Introduction
Atherosclerosis (AS) is a common and detrimental
disease characterized by lipid deposition in arterial intima,
abnormal proliferation of vascular smooth muscle cells (VSMCs) and
connective tissue hyperplasia, leading to intimal focal fibrous
thickening and atherosclerotic plaque formation. Hardening and
narrowing of the arteries are frequently observed among patients
with AS. AS may lead to cardiovascular, cerebrovascular and
thromboembolic diseases. Proliferation and angiogenesis of VSMCs
are important biological processes in the development of AS.
Therefore, inhibition of excessive proliferation and induction of
apoptosis of VSMCs could be an effective method for the treatment
and control of AS (1). Although
there were a number of studies investigating the etiology of AS
(2-4), the exact mechanism remains to be
elucidated.
Long noncoding (lnc)RNAs are a group of RNA
molecules that lack protein-coding function and are >200
nucleotides in length (5). It has
been demonstrated that lncRNAs are involved in the regulation of
various biological processes in cells, including proliferation,
migration, apoptosis and differentiation (6). Although the function and mechanism
of lncRNAs remain to be completely elucidated, previously published
studies indicated that lncRNAs are closely associated with AS and
development of other cardiovascular diseases. LncRNAs associated
with AS have been studied by researchers due to their regulatory
roles in lipid metabolism, proliferation, migration, apoptosis,
adhesion and inflammatory response (7). It has been previously demonstrated
that the expression levels of long intergenic noncoding
(linc)RNA-p21 decreased in AS-prone apolipoprotein E
(ApoE)-deficient atherosclerotic plaques and lincRNA-p21 repressed
cell proliferation and induced apoptosis of VSMCs by enhancing
transcriptional activity of p53 (2). Smooth muscle induced lncRNA,
enhancer of proliferation and metastasis associated lung
adenocarcinoma transcript 1 have been reported to be involved in
controlling vascular cell proliferation (8), endothelial cell (EC) sprouting
(9) and phenotypic transition
(10). Long intergenic
non-protein coding RNA 599 silencing contributed to EC and VSMC
dysfunction, and aggravated AS (11). Maternally expressed gene 3 (MEG3)
is an imprinted gene with a length of ~1.6 kb, and its
transcriptional deficiency may induce the occurrence and
development of multiple tumors, including non-small cell lung
cancer (12), gastric cancer
(13) and liver cancer (14). Our recent study indicated that the
expression of MEG3 was decreased in pulmonary arteries of patients
with pulmonary hypertension resulting in increased human pulmonary
artery smooth muscle cell proliferation and migration through the
p53 signaling pathway (15).
However, the function and mechanism of MEG3 in AS remain to be
elucidated, and, therefore, it is necessary to further investigate
the molecular mechanism of MEG3 in the development of AS.
Scutellaria baicalensis Georgi is a commonly
used herbal medicine, which exhibits a variety of therapeutic
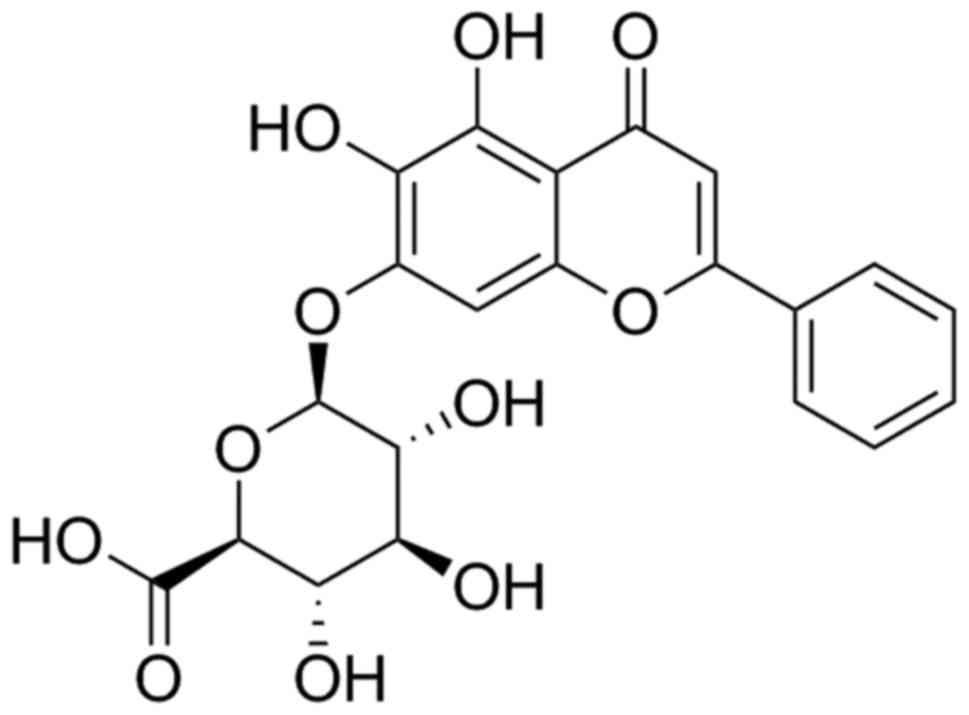
effects in traditional Chinese Medicine formulations (16). Baicalin (Fig. 1) (17), the main active component of
Scutellaria baicalensis Georgi, is a flavonoid compound
extracted from the dry roots exhibiting biological activity
(18). Previous studies indicated
that baicalin may induce numerous pharmacological effects,
including anti-oxidative (19),
antitumor (20),
anti-inflammatory (21) and
antiproliferative (17)
functions. Baicalin inhibited the activation of nuclear factor-κB,
decreased the expression of pro-inflammatory mediators and
prevented renal dysfunction in ApoE knock out mice on
high-cholesterol diets, which served an important role in the
prevention of AS (22,23). A number of studies reported that
p53 serves an important role in the pathogenesis of AS (2,24,25). Furthermore, p53 regulates cell
cycle and apoptosis (15,25). Wu et al (2) hypothesized that lincRNA-p21 may
regulate vascular smooth muscle cell proliferation and apoptosis by
enhancing the activity of p53 in AS (2). Our previous studies suggested that
MEG3 may increase the activity of p53 in pulmonary artery smooth
muscle cells (15). The current
results indicated that the expression levels of MEG3 decreased in
serum samples from patients with AS and oxidized low-density
lipoprotein (ox-LDL)-stimulated human aorta vascular smooth muscle
cells (HA-VSMCs) compared with the control samples. Treatment with
baicalin promoted the expression of MEG3 and inhibited the
proliferation of HA-VSMCs induced by MEG3 knockdown. MEG3 knockdown
increased the expression of proliferating cell nuclear antigen
(PCNA), cyclin A and E. However, following treatment of HA-VSMCs
with different concentrations baicalin, expression of PCNA, cyclin
A and E was inhibited in cells with MEG3 knockdown. The p53
signaling pathway components are expressed in the nucleus under
normal conditions (26); the
expression of p53 was detected in the cytoplasm after MEG3
knockdown. When HA-VSMCs were treated with different concentrations
of baicalin, p53 expression was detected in the nucleus. In
addition, the protein expression level of p53 decreased compared
with the NC group after MEG3 knockdown. Baicalin could increase p53
protein expression after MEG3 knockdown in ox-LDL-treated HA-VSMCs.
In conclusion, the current study aimed to further investigate the
underlying roles and molecular basics of baicalin and MEG3/p53 in
the progression to AS, implicating the potential values of baicalin
and MEG3 in AS therapy.
Materials and methods
Reagents and antibodies
Baicalin (purity, >99.0%; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was dissolved in dimethylsulfoxide. The
antibodies against PCNA (cat. no. 10205-2-AP; 1:1,000), cyclin A
(cat. no. 13295-1-AP; 1:2,000), cyclin E (cat. no. 11554-1-AP;
1:2,000) and β-actin (cat. no. 60008-1-1g; 1:5,000) were purchased
from ProteinTech Group, Inc. (Chicago, IL, USA). CycleTEST™ PLUS
DNA Reagent kit was obtained from BD Biosciences (Franklin Lakes,
NJ, USA). The bromodeoxyuridine (BrdU) proliferation kit was
purchased from EMD Millipore (Billerica, MA, USA). Ox-LDL, VicFemto
enhanced chemiluminesence (ECL) kit and PBS were purchased from
Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China).
Clinical samples
The present study was approved by the Ethics
Committee for the use of human samples of the First People’s
Hospital of Lianyungang, which was in accordance with the code of
ethics of the Declaration of Helsinki developed by the World
Medical Association. Each individual provided written informed
consent prior to their participation. Serum samples were collected
from 40 patients with AS (age, 60±10 years; 18 male and 22 female)
and 40 healthy volunteers (age, 58±9 years; 21 male and 19 female)
from the Department of Cardiovascular Medicine, the First People’s
Hospital of Lianyungang (Lianyungang, China) between January 2015
and January 2016.
Clinical inclusion and exclusion
criteria
The AS group included patients with >80% coronary
artery stenosis confirmed by coronary angiogram. The control group
included patients without clinically significant coronary artery
occlusion diagnosed by coronary angiogram. Patients with vascular
diseases, including hypertension and type I/II diabetes were
excluded. Total RNA was extracted from peripheral blood monocytes
of patients and controls.
Cell culture
HA-VSMCs were obtained from American Type Culture
Collection (Manassas, VA, USA) and cultured in F-12K medium
(American Type Culture Collection) containing 10% heat-inactivated
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Other nutrients added to the HA-VSMC culture
were based on previously published literature (27). Cells were incubated at 37˚C with
5% CO2.
Cell transfection and treatment
The sequences of siRNA targeting lncRNA MEG3 and
scrambled negative control siRNA (si-NC) were designed and
synthesized by GenePharma (Shanghai GenePharma Co., Ltd., Shanghai,
China) according to the previously published protocol (15). pcDNA-MEG3 and empty vector (pcDNA)
were synthesized by GenePharma (Shanghai GenePharma Co., Ltd.). At
60% confluence, HA-VSMCs were cultured in Dulbecco’s modified
Eagle’s medium (Gibco; Thermo Fisher Scientific, Inc.) at 37°C with
5% CO2 for 24 h. HA-VSMCs were transfected with si-NC,
si-MEG3 at a final concentration of 20 nmol/l in accordance with
the manufacturer’s protocol. Furthermore, HA-VSMCs were transfected
with pcDNA-MEG3 at a final concentration of 2 mg/l using. The
following sequences were used: si-lncRNA MEG3,
5′-GGGCTTCTGGAATGAGCAT-3′; si-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′
(15). HA-VSMCs were transfected
using Lipofectamine® 2000 on 6-well plates (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer’s
protocol. Cells were harvested for the subsequent experiments 24 h
after transfection.
DNA BrdU incorporation assay
Cell proliferation analysis was performed with BrdU.
Transfected and untransfected cells were seeded into 96-well plates
at a density of 1×104 cells/well and treated with
control (no treatment) and different concentrations of ox-LDL (25,
50 or 75 µg/ml) and baicalin (5, 10 or 20 µmol/l) in DMEM
containing 2% FBS at 37°C with 5% CO2 and BrdU was added
to the proliferating cells 2-24 h before the end of the test
reagent incubation. The following steps were performed according to
the manufacturer’s protocol and as previously described (28). Finally, the absorbance was
measured at a dual wavelength of 450/550 nm.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from serum samples and
HA-VSMCs using the TRIzol reagent according to the manufacturer’s
protocol (Thermo Fisher Scientific, Inc.). RT reaction and qPCR
were performed as previously described (15). cDNA was reverse transcribed from
100 ng of total RNA using 4 µl of miScript HiSpec buffer, 2 µl of
Nucleics Mix, 2 µl of miScript Reverse Transcriptase mix (Qiagen
GmbH, Hilden,
Germany) and sterilized distilled water up to a
total volume of 20 µl. The RT reaction was performed at 37°C for 60
min followed by heat inactivation at 95°C for 5 min. The qPCR
reactions were performed in triplicate using 7500 Fast Real-Time
PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Reaction mixtures had a total volume of 25 µl, including 2
µl of cDNA, 12.5 µl of QuantiTect SYBR-Green PCR
master mix (Qiagen GmbH), 2.5 µl of universal primers
(Qiagen GmbH), 2.5 µl of MEG3 or GAPDH-specific primers and
5.5 µl of nuclease-free water. Reactions were incubated in
96-well optical plates (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using the following thermocycling conditions:
Initial denaturation at 95°C for 10 min; 40 cycles of 95°C for 10
sec and 60°C for 30 sec. At the end of the PCR cycles, melting
curve analysis was performed to validate the specific generation of
the expected PCR products. Primers were designed using Applied
Biosystems Primer Express 3.0 (Applied Biosystems; Thermo Fisher
Scientific, Inc). GAPDH was used as internal reference and the
2−ΔΔCq method was used to calculate the expression of
lncRNA MEG3 in samples from serum and cells (29). The following primer sequences were
used for the qPCR: MEG3: 5′-CTGCCCATCTACACCTCACG-3′ (forward) and
5′-CTCTCCGCCGTCTGCGCTAGG GGCT-3′ (reverse); GAPDH:
5′-GAAGGTGAAGGTCGGAGTC-3′ (forward) and 5′-GAAGATGGTGATGGGATTTC-3′
(reverse).
Western blotting
A total of 24 h after transfection, cells were
washed 3 times with PBS and incubated with radioimmunoprecipitation
assay lysis buffer (cat. no. KGP702-100; Nanjing KeyGen Biotech
Co., Ltd.) for 30 min on ice. After 30 min of cell lysis, cell
lysates were sonicated with 20 KHz frequency on ice for 1 min and
centrifuged at 14,000 × g for 15 min at 4°C, and the protein
concentration was quantified using bicinchoninic acid protein
concentration kit (Nanjing KeyGen Biotech Co., Ltd.). Samples
containing 20 µg total protein were analyzed using SDS-PAGE
(10% gel) using an electrophoresis instrument (Bio-Rad
Laboratories, Inc., Hercules, CA, USA), and western blotting was
performed according to the previously published protocol (28). Antibodies against PCNA, cyclin A
and E, p53 and β-actin were used overnight at 4°C and washed 6
times with tris buffered saline with Tween-20 (TBST) at room
temperature (5 min/wash). Subsequently, the membrane was treated
with horseradish peroxidase labeled goat-anti-rabbit immunoglobulin
G (IgG) secondary antibody (cat. no. VA002; 1:5,000; Xuzhou VICMED
Biological Technology Co., Ltd., Xuzhou, China; www.vicmed.cn) or goat-anti-mouse IgG secondary
antibody (cat. no. VA001; 1:5,000; Xuzhou VICMED Biological
Technology Co., Ltd.), shaken and incubated at room temperature for
1 h. Following washing with TBST 6 times at room temperature (5
min/wash), the membranes were incubated with ECL solution for 5
min. Bands were imaged using VersaDoc™ MP 4000 (Bio-Rad
Laboratories, Inc.) and analyzed by using PDQuest Advanced 2D
analysis software (version 8.0, Bio-Rad Laboratories, Inc.).
Flow cytometry analysis of cell
cycle
The transfected HA-VSMCs were cultured in 6-well
plates. To perform cell cycle analysis, the cells were digested
with trypsin and fixed with 70% ethanol for 24 h at 4°C. The cells
were stained according to the manufacturer’s protocol of CycleTEST
PLUS DNA Reagent kit (BD Biosciences). The fixed cells were washed
3 times with PBS and centrifuged at 300 × g for 5 min in room
temperature. The supernatant was subsequently discarded. Cells were
incubated with propidium iodide (PI) stain buffer for 10 min and
subsequently treated with 200 µl of PBS, 200 µl of
RNase A and 200 µl of PI for 10 min at 4°C in the dark.
Finally, the cells were screened by 400-mesh sieves and analyzed by
flow cytometry (FACSCanto II; BD Biosciences). The results obtained
were analyzed using the ModFit software (version 4.1; Verity
Software House, Inc., Topsham, ME, USA).
Wound healing assay
For the wound healing assay, the following groups of
HA-VSMCs were cultured in 6-wellplates until 60% confluence:
control, si-NC, si-lncRNA MEG3, si-lncRNA MEG3 + baicalin (5
µmol/l), si-lncRNA MEG3 + baicalin (10 µmol/l),
si-lncRNA MEG3 + baicalin (20 µmol/l). Cells were scratched
with pipette tips and washed three times with PBS (15). Images of scratch areas were
captured at 0 and 24 h. Each experiment was repeated five times
independently.
Mitochondrial depolarization assay
The potential of mitochondrial membrane was analyzed
as previously described using JC-1 staining (30). Mitochondrial depolarization is
demonstrated by increased green/red fluorescence intensity ratio.
When mitochondrial membrane potential level is low, green
fluorescence is primarily observed. When the mitochondrial membrane
potential increases, a polymer is formed and red fluorescence is
observed (31). Mitochondrial
membrane potentials were detected using a fluorescent microscope
with 488 nm excitation wavelength.
5-Ethynyl-2′-deoxyuridine (EdU)
staining
Cell proliferation was analyzed using kFluor647
Click-iT EdU kit (Nanjing KeyGen Biotech Co., Ltd.) according to
the manufacturer’s protocol. HA-VSMCs were seeded into 96-well cell
culture plates at a density of 1×104 cells/well.
Following transfection and treatment with baicalin, HA-VSMCs were
incubated with 10 µmol/l EdU for 4 h at 37°C. Cells were
permeabilized for 20 min with 0.5% Triton X-100 after fixing with
4% formaldehyde for 30 min at room temperature. Cells in each well
were washed three times with 0.1 ml 3% bovine serum albumin (cat.
no. VIC018; Xuzhou VICMED Biological Technology Co., Ltd.) in PBS
and subsequently incubated with 1X Click-iT EdU reaction buffer at
room temperature for 30 min in the dark. HA-VSMC nuclei stained by
Hoechst were used to count cells and visualization was performed
using a fluorescence microscope as described below (Nikon
Corporation, Tokyo, Japan).
Hoechst staining
Treated HA-VSMCs were cultured on 24-well cell
culture plates. Subsequently, 1 µl of Hoechst 33342 (5
mg/ml; Sigma-Aldrich; Merck KGaA) in 200 µl of PBS was added
to HA-VSMCs and incubated for 20 min in room temperature. The
stained cells were washed three times with PBS, and cell morphology
was observed under a fluorescence microscope.
Immunofluorescence
HA-VSMCs were fixed with 4% paraformaldehyde for 15
min at room temperature, and washed in cold PBS 3 times for 5 min.
Permeabilization was performed in 0.4% Triton X-100 (Sigma-Aldrich;
Merck KGaA) for 10 min at room temperature. For immunofluorescence
staining, the experiment was conducted as previously described
(15).
Cell viability assay
Cell viability was assayed by the MTT assay, as
previously described (28).
HA-VSMCs were cultured in 96-well microtitration plates and then
the cells were subjected to growth arrest in DMEM without serum for
24 h. Following transfection and treatment, HA-VSMCs were incubated
for 4 h in a medium containing 0.5% MTT. Thereafter, the
supernatant was removed, and dimethylsulfoxide (150 µl/well)
was added. The plates were then agitated on a plate shaker for 10
min at room temperature. The absorbance was measured at a
wavelength of 490 nm in a spectrophotometer.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Comparisons between two groups were performed using
unpaired Student’s t-test and three or more groups were compared
using one-way analysis of variance, followed by Dunnett’s test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression level of MEG3 decreases in
serum samples from patients with AS and ox-LDL-treated HA-VSMCs,
and baicalin increases the expression levels of MEG3 in
ox-LDL-treated HA-VSMCs
The current study determined the expression levels
of MEG3 in serum samples from patients with AS and HA-VSMCs treated
with ox-LDL. RT-qPCR assay indicated that the expression level of
MEG3 significantly decreased in patients with AS compared with
healthy controls (Fig. 2A). A
previous study reported that ox-LDL serves an important role in the
progression of AS (32). Ox-LDL
promotes osteogenic differentiation and proliferation of VSMCs
(33), and has been widely used
to induce a model of AS (27,32,33). Consistent with these results, the
current study indicated that ox-LDL promoted cell proliferation in
a concentration dependent manner (Fig. 2B and C). In addition, the
expression levels of MEG3 were detected in HA-VSMCs after treatment
with ox-LDL. The expression of MEG3 decreased following stimulation
with ox-LDL. The results indicated that the expression level of
MEG3 decreased in a concentration-dependent manner. The greatest
inhibition of MEG3 expression was achieved using a dose of 75
µg/ml ox-LDL, and, therefore, this dose was used in
subsequent experiments (Fig. 2D).
These results indicated that MEG3 may be a mediator in AS
progression. HA-VSMCs treated with ox-LDL were used as an in
vitro model of AS and subsequently treated with baicalin. The
present study aimed to determine whether different concentrations
of baicalin affected the expression of MEG3. Data indicated that
different concentrations of baicalin did not affect the expression
of MEG3 in the absence of oxLDL treatment (Fig. 2E), suggesting that this drug did
not affect the expression of MEG3 in normal cells. However, in
ox-LDL-treated HA-VSMCs, incubation with ox-LDL resulted in a
significant decrease in MEG3 expression compared with the control.
Baicalin reversed this effect in a dose-dependent manner (Fig. 2F).
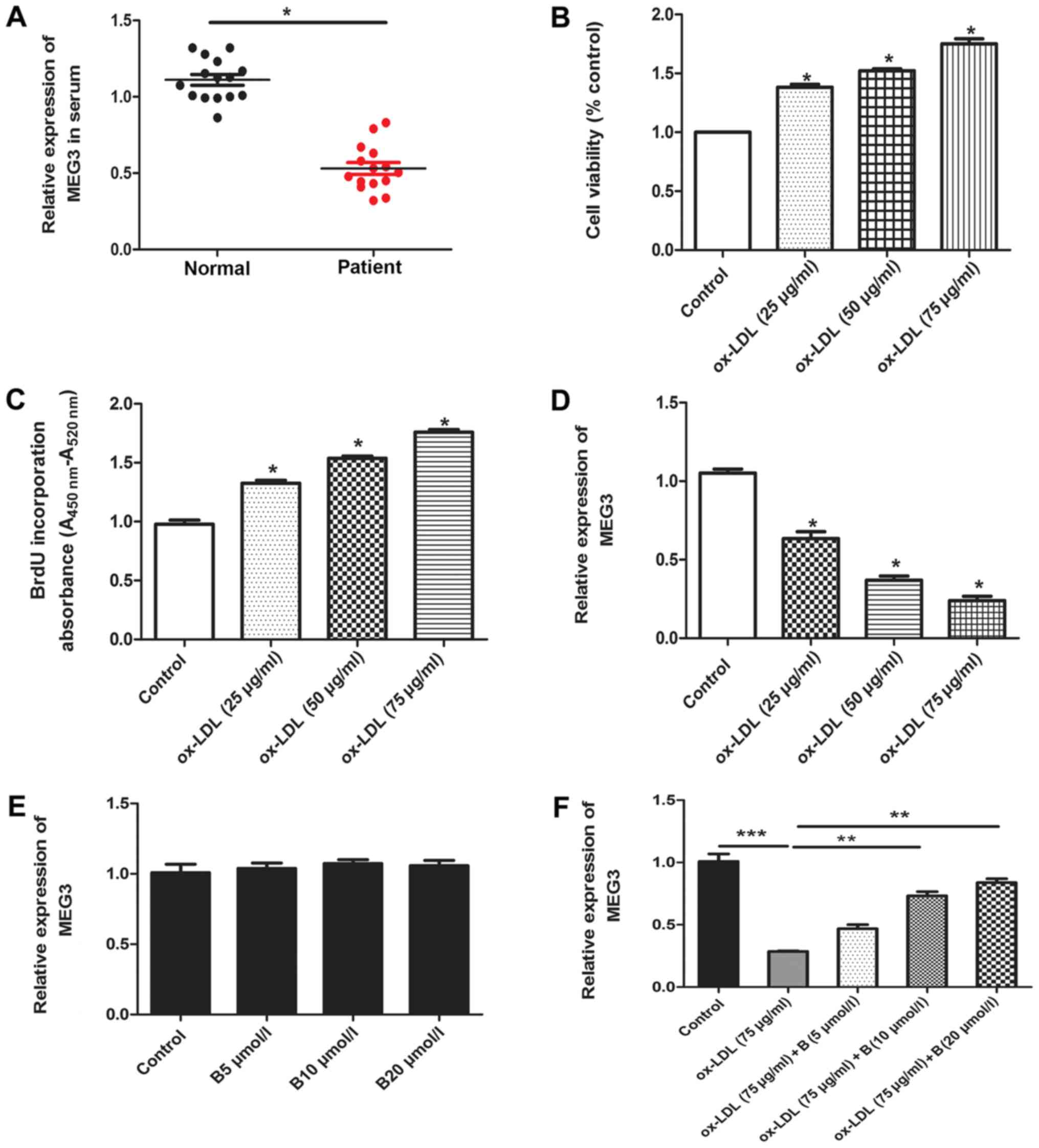 | Figure 2Expression of MEG3 in serum samples
and ox-LDL-treated HA-VSMCs, and the effect of treatment with
baicalin. (A) The expression of MEG3 in serum samples from patients
and controls. n=40. *P<0.05, as indicated. (B) MTT
assay was performed to determine the HA-VSMC viability following
treatment with different concentrations of ox-LDL (0, 25, 50 and 75
µg/ml). *P<0.05 vs. the control group. n=6.
(C) BrdU assay was performed to determine DNA synthesis following
treatment with different concentrations of ox-LDL (0, 25, 50 and 75
µg/ml). n=6. *P<0.05 vs. the control group.
(D) HA-VSMCs were treated with different concentrations of ox-LDL
(0, 25, 50 and 75 µg/ml) for 24 h, followed by the detection
of MEG3 expression. n=6. *P<0.05 vs. the control
group. (E) Effect of baicalin on MEG3 expression. (F) Effect of
baicalin on MEG3 expression in cells treated with ox-LDL. n=6.
**P<0.01 and ***P<0.001, as indicated.
MEG3, maternally expressed gene 3; B, baicalin; ox-LDL, oxidized
low-density lipoprotein; HA-VSMC, human aorta vascular smooth
muscle cell; BrdU, bromodeoxyuridine. |
Baicalin inhibits MEG3 silencing-induced
proliferation of ox-LDL- treated HA-VSMCs
To verify the function of MEG3 in the progression of
AS, the current study determined the effects of MEG3 silencing on
proliferation of ox-LDL-treated HA-VSMCs. HA-VSMCs treated with 75
µg/ml ox-LDL were transfected with si-MEG3 and treated with
different concentration of baicalin (5, 10 and 20 µmol/l),
followed by measurement of cell proliferative ability. The
knockdown of MEG3 was confirmed by qPCR (Fig. 3A). BrdU assay indicated that
downregulation of MEG3 caused a significant increase in cell
proliferation compared with the si-NC group in ox-LDL-treated
HA-VSMCs. However, cell proliferation was inhibited following
treatment with different concentrations of baicalin (Fig. 3B). To determine whether baicalin
affected cell cycle progression by increasing MEG3 expression in
the presence of ox-LDL, flow cytometry was used to detect the
number of cells at different stages of the cell cycle. MEG3
knockdown increased the percentage of cells at the
G2/M+S phase compared with the si-NC group. Baicalin
inhibited cell cycle progression and increased the number of
HA-VSMCs at the G0/G1 phase (Fig. 3C and D). EdU staining analysis
indicated that the number of proliferating cells increased
following knockdown of MEG3 compared with the si-NC group (Fig. 3E and F). This effect was reversed
in the presence of baicalin at a concentration of 10 or 20
µmol/l. MEG3 is a tumor suppressor gene, and its ectopic
expression inhibited cell proliferation and promoted cell apoptosis
in human glioma cell line (34).
Our previous study indicated that MEG3 knockdown may trigger human
pulmonary artery smooth muscle cell proliferation and migration
(15). In the present study, MEG3
knockdown promoted the proliferation of HA-VSMCs in the absence of
ox-LDL, compared with the si-NC group. However, compared with the
MEG3 knockdown group untreated with ox-LDL, proliferation increased
in cells transfected with si-lncRNA MEG3 and treated with ox-LDL
(Fig. 4A). In the absence of
ox-LDL, baicalin inhibited the proliferation of HA-VSMCs
transfected with si-lncRNA (Fig.
4B). The above results indicated that baicalin inhibited the
proliferation induced by MEG3 knockdown and stimulation with
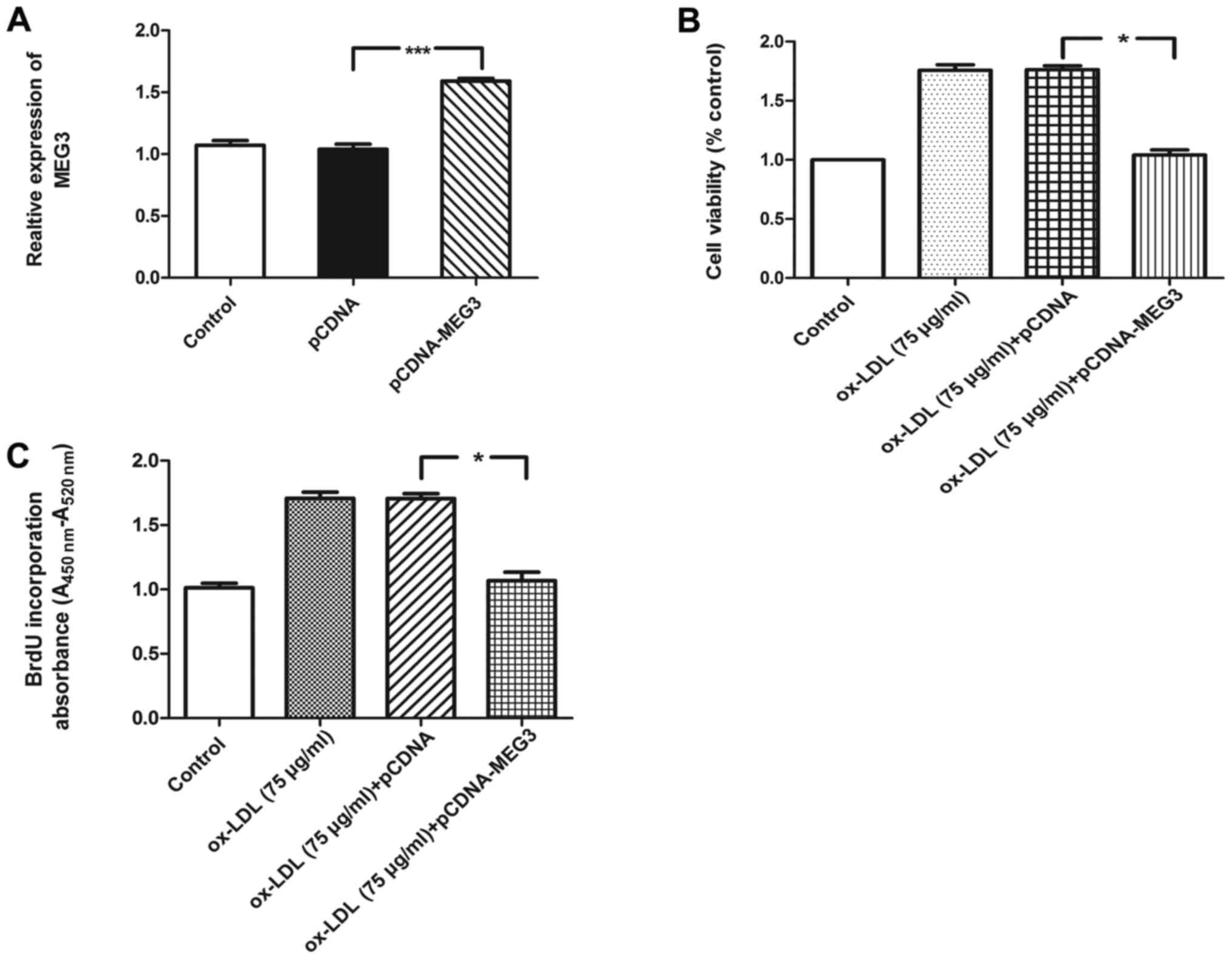
ox-LDL. Furthermore, RT-qPCR analysis of MEG3 expression was
performed following transfection with pCDNA-MEG-3. Compared with
the pCDNA group, the expression level of MEG3 increased by
1.59-fold in the pCDNA-MEG3 group (Fig. 5A). Furthermore, overexpression of
MEG3 suppressed the viability and proliferation in ox-LDL-treated
HA-VSMCs compared with the ox-LDL + pCDNA group (Fig. 5B and C).
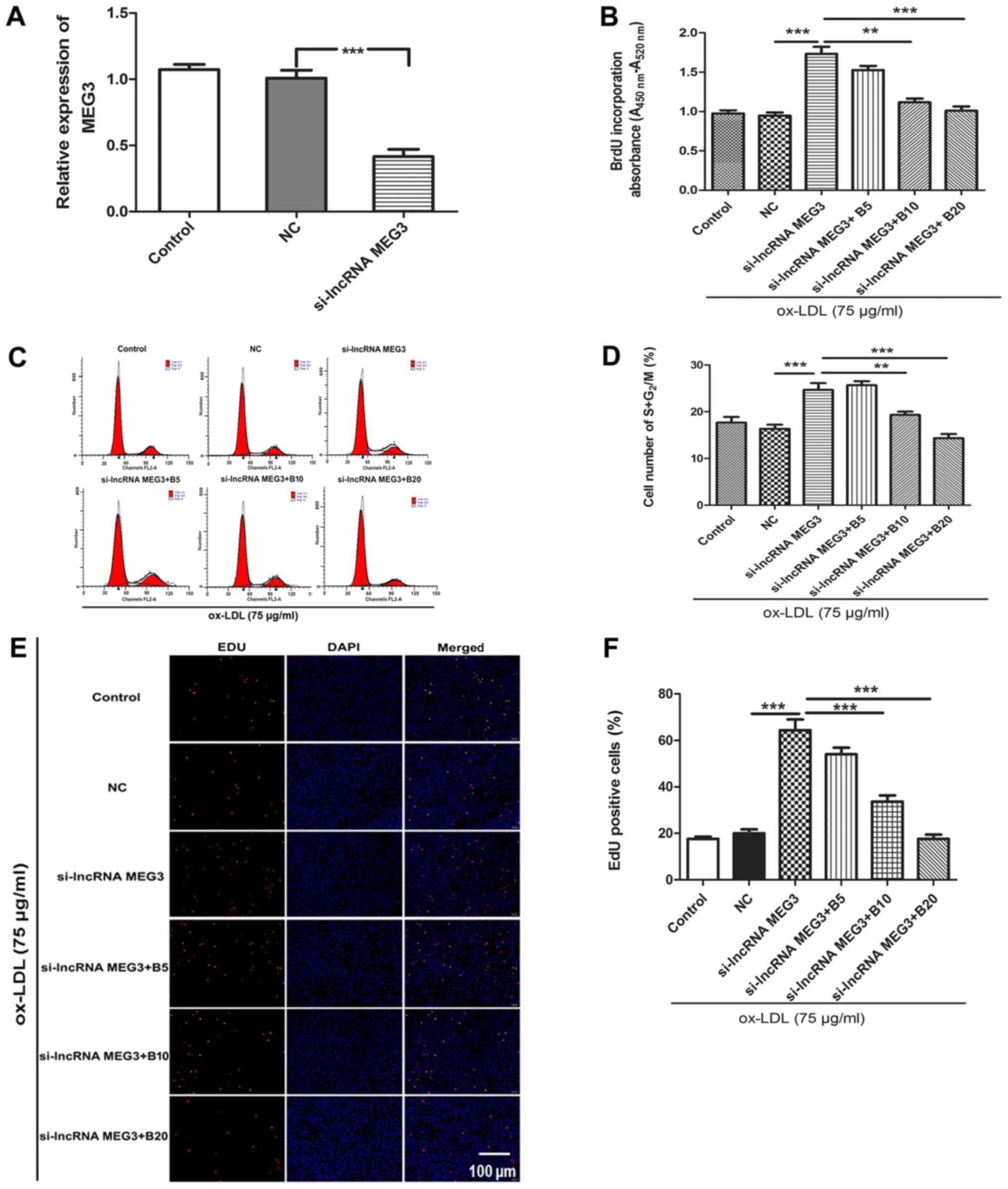 | Figure 3Baicalin reverses MEG3
knockdown-induced proliferation in ox-LDL-treated HA-VSMCs. (A)
Reverse transcription-quantitative polymerase chain reaction
verified MEG3 knockdown efficiency. (B) BrdU analysis of DNA
synthesis. (C) Flow cytometry plots. (D) Quantitative analysis of
flow cytometry results on cell cycle progression. (E) EdU staining
to study cell proliferation. (F) Quantitative analysis of EdU
staining results. n=6. **P<0.01 and
***P<0.001, as indicated. si, small interfering; lnc,
long noncoding; MEG3, maternally expressed gene 3; B, baicalin;
ox-LDL, oxidized low-density lipoprotein; HA-VSMC, human aorta
vascular smooth muscle cell; BrdU, bromodeoxyuridine; NC, negative
control; 5, 10 and 20 refer to the concentration of baicalin in
µmol/l; EdU, 5-ethynyl-2′-deoxyuridine. |
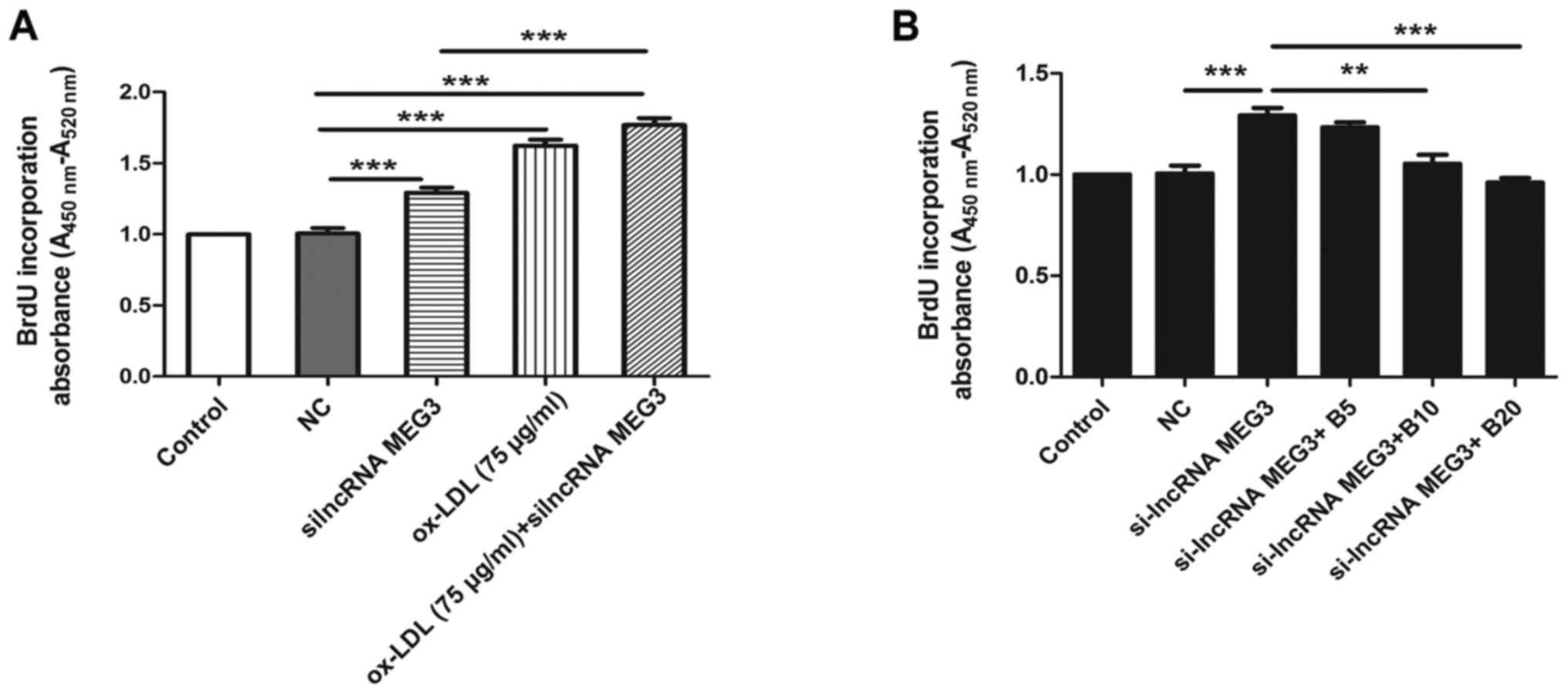 | Figure 4Effect of MEG3 silencing on cell
proliferation. (A) BrdU assay was used to analyze the effect of
MEG3 knockdown in the proliferation of HA-VSMCs in the absence and
presence of ox-LDL. (B) BrdU assay was used to analyze the effect
of baicalin on the proliferation of HA-VSMCs in the absence of
ox-LDL. **P<0.01 and ***P<0.001, as
indicated. n=6. BrdU, bromodeoxyuridine; si, small interfering;
lnc, long noncoding; MEG3, maternally expressed gene 3; B,
baicalin; ox-LDL, oxidized low-density lipoprotein; HA-VSMC, human
aorta vascular smooth muscle cell; NC, negative control; 5, 10 and
20 refer to the concentration of baicalin in µmol/l. |
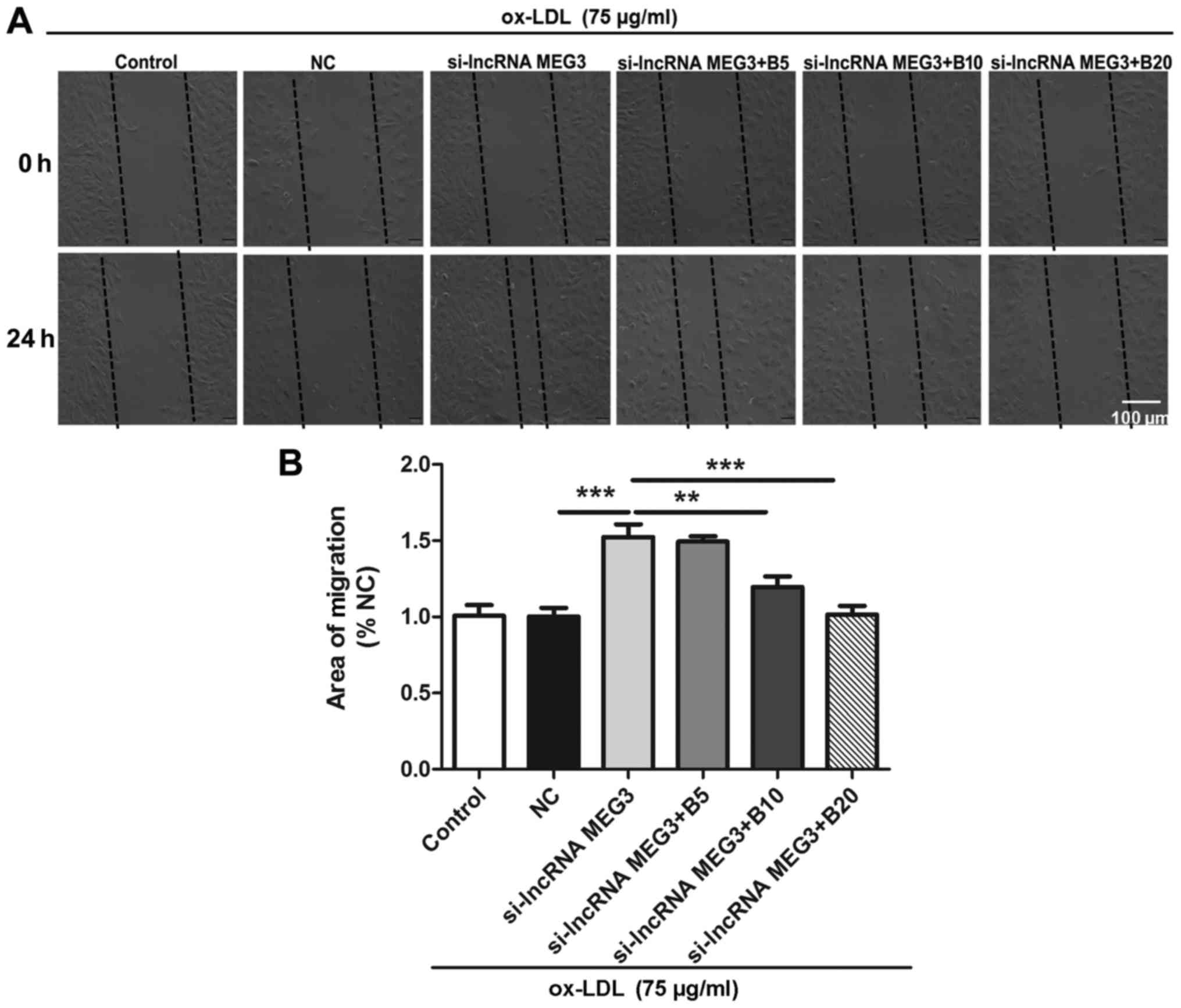
Baicalin inhibits MEG3 knockdown-induced
migration in ox-LDL- treated HA-VSMCs
It has been reported that migration of HA-VSMCs
contributed to vascular remodeling in AS (35). The current study examined the
effect of baicalin on MEG3 silencing-induced migration of HA-VSMCs
using a wound healing assay. Ox-LDL- treated HA-VSMCs were treated
with control, si-NC, si-lncRNA MEG3, si-lncRNA MEG3 + baicalin (5
µmol/l), si-lncRNA MEG3 + baicalin (10 µmol/l) and
si-lncRNA MEG3 + baicalin (20 µmol/l). A scratch was made
using a pipette tip and images were captured at 0 and 24 h to
analyze the degree of migration in different treatment groups. The
results indicated that knockdown of MEG3 increased the migration of
HA-VSMCs, compared with the si-NC group; however, treatment with
baicalin at a dose of 10 and 20 µmol/l significantly
reversed the effect of MEG3 knockdown (Fig. 6A and B).
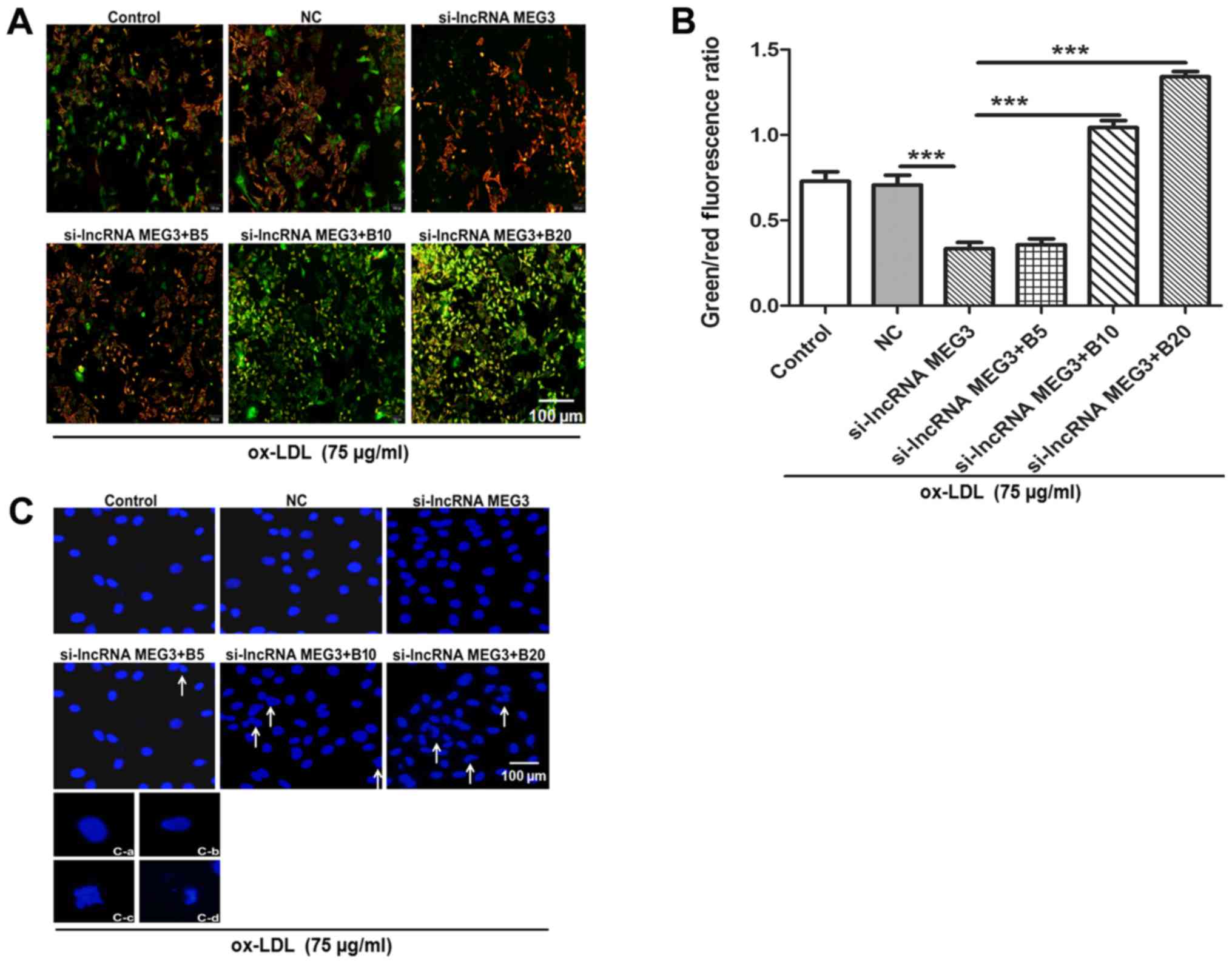
Baicalin reverses the effect of MEG3
silencing on cell proliferation and promotes apoptosis in
ox-LDL-induced HA-VSMCs
In order to further investigate whether baicalin
could promote apoptosis in AS in vitro, the expression of
MEG3 was decreased in HA-VSMCs through pre-treatment with ox-LDL.
Alterations in mitochondrial function were initially investigated.
According to previous studies, disruption of mitochondrial membrane
potential is an early event of apop-tosis (31,36,37). The alterations of mitochondrial
membrane potential were verified by JC-1 staining. Following MEG3
knockdown the green/red fluorescence ratio was reduced compared
with the si-NC group, suggesting an increase in mitochondrial
membrane potential. By contrast, the mitochondrial membrane
potential decreased following treatment with baicalin, as evidenced
by increased ratio of green to red fluorescence (Fig. 7A and B). Furthermore, chromatin
condensation and cell nuclear morphology were observed by staining
with Hoechst 33342. The MEG3 knockdown group exhibited the same
complete nuclear morphology as the NC group in cells, while
treatment with baicalin resulted in alterations in chromatin
morphology including crenation, condensation and fragmentation
(Fig. 7C).
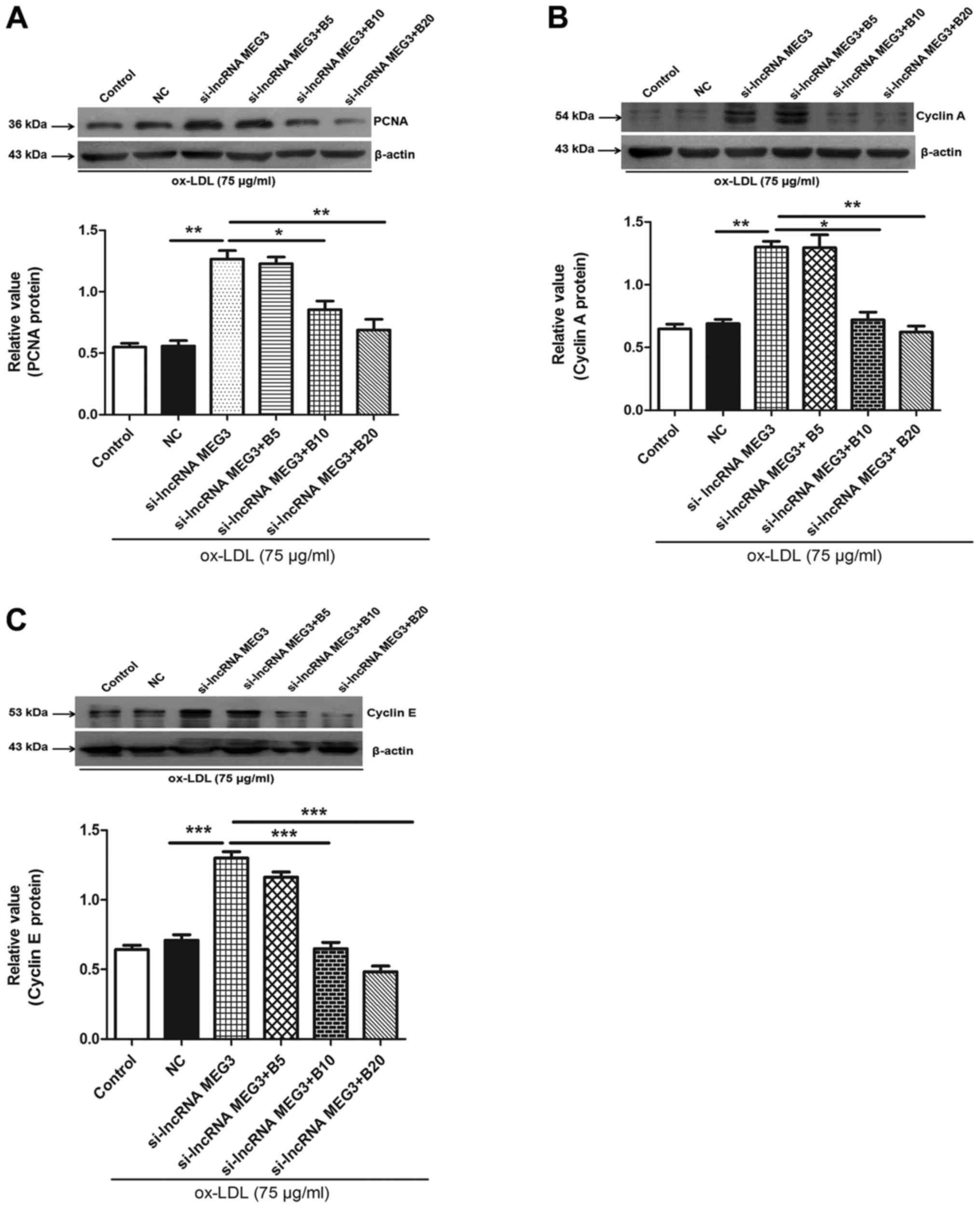
Baicalin inhibits the expression of PCNA,
cyclin A and E in HA-VSMCs
To further determine the mechanism underlying
baicalin-mediated inhibition of MEG3 knockdown-induced
proliferation, the expression of PCNA was determined by western
blotting. The results indicated that MEG3 knockdown increased the
expression of PCNA, which was reversed in the presence of different
concentration of baicalin (Fig.
8A). Cyclin A and E serve roles in cell cycle progression, and,
therefore, expression levels of these proteins were determined in
ox-LDL-stimulated HA-VSMCs. The results indicated that knockdown of
MEG3 increased the expression of cyclin A and E; however, this
effect was reversed by treatment with baicalin (Fig. 8B and C). These results indicated
that baicalin may serve a role in regulating cell cycle progression
and inhibiting cell proliferation.
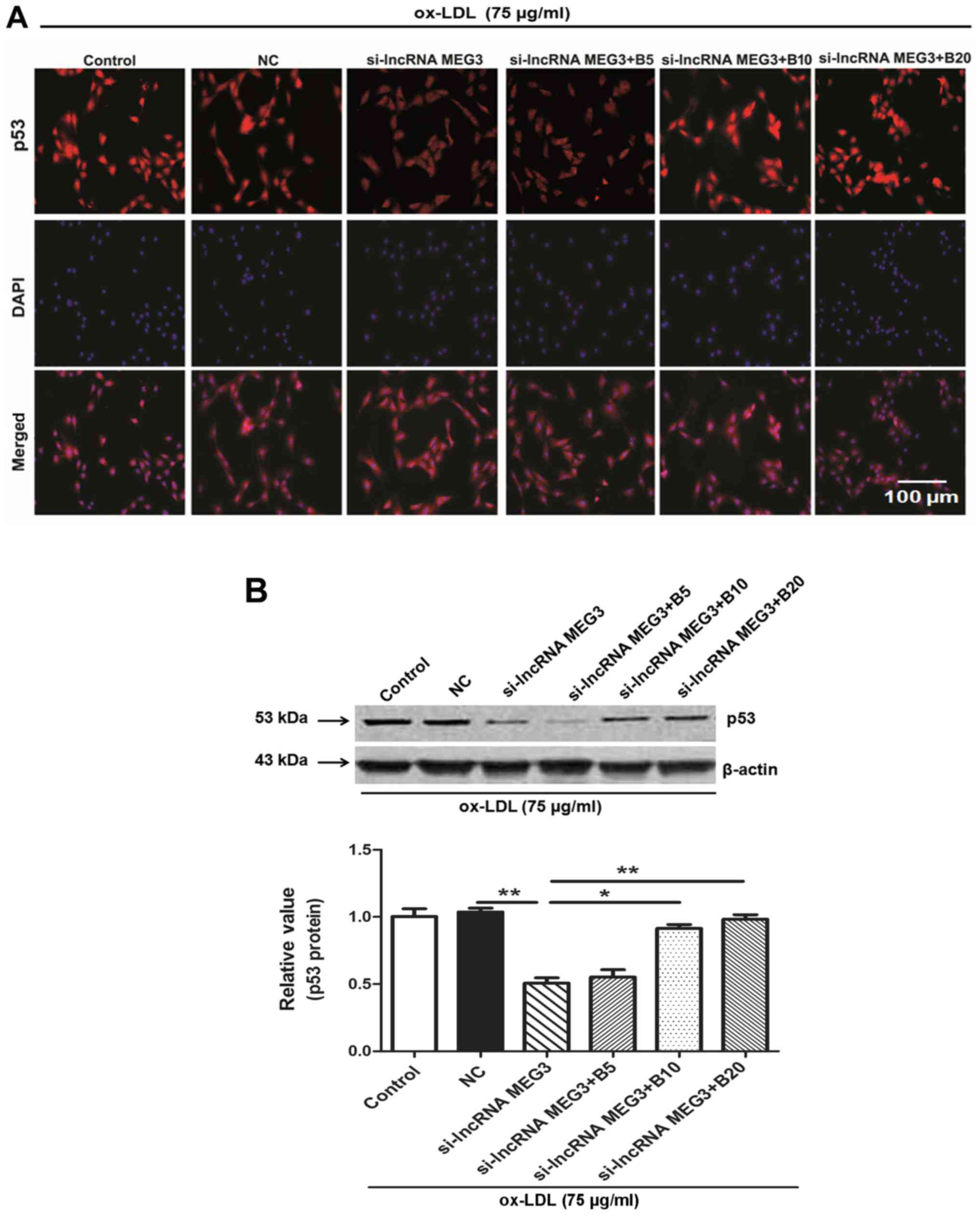
Baicalin suppresses the ox-LDL-stimulated
cell growth via modulation of the MEG3/p53 pathway in HA-VSMCs
Previous studies demonstrated that the p53 signaling
pathway serves a role in AS (38,39). To further examine the mechanism
underling baicalin-mediated inhibition of cell growth, p53
immunofluorescence and western blotting were performed in
ox-LDL-treated HA-VSMCs. Fluorescence data indicated that p53 was
expressed in the nucleus under normal conditions; however,
cytoplasmic expression was detected following knockdown of MEG3.
Following treatment with different concentrations of baicalin, p53
was activated and returned to nuclear expression (Fig. 9A). Western blotting further
indicated that the low expression of p53 following MEG3 knockdown
gradually increased following administration of different
concentrations of baicalin (Fig.
9B). The above data indicated that baicalin inhibited the
growth of HA-VSMCs possibly through the p53 signaling pathway.
Discussion
At present, AS is one of the most common
cardiovascular disorders, even though there have been significant
improvements in clinical diagnosis and control of symptoms
(2,7,40).
AS remains a major health concern and exhibits high morbidity and
mortality rates worldwide (41).
A number of studies indicated that lncRNAs serve important roles in
the progression of AS (2,27,42). The current study demonstrated that
lncRNA MEG3 may be an important regulator of cell proliferation and
apoptosis in vitro. MEG3 knockdown promoted the
proliferation of HA-VSMCs in the absence of ox-LDL. Furthermore, in
the presence of ox-LDL, MEG3 knockdown increased the proliferation
even further. Ox-LDL can induce AS via multiple mechanisms,
including promotion of EC activation and dysfunction, VSMC
proliferation and migration, and foam cell formation (32,43). VSMCs, the major cell type in blood
vessels, participate in remodeling of the arterial wall in order to
maintain blood flow in vessels affected by AS (3,44).
MEG3 is a tumor suppressor gene and its ectopic expression
inhibited cell proliferation and promoted cell apoptosis in human
glioma cell line(34). Therefore,
the present study aimed to investigate whether MEG3 was involved in
the regulation of AS progression in ox-LDL-stimulated HA-VSMCs. The
results indicated that the expression of MEG3 was reduced in serum
samples from patients with AS and ox-LDL-induced HA-VSMCs, compared
with normal control samples and cells untreated with ox-LDL,
respectively. Further experiments indicated that the decrease in
MEG3 expression was reversed by treatment with baicalin in a
concentration-dependent manner. Knockdown of endogenous MEG3
promoted proliferation and migration, and inhibited apoptosis in
HA-VSMCs. Baicalin reversed the effects of MEG3 knockdown on
proliferation, migration and apoptosis in ox-LDL-treated HA-VSMCs.
We previously reported that MEG3 knockdown triggered human
pulmonary artery smooth muscle cell proliferation and migration
(15). The current study
demonstrated increased expression levels of cyclin A and E
following MEG3 knockdown. However, following treatment of HA-VSMCs
with different concentrations of baicalin, the MEG3
knockdown-induced effect on the expression levels of cyclin A and E
was reversed. Baicalin inhibited proliferation and facilitated
apoptosis through activating the MEG3/p53 expression. This result
suggests a regulatory activity of lncRNA MEG3 in AS and this
molecule may be a novel therapeutic target for the treatment of
human cardiovascular disease.
Abnormal proliferation, apoptosis and migration of
VSMCs are considered key factors in the development of AS (4,45).
Previous studies have demonstrated that lncRNAs serve important
roles in the development of AS (2,27,46). Numerous studies demonstrated the
potential role of lncRNAs in proliferation and migration of smooth
muscle cells. A recent study suggested that lncRNA-H19 knockdown
inhibited proliferation and induced apoptosis of HA-VSMCs by
inactivating microRNA-148b/WNT/β-catenin signaling pathway,
suggesting the potential application of H19 in the prevention of AS
(27), which could be used as a
potential target for AS. Another study reported that lncRNA
antisense RNA in the INK4 locus (ANRIL) was closely associated with
the severity of AS (46). ANRIL
increased the proliferation of VSMCs by reducing the expression of
cyclin dependent kinase inhibitor 2A and B (47). LincRNA-p21, a novel regulatory
factor, has been revealed to regulate neointima formation,
proliferation, apoptosis and AS in VSMCs by increasing the activity
of p53 (2). To the best of our
knowledge, the current study is the first to identify an
interaction between MEG3 and AS in vitro. In serum samples
from patients with AS, the expression of MEG3 was significantly
reduced compared with the normal control group. Furthermore,
silencing of MEG3 significantly accelerated proliferation and
migration, and inhibited apoptosis of HA-VSMCs, suggesting that
MEG3 silencing may suppress AS progression.
Baicalin has been reported to induce
anti-inflammatory (21) and
antiproliferative effects (17).
Baicalin is widely used in the treatment of inflammatory diseases
(21,48) and dysfunction of VSMCs and ECs
(17,49). Numerous studies have confirmed
that baicalin serves anti-atherosclerotic roles by inhibiting
inflammation (21) and reducing
oxidative stress (19). In
addition, baicalin can protect chronic hypoxia-induced pulmonary
hypertension and cor pulmonale by reducing the activity of the p38
mitogen-activated protein kinase pathway and matrix
matalloprotease-9 (50). In a
previous study, baicalin and geniposide inhibited the progression
of AS by enhancing the expression of Wnt family member 1 and
attenuating the expression of dickkopf-related protein 1 (51). In a high-cholesterol diet,
baicalin decreased the expression of monocyte chemoattractant
protein 1 and interleukin-6 in the kidneys of ApoE knock out mice
(23). Although baicalin has been
demonstrated to exhibit a number of biological functions, the
underlying mechanism of baicalin-mediated prevention of AS
progression by altering lncRNA expression is not well documented.
The present study indicated that baicalin at a dose of 10 and 20
µmol/l significantly inhibited the proliferation and
migration, and promoted apoptosis of HA-VSMCs treated with xo-LDL.
The antiproliferative effect of baicalin was associated with cell
cycle arrest at the G0/G1 stage. Cell cycle
progression is regulated by a complex regulatory molecular network,
including cyclins (52). Cyclin A
and E are the main mediators during the cell cycle progression from
the G0/G1 to the S phase (53). In the present study, baicalin
arrested the cell cycle at the G0/G1 phase
and decreased the expression of cell cycle regulatory proteins
PCNA, cyclin A and E in ox-LDL-treated HA-VSMCs with MEG3
knockdown. It may be hypothesized that baicalin inhibited cell
proliferation via downregulation of expression of cell cycle
regulatory proteins, which resulted in cell cycle arrest at the
G0/G1 phase.
The p53 signaling pathway has been reported to be
involved in vascular remodeling and atherosclerotic plaque
formation (15,54,55). In a previous study, the expression
of p53 in atherosclerotic plaques was negatively correlated with
cell proliferation (56). In the
rabbit carotid artery, overexpression of p53 significantly
inhibited cell proliferation (57). In addition, high expression of p53
may inhibit monocyte/macrophage cell division and promote the
apoptosis of macrophages, thus preventing the development of AS
(25). The current study
confirmed that baicalin may increase the expression of p53 and
promote its transport from the cytoplasm to nucleus following MEG3
silencing. These results suggest that baicalin may ameliorate AS
and the MEG3/p53 signaling pathway may be involved in this
process.
In conclusion, baicalin inhibited proliferation and
promoted apoptosis in ox-LDL-treated HA-VSMCs by activating the
expression of MEG3/p53. Baicalin may be used to increase the
expression of MEG3/p53 and prevent AS. However, in future clinical
trials, the administration of baicalin to patients with AS may
provide understanding of disease pathways and further clarify the
therapeutic potential of baicalin.
Funding
The study is supported by the Open Fund of Key
Laboratory of Minstry of Education for TCM Viscera State Theory and
Applications, Liaoning University of Traditional Chinese Medicine
(grant no. zyzx1605), National Natural Science Foundation of China
(grant no. 31871155), Natural Science Foundation of Jiangsu
Province (grant no. BK20161297), the Social Fund for Development of
Lianyungang Science and Technology Bureau (grant no. SH1612),
project of Jiangsu Provincial Commission of Health and Family
Planning (grant no. QNRC2016505), and Six Big Talent Peak C Project
(grant no. YY-110).
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors’ contributions
YL designed the present study and the experiments,
and wrote the manuscript. LJ conducted western blotting and RT-qPCR
experiments. DM performed BrdU, EdU, JC-1 and flow cytometry
assays. YX performed cell culture, cell transfection and wound
healing assay. JZ performed patient blood sample collection and
Hoechst staining. ZS performed immunofluo-rescence assay. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee for the use of human samples of the First People’s
Hospital of Lianyungang, which was in accordance with the code of
ethics of the Declaration of Helsinki developed by the World
Medical Association. Each individual provided written informed
consent prior to their participation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Antoniades C, Antonopoulos AS, Bendall JK
and Channon KM: Targeting redox signaling in the vascular wall:
from basic science to clinical practice. Curr Pharm Des.
15:329–342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen
C, Cai Y, Huang H, Yang Y, Liu Y, et al: LincRNA-p21 regulates
neointima formation, vascular smooth muscle cell proliferation,
apoptosis, and atherosclerosis by enhancing p53 activity.
Circulation. 130:1452–1465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chistiakov DA, Orekhov AN and Bobryshev
YV: Vascular smooth muscle cell in atherosclerosis. Acta Physiol
(Oxf). 214:33–50. 2015. View Article : Google Scholar
|
|
4
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao XY and Lin JD: Long Noncoding RNAs: a
new regulatory code in metabolic control. Trends Biochem Sci.
40:586–596. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Zheng L, Wang Q and Hu YW: Emerging
roles and mechanisms of long noncoding RNAs in atherosclerosis. Int
J Cardiol. 228:570–582. 2017. View Article : Google Scholar
|
|
8
|
Ballantyne MD, Pinel K, Dakin R, Vesey AT,
Diver L, Mackenzie R, Garcia R, Welsh P, Sattar N, Hamilton G, et
al: Smooth muscle enriched long noncoding RNA (SMILR) regulates
cell proliferation. Circulation. 133:2050–2065. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li
YJ, Yan B and Jiang Q: Pathogenic role of lncRNA-MALAT1 in
endothelial cell dysfunction in diabetes mellitus. Cell Death Dis.
5:e15062014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Michalik KM, You X, Manavski Y,
Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W,
Uchida S, et al: Long noncoding RNA MALAT1 regulates endothelial
cell function and vessel growth. Circ Res. 114:1389–1397. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shan K, Jiang Q, Wang XQ, Wang YN, Yang H,
Yao MD, Liu C, Li XM, Yao J, Liu B, et al: Role of long non-coding
RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell
Death Dis. 7:e22482016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu
WQ, Xie WP and Hou YY: Long non-coding RNA MEG3 inhibits NSCLC
cells proliferation and induces apoptosis by affecting p53
expression. BMC Cancer. 13:4612013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cui HB, Ge HE, Wang YS and Bai XY:
MiR-208a enhances cell proliferation and invasion of gastric cancer
by targeting SFRP1 and negatively regulating MEG3. Int J Biochem
Cell Biol. 102:31–39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng Q, Lin Z, Xu J, Lu Y, Meng Q, Wang
C, Yang Y, Xin X, Li X, Pu H, et al: Long noncoding RNA MEG3
suppresses liver cancer cells growth through inhibiting β-catenin
by activating PKM2 and inactivating PTEN. Cell Death Dis.
9:2532018. View Article : Google Scholar
|
|
15
|
Sun Z, Nie X, Sun S, Dong S, Yuan C, Li Y,
Xiao B, Jie D and Liu Y: Long non-coding RNA MEG3 downregulation
triggers human pulmonary artery smooth muscle cell proliferation
and migration via the p53 signaling pathway. Cell Physiol Biochem.
42:2569–2581. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li C, Lin G and Zuo Z: Pharmacological
effects and pharmacokinetics properties of Radix Scutellariae and
its bioactive flavones. Biopharm Drug Dispos. 32:427–445. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong LH, Wen JK, Miao SB, Jia Z, Hu HJ,
Sun RH, Wu Y and Han M: Baicalin inhibits PDGF-BB-stimulated
vascular smooth muscle cell proliferation through suppressing
PDGFRβ-ERK signaling and increase in p27 accumulation and prevents
injury-induced neointimal hyperplasia. Cell Res. 20:1252–1262.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu W, Jin Z, Yu J, Liang J, Yang Q, Li F,
Shi X, Zhu X and Zhang X: Baicalin ameliorates experimental
inflammatory bowel disease through polarization of macrophages to
an M2 phenotype. Int Immunopharmacol. 35:119–126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Waisundara VY, Siu SY, Hsu A, Huang D and
Tan BK: Baicalin upregulates the genetic expression of antioxidant
enzymes in type-2 diabetic Goto-Kakizaki rats. Life Sci.
88:1016–1025. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Z, Ma L, Su M, Zhou Y, Mao K, Li C,
Peng G, Zhou C, Shen B and Dou J: Baicalin induces cellular
senescence in human colon cancer cells via upregulation of DEPP and
the activation of Ras/Raf/MEK/ERK signaling. Cell Death Dis.
9:2172018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang X, Zhang Q, Gao Z, Yu C and Zhang L:
Baicalin alleviates IL-1β-induced inflammatory injury via
down-regulating miR-126 in chondrocytes. Biomed Pharmacother.
99:184–190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ku SK and Bae JS: Baicalin, baicalein and
wogonin inhibits high glucose-induced vascular inflammation in
vitro and in vivo. BMB Rep. 48:519–524. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu L, Liao P, Wang B, Fang X, Li W and
Guan S: Baicalin inhibits the expression of monocyte
chemoattractant protein-1 and interleukin-6 in the kidneys of
apolipoprotein E-knockout mice fed a high cholesterol diet. Mol Med
Rep. 11:3976–3980. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guevara NV, Kim HS, Antonova EI and Chan
L: The absence of p53 accelerates atherosclerosis by increasing
cell proliferation in vivo. Nat Med. 5:335–339. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mercer J, Figg N, Stoneman V, Braganza D
and Bennett MR: Endogenous p53 protects vascular smooth muscle
cells from apoptosis and reduces atherosclerosis in ApoE knockout
mice. Circ Res. 96:667–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
el-Deiry WS, Tokino T, Velculescu VE, Levy
DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and
Vogelstein B: WAF1, a potential mediator of p53 tumor suppression.
Cell. 75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang L, Cheng H, Yue Y, Li S, Zhang D and
He R: H19 knockdown suppresses proliferation and induces apoptosis
by regulating miR-148b/WNT/β-catenin in ox-LDL -stimulated vascular
smooth muscle cells. J Biomed Sci. 25:112018. View Article : Google Scholar
|
|
28
|
Liu Y, Ma C, Zhang Q, Yu L, Ma J, Zhang L,
Hao X, Cao F, Wang L and Zhu D: The key role of transforming growth
factor-beta receptor I and 15-lipoxygenase in hypoxia-induced
proliferation of pulmonary artery smooth muscle cells. Int J
Biochem Cell Biol. 44:1184–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Liu Y, Cao Y, Sun S, Zhu J, Gao S, Pang J,
Zhu D and Sun Z: Transforming growth factor-beta1 upregulation
triggers pulmonary artery smooth muscle cell proliferation and
apoptosis imbalance in rats with hypoxic pulmonary hypertension via
the PTEN/AKT pathways. Int J Biochem Cell Biol. 77(Pt A): 141–154.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma J, Liang S, Wang Z, Zhang L, Jiang J,
Zheng J, Yu L, Zheng X, Wang R and Zhu D: ROCK pathway participates
in the processes that 15-hydroxyeicosatetraenoic acid (15-HETE)
mediated the pulmonary vascular remodeling induced by hypoxia in
rat. J Cell Physiol. 222:82–94. 2010. View Article : Google Scholar
|
|
32
|
Pirillo A, Norata GD and Catapano AL:
LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm.
2013:1527862013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang Y, Xu Q, Peng H, Liu Z, Yang T, Yu Z,
Cheng G, Li X, Zhang G and Shi R: The role of vascular peroxidase 1
in ox-LDL-induced vascular smooth muscle cell calcification.
Atherosclerosis. 243:357–363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang P, Ren Z and Sun P: Overexpression of
the long non-coding RNA MEG3 impairs in vitro glioma cell
proliferation. J Cell Biochem. 113:1868–1874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Goncharova EA, Ammit AJ, Irani C, Carroll
RG, Eszterhas AJ, Panettieri RA and Krymskaya VP: PI3K is required
for proliferation and migration of human pulmonary vascular smooth
muscle cells. Am J Physiol Lung Cell Mol Physiol. 283:L354–L363.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Salido M, Gonzalez JL and Vilches J: Loss
of mitochondrial membrane potential is inhibited by bombesin in
etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol
Cancer Ther. 6:1292–1299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Z, Tang X, Li Y, Leu C, Guo L, Zheng
X and Zhu D: 20-Hydroxyeicosatetraenoic acid inhibits the apoptotic
responses in pulmonary artery smooth muscle cells. Eur J Pharmacol.
588:9–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xiong Y, Yu Y, Montani JP, Yang Z and Ming
XF: Arginase-II induces vascular smooth muscle cell senescence and
apoptosis through p66Shc and p53 independently of its l-arginine
urea-hydrolase activity: implications for atherosclerotic plaque
vulnerability. J Am Heart Assoc. 2:e0000962013. View Article : Google Scholar
|
|
39
|
Leeper NJ, Raiesdana A, Kojima Y, Kundu
RK, Cheng H, Maegdefessel L, Toh R, Ahn GO, Ali ZA, Anderson DR, et
al: Loss of CDKN2B promotes p53-dependent smooth muscle cell
apoptosis and aneurysm formation. Arterioscler Thromb Vasc Biol.
33:e1-e102013. View Article : Google Scholar :
|
|
40
|
Li W, Huang H, Li L, Wang L, Li Y, Wang Y,
Guo S, Li L, Wang D, He Y, et al: The pathogenesis of
atherosclerosis based on human signaling networks and stem cell
expression data. Int J Biol Sci. 14:1678–1685. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mozaffarian D, Benjamin EJ, Go AS, Arnett
DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP,
Fullerton HJ, et al Writing Group Members: American Heart
Association Statistics Committee; Stroke Statistics Subcommittee:
Heart disease and stroke statistics-2016 update: a report from the
american heart association. Circulation. 133:e38–e360. 2016.
|
|
42
|
Li H, Zhu H and Ge J: Long noncoding RNA:
recent updates in atherosclerosis. Int J Biol Sci. 12:898–910.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nakajima K, Nakano T and Tanaka A: The
oxidative modification hypothesis of atherosclerosis: the
comparison of atherogenic effects on oxidized LDL and remnant
lipoproteins in plasma. Clin Chim Acta. 367:36–47. PubMed/NCBI
|
|
44
|
Johnson JL: Emerging regulators of
vascular smooth muscle cell function in the development and
progression of atherosclerosis. Cardiovasc Res. 103:452–460. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang P, Xu TY, Guan YF, Zhao Y, Li ZY, Lan
XH, Wang X, Yang PY, Kang ZM, Vanhoutte PM, et al: Vascular smooth
muscle cell apoptosis is an early trigger for hypothyroid
atherosclerosis. Cardiovasc Res. 102:448–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Holdt LM, Beutner F, Scholz M, Gielen S,
Gäbel G, Bergert H, Schuler G, Thiery J and Teupser D: ANRIL
expression is associated with atherosclerosis risk at chromosome
9p21. Arterioscler Thromb Vasc Biol. 30:620–627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Congrains A, Kamide K, Oguro R, Yasuda O,
Miyata K, Yamamoto E, Kawai T, Kusunoki H, Yamamoto H, Takeya Y, et
al: Genetic variants at the 9p21 locus contribute to
atherosclerosis through modulation of ANRIL and CDKN2A/B.
Atherosclerosis. 220:449–455. 2012. View Article : Google Scholar
|
|
48
|
Hou J, Wang J, Zhang P, Li D, Zhang C,
Zhao H, Fu J, Wang B and Liu J: Baicalin attenuates proinflammatory
cytokine production in oxygen-glucose deprived challenged rat
microglial cells by inhibiting TLR4 signaling pathway. Int
Immunopharmacol. 14:749–757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim DH, Cho KH, Moon SK, Kim YS, Kim DH,
Choi JS and Chung HY: Cytoprotective mechanism of baicalin against
endothelial cell damage by peroxynitrite. J Pharm Pharmacol.
57:1581–1590. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yan S, Wang Y, Liu P, Chen A, Chen M, Yao
D, Xu X, Wang L and Huang X: Baicalin attenuates hypoxia-induced
pulmonary arterial hypertension to improve hypoxic cor pulmonale by
reducing the activity of the p38 MAPK signaling pathway and MMP-9.
Evid Based Complement Alternat Med. 2016:25464022016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang B, Liao PP, Liu LH, Fang X, Li W and
Guan SM: Baicalin and geniposide inhibit the development of
atherosclerosis by increasing Wnt1 and inhibiting dickkopf-related
protein-1 expression. J Geriatr Cardiol. 13:846–854.
2016.PubMed/NCBI
|
|
52
|
Bendris N, Lemmers B and Blanchard JM:
Cell cycle, cytoskeleton dynamics and beyond: The many functions of
cyclins and CDK inhibitors. Cell Cycle. 14:1786–1798. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Braun-Dullaeus RC, Mann MJ, Sedding DG,
Sherwood SW, von der Leyen HE and Dzau VJ: Cell cycle-dependent
regulation of smooth muscle cell activation. Arterioscler Thromb
Vasc Biol. 24:845–850. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mayr U, Mayr M, Li C, Wernig F, Dietrich
H, Hu Y and Xu Q: Loss of p53 accelerates neointimal lesions of
vein bypass grafts in mice. Circ Res. 90:197–204. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sanz-González SM, Barquín L, García-Cao I,
Roque M, González JM, Fuster JJ, Castells MT, Flores JM, Serrano M
and Andrés V: Increased p53 gene dosage reduces neointimal
thickening induced by mechanical injury but has no effect on native
atherosclerosis. Cardiovasc Res. 75:803–812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ihling C, Menzel G, Wellens E, Mönting JS,
Schaefer HE and Zeiher AM: Topographical association between the
cyclin-dependent kinases inhibitor P21, p53 accumulation, and
cellular proliferation in human atherosclerotic tissue.
Arterioscler Thromb Vasc Biol. 17:2218–2224. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Scheinman M, Ascher E, Levi GS, Hingorani
A, Shirazian D and Seth P: p53 gene transfer to the injured rat
carotid artery decreases neointimal formation. J Vasc Surg.
29:360–369. 1999. View Article : Google Scholar : PubMed/NCBI
|