T,
Kilinc K, Sargon MF and Kaptanoglu E: Neuroprotective effects of
propofol, thiopental, etomidate, and midazolam in fetal rat brain
in ischemia-reperfusion model. Childs Nerv Syst. 28:1055–1062.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pfeilschifter W, Czech-Zechmeister B,
Sujak M, Mirceska A, Koch A, Rami A, Steinmetz H, Foerch C, Huwiler
A and Pfeilschifter J: Activation of sphingosine kinase 2 is an
endogenous protective mechanism in cerebral ischemia. Biochem
Biophys Res Commun. 413:212–217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shuhua X, Ziyou L, Ling Y, Fei W and Sun
G: A role of fluofide on free radical generation and oxidative
stress in BV-2 microglia cells. Mediators Inflamm. 2012:1029542012.
View Article : Google Scholar
|
|
39
|
Brites D and Femandes A: Neuroinflammation
and depression: Microglia activation, extracellular mierovesicles
and microRNA dysregulation. Front Cell Neurosci. 9:4762015.
View Article : Google Scholar
|
|
40
|
Zheng X, Huang H, Liu J, Li M, Liu M and
Luo T: Propofol attenuates inflammatory response in LPS-activated
microglia by regulating the miR-155/SOCS1 pathway. Inflammation.
41:11–19. 2018. View Article : Google Scholar
|
|
41
|
Hagiwara S, Iwaska H, Hasegawa A, Hidaka
S, Uno A, Kaori U, Uchida T and Noguchi T: Continuous
hemodiafiltration therapy ameliorates LPS-induced systemic
inflammation in a rat model. J Surg Res. 171:791–796. 2011.
View Article : Google Scholar
|
|
42
|
Tang J, Chen X, Tu W, Guo Y, Zhao Z, Xue
Q, Lin C, Xiao J, Sun X, Tao T, et al: Propofol inhibits the
activation of p38 through up-regulating the expression of annexin
A1 to exert its anti-inflammation effect. PLoS One. 6:e278902011.
View Article : Google Scholar : PubMed/NCBI
|